

Abstract
We have previously shown that differentiation of hypertrophic chondrocytes is delayed in mice expressing a mutated PTH/PTHrP receptor (PTHR) (called DSEL here) that stimulates adenylyl cyclase normally but fails to activate phospholipase C (PLC). To better understand the role of PLC signaling via the PTHR in skeletal and mineral homeostasis, we examined these mice fed a normal or calcium-deficient diet. On a standard diet, DSEL mice displayed a modest decrease in bone mass. Remarkably, when fed a low-calcium diet or infused with PTH, DSEL mice exhibited strikingly curtailed peritrabecular stromal cell responses and attenuated new bone formation when compared with Wt mice. Attenuated in vitro colony formation was also observed in bone marrow cells derived from DSEL mice fed a low-calcium diet. Furthermore, PTH stimulated proliferation and increased mRNAs encoding cyclin D1 in primary osteoblasts derived from Wt but not from DSEL mice. Our data indicate that PLC signaling through the PTHR is required for skeletal homeostasis.
Normal proliferative responses to PTH in mouse bone in vivo require phospholipase C activation.
The PTH/PTHrP receptor (PTHR) is a class II G protein-coupled receptor that, like many other G protein-coupled receptors, activates several signaling pathways, including the Gsα-linked adenylyl cyclase (AC)-protein kinase A (PKA) signaling pathway and the Gq/11-linked phosphatidylinositol-specific phospholipase C (PI-PLC, named PLC here)-protein kinase C (PKC) signaling pathway in studies in cell culture (1,2). As with other such receptors, however, few studies establish whether the activation of multiple G proteins actually occurs in vivo and has importance in the action of such receptors. To explore this possibility, we have generated knock-in mice with a mutated PTHR, named DSEL (3). The DSEL receptors, with four mutated residues in the receptor’s second intracellular loop, can activate AC normally but cannot activate PLC (1,3). In initial studies, we explored the role of signaling via multiple G proteins by the PTHR in the chondrocytes of growing bones. Hypertrophic differentiation of chondrocytes is modestly delayed in DSEL mice, indicating that the PLC signaling pathway via the PTHR is essential for normal chondrocyte differentiation (3). The PTHR is important not only for chondrocyte differentiation but also for bone formation (4,5,6,7) and calcium/phosphate homeostasis. Here we address the roles of signaling by multiple signaling pathways in the role of the PTHR in regulating skeletal and mineral ion homeostasis.
Intermittent PTH administration has been shown to increase bone density, improve skeletal architecture, enhance biomechanical strength, and reduce fracture risk (8,9,10,11), whereas continuous infusion of PTH causes pathological changes similar to those seen in clinical hyperparathyroidism, including accumulation of peritrabecular stromal cells, increased bone resorption, and hypercalcemia (12,13). Despite these striking differences, both intermittent and continuous PTH administration increase both bone formation and bone resorption; the balance of these two responses and perhaps their mechanisms differ. The mechanistic basis for these varying responses to PTH has been extensively explored but is yet to be fully identified (14,15,16). Several lines of evidence suggest that intermittent PTH treatment results in increased osteoblast number and activity via increased differentiation and survival of osteoblasts (17,18,19), whereas continuous PTH treatment leads to increased osteoclast differentiation and activity through effects on cells of the osteoblast lineage (20,21).
The roles of distinct signaling pathways activated by the PTHR in generating these skeletal responses to PTH are not fully understood. Studies using amino-truncated PTH analogs that cannot stimulate cAMP production suggested that such stimulation was required for the anabolic actions of PTH (22,23), although subsequent studies suggested that such analogs are defective in PLC activation as well (11). Recently, PTHR PLC signaling stimulated by administration of signal-specific PTH analogs has been shown not to be required for the anabolic effect of PTH on bone (11), but other evidence suggests that the cAMP/PKA signaling pathway alone may not be sufficient to elicit a full anabolic response (24,25). Of course, in these studies using PTH analogs given to normal rodents, the possible importance of signaling in response to endogenous PTH and PTHrP, expected to activate both cAMP/PKA and PLC, could not be evaluated. Clarification of the roles of distinct signaling pathways downstream from the PTHR may further understanding of the varying actions of PTH on bone and could facilitate the design of suitable agents for the treatment of bone diseases such as osteoporosis.
To better understand the role of PLC signaling via the PTHR in bone modeling and remodeling and also in calcium/phosphate homeostasis, we examined the bone phenotype and serum calcium/phosphate in mutant mice fed either a standard diet or a low-calcium diet designed to provide a model of secondary hyperparathyroidism. Here, we demonstrate that in the basal state, DSEL mice displayed low bone mass perhaps partly due to increased osteoclast activity in the primary spongiosa, whereas the mutant mice, when fed a low-calcium diet or infused with PTH, exhibited strikingly curtailed peritrabecular stromal cell responses and attenuated new bone formation despite elevated serum PTH. Our data indicate that PLC signaling through the PTHR is essential for normal bone turnover.
Materials and Methods
Animals
The DSEL mice used in the present experiment were backcrossed to the C57/B6 background for more than 10 generations. All animals were maintained in facilities operated by the Center for Comparative Research of the Massachusetts General Hospital, and all animal experimental procedures were approved by the institution’s Subcommittee on Research Animal Care.
Sample preparation and histological analysis
Both wild-type (Wt) and DSEL homozygous mice were fed with a standard diet. To induce secondary hyperparathyroidism, 20-d-old mice from both Wt and DSEL homozygous mice were fed with a low-calcium diet (0.02% calcium and 0.4% phosphorus) or a control diet (0.6% calcium and 0.4% phosphorus) for 3 wk (low-calcium diet TD 02279 and control diet TD 97191 were purchased from Harlan Teklad, Madison, WI). For histological analysis, tissues from Wt and DSEL homozygous mice were fixed with 10% formalin and 70% ethanol. In selected cases, hind limbs were decalcified with 20% EDTA, and paraffin blocks were prepared by standard histological procedures. For selected samples, tartrate-resistant acid phosphatase staining was performed using a Sigma Chemical Co. (St. Louis, MO) acid phosphatase detection kit.
Serum biochemistry
Blood was collected by orbital sinus puncture before killing for serum biochemistry. Serum calcium and inorganic phosphorus were assayed with the UV determination kits (Stanbio Laboratory, Boerne, TX). Mouse intact PTH(1-84) was measured in duplicate using an ELISA kit (Immutopics Inc., San Clemente, CA). Serum N-terminal propeptide of type I procollagen (P1NP) and C-terminal telopeptide α1 chain of type I collagen (CTX) were measured with ELISA kits from Immunodiagnostic Systems (Fountain Hills, AZ).
Microcomputed tomography (μCT)
μCT analysis of the distal femur was performed using a desktop microtomographic image system, as described previously (11).
