

Abstract
Despite the increasing knowledge about the genetic alterations and molecular pathways involved in gliomas, few studies have investigated the association between the gene expression profiles (GEP) and both cytogenetics and histopathology of gliomas. Here, we analyzed the GEP (U133Plus2.0 chip) of 40 gliomas (35 astrocytic tumors, 3 oligodendrogliomas, and 2 mixed tumors) and their association with tumor cytogenetics and histopathology. Unsupervised and supervised analyses showed significantly different GEP in low- vs high-grade gliomas, the most discriminating genes including genes involved in the regulation of cell proliferation, apoptosis, DNA repair, and signal transduction. In turn, among glioblastoma multiforme (GBM), 3 subgroups of tumors were identified according to their GEP, which were closely associated with the cytogenetic profile of their ancestral tumor cell clones: (i) EGFR amplification, (ii) isolated trisomy 7, and (iii) more complex karyotypes. In summary, our results show a clear association between the GEP of gliomas and tumor histopathology; additionally, among grade IV astrocytoma, GEP are significantly associated with the cytogenetic profile of the ancestral tumor cell clone. Further studies in larger series of patients are necessary to confirm our observations.
Keywords: cytogenetics, FISH, gene expression, glioblastoma, glioma, histopathology, microarray
In the last decades an increasingly high amount of information has accumulated about the cytogenetic characteristics of human gliomas.1–3 Although different and unique cytogenetic profiles have been described in low- vs high-grade gliomas, no clearly defined cytogenetic subgroups have been further defined among the most frequent tumor subtypes—eg, glioblastoma multiforme (GBM). Because of this, more recent attempts have been made to search for new relevant molecular markers that could be of help for a better subclassification of low- and high-grade gliomas and the identification of putative new therapeutic targets.4–6 Until now, such efforts have mainly concentrated on genes that are specifically altered in a significant fraction of all gliomas, particularly among high-grade tumors (eg, EGFR, PTEN, TP53, CDKN2A, MDM2, and PDGFA genes) and their potential association with specific intracellular signaling pathways such as those involving STAT3, MAPK, and AKT;7–10 however, these studies have only been partially successful.11–13
In recent years, the availability of large-scale genomic approaches and new bioinformatic tools has fostered the identification of molecular profiles that could be characteristic of human gliomas and define specific subgroups of GBM. Thus, it has been shown that the patterns of gene expression of gliomas correlate with tumor histopathology14–18 and patient survival19,20 and gene expression profiles (GEP) emerge in some studies as an independent prognostic factor among high-grade gliomas.20 According to these studies, most informative genes include genes involved in cell proliferation, RNA processing, signal transduction, and the proteasome functional activity.18 However, no clear association between the GEP of gliomas and tumor cytogenetics has been clearly established so far, which could help on the understanding of the various GEP described.
In a recent paper21 using multicolor interphase fluorescence in situ hybridization (iFISH), we showed the existence of highly variable and heterogeneous intratumoral patterns of cytogenetic abnormalities in human gliomas; such cytogenetic profiles are typically characterized by complex karyotypes consisting of combined gains of chromosome 7, del(9p21)/null 9p, and/or del(10q23) in association with a variable number of other more heterogeneous cytogenetic changes; interestingly, the acquisition of these cytogenetic lesions followed different clonal pathways.21 In this regard, it should be noted that different pathways of intratumoral cytogenetic evolution were found in low- vs high-grade gliomas, which further suggests that the latter tumors might not always correspond to more advanced stages of histologically low-grade gliomas. Even more interestingly, different recent pathways of intratumoral clonal evolution were defined within GBM.21 On the basis of these results, the potential impact of these unique cytogenetic pathways in tumor behavior through the expression of different GEP remains to be elucidated.
The aim of the present study was to identify whether the reported cytogenetic heterogeneity of human gliomas—particularly that of high-grade tumors―translates into the existence of different GEP, which could contribute to a better understanding of the behavior of the tumor and patient outcome. Overall, our results based on a pilot series of 40 gliomas confirm the existence of a clear association between GEP of gliomas and tumor histopathology; in addition, for the first time we show that among grade IV astrocytomas, GEP are significantly associated with the cytogenetic profile of the ancestral tumor cell clone, further supporting its potential impact on the outcome of the disease.
Materials and Methods
Patients and Samples
A total of 40 consecutive adult patients—22 males (55%) and 18 females (45%)—diagnosed with primary gliomas were studied. Mean age at diagnosis was 57 ± 17 years (range: 21– 84 years). In all cases, resected tumor samples were obtained by conventional surgical procedures, after informed consent was given by the patient, according to the recommendations of the local Ethics Committee of the University Hospital of Coimbra (Coimbra, Portugal). For each case, the most relevant histopathological, clinical, and biological features of the disease were recorded, including patient age, gender, performance status (Karnofsky index), and tumor localization (Table 1). At the moment of closing this study, 24 patients (60%) had died and 7 had relapsed after a median follow-up of 15 and 18 months, respectively.
Table 1.