Tibial trabecular bone histomorphometry
For dynamic histomorphometry, 10-wk-old animals were injected ip with fluorochromes calcein (2 mg/ml; Sigma) and demeclocycline (2 mg/ml; Sigma) 3 and 10 d before killing, respectively. Tibiae and vertebrae were collected for histomorphometric analysis. Bones were fixed with 10% formalin overnight and then 70% ethanol and embedded in methyl methacrylate resin. Five-micrometer sections were stained with toluidine blue and von Kossa, and all measurements were performed on the trabecular area of proximal tibia in a region between 0.5 and 2.5 fields (at ×200) distal to the growth plate using a digitizing image analysis system and a morphometric program, Osteomeasure (Osteometrics Inc., Atlanta, GA). Fibrosis volume was measured as peritrabecular stromal cell response and determined by measurement of fibroblastic volume/bone volume in the primary and secondary spongiosa regions. Fibrosis was defined as multiple layers of elongated or fusiform cells surrounded by extracellular matrix that line the trabecular bone surface.
In situ hybridization
In situ hybridizations were performed on paraffin sections using complementary 35S-labeled riboprobes (cRNAs) transcribed from the plasmids, kindly provided by the Massachusetts General Hospital Endocrine Unit histology core facility (26).
Quantitative real-time PCR
RNA was extracted from bone and cultured primary osteoblastic cells using Trizol (Invitrogen, Carlsbad, CA) and was reverse transcribed into cDNA with SuperscriptRT II (Invitrogen). SYBRGreen Universal Master Mix (Applied Biosystems, Foster City, CA) was used for quantitative, real-time PCR, as described previously (27). The PCR primer sequences used are as follows: cyclin D1 forward 5′-gcaaagaggaaggagccagcc-3′ and reverse 5′-ggtgatgcagattctatctct-3′ and Gapdh forward 5′-tggagtggtgtcttcactact-3′ and reverse 5′-aagcagttggtggtgcaggat-3′. Relative expression was calculated for each gene by the 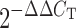 method with Gapdh for normalization.
method with Gapdh for normalization.
Effect of low-calcium diet on colony formation in vitro
Tibiae were dissected from 4-wk-old Wt and DSEL mice on the low-calcium diet or control diet for 1 wk. Epiphyses were removed and the diaphysis flushed with 15 ml α-MEM using a 20-ml syringe fitted with a 23-gauge needle. Flushed bone marrow cells were plated in 12-well plates at 5 × 105 cells per well and cultured for 14 d with α-MEM supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin at 37 C in a humidified atmosphere of 95% air and 5% carbon dioxide. On d 14, the cells were fixed with 10% neutral buffered formalin in PBS for 10 min. Total number of colonies [colony-forming units (CFU)-F] and colonies positive for alkaline phosphatase (ALP) (CFU-ALP) were determined by staining with methylene blue or with ALP staining buffer (Sigma), respectively. Colonies large enough to be seen with the naked eye were counted, and counts were performed blind on coded plates.
Primary osteoblast cultures
Six-week-old tibiae were dissected from Wt and DSEL mice. After bone marrow was flushed, the dissected bones were minced and cultured in six-well plates with α-MEM containing 10% FBS for 5 d, and then the growing primary osteoblastic cells were plated in slide chambers for proliferation and apoptosis assays or grown in six- and 12-well plates for in vitro determination of osteoblast differentiation. Cell proliferation was quantified by bromodeoxyuridine (BrdU) assay with an in situ cell proliferation kit (Roche, Indianapolis, IN), and apoptosis was determined by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay with an in situ cell death kit (Roche). For cell proliferation experiment, primary osteoblasts were cultured in an eight-well slide chamber with α-MEM containing 10% FBS. Cells at 70% confluence were treated in various ways for 24 h and labeled with BrdU (0.5 mg/ml) for 2 h. To examine apoptosis, primary osteoblasts were plated in an eight-well slide chamber with α-MEM containing 10% FBS. Cells at 90% confluence were cultured with α-MEM containing 0.1% FBS for 24 h and then treated with vehicle or PTH for an additional 24 h. For in vitro determination of osteoblast differentiation, primary osteoblasts were cultured in differentiation medium containing 10% FBS, β-glycerophosphate (0.2 mm), and ascorbic acid (50 μg/ml), and after confluence, cells were refed every 48 h with differentiation medium containing PTH (100 nm) (so-called continuous PTH treatment), or cells were treated with PTH in a 4 h/48 h schedule (so-called intermittent PTH treatment), whereby cells were treated with PTH for 4 h in differentiation medium and then the medium with peptide was aspirated and cells were rinsed with α-MEM twice before being refed with fresh differentiation medium and incubated for another 44 h before the procedure was repeated. Osteoblastic differentiation was assessed by von Kossa stain, ALP activity, and calcium content measurements, as described previously (28).
cAMP and inositol triphosphate (IP3) measurements
Intracellular cAMP accumulation and IP3 stimulation in primary osteoblastic cells were measured as described previously (28).
In vitro intracellular calcium assay
We used a Fluo-4 direct calcium assay kit from Invitrogen (catalog item F10471) to measure the intracellular calcium response to PTH in primary osteoblastic cells. Primary osteoblastic cells isolated from 6-wk-old tibiae as described above were cultured in a 96-well plates with α-MEM containing 10% FBS. After confluence, primary cells were treated with human PTH (hPTH), ionomycin, phorbol 12-myristate 13-acetate (PMA), or forskolin and incubated with Fluo-4 direct calcium reagent loading solution for 40 min. After incubation, the fluorescence signal was immediately measured using a fluorescence microplate reader at excitation and emission of 509 and 516 nm, respectively.
PTH infusion
PTH was infused in mice at 4 wk of age. Human PTH(1-34) was reconstituted in a solution containing 150 mm NaCl, 1 mm HCl, and 2% heat-inactivated mouse serum and loaded into Alzet osmotic minipumps (model 1002; Durect Corp., Cupertino, CA). The pumps were equilibrated in 0.9% NaCl overnight at 37 C and then implanted into an interscapular sc pocket under Avertin anesthesia. Wt or DSEL mice were infused with vehicle (control) or PTH at a dose of 80 μg/kg · d for 14 d, with six mice in each group.
Statistics
All values in the paper are expressed as mean ± sd or sem. Comparisons between groups were made by unpaired two-tailed Student’s t test for data from in vitro and serum biochemistry assays and by ANOVA (two-way ANOVA) with a post hoc Fisher’s test for data from μCT measurements and histomorphometry. P values smaller than 0.05 were considered statistically significant.
Results
PTHR signaling in DSEL osteoblastic cells
To verify that the signaling properties of the PTHR in osteoblastic cells in the DSEL mouse resembled those of this mutant receptor in other settings (1,3), we measured cAMP accumulation as an indication of activation of Gs, and intracellular calcium and IP3 stimulation as indications of activation of Gq/11. As expected, PTH stimulated cAMP production in DSEL-derived primary osteoblastic cells with a dose dependence similar to that seen in Wt primary osteoblastic cells, whereas PTH induced a calcium response and IP3 stimulation only in Wt-derived osteoblasts but not in DSEL-derived osteoblasts (Fig. 1, A–C). These results indicate that the DSEL mutation of the PTHR blocks PTH signaling through receptor-Gq/11 coupling but not through receptor-Gs coupling in osteoblastic cells.
Figure 1.

PTHR signaling and bone mass in DSEL mice. A, Dose-dependent stimulation of cAMP by PTH in DSEL-derived primary osteoblastic cells. Primary osteoblastic cells isolated from Wt (white bars) and D/D (black bars) mice were cultured in 24-well plates, and after confluence, cells were incubated with cAMP assay buffer containing hPTH(1-34) (∼0–1000 nm) or forskolin (10 μm) for 20 min before cAMP measurement. B, PTH fails to induce intracellular calcium response in DSEL-derived primary osteoblastic cells. Primary osteoblastic cells (white bars for Wt cells and black bars for D/D cells) were cultured in 96-well plates, and after confluence, cells were incubated with Fluo-4 direct calcium buffer containing hPTH (∼0–1000 nm), ionomycin (Ionom) (10 μm), PMA (10 nm), or forskolin (Fsk) (10 μm) for 40 min before fluorescence signal measurement. Data are expressed as relative fluorescence units (RFU). C, No PLC activation by PTH in DSEL-derived osteoblastic cells. Primary osteoblastic cells from Wt and D/D mice were cultured in six-well plates. At 80% confluence, cells were labeled with [3H]myoinositol for 48 h and then stimulated with hPTH (1000 nm) (black bars) or vehicle (white bars) for 40 min in the presence of 20 mm LiCl. The cellular content of radioactive IP3 was determined. Values are mean ± sd of triplicate wells. *, P < 0.05 vs. vehicle. Each experiment was repeated twice with similar results. D, Representative micrographs of plastic sections of tibiae from sex-matched Wt and DSEL homozygous (D/D) male mice at the age of 10 wk, stained with von Kossa. E, μCT analysis of 10-wk-old distal femur from both male and female Wt (white bars) and D/D (black bars) mice. F, Serum P1NP and CTX were measured in both Wt and D/D mice at the age of 10 wk. Error bars in E and F represent se (n = 8). *, P < 0.05 vs. Wt mice. BV/TV, Bone volume/total volume; Tb. N, trabecular number; Tb. Sp, trabecular spacing.
Bone phenotype in postnatal DSEL mice
To determine the role of the PLC signaling pathway via the PTHR in postnatal bone development, tibiae at age 10 wk from both Wt and DSEL (D/D) mice were examined. DSEL mice displayed a significant decrease in the amount of trabecular bone with little alteration in the cortical bone (Fig. 1D). Histomorphometry of proximal tibial trabecular bone at 10 wk of age demonstrated a significant decrease (by 25%) in trabecular volume in the secondary spongiosa that was associated with reduced trabecular number and thickness and increased trabecular spacing (Table 1). The decreased trabecular bone volume associated with decreased trabecular number and increased trabecular spacing in the mutant mice was confirmed by μCT analysis of 10-wk-old distal femurs (Fig. 1E). To determine whether the decreased trabecular bone in the mutant mice was partly caused by reduced osteoblast activity, osteoblast number and dynamic parameters of bone formation were examined. The histomorphometric analysis showed that the number of trabecular osteoblasts, mineral apposition rate, and bone formation rate in the mutant mice were not significantly different from those observed in the Wt control mice (Table 1). No significant difference in osteoclast number was observed by histomorphometric analysis of 10-wk-old proximal tibial trabecular bone in the secondary spongiosa (Table 1). Serum P1NP was modestly decreased in DSEL mice, whereas levels of serum CTX were not significantly different between the two groups (Fig. 1F).
Table 1.
Histomorphometry of proximal tibial metaphyseal trabecular bone
| Measurements | 10 wk old
|
6 wk old
|
||
|---|---|---|---|---|
| Wt | D/D | Wt | D/D | |
| Structural parameters | ||||
| BV/TV (%) | 11.66 ± 1.0 | 8.82 ± 0.66a | 11.34 ± 0.95 | 8.08 ± 0.80a |
| Tb.Th (μm) | 33.05 ± 1.37 | 28.91 ± 0.92a | 22.92 ± 1.55 | 21.07 ± 1.19 |
| Tb.Sp (μm) | 256.7 ± 19.8 | 313.9 ± 18.6a | 186.8 ± 18.7 | 238.4 ± 30.5a |
| Tb.N (mm−1) | 3.65 ± 0.23 | 3.02 ± 0.17a | 5.11 ± 0.61 | 3.75 ± 0.32a |
| Static parameters | ||||
| Ob/TA (no./mm2) | 191.4 ± 19.5 | 170.2 ± 8.1 | 254.1 ± 24.1 | 204.6 ± 18.5a |
| N.Ob/BS (no./μm) | 27.4 ± 1.3 | 26.1 ± 1.1 | 25.33 ± 1.12 | 25.34 ± 1.14 |
| Oc/TA (no./mm2) | 84.1 ± 10.2 | 72.5 ± 6.1 | 27.07 ± 3.48 | 31.52 ± 5.8 |
| N.Oc/BS (no./mm) | 10.6 ± 1.1 | 11.6 ± 0.41 | 2.61 ± 0.34 | 3.71 ± 0.36a |
| Dynamic parameters | ||||
| MAR (μm/yr) | 564.8 ± 62.4 | 526.5 ± 34.6 | ||
| BFR/BS (μm3/μm2 · yr) | 183.2 ± 19.2 | 193.0 ± 16.5 | ||
Data are presented as group mean ± sem. BFR, Bone formation rate; BS, bone surface; BV, bone volume; MAR, mineral apposition rate; MS, mineralizing surface; N, number; N.Ob/BS, osteoblast number per bone surface; N.Oc/BS, osteoclast number per bone surface; Ob, osteoblast; Ob.S/BS, osteoblast surface per bone surface; Oc, osteoclast; Oc.S/BS, osteoclast surface per bone surface; Sp, spacing; TA, total area; Tb, trabecular; Th, thickness; TV, total volume.
P < 0.05 vs. Wt (n = 8 per group).