Clinicobiological characteristics of glioma patients and their corresponding tumors analyzed by both interphase FISH and DNA oligonucleotide expression arrays (n = 40)
| Case ID | Histology | WHO grade | Ancestral tumor cell clone | Age | Gender | Karnofsky index (%) | Patient status | Overall survival (months)b |
|---|---|---|---|---|---|---|---|---|
| 1 | Pilocytic astrocytoma | I | Tetraploidy | 21 | F | 80 | Alive | 24 |
| 2 | Pilocytic astrocytoma | I | Tetraploidy | 50 | F | 90 | Alive | 22 |
| 3 | Pilocytic astrocytoma | I | +7+10q | 34 | F | 90 | Alive | 10 |
| 4 | Difuse astrocytoma | II | +7p | 67 | F | 70 | Dead | 11 |
| 5 | Difuse astrocytoma | II | Tetraploidy | 38 | M | 90 | Alive | 27 |
| 6 | Anaplastic astrocytoma | III | +7+10q | 30 | M | 80 | Dead | 4 |
| 7 | Anaplastic astrocytoma | III | Tetraploidy | 27 | M | 90 | Alive | 28 |
| 8 | GBM | IV | Amp 7p | 67 | F | 60 | Dead | 15 |
| 9 | GBM | IV | Amp 7p | 50 | F | 80 | Dead | 13 |
| 10 | GBM | IV | Amp 7p | 71 | F | 60 | Dead | 8 |
| 11 | GBM | IV | Amp 7p | 70 | M | 80 | Alive | 18 |
| 12a | GBM | IV | Amp 7p | 70 | F | 70 | Alive | 17 |
| 13a | GBM | IV | Amp 7p | 45 | F | 60 | Alive | 15 |
| 14 | GBM | IV | Amp 7p | 48 | M | 90 | Dead | 13 |
| 15 | GBM | IV | Amp 7p | 74 | M | 60 | Alive | 8 |
| 16 | GBM | IV | +7 | 69 | F | 60 | Dead | 1 |
| 17 | GBM | IV | +7 | 79 | M | 80 | Dead | 4 |
| 18 | GBM | IV | +7 | 62 | M | 60 | Dead | 3 |
| 19 | GBM | IV | +7 | 67 | M | 60 | Dead | 2 |
| 20a | GBM | IV | +7q | 70 | F | 80 | Dead | 18 |
| 21 | GBM | IV | +7q | 35 | F | 80 | Dead | 14 |
| 22a | GBM | IV | +7q | 39 | F | 80 | Dead | 19 |
| 23a | GBM | IV | +7p | 30 | F | 80 | Alive | 62 |
| 24 | GBM | IV | +7p | 69 | M | 60 | Dead | 5 |
| 25 | GBM | IV | −9p | 67 | F | 70 | Dead | 9 |
| 26 | GBM | IV | −9p | 50 | F | 50 | Dead | 2 |
| 27 | GBM | IV | −9p | 73 | M | 70 | Dead | 2 |
| 28 | GBM | IV | +7p–9p | 66 | M | 80 | Dead | 25 |
| 29 | GBM | IV | +7p–9p | 74 | M | 70 | Dead | 1 |
| 30 | GBM | IV | +7p–9p | 84 | M | 70 | Alive | 10 |
| 31 | GBM | IV | +7p–9p | 56 | M | 80 | Alive | 8 |
| 32 | GBM | IV | −10q | 76 | F | 60 | Alive | 11 |
| 33 | GBM | IV | −10q | 60 | M | 60 | Dead | 2 |
| 34 | Gliosarcoma | IV | +17p+19q | 59 | M | 70 | Dead | 8 |
| 35a | Gliosarcoma | IV | +7+10q+17p | 39 | M | 80 | Dead | 21 |
| 36 | OL | II | −1p−19q | 37 | M | 90 | Alive | 11 |
| 37 | AO | III | −9 | 64 | M | 70 | Dead | 5 |
| 38 | AO | III | +7p–9p | 53 | M | 70 | Alive | 11 |
| 39 | Mixed AOA | III | +7–9p | 81 | F | 70 | Dead | 3 |
| 40a | Mixed AOA | III | −9p | 65 | M | 70 | Alive | 17 |
Abbreviations: FISH, fluorescence in situ hybridization; F, female; M, male; GBM, glioblastoma multiforme; OL, oliodendroglioma; AO, anaplastic oligodendroglioma; AOA, anaplastic oligoastrocytoma.
aRecurrent tumor.
bOverall survival defined from diagnosis to the last visit or date of death.
Tumor Histopathology
Histopathological studies included assessment of both the histological and morphological features of the disease evaluated at the moment of the diagnosis by an expert neuropathologist (eg, type of cell of origin, atypia, variation in nuclear shape or size accompanying hyperchromasia, cell size, cell shape, cellular organization, endothelial proliferation, mitotic activity, overall [eg, increased] cellularity, necrosis, and/or microvascular proliferation).
Histological diagnosis and classification of the tumors were performed according to the WHO criteria22 and patients were distributed as follows: 35 cases (87.5%) were astrocytomas (grade I pilocytic astrocytomas, 3 cases; grade II diffuse astrocytomas, 2; grade III anaplastic astrocytomas, 2 patients; grade IV glioblastomas, 26, and grade IV gliosarcomas, 2 cases); 3 patients (7.5%) had oligodendrogliomas (1 grade II oligodendroglioma, and 2 grade III anaplastic oligodendrogliomas), and the remaining 2 cases (5%) corresponded to mixed oligoastrocytomas (both grade III anaplastic oligoastrocytomas).
In all cases, additional tumor samples were obtained at diagnostic surgery, cut into several pieces, and freshly frozen to be used afterwards for genetic and genomic studies. Only those parts of the tumor showing both macroscopical and microscopical infiltration were used for iFISH analyses as well as to extract RNA. The remaining tumor sample was fixed in formalin and embedded in paraffin. Infiltration by tumor cells in tissue areas mirror-cut to those used for GEP was constantly >65%, as assessed by microscopical analysis of hematoxylin and eosin-stained tissue specimens.
iFISH Studies
In all cases, iFISH analyses were performed on freshly frozen single tumor cell suspensions obtained by mechanical disaggregation of tumor samples, after fixation in methanol/acetic 3/1 (v/v) (Panreac). For iFISH analyses the following commercially available dual color probes (DCP)—all obtained from Vysis, Inc, except the 7p12/alphasatellite 7 DNA DCP that was obtained from Q-BIOgene—were systematically used in double-stainings, for the detection of numerical abnormalities of 16 different chromosome regions that have been reported as frequently altered in gliomas (Supplementary Material, Table S1): (i) for chromosome 1, the LSI 1p36/LSI 1q25 DCP was used; (ii) for chromosome 19, the LSI 19q13/LSI 19p13 DCP was employed; (iii) for chromosome 7, the LSI ELN/LSI 7q31 DCP was applied; (iv) for chromosomes 9 and 22, the LSI bcr/abl ES DCP was used; (v) in addition, for chromosome 9, the LSI 9p21/CEP-9 DCP was also employed, and (vi) for chromosome 10, the LSI PTEN/CEP-10 DCP was applied. For chromosomes 13 and 17, the LSI Rb1 and LSI p53 probes conjugated with spectrum orange were used, respectively.
An Axioscope fluorescence microscope equipped with a 100× oil objective (Zeiss) was used to count the number of hybridization spots per nuclei (n ≥ 200) for each slide. Only those spots with a similar size, intensity, and shape in nonoverlapping nuclei were evaluated; doublet signals were considered as single spots. The extensive description of the iFISH techniques and the cut-offs used to define the presence of numerical abnormalities, for each individual chromosome, have been previously described in detail.23 The definition of the major tumor cell clones present in each sample was arbitrarily based on the presence of ≥10% nuclei carrying identical numbers of hybridization spots for all probes analyzed in that tumor sample.23,24 On the basis of iFISH results a clonal hierarchy was defined at the intratumoral cell level within each tumor. Accordingly, for each tumor the ancestral tumor cell clone was defined as being formed by those cells carrying cytogenetic changes shared by all tumor cells in the sample. In turn, secondary tumor cell clones were identified as those carrying genetic abnormalities, which were only present in a fraction of the tumor cells in the same sample, as previously defined.21
Isolation of Tumoral RNA and Microarray Analysis of GEP
Frozen tissue samples stored at −80°C were used for GEP studies. After thawing, tumor tissue samples were homogenized using a Potter-‘S’-Elvehjem homogenizer (Uniform). Total RNA was isolated in 2 steps using TRIzol (Invitrogen Life Technologies) and the RNeasy Mini Kit (QIAGEN). The integrity and purity of the extracted RNA were determined using a microfluidic electrophoretic system (Agilent 2100 Bioanalyzer, Agilent Technologies). GEP were analyzed with the Gene Chip Human Genome U133Plus2.0 Array (Affymetrix Inc.) according to the manufacturer's instructions, using the 1-cycle cDNA synthesis kit and the Poly-A RNA gene chip control kit (Affymetrix, Inc.).
Analysis of Microarray Data
Owing to the relatively limited number of tumors analyzed, a carefully defined statistical plan was established prior to data analysis. First, the whole set of probes contained in the array was used in a prospective unsupervised exploratory analysis of all tumor samples to identify the most significant differences in GEP among the distinct subtypes of gliomas. Subsequently, a classification approach was attempted, aimed at identifying the best combination of genes to define the specific GEP subgroups identified in the first step. Special emphasis was placed in the investigation of the associations existing between the GEP and both tumor histopathology and cytogenetics.
Briefly, data files containing expression levels for the 40 tumors―obtained through the analysis of the Affymetrix oligonucleotide arrays―were normalized using the Robust Multi-array Average (RMA) expression measure25 and analyzed using the R (version 2.7.1; http://www.r-project.org) and Bioconductor (http://www.bioconductor.org) software tools. In a first step, a prospective unsupervised exploratory analysis was performed to identify the structure of the data at the gene expression level, using all probe sets contained in the array, for which RNA expression was quantified, except the Affymetrix control sequences (total of 54 613 out of 54 675 probe sets); then, a heatmap with a hierarchical cluster was built using euclidean distances and complete linkage. A multidimensional scaling (MDS) analysis—a powerful statistical tool for the multivariate identification of those variables associated with the highest contribution in explaining differences (separability) among individual samples or groups of samples—including those 6000 probes showing the highest level of variation among samples was performed to identify the most relevant differences between groups of tumors.
In a second step, a classification process was performed in order to derive a quantitative measure of class predictability and select the most relevant genes related to WHO tumor grade. An iterative classification process based on random forest algorithms was used26 to rank gene classification power. This approach was selected because it is particularly well-suited for the analysis of a relatively limited number of cases (as it applies for the 40 tumors analyzed in our study); all cases were used for the initial test set. In brief, the first iteration started with the whole collection of probe sets, and a classifier was built taking all of them into consideration. In each of the subsequent iterations, those 90% genes showing the greatest classification power in the previous iteration were used to build the new classifier. In each of the following steps, in order to identify the family of genes with the greatest classification power, 100 different iterations with a data set containing all cases were performed, and a random forest was built and tested on them; all except the 2 gliosarcoma cases were considered in this part of the analysis (n = 38 tumors). The percentage of error obtained in the 3800 (38 testing cases × 100 iterations) classification trials was used to measure the overall classification power of each family of genes. The most informative genes were chosen to be those from the selected family of genes associated with the lowest error (false discovery rate, FDR). The process was only stopped when there were no more genes available. In order to assure the reproducibility of the classification obtained, only those genes with very low FDR (P < 0.001) were selected into the model.
A similar unsupervised analysis to that described above was specifically repeated for the 26 GBM tumors and new significance and class prediction analyses were performed using the R, SAM, and PAM software programs (Significance Analysis of Microarrays software version 2.23 and Prediction Analysis of Microarrays software version 2.1, Tibshirani Lab, Department of Statistics, University of Stanford), respectively.
Other Statistical Methods
In order to assess the statistical significance of differences observed between groups of patients, the Student's t-test and the Mann–Whitney U test were used for parametric and nonparametric (continuous) variables, respectively; for qualitative variables, the χ2 test was applied (SPSS software, SPSS 15.0, SPSS Inc.). Survival curves were plotted according to the method of Kaplan and Meier and the log-rank test was used to assess the statistical significance of differences observed in survival between distinct groups of patients (SPSS software). P-values ≤0.05 were considered to be associated with statistical significance.