To examine whether PLC signaling via the PTHR is more important for PTH-regulated bone turnover during rapid growth, histomorphometric analysis was also performed in 6-wk-old tibiae (Table 1). Decreased trabecular bone associated with lowered trabecular number and increased trabecular spacing was consistent with that seen in the bones from 10-wk-old mice. Interestingly, unlike the nonsignificant change in trabecular bone cellularity observed in 10-wk-old tibiae, the mutant trabecular bone at age of 6 wk exhibited a significant decrease in osteoblast number per total area and a significant increase in osteoclast number per millimeter of bone surface, suggesting that the PLC signaling pathway via the PTHR may be required for generation of normal numbers of osteoblasts and osteoclasts in young growing bone.
Effect of low-calcium diet on bone remodeling
We have previously shown that activation of PLC by the PTHR requires much higher concentrations of PTH than activation of AC requires (29). We hypothesized that physiological stresses that lead to high PTH levels might bring out the actions of PTHR-stimulated PLC in bone. Serum PTH can be increased by either continuous infusion of PTH to mimic primary hyperparathyroidism or by feeding mice a low-calcium diet that causes secondary hyperparathyroidism (12,30). To evaluate the role of PTH-induced PLC signaling in bone turnover, bone responses were examined in a model of secondary hyperparathyroidism caused by a low-calcium diet. Twenty-day-old mice were placed on either a control diet or a low-calcium diet for 3 wk. As expected, blood calcium levels fell and PTH levels rose similarly in both genotypes of mice (Table 2). Interestingly, the blood phosphate fell in the Wt mice but increased in the DSEL mice (Table 2). These differing phosphate responses may primarily reflect differences in the renal response to PTH in these mice. The low-calcium diet caused a similar loss of body weight (decreased by approximately 20% compared with control diet) in both Wt and D/D mice. When stained for phosphate through von Kossa staining, both the Wt and DSEL mice exhibited dramatically lowered levels of bone mineral on the low-calcium diet (Fig. 2A). The low-calcium diet caused extensive lowering of cortical bone mass in both Wt and DSEL mice (Fig. 2, B and C), whereas increased trabecular bone volume observed in Wt mice fed a low-calcium diet was attenuated in D/D mice (Fig. 2C). Interestingly, the peritrabecular space, normally occupied by hematopoietic cells, in the Wt mice on the low-calcium diet was filled with the fibroblast-like stromal cells, most striking in the metaphyseal region, whereas such a peritrabecular stromal cell response to the low-calcium diet was not present in the DSEL mice (Figs. 2A and 3A). Substantial amounts of disorganized bone containing a high density of osteocytes and unmineralized matrix, as shown in red by Masson-Goldner trichrome staining (Fig. 3A), were observed only in the Wt but not in the DSEL mice fed the low-calcium diet. Such irregular bones are very similar to woven bone and probably represent newly formed bone; this finding suggests more rapid bone formation in the Wt mice.
Table 2.
Effect of low-calcium diet on serum parameters
| Measurements | Male mice
|
Female mice
|
||
|---|---|---|---|---|
| Wt | D/D | Wt | D/D | |
| Serum intact PTH (pg/ml) | ||||
| Control diet | 59 ± 12.4 | 106.2 ± 22.3 | 83.1 ± 22.1 | 145.5 ± 29.1 |
| Low-calcium diet | 591.5 ± 65.2a | 504.7 ± 55.5a | 607.1 ± 59.4a | 607.6 ± 90.3a |
| Total serum calcium (mg/dl) | ||||
| Control diet | 8.39 ± 0.36 | 8.95 ± 0.41 | 8.04 ± 0.19 | 7.3 ± 0.5 |
| Low-calcium diet | 7.54 ± 0.2a | 6.65 ± 0.48a | 7.01 ± 0.33a | 5.95 ± 0.43 |
| Serum inorganic phosphorus (mg/dl) | ||||
| Control diet | 8.13 ± 0.37 | 9.54 ± 0.62 | 9.03 ± 0.54 | 9.44 ± 0.26 |
| Low-calcium diet | 6.22 ± 0.24a | 13.36 ± 1.3a | 7.84 ± 0.51a | 12.08 ± 1.01a |
Data are presented as group mean ± sem.
P < 0.01 vs. control diet (n = 8 per group).
Figure 2.

Effect of calcium-deficient diet on bone turnover. A, Representative micrographs of proximal tibiae; B, diaphyseal cortical bone from both Wt and D/D mice fed a low-calcium diet or a corresponding control diet for 3 wk after weaning, stained with von Kossa for mineral, hematoxylin and eosin (H&E) for histology, and Masson-Goldner trichrome for bone matrix, as indicated. Both groups of mice on the low-calcium diet showed a similar dramatic loss of bone mineral and cortical bone mass, but H&E stain demonstrated a striking difference (indicated by insets) in metaphyseal trabecular bone turnover between Wt and D/D mice on the low-calcium diet. C, Histomorphometric measurements of diaphyseal cortical bone thickness and metaphyseal trabecular bone volume in 6-wk-ld Wt and D/D mice fed low-calcium and control diet, as indicated. Error bars represent se (n = 6); *, P < 0.05 vs. control diet; #, P < 0.05 vs. Wt.
Figure 3.

DSEL mice on the low-calcium diet exhibited strikingly curtailed peritrabecular stromal cell accumulation, new bone formation, and hypertrophic expansion. A, Representative micrographs of proximal tibial metaphyseal trabecular bone from mice fed a low-calcium diet or a corresponding control diet for 3 wk after weaning, stained with hematoxylin and eosin (H&E) for histology (×20 magnification), showing expanded hypertrophic chondrocytes (as indicated by the black bracket) and accumulation of peritrabecular fibroblast-like stromal cells (indicated by arrows) in Wt but not in D/D mice fed the low-calcium diet, and stained with Masson-Goldner trichrome (×40 magnification), demonstrating massive disorganized newly formed undermineralized bone, shown in red, in Wt but not in D/D mice fed the low-calcium diet. B, Measurements of peritrabecular fibrosis. Fb.V/TV, Fibrosis volume/total volume. *, P < 0.05 vs. control diet; n = 6. C, Osteopontin in situ in proximal tibiae on the low-calcium diet for 3 wk.
Peritrabecular fibroblast-like stromal cells are diminished in DSEL mice
Continuous PTH treatment in rats causes the accumulation of peritrabecular stromal cells, increased bone resorption, and accumulation of poorly mineralized extracellular matrix on bone surfaces (10,30). Primary hyperparathyroidism is associated with continuously increased PTH levels, and this disease is characterized by accumulation of osteoid, focal bone resorption, increased bone formation, and peritrabecular marrow fibrosis (peritrabecular stromal cell response). To demonstrate the extent of the peritrabecular stromal cell response to the low-calcium diet, we measured the fibrosis volume in the tibial metaphyseal region. No peritrabecular fibrosis is present in the normal bone. Surprisingly, little peritrabecular fibrosis was observed in the mutant mice on the low-calcium diet, whereas the Wt mice on the low-calcium diet displayed extensive peritrabecular fibrosis, most strikingly in the metaphyseal region (Fig. 3B). These peritrabecular stromal cells strongly express osteopontin and collagen a1(I) mRNAs (Fig. 3C), suggesting that they may be preosteoblasts (30).
Diminished stromal cell response and bone formation in PTH-infused DSEL mice
Similar phenomena were also observed in mice with continuous PTH infusion. In Wt mice, continuous PTH infusion caused both peritrabecular stromal cell responses and disorganized bone formation similar to those observed in Wt mice fed a low-calcium diet, whereas such stromal cell responses and disorganized bone formation were dramatically attenuated in DSEL mice with continuous PTH infusion (Fig. 4A). Consistent with the observed alterations in metaphyseal trabecular bone after PTH infusion, levels of serum P1NP, a marker of bone formation, were significantly increased in Wt but not in DSEL mice receiving continuous PTH administration, whereas levels of serum CTX, a bone resorption marker, were elevated in both groups of mice (Fig. 4B). Continuous PTH infusion caused similarly undetectable serum intact PTH and elevated serum calcium in both Wt and D/D mice, whereas serum phosphate was significantly decreased in Wt but not in D/D mice after PTH infusion (Fig. 4B). At the end of PTH infusion, both Wt and D/D mice showed a similar decrease in body weight (by approximately 15% compared with vehicle-infused control mice) and appeared to be sick, but the level of serum creatinine in both Wt and D/D mice with PTH infusion was not significantly increased compared with vehicle-infused control mice (data not shown), suggesting that both mice after PTH infusion were not in severe renal failure.
Figure 4.

DSEL mice with PTH infusion exhibited curtailed peritrabecular stromal cell responses and bone formation. A, Representative micrographs of proximal tibia from 4-wk-old mice receiving continuous infusion of hPTH (80 μg/kg · d) or vehicle through sc implantation of Alzet mini-osmotic pumps for 2 wk, stained with hematoxylin and eosin (H&E) for histology (×40 magnification), showing dramatic accumulation of peritrabecular fibroblast-like stromal cells (indicated by arrows) in Wt but not in D/D mice, and stained with Masson-Goldner trichrome and von Kossa (×40 magnification) demonstrating massive disorganized bone formation in Wt but not in D/D mice. B, Serum P1NP, CTX, intact PTH(1-84) (PTH), total calcium, and phosphorus (Pi) were measured in Wt and D/D mice after infusion with vehicle (white bars) and PTH (black bars). Error bars represent se (n = 6). a, P < 0.05 vs. vehicle; b, P < 0.05 vs. Wt PTH; c, P < 0.05 vs. Wt vehicle.
Attenuated colony formation and proliferation in DSEL-derived bone marrow cells and primary osteoblasts
To examine the effect of the DSEL mutation on osteoblast progenitors, bone marrow cells from mice on the normal or low-calcium diets for 1 wk were plated in culture for 2 wk. Interestingly, on the low-calcium diet, Wt mice increased the numbers of CFU-F and CFU-ALP, whereas the DSEL mice failed to increase the numbers of both kinds of colonies (Fig. 5A), presumably reflecting a failure of these mice to generate the colony-forming cells in vivo. Activation of PKC by PTH signaling has been implicated in the mitogenic action of PTH on osteoblastic cells (31,32). To determine whether osteoblastic cells from the DSEL mouse exhibited abnormalities in cellular proliferation in response to PTH, we stimulated primary osteoblastic cells derived from both Wt and DSEL mice with PTH. PTH treatment significantly increased the number of BrdU-positive cells in osteoblastic cells derived from Wt but not from DSEL mice. Furthermore, the effect of PTH to increase cell proliferation in the Wt primary osteoblastic cells was not mimicked by treatment with [G1,R19]hPTH(1-28), a signal-selective peptide with defective activation of PLC/PKC signaling by the PTHR (28,33). In contrast, basic fibroblast growth factor, a potent mitogenic stimulator in osteoblastic cells (34), had a similar stimulatory effect on cell proliferation in primary osteoblastic cells from both Wt and DSEL mice (Fig. 5B).
Figure 5.

Decreased colony formation and proliferation in DSEL-derived primary bone and bone marrow cells. A, Decreased colony formation in DSEL-derived bone marrow cells. Bone marrow cells from 4-wk-old tibiae on either a low-calcium diet (Ca diet) or a control diet for 1 wk were cultured in12-well plates for 14 d. The cultured plates were stained with methylene blue for total number of colonies (CFU-F) or with ALP staining solution to count positive colonies for ALP (CFU-ALP). Counts of colonies were performed blind on coded plates. *, P < 0.05 vs. Wt control diet; #, P < 0.05 vs. Wt low-calcium diet (n = 4). B, In vitro osteoblast proliferation. Primary osteoblasts were treated with vehicle (Veh), 100 nm hPTH(1-34) (PTH), 100 nm [G1,R19]hPTH(1-28) (1-28), or 10 ng/ml basic fibroblast growht factor (bFGF) for 24 h and labeled with BrdU for 2 h. In situ anti-BrdU fluorescein (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue) stain were performed and BrdU-positive cells were counted, as shown in bar graph (two wells per treatment in three independent experiments). *, P < 0.05 vs. vehicle. C and D, Real-time PCR detection of cyclin D1 mRNA expression in primary osteoblastic cells isolated from Wt and D/D mice (C) or from D/D mice (D). Primary osteoblasts were treated with 100 nm hPTH(1-34) (PTH) or [G1,R19]hPTH(1-28) (1-28) (C) or with 10 nm PMA and 100 μm 8-bromo-cAMP (8Brom) (D) for 0–24 h, as indicated. Transcript expression of the cultured cells was determined by quantitative RT-PCR with primers specific for cyclin D1. Data are expressed as percent basal levels, and similar results were obtained in three independent experiments.
PTH fails to induce cyclin D1 mRNA expression in the primary osteoblastic cells derived from DSEL mice
Increased expression of cyclin D1 leads to increased cell proliferation in many tissues, and recently, cyclin D1 has been implicated as a primary target of mitogenic signals in osteoblastic cells (35,36,37,38,39). To determine whether failure of PTH to stimulate proliferation in DSEL-derived primary osteoblasts is in part due to curtailed induction of cyclin D1 by PTH, we next examined cyclin D1 mRNA expression in the primary cultures of osteoblastic cells after PTH treatment. In the primary osteoblasts derived from Wt mice, PTH treatment significantly increased cyclin D1 mRNA levels in a time-dependent manner, whereas such induction of cyclin D1 by PTH was not seen in the DSEL-derived primary osteoblasts (Fig. 5C). Furthermore, PTH-mediated induction of cyclin D1 mRNA was mimicked by treatment with PMA but not by treatment with either [G1,R19]hPTH(1-28) or 8-bromo-cAMP (Fig. 5C), indicating that activation of the PLC/PKC pathway by the PTHR is essential for PTH to induce cyclin D1 expression and to stimulate proliferation of osteoblastic cells.