Results
GEP of Gliomas
GEP of gliomas as measured by DNA oligonucleotide microarrays were highly heterogeneous and were associated with tumor grade and histopathology (Fig. 1). Unsupervised hierarchical clustering of all tumors analyzed (n = 40) according to their GEP, showed that gliosarcomas (n = 2) were those tumors that more significantly differed from all other tumors; in turn, the latter clustered into 2 major groups, one of which included all GBM (Fig. 1A and B). Among those 72 genes showing the highest discriminating power between gliosarcomas and other gliomas, 18 were overexpressed in the former group, and 54 were underexpressed. Of note, overexpressed genes included KRT80 gene (keratin 80) coded at 12q13.13 and the BAIAP2L1 gene (BAI1-associated protein 2-like 1) coded at 7q21.3, whereas the PTPRZ1 gene (protein tyrosine phosphatase, receptor-type, Z polypeptide 1) coded at 7q31.3 and the GFAP gene (glial fibrillary acidic protein) coded at 17q21 were among the most relevant underexpressed genes. A more detailed description of these and other relevant genes is shown in Supplementary Material, Table S2.
Fig. 1.

Unsupervised and supervised classification of glioma samples based on their GEP. In (A) unsupervised classification of the tumors according to the 54 613 probes included in the Gene Chip Human Genome U133 Plus 2.0 array are shown; in (A) different histopathological subtypes of gliomas (n = 40) are identified by a distinct color code: pilocytic tumors are coded black; diffuse astrocytomas are identified as green; anaplastic astrocytomas, blue; glioblastoma multiforme, red; gliosarcomas, purple; oligodendrogliomas, yellow; anaplastic oligodendrogliomas, grey, and mixed oligoastrocytomas correspond to those cases depicted as orange. In (B) an MDS plot corresponding to the same 40 gliomas (identified according to their histopathologic features with the same color code as described for [A]) classified according to those 6000 genes showing the highest variability in their expression among the different tumors. In (C) is a heat map representation of the results reflecting a hierarchical cluster analysis based on the expression of those 71 genes showing the highest classification power for tumor grade/histopathology; in (C) the different histopathological subtypes of gliomas are also identified by the same color code as described for (A).
A supervised hierarchical clustering analysis (Fig. 1C) with a classifier built on those 71 genes that would mostly contribute to discriminate between low-grade—grade I and II—vs high-grade—grade III and IV—gliomas (after excluding gliosarcomas), showed a high classification power with only 6% of the cases being misclassified (Supplementary Material, Table S3). Interestingly, one-fourth of the 71 selected genes corresponded to genes coded in chromosome 10 (21/71), 7/71 genes were coded in chromosome 17, and 3 genes were coded in chromosomes 7 and 22, each. The remaining 36 genes were spread among the other chromosomes, with the exception of chromosomes 2, 3, 16, 18, and 21, which did not contain informative genes differentially expressed in the 2 groups of patients. The 6 most informative genes included the GRAMD1B gene (GRAM domain containing 1B) coded at 11q24.1, the PITPNC1 gene (phosphatidylinositol transfer protein, cytoplasmic 1) localized at chromosome 17q24.2, the C1orf21 gene (chromosome 1 open reading frame 21) at 1q25, the VPS26A gene (vacuolar protein sorting 26 homolog A) at 10q21.1, the MSRB2 gene (methionine sulfoxide reductase B2) at 10p12, and the MGEA5 (meningioma expressed antigen 5) coded at 10q24.1–q24.3. Interestingly, 19 of these 71 genes had been either previously specifically related to gliomas or generally involved in carcinogenesis through their participation in the regulation of cell proliferation, apoptosis, DNA repair, transcription, and signal transduction: EXO1 (1q42–q43), CBX8 (17q25.3), NCOA4 (10q11.2), SGMS1 (10q11.2), ZYX (7q32), TK1 (17q23.2-q25), SCAPER (15q24), CENPE (4q24–q25), BLOC1S2 (10q24.31), DLG7 (14q22.3), CDC45L (22q11.21), AURKB (17p13.1), CDCA5 (11q12.11), CHAF1A (19p13.3), RECQL4 (8q24.3), CCNA2 (4q25–q31), BIRC5 (17q25), GTSE1 (22q13.2–q13), and UBE2C (20q13.12) genes.
The clinico-biological and cytogenetic characteristics of the 2 groups of gliomas distributed according to tumor grade are summarized in Table 2. As displayed in it, low-grade gliomas more frequently occurred in younger adults (<40 years) (P = 0.01) and they showed both a higher Karnofsky index (P = 0.002) and a better outcome (P = 0.03); in addition, they displayed a lower frequency of both multilobular and temporal tumors (P < 0.001), lower number of neoplastic cell clones per tumor (P = 0.04), and a higher frequency of tetraploid ancestral tumor cell clones (P = 0.008).
Table 2.
Clinical and cytogenetic characteristics of glioma tumors (n = 40) grouped according to the WHO histological grade
| Low-grade (I–II) (n = 6) | High-grade (III–IV) (n = 34) | P-value | |
|---|---|---|---|
| Age | |||
| <40 years | 4/6 | 6/34 | 0.01 |
| >40 years | 2/6 | 28/34 | |
| Gender | |||
| Male | 2/6 | 20/34 | 0.25 |
| Female | 4/6 | 14/34 | |
| Karnofsky indexa | 85 ± 8 (70–90) | 71 ± 10 (50–90) | 0.002 |
| Tumor localization | |||
| Frontal | 2/6 | 12/34 | <0.001 |
| Temporal | 0/6 | 12/34 | |
| Parietal | 1/6 | 0/34 | |
| Occipital | 1/6 | 0/34 | |
| Posterior fossa | 1/6 | 0/34 | |
| Cerebellum | 1/6 | 0/34 | |
| Multilobular | 0/6 | 10/34 | |
| Ancestral tumor cell clone | |||
| Amp7 | 0/6 | 8/34 | 0.008 |
| +7q or +7p | 1/6 | 5/34 | |
| +7 | 0/6 | 4/34 | |
| −9p/−9p | 0/6 | 5/34 | |
| −10q | 0/6 | 2/34 | |
| +7p–9p | 0/6 | 6/34 | |
| +7+10q | 1/6 | 2/34 | |
| +17p+19q | 0/6 | 1/34 | |
| Tetraploidy | 3/6 | 1/34 | |
| −1p−19q | 1/6 | 0/34 | |
| Number of tumor cell clonesa (by iFISH) | 3 ± 1 (2–4) | 5 ± 2 (2–9) | 0.04 |
| Number of relapses | 0/6 | 7/34 | 0.22 |
| Number of deaths | 1/6 | 22/34 | 0.03 |
Abbreviation: WHO, World Health Organization. Results expressed as number (N) of cases/total cases.
aMean ± one standard deviation (range).