PTH efficiently prevents apoptosis and regulates osteoblast differentiation in DSEL-derived primary osteoblastic cells
An antiapoptotic action of PTH has been proposed to be a mechanism underlying the increased osteoblast number associated with the anabolic response of bone to intermittent PTH administration (16,18). To determine whether the antiapoptotic action of PTH might be dampened in bones of DSEL mice, we next examined the effect of PTH on apoptosis of primary osteoblasts derived from both Wt and DSEL tibiae. The proportion of osteoblastic cells undergoing apoptosis, as determined by TUNEL labeling, was significantly decreased by PTH in primary osteoblastic cells from both Wt and DSEL mice (Fig. 6A). This normal antiapoptotic response in DSEL-derived primary osteoblasts suggests that PLC/PKC signaling through the PTHR is not critical for osteoblast survival and that the low bone mass in the DSEL mice probably does not result from altered antiapoptotic actions of PTH in osteoblasts in DSEL mice.
Figure 6.

Effect of PTH on apoptosis and differentiation in primary osteoblastic cells. A, In vitro osteoblast apoptosis was quantified by TUNEL assay. Primary osteoblasts cultured in an eight-well slide chamber, were treated with vehicle or hPTH(1-34) (100 nm) for 24 h, and then in situ fluorescein (TUNEL, green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue) stain were performed. TUNEL-positive cells were counted as apoptotic cells (percent, as shown in bar graph). Data are represented as mean ± se (two wells per slide in three independent experiments). *, P < 0.05 vs. vehicle (t test). B, Effect of PTH on osteoblast differentiation in vitro. Primary osteoblastic cells isolated from 6-wk-old tibiae were cultured in six-well or 12-well plates with α-MEM containing 10% FBS, and after confluence, cells were cultured with differentiation medium and treated continuously (freshly adding PTH every 48 h) or intermittently (adding PTH for 4 h in every 48 h) with 100 nm hPTH(1-34) for 4–6 wk. Mineralization was assessed by von Kossa stain and calcium (Ca) content (bar graph) was measured in acid extracts of the cultures (bar graph). ALP activity was determined by stain and also measured enzymatically (bar graph) (28).
To assess whether PLC signaling through the PTHR is important for PTH-mediated differentiation of osteoblastic cells, primary osteoblasts were isolated from 6-wk-old tibiae of Wt and DSEL mice and treated with PTH continuously or intermittently to determine the effect of PTH on osteoblast differentiation. In cells from both Wt and DSEL mice, continuous treatment with PTH similarly suppressed ALP activity, as determined by staining cells for enzyme activity or by measuring enzymatic activity in cell extracts. PTH also similarly inhibited mineralization, shown by von Kossa stain of cells for phosphate and by measuring calcium content in the cell layer, in both types of primary osteoblastic cells. Furthermore, intermittent PTH administration dramatically increased ALP activity and mineralization in primary osteoblastic cells from both Wt and DSEL mice (Fig. 6B). The similar differentiation responses to PTH in cells from the Wt and DSEL mice suggests that PLC signaling through the PTHR is not required for PTH-mediated osteoblast differentiation at least in vitro.
Discussion
Although the PTHR signaling pathways have been extensively studied for decades (40,41,42,43,44), the role of PLC signaling via the PTHR in vivo remains incompletely understood, especially regarding the regulation of bone modeling and remodeling. To examine the roles of distinct signaling pathways in vivo, we have generated a knock-in mutant mouse that expresses the PTHR with the DSEL mutation instead of the Wt receptor (3). The DSEL receptor stimulates AC normally but fails to activate PLC (1,2). The DSEL mice exhibit a mild delay in hypertrophic differentiation of chondrocytes during embryonic development, indicating that PLC signaling through the PTHR is important for embryonic chondrocyte differentiation (3). In the present study, we demonstrate that in the basal state, the DSEL mice at 10 wk of age displayed reduced trabecular bone mass in both tibiae and femurs. However, the mechanism underlying the low bone mass in the mutant mice in the basal state is difficult to analyze, because histomorphometric parameters of trabecular bone cellularity and bone formation rate were not significantly altered in the secondary spongiosa of tibiae from 10-wk-old DSEL mice. The decreased bone mass may reflect abnormal modeling early in life. The mutant mice exhibit a modest decrease in osteoblast number and increase in osteoclast surface density at 6 wk of age. Those changes during growth, combined with possible indirect consequences of the growth plate abnormality, might explain the decreased bone mass at 10 wk of age. Alternatively, the modest decrease in serum P1NP, a marker of bone formation, at 10 wk in the DSEL mice may reflect a decrease in bone formation at that age missed in the histomorphometric analysis of the tibial trabecular secondary spongiosa.
DSEL mice at age of 6 wk, when fed a low-calcium diet, developed secondary hyperparathyroidism with elevated levels of serum PTH and substantial cortical bone loss but failed to develop peritrabecular stromal cell and woven bone responses, whereas Wt mice exhibited disorganized bone and extensive peritrabecular fibroblast-like stromal cell accumulation in response to the low-calcium diet. These stromal cells strongly express mRNAs encoding a1(I) collagen and osteopontin, markers of the osteoblast lineage, suggesting that they are preosteoblasts, and this observation is very similar to that seen in rat continuously infused with PTH (30). Lotinun et al. (30) pulse labeled these stromal cells with thymidine and showed that, after stopping the PTH infusion, these cells disappeared, and radioactive thymidine appeared rapidly in mature osteoblasts. These data suggest that at least some of the stromal cells are osteoblast precursors. The data here suggest that the accumulation of these stromal cells in response to high levels of PTH requires PLC signaling through the PTHR. Such accumulation could be explained by a variety of mechanisms. Here we show that the Wt mouse responds to continuously elevated levels of PTH by increasing the number of cells capable of forming colonies (CFU-F and CFU-ALP) in vitro and that the DSEL mouse fails to demonstrate this response. This finding suggests that one locus of action of the PTHR that requires PLC activation involves early cells in the mesenchymal lineage, directly or indirectly. In UMR106 and primary osteoblastic cells, PTH stimulates proliferation, and such a stimulatory effect was shown to be dependent on activation of PKC (31,45,46). Interestingly, PTH failed to increase proliferation in the DSEL-derived primary osteoblastic cells, whereas PTH treatment significantly stimulated proliferation in the Wt-derived primary osteoblastic cells, and such a stimulatory effect of PTH is not mimicked by a PLC/PKC-defective peptide [G1,R19]hPTH(1-28). Furthermore, PTH induced expression of cyclin D1 mRNA, an important part of the mitogenic response in the Wt primary osteoblastic cells but failed to increase cyclin D1 mRNA in cells from the DSEL mice. Furthermore, treatment with a PKC activator stimulates cyclin D1 mRNA in cells from DSEL mice. Our data indicate that PLC signaling by the PTHR is important for the stimulatory effect of PTH on cyclin D1 expression and for proliferation in osteoblastic cells. Interestingly, PTH treatment in vitro has a similar effect on apoptosis and on differentiation in primary cultured bone cells derived from both Wt and DSEL tibiae. Taken together, these findings suggest that the stromal cell accumulation in response to high levels of PTH in Wt mice results from a proliferative response to PTH that requires activation of PLC.
In summary, our findings in vivo indicate that the PLC signaling pathway via the PTHR is essential for normal bone modeling and remodeling and for the stromal cell response to elevations of PTH levels. This in vivo evidence for roles of PLC signaling on bone in response to PTH broadens the possible strategies for mimicking or enhancing the effects of PTH in diseases such as osteoporosis.
Acknowledgments
We thank Xiang Zheng for valuable help with the fluorescence microplate reader.
Footnotes
This work was supported by National Institutes of Health Grant DK11794 (to H.M.K.).
Present address for D.Y.: Department of Spinal and Orthopedic Surgery, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China.
Disclosure Summary: None of the authors have conflicts of interest in this report.
First Published Online May 25, 2010
Abbreviations: AC, Adenylyl cyclase; ALP, alkaline phosphatase; BrdU, bromodeoxyuridine; CFU, colony-forming units; μCT, microcomputed tomography; CTX, C-terminal telopeptide α1 chain of type I collagen; FBS, fetal bovine serum; hPTH, human PTH; IP3, inositol triphosphate; PKA, protein kinase A; PKC, protein kinase C; PLC, phosphatidylinositol-specific phospholipase C; PMA, phorbol 12-myristate 13-acetate; P1NP, N-terminal propeptide of type I procollagen; PTHR, PTH/PTHrP receptor; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling; Wt, wild type.
References
- Iida-Klein A, Guo J, Takemura M, Drake MT, Potts Jr JT, Abou-Samra A, Bringhurst FR, Segre GV 1997 Mutations in the second cytoplasmic loop of the rat parathyroid hormone (PTH)/PTH-related protein receptor result in selective loss of PTH-stimulated phospholipase C activity. J Biol Chem 272:6882–6889 [DOI] [PubMed] [Google Scholar]
- Guo J, Liu BY, Bringhurst FR 1997 Mechanisms of homologous and heterologous desensitization of PTH/PTHrP receptor signaling in LLC-PK1 cells. Am J Physiol 273:E383–E393 [DOI] [PubMed] [Google Scholar]
- Guo J, Chung UI, Kondo H, Bringhurst FR, Kronenberg HM 2002 The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev Cell 3:183–194 [DOI] [PubMed] [Google Scholar]
- Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LH, Ho C, Mulligan RC, Abou-Samra AB, Jüppner H, Segre GV, Kronenberg HM 1996 PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 273:663–666 [DOI] [PubMed] [Google Scholar]
- Lee K, Lanske B, Karaplis AC, Deeds JD, Kohno H, Nissenson RA, Kronenberg HM, Segre GV 1996 Parathyroid hormone-related peptide delays terminal differentiation of chondrocytes during endochondral bone development. Endocrinology 137:5109–5118 [DOI] [PubMed] [Google Scholar]
- Miao D, He B, Jiang Y, Kobayashi T, Sorocéanu MA, Zhao J, Su H, Tong X, Amizuka N, Gupta A, Genant HK, Kronenberg HM, Goltzman D, Karaplis AC 2005 Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J Clin Invest 115:2402–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Chung UI, Schipani E, Starbuck M, Karsenty G, Katagiri T, Goad DL, Lanske B, Kronenberg HM 2002 PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development 129:2977–2986 [DOI] [PubMed] [Google Scholar]
- Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH 2001 Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344:1434–1441 [DOI] [PubMed] [Google Scholar]
- Dempster DW, Cosman F, Kurland ES, Zhou H, Nieves J, Woelfert L, Shane E, Plavetiæ K, Müller R, Bilezikian J, Lindsay R 2001 Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. J Bone Miner Res 16:1846–1853 [DOI] [PubMed] [Google Scholar]
- Zhou H, Shen V, Dempster DW, Lindsay R 2001 Continuous parathyroid hormone and estrogen administration increases vertebral cancellous bone volume and cortical width in the estrogen-deficient rat. J Bone Miner Res 16:1300–1307 [DOI] [PubMed] [Google Scholar]
- Yang D, Singh R, Divieti P, Guo J, Bouxsein ML, Bringhurst FR 2007 Contributions of parathyroid hormone (PTH)/PTH-related peptide receptor signaling pathways to the anabolic effect of PTH on bone. Bone 40:1453–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobnig H, Turner RT 1997 The effects of programmed administration of human parathyroid hormone fragment (1-34) on bone histomorphometry and serum chemistry in rats. Endocrinology 138:4607–4612 [DOI] [PubMed] [Google Scholar]
- Marx SJ 2000 Hyperparathyroid and hypoparathyroid disorders. N Engl J Med 343:1863–1875 [DOI] [PubMed] [Google Scholar]
- Ishizuya T, Yokose S, Hori M, Noda T, Suda T, Yoshiki S, Yamaguchi A 1997 Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest 99:2961–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locklin RM, Khosla S, Turner RT, Riggs BL 2003 Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone. J Cell Biochem 89:180–190 [DOI] [PubMed] [Google Scholar]
- Jilka RL 2007 Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40:1434–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobnig H, Turner RT 1995 Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology 136:3632–3638 [DOI] [PubMed] [Google Scholar]
- Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC 1999 Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest 104:439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellido T, Ali AA, Plotkin LI, Fu Q, Gubrij I, Roberson PK, Weinstein RS, O'Brien CA, Manolagas SC, Jilka RL 2003 Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem 278:50259–50272 [DOI] [PubMed] [Google Scholar]
- Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, Chandrasekhar S, Martin TJ, Onyia JE 2001 Catabolic effects of continuous human PTH(1–38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology 142:4047–4054 [DOI] [PubMed] [Google Scholar]
- Nakchbandi IA, Mitnick MA, Masiukiewicz US, Sun BH, Insogna KL 2001 IL-6 negatively regulates IL-11 production in vitro and in vivo. Endocrinology 142:3850–3856 [DOI] [PubMed] [Google Scholar]
- Hilliker S, Wergedal JE, Gruber HE, Bettica P, Baylink DJ 1996 Truncation of the amino terminus of PTH alters its anabolic activity on bone in vivo. Bone 19:469–477 [DOI] [PubMed] [Google Scholar]