GEP of Grade IV Astrocytomas
Owing to the relatively high heterogeneity of the GEP of grade IV astrocytic tumors (Fig. 1B and C), we further investigated the potential association between the gene expression signatures and the genetic, clinical, and biological features of the disease, specifically among grade IV astrocytomas/GBM. Unsupervised hierarchical clustering analysis of the GEP of the 26 GBM showed 3 clearly distinct groups of tumors for the 54 613 probes analyzed (Fig. 2A).
Fig. 2.
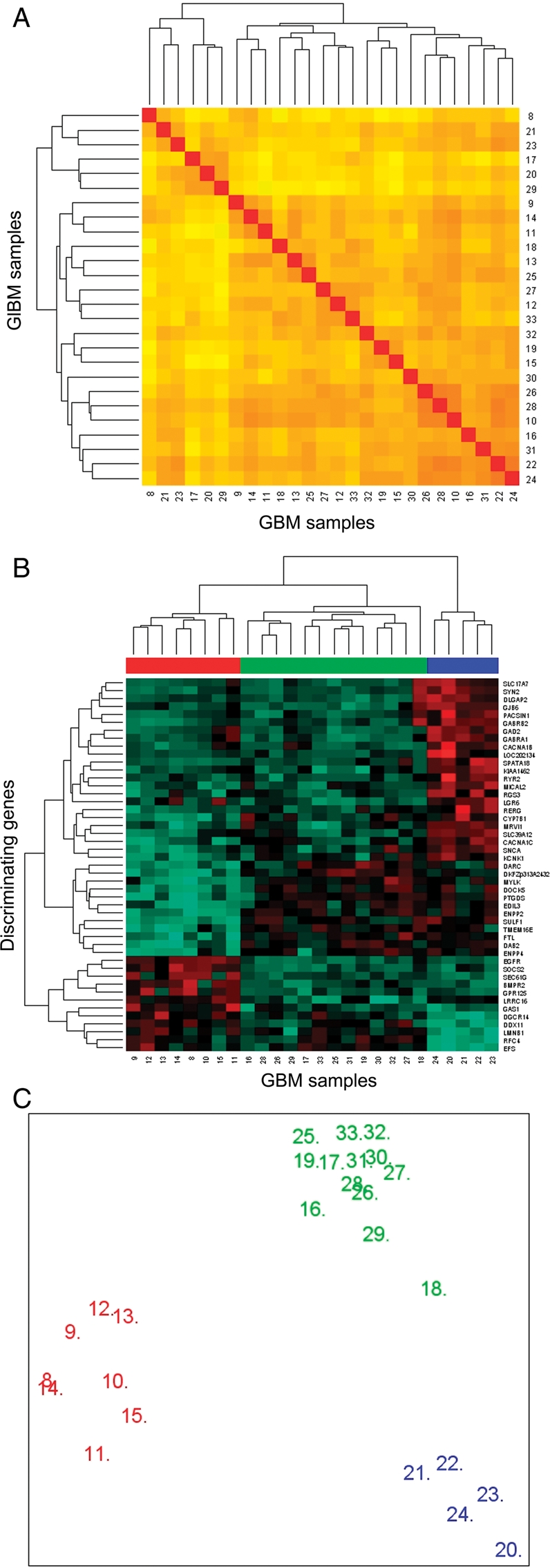
Unsupervised and supervised classification of GBM (n = 26) according to their GEP. In (A) unsupervised classification based on the 54 613 probes included in the GeneChip U133A plus 2.0 array is shown. In turn, in (B) hierarchical clustering based on those 47 genes with the highest discrimination power as identified by significance microarray analysis (SAM) shows 3 groups of tumors with distinct GEP (GEP groups: GEP 1, GEP 2, and GEP 3 are colored red, blue, and green, respectively) is displayed. In (C) a MSD plot corresponding to the same tumors classified according to their GEP and identified with the same color code as described above.
Overall, 47 genes were identified, which allowed for a clear discrimination among the 3 groups of GBM showing different GEP (Fig. 2B and Table 3). Four of these 47 genes have been previously shown to be involved in the pathogenesis of gliomas acting either as tumor suppressor genes, or being involved in signal transduction: EGFR (7p12), ENPP2 (8q24.1), GAS1 (9q21.3–q22), and FTL (19q13.3–q13). Further analyses searching for the best combination of genes to predict for the classification of GBM into the three GEP groups identified a combination of 79 informative genes (Fig. 3A and Supplementary Material, Table S4). These genes were coded in 16 different chromosomes: 8 genes in chromosomes 7 and 5 each; 7 in chromosome 4; 6 in chromosomes 1, 3, and 12; 5 in chromosome 8; 4 genes in chromosomes 10 and 11, and either 3 or 2 genes localized in chromosomes 2, 6, 9, 13, 14, 15, 17, 18, 19, 20, and 22. Once again, among the most discriminating genes, the EGFR (epidermal growth factor receptor) gene was selected, together with the SOCS2 (suppressor of cytokine signaling-2) (both overexpressed in GEP 1 cases), SPATA18 (spermatogenesis/phosphodiesterase), SLC39A12 (solute carrier family 39, zinc transporter) (overexpressed in GEP 2 tumors), ENPP2 (ectonucleotide pyrophosphatase/phosphodiesterase), and SULF1 (sulfatase-1) (underexpressed in GEP 1 and overexpressed in both GEP 2 and GEP 3 cases) genes localized at the 7p12, 12q, 4q12, 10p12.33, 8q24.1, and 8q13.2–q13.3 chromosome regions, respectively (Fig. 3A and Supplementary Material, Table S4).
Table 3.
Genes (n = 47) differentially expressed in the three groups of GBM identified by their GEP
| Gene name | Gene symbol | Chromosomal localization | GEP tumor groups |
||
|---|---|---|---|---|---|
| GEP 1 | GEP 2 | GEP 3 | |||
| Duffy blood group, chemokine receptor | DARC | 1q21–q22 | − | + | + |
| Leucine-rich repeat containing G protein-coupled receptor 6 | LGR6 | 1q32.1 | − | + | − |
| Ryanodine receptor 2 (cardiac) | RYR2 | 1q42.1–q43 | − | + | − |
| Potassium channel, subfamily K, member 1 | KCNK1 | 1q42–q43 | − | + | − |
| Bone morphogenetic protein receptor, type II | BMPR2 | 2q33–q34 | + | − | − |
| Synapsin II | SYN2 | 3p25 | − | + | − |
| Myosin, light chain kinase | MYLK | 3q21 | − | + | + |
| Replication factor C (activator 1) 4, 37 kDa | RFC4 | 3q27 | + | − | + |
| G protein-coupled receptor 125 | GPR125 | 4p15.31 | + | − | − |
| Spermatogenesis associated 18 homolog | SPATA18 | 4q12 | − | + | − |
| Synuclein, alpha | SNCA | 4q21 | − | + | − |
| Disabled homolog 2, mitogen-responsive phosphoprotein | DAB2 | 5p13 | − | + | + |
| EGF-like repeats and discoidin I-like domains 3 | EDIL3 | 5q14 | − | + | + |
| Lamin B1 | LMNB1 | 5q23.3–q31.1 | + | − | + |
| Gamma-aminobutyric acid (GABA) A receptor, beta 2 | GABRB2 | 5q34 | − | + | − |
| Gamma-aminobutyric acid (GABA) A receptor, alpha 1 | GABRA1 | 5q34–q35 | − | + | − |
| Hypothetical protein LOC202134 | LOC202134 | 5q35.2 | − | + | − |
| Ectonucleotide pyrophosphatase/phosphodiesterase 4 | ENPP4 | 6p12.3 | − | + | + |
| Protein kinase C and casein kinase substrate in neurons 1 | PACSIN1 | 6p21.3 | − | + | − |
| Leucine rich repeat containing 16 | LRRC16 | 6p22.2 | + | + | − |
| Sec61 gamma subunit | SEC61G | 7p11.2 | + | − | − |
| Epidermal growth factor receptor | EGFR | 7p12 | + | − | − |
| Dedicator of cytokinesis 5 | DOCK5 | 8p21.2 | − | + | + |
| Discs, large (Drosophila) homolog-associated protein 2 | DLGAP2 | 8p23 | − | + | − |
| Sulfatase 1 | SULF1 | 8q13.2–q13.3 | − | + | + |
| Cytochrome P450, family 7, subfamily B, polypeptide 1 | CYP7B1 | 8q21.3 | − | + | + |
| Ectonucleotide pyrophosphatase/phosphodiesterase 2 | ENPP2 | 8q24.1 | − | + | + |
| Growth arrest-specific 1 | GAS1 | 9q21.3–q22 | + | − | − |
| Regulator of G protein signaling 3 | RGS3 | 9q32 | − | + | − |
| Calcium channel, voltage-dependent, N type, alpha 1B subunit | CACNA1B | 9q34 | − | + | − |
| Prostaglandin D2 synthase | PTGDS | 9q34.2–q34.3 | − | + | + |
| Glutamate decarboxylase 2 | GAD2 | 10p11.23 | − | + | − |
| KIAA1462 | KIAA1462 | 10p11.23 | − | + | − |
| Solute carrier family 39 (zinc transporter), member 12 | SLC39A12 | 10p12.33 | − | + | − |
| Hypothetical protein DKFZp313A2432 | DKFZp313A2432 | 11p14.2 | − | + | + |
| Transmembrane protein 16E | TMEM16E | 11p14.3 | − | + | + |
| Murine retrovirus integration site 1 homolog | MRVI1 | 11p15 | − | + | − |
| Microtubule associated monoxygenase, calponin and LIM 2 | MICAL2 | 11p15.3 | − | + | − |
| DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 11 | DDX11 | 12p11 | + | − | + |
| RAS-like, estrogen-regulated, growth inhibitor | RERG | 12p12.3 | − | + | − |
| Calcium channel, voltage-dependent, L type, alpha 1C | CACNA1C | 12p13.3 | − | + | − |
| Suppressor of cytokine signaling 2 | SOCS2 | 12q | + | − | − |
| Gap junction protein, beta 6 | GJB6 | 13q11–q12.1 | − | + | − |
| Embryonal Fyn-associated substrate | EFS | 14q11.2–q12 | + | − | + |
| Solute carrier family 17, member 7 | SLC17A7 | 19q13 | − | + | − |
| Ferritin, light polypeptide | FTL | 19q13.3–q13.4 | − | + | + |
| DiGeorge syndrome critical region gene 14 | DGCR14 | 22q11.21 | + | − | + |
Abbreviations: GBM, glioblastoma multiforme; GEP, gene expression profiles.
Fig. 3.

Detailed description of those genes (n = 79) showing a significant independent value for predicting the GEP of GBM (n = 26) and impact of the different GEP on overall patient survival. In (A) the size of horizontal lines reflects variation on the expression of individual genes within each group of tumors showing different GEP, according to the mean overall expression of that gene in all GBM tumors (n = 26); horizontal bars to the right of vertical lines indicate higher expression than the mean, whereas bars to the left of vertical lines reflect gene expression levels lower than the mean. In (B) overall survival curves of GBM patients grouped according to their GEP are displayed.