- Rixon RH, Whitfield JF, Gagnon L, Isaacs RJ, Maclean S, Chakravarthy B, Durkin JP, Neugebauer W, Ross V, Sung W, Willick GE 1994 Parathyroid hormone fragments may stimulate bone growth in ovariectomized rats by activating adenylyl cyclase. J Bone Miner Res 9:1179–1189 [DOI] [PubMed] [Google Scholar]
- Mohan S, Kutilek S, Zhang C, Shen HG, Kodama Y, Srivastava AK, Wergedal JE, Beamer WG, Baylink DJ 2000 Comparison of bone formation responses to parathyroid hormone(1-34), (1–31), and (2–34) in mice. Bone 27:471–478 [DOI] [PubMed] [Google Scholar]
- Murrills RJ, Matteo JJ, Samuel RL, Andrews JL, Bhat BM, Coleburn VE, Kharode YP, Bex FJ 2004 In vitro and in vivo activities of C-terminally truncated PTH peptides reveal a disconnect between cAMP signaling and functional activity. Bone 35:1263–1272 [DOI] [PubMed] [Google Scholar]
- Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, Schipani E 2001 Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest 107:277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Chung UI, Yang D, Karsenty G, Bringhurst FR, Kronenberg HM 2006 PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev Biol 292:116–128 [DOI] [PubMed] [Google Scholar]
- Yang D, Guo J, Divieti P, Bringhurst FR 2006 Parathyroid hormone activates PKC-δ and regulates osteoblastic differentiation via a PLC-independent pathway. Bone 38:485–496 [DOI] [PubMed] [Google Scholar]
- Guo J, Iida-Klein A, Huang X, Abou-Samra AB, Segre GV, Bringhurst FR 1995 Parathyroid hormone (PTH)/PTH-related peptide receptor density modulates activation of phospholipase C and phosphate transport by PTH in LLC-PK1 cells. Endocrinology 136: 3884–3891 [DOI] [PubMed] [Google Scholar]
- Lotinun S, Sibonga JD, Turner RT 2005 Evidence that the cells responsible for marrow fibrosis in a rat model for hyperparathyroidism are preosteoblasts. Endocrinology 146:4074–4081 [DOI] [PubMed] [Google Scholar]
- Swarthout JT, Doggett TA, Lemker JL, Partridge NC 2001 Stimulation of extracellular signal-regulated kinases and proliferation in rat osteoblastic cells by parathyroid hormone is protein kinase C-dependent. J Biol Chem 276:7586–7592 [DOI] [PubMed] [Google Scholar]
- Somjen D, Tordjman K, Katzburg S, Knoll E, Sharon O, Limor R, Naidich M, Naor Z, Hendel D, Stern N 2008 Lipoxygenase metabolites are mediators of PTH-dependent human osteoblast growth. Bone 42:491–497 [DOI] [PubMed] [Google Scholar]
- Takasu H, Gardella TJ, Luck MD, Potts JT Jr, Bringhurst FR 1999 Amino-terminal modifications of human parathyroid hormone (PTH) selectively alter phospholipase C signaling via the type 1 PTH receptor: implications for design of signal-specific PTH ligands. Biochemistry 38:13453–13460 [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim SY, Kwon CH, Kim YK 2007 Differential effect of FGF and PDGF on cell proliferation and migration in osteoblastic cells. Growth Factors 25:77–86 [DOI] [PubMed] [Google Scholar]
- Datta NS, Pettway GJ, Chen C, Koh AJ, McCauley LK 2007 Cyclin D1 as a target for the proliferative effects of PTH and PTHrP in early osteoblastic cells. J Bone Miner Res 22:951–964 [DOI] [PubMed] [Google Scholar]
- Zhang X, Yu S, Galson DL, Luo M, Fan J, Zhang J, Guan Y, Xiao G 2008 Activating transcription factor 4 is critical for proliferation and survival in primary bone marrow stromal cells and calvarial osteoblasts. J Cell Biochem 105:885–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Abe T, Nakamoto N, Tomaru Y, Koshikiya N, Nojima J, Kokabu S, Sakata Y, Kobayashi A, Yoda T 2008 Nicotine induces cell proliferation in association with cyclin D1 up-regulation and inhibits cell differentiation in association with p53 regulation in a murine pre-osteoblastic cell line. Biochem Biophys Res Commun 377:126–130 [DOI] [PubMed] [Google Scholar]
- Liu B, Yu HM, Hsu W 2007 Craniosynostosis caused by Axin2 deficiency is mediated through distinct functions of β-catenin in proliferation and differentiation. Dev Biol 301:298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schütz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, Mann JJ, Hen R, Ducy P, Karsenty G 2008 Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell 135:825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge NC, Kemp BE, Veroni MC, Martin TJ 1981 Activation of adenosine 3′,5′-monophosphate-dependent protein kinase in normal and malignant bone cells by parathyroid hormone, prostaglandin E2, and prostacyclin. Endocrinology 108:220–225 [DOI] [PubMed] [Google Scholar]
- Abou-Samra AB, Jüppner H, Force T, Freeman MW, Kong XF, Schipani E, Urena P, Richards J, Bonventre JV, Potts Jr JT 1992 Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci USA 89:2732–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Lanske B, Liu BY, Divieti P, Kronenberg HM, Bringhurst FR 2001 Signal-selectivity of parathyroid hormone (PTH)/PTH-related peptide receptor-mediated regulation of differentiation in conditionally immortalized growth-plate chondrocytes. Endocrinology 142:1260–1268 [DOI] [PubMed] [Google Scholar]
- Radeff JM, Singh AT, Stern PH 2004 Role of protein kinase A, phospholipase C and phospholipase D in parathyroid hormone receptor regulation of protein kinase Cα and interleukin-6 in UMR-106 osteoblastic cells. Cell Signal 16:105–114 [DOI] [PubMed] [Google Scholar]
- Chase LR, Aurbach GD 1967 Parathyroid function and the renal excretion of 3′5′-adenylic acid. Proc Natl Acad Sci USA 58:518–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini M, Lesur C, Pacherie M, Pastoureau P, Kucharczyk N, Fauchère JL, Bonnet J 1996 Effects of parathyroid hormone and agonists of the adenylyl cyclase and protein kinase C pathways on bone cell proliferation. Bone 18:59–65 [DOI] [PubMed] [Google Scholar]
- Miao D, Tong XK, Chan GK, Panda D, McPherson PS, Goltzman D 2001 Parathyroid hormone-related peptide stimulates osteogenic cell proliferation through protein kinase C activation of the Ras/mitogen-activated protein kinase signaling pathway. J Biol Chem 276:32204–32213 [DOI] [PubMed] [Google Scholar]