Interestingly, a significant (P = 0.001) association was found between the GEP and the cytogenetic patterns of the ancestral tumor cell clones of GBM (Table 4). Accordingly, GEP 1 and GEP 2 cases systematically showed isolated chromosome 7 abnormalities in the ancestral tumor cell clone: although GEP 1 tumors typically showed amplification of the EGFR gene in chromosome 7p, GEP 2 cases displayed partial gains of either the long- or the short arm of chromosome 7 in their ancestral tumor cell clone. In turn, cases included in the GEP 3 group had more variable cytogenetic profiles in their ancestral tumor cell clones, mainly consisting of isolated trisomy 7 (31%), del(10q) (15%), and del(9p)/null 9p alone (23%) or in combination with gains of chromosome 7 (31%).
Table 4.
Chromosomal aberrations of GBM grouped according to their GEP
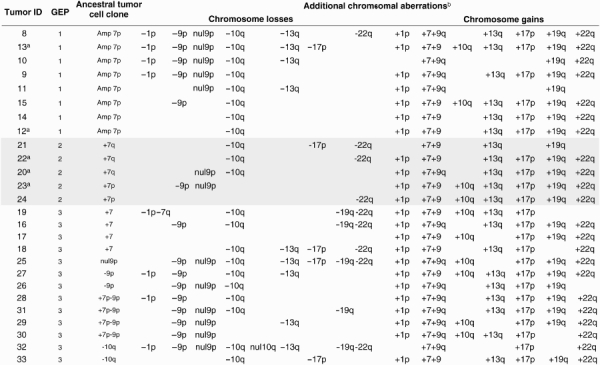 |
Abbreviations: GBM, glioblastoma multiforme; GEP, gene expression profiles.
aRecurrent tumors.
bPresent in tumor cell clones other than the ancestral tumor cell clone (ie, not common to all altered tumor cells in the sample).
Noteworthy, once GBM were grouped according to their GEP, a significant association was found with patient age (P = 0.001) and disease outcome (P = 0.05) (Fig. 3B and Table 5). Accordingly, GEP 2 was more frequently found in younger patients; in turn, GEP 3 cases displayed a shorter overall survival rate (P = 0.05) vs the other two groups (Table 5). In addition, GEP 1 tumors that systematically displayed EGFR amplification in their ancestral tumor cell clone tended to have a higher number of clones per tumor sample, as defined by iFISH (P = 0.09). In contrast, no significant differences were found between the 3 groups of GBM with different GEP as regards patient gender, Karnofsky index, and tumor localization.
Table 5.
Clinical and cytogenetic characteristics of glioblastoma multiforme (GBM) patients grouped according to their tumoral gene expression profiles (GEP)
| GEP group |
P-value | |||
|---|---|---|---|---|
| GEP 1 (n = 8) | GEP 2 (n = 5) | GEP 3 (n = 13) | ||
| Age | ||||
| <40 years | 0/8 | 3/5 | 0/13 | 0.001 |
| >40 years | 8/8 | 2/5 | 13/13 | |
| Gender | ||||
| Male | 3/8 | 1/5 | 9/13 | 0.12 |
| Female | 5/8 | 4/5 | 4/13 | |
| Karnofsky indexa | 70 ± 12 (60–90) | 76 ± 9 (60–80) | 67 ± 10 (50–80) | 0.26 |
| Tumor localization | ||||
| Frontal | 3/8 | 1/5 | 4/13 | 0.50 |
| Temporal | 1/8 | 3/5 | 5/13 | |
| Multilobular | 4/8 | 1/5 | 4/13 | |
| Ancestral tumor cell clone | ||||
| Amp7 | 8/8 | 0/0 | 0/0 | <0.001 |
| +7q or +7p | 0/0 | 5/5 | 0/0 | |
| +7 | 0/0 | 0/0 | 4/13 | |
| −9p/−9p | 0/0 | 0/0 | 3/13 | |
| −10q | 0/0 | 0/0 | 2/13 | |
| +7p–9p | 0/0 | 0/0 | 4/13 | |
| Number of tumor cell clonesa (by iFISH) | 6 ± 2 (4–9) | 4 ± 1 (3–5) | 4 ± 1 (3–7) | 0.09 |
| Median OS (months)b | 13 ± 4 (8–18) | 16 ± 8 (5–25) | 6 ± 7 (0–25) | 0.05 |
| Number of relapses | 2/8 | 3/5 | 0/13 | 0.01 |
| Number of deaths | 3/8 | 4/5 | 10/13 | 0.14 |
Abbreviation: OS, overall survival. Results expressed as number (N) of cases/total cases.
aMean ± one standard deviation (SD).
bMedian and range between brackets.
Discussion
In this study, we describe for the first time the existence of unique GEP associated with both tumor histopathology and, among GBM, also with tumor cytogenetics. Although these results should be regarded with caution because of the limited number of the cases analyzed, our pilot study based on a series of 40 samples points out several new lines of research related to the identification of new potentially useful diagnostic markers, and clinically and biologically relevant cytogenetic profiles. In this regard, it should be emphasized that despite the relatively low number of cases studied they all corresponded to primary diagnostic tumor samples from previously untreated patients, which were freshly obtained and immediately processed with a proven high-quality RNA (as assessed by microfluidic electrophoresis; Agilent 2100 Bioanalyzer, Agilent Technologies), and a high tumor cell content (systematically >70% of the cells contained in each tumor sample were tumor cells); in addition, detailed information about the intratumoral patterns of clonal evolution was also obtained for each tumor sample, making this series of gliomas unique.
Regarding tumor histopathology, gliosarcomas were found to display unique GEP that were clearly different from all other subtypes of gliomas. This could be due to the fact that gliosarcomas are a unique histopathologic subtype of GBM characterized by the presence of tumor cells with both gliomatous and sarcomatous differentiation; the latter component is absent in all other subtypes of gliomas and has been associated with aberrant mesenchymal differentiation in highly malignant astrocytic neoplasms.27 In line with these findings, structural genes, such as cytokeratin 80 (KRT80), collagen type X alpha 1 (COL10A1) (overexpressed), and both glial fibrillary acidic protein (GFAP) and peripheral myelin protein 2 (PMP2) (decreased expression), were included among the most informative genes for the discrimination between gliosarcomas and other gliomas. Of note, gliosarcomas also showed overexpression of the GATA3 transcription factor and downregulation of the Rho guanine nucleotide exchange factor (ARHGEF4) signaling molecule and other several cytoplasmic membrane receptors and chromosome ORF genes. These findings point out the expression of unique structural and functional marker profiles in gliosarcomas that in addition to GFAP28 could contribute to a more refined histopathological classification of gliosarcomas vs other glioblastomas. However, these results require further confirmation in larger series of patients from brain tumor trial consortia.
Interestingly, upon excluding gliosarcomas, hierarchical clustering analysis of GEP provided a classifier for low- vs high-grade gliomas based on the expression of 71 different genes; the most informative genes included the GRAMD1B, PITPNC1, C1orf21, VPS26A, MSRB2, and MGEA5 genes, together with another 23 genes that had been previously related to gliomas and/or involved in oncogenetic events in gliomas and other tumors, DNA repair, regulation of transcription, cell division, cell proliferation, tumor cell growth, and metastasis.29–34 Overall, high-grade tumors were associated with GEP characterized by greater expression of genes associated with increased cell proliferation and survival.31,34,35 Among other genes these included the DLG7, BIRC5 (survivin), and UBE2C (or UBCH10) genes coded at 14q22.3, 17q25, and 20q13.12, which have been involved in cell cycle regulation and stem cellness (eg, DLG7), cell division and suppression of apoptosis (eg, survivin), and regulation of cell cycle progression and the ontogenesis of astrocytic tumors (eg, UBEC2C).29,35–37 Of note, the STXBP6 (syntaxin binding protein 6-amisyn) gene encoded at 14q13 was the individual gene showing the highest differential expression between high- vs low-grade gliomas; despite the fact that the protein encoded by this gene is known to be involved in vesicle-mediated intracellular transport, the precise functional role and pathways related to this protein remain largely unknown.
Interestingly, low- vs high-grade tumors did not only show different GEP but they were also associated with different cytogenetic patterns. Accordingly, high-grade gliomas displayed a greater intratumoral cytogenetic heterogeneity (eg, they had a higher number of tumor cell clones), which probably reflects a greater genetic instability associated with a higher rate of accumulation of genetic aberrations.38 This could be related at least to a certain extent to the differential expression in low- vs high-grade gliomas of genes involved in DNA repair (CHAF1A and RECQL4),39 cell cycle and cell division (TK1, CENPE, CDC45L, AURKB, CDCA5, CCNA2, and GTSE1)32,40 and apoptosis (BLOC1S2, SGSM1, and BIRC5).34,35,41 Alternatively, this could be also associated with the pattern of cytogenetic abnormalities detected in the ancestral tumor cell clone since some genetic abnormalities leading to tumor progression (eg, amplification of EGFR) were exclusively found among high-grade tumors whereas tetraploidy, a genetic abnormality typically associated with a better outcome,42,43 was restricted to low-grade astrocytomas.
In line with this latter hypothesis, 3 subgroups of GBM could be established according to their GEP, which in turn were tightly associated with the cytogenetic profile of their ancestral tumor cell clones. Accordingly, EGFR amplification and overexpression emerged as typically associated with one of these GEP (GEP1) detected among GBM.44–46 In addition, GEP1 GBM patients also showed significantly higher expression of SOCS2, a gene whose methylation has been involved in the pathogenesis of GBM where it is associated with resistance to conventional therapy and patient outcome.47
In contrast to GEP1 GBM, cases classified as GEP 2 typically showed isolated gains of one arm of chromosome 7 in their ancestral tumor cell clones in association with overexpresion of the SPATA18, SLC39A12, and ENPP2 genes. Although the former 2 genes are poorly characterized genes, they have been associated with cell differentiation and metal ion transport, respectively. In turn, ENPP2 encodes for an autotaxin reported to be overexpresed in GBM48 and promote tumor invasion49 and was highly expressed in both GEP2 and GEP3 gliomas. Upon comparison with GEP1 and GEP2 tumors, GEP3 GBM typically showed more heterogeneous cytogenetic profiles in their ancestral tumor cell clones, which could contribute to explain their relatively more variable GEP. Of note, further cytogenetic evolution of tumor cell clones was frequently associated in all these GEP subgroups of GBM with further gains of chromosome 7, del(9p), and del(10q) suggesting the simultaneous occurrence of activation of wild-type and/or mutated EGFR and loss of both Ink4A/Arf and PTEN tumor suppressor genes, which has been recently shown to induce a fully penetrant and rapid-onset, high-grade malignant glioma phenotype with prominent pathological and molecular resemblance to human GBM, in the CNS of adult mice.46 Altogether, these observations suggest that the sequence of appearance of different genetic abnormalities in GBM could have a relevant impact on the GEP and the behavior of the tumor, independently of the specific combination of genetic abnormalities accumulated in secondary tumor cell clones. In other words, the sequence of appearance in a tumor of different genetic abnormalities will have a specific influence on the GEP of tumor cells, over the end result.
Interestingly, GEP of GBM also showed a clear association with patient age, the number of different tumor cell clones defined by iFISH, and survival, which points out the potential impact of the GEP of GBM in determining tumor behavior at both the cytogenetic and clinical levels.
In conclusion, in this pilot study, we show that GEP of human gliomas are significantly associated with tumor histopathology, unique GEP being found in low- vs high-grade gliomas. Most interestingly, among GBM 3, distinct GEP were found that appear to be associated with early genetic tumor changes. Further studies in independent and larger series of patients are required to confirm these results.
Supplementary Material
Supplementary Material is available at Neuro-Oncology online.
Conflict of interest statement. None declared.
Funding
FCG (Portuguese Calouste Gulbenkian Foundation, Project Ref. 68708), FCT (Portuguese Foundation for Science and Technology) FCT: PIC/IC/83108/2007—PhD fellowship (SFRH/BD/11820/2003).
References
- 1.Korshunov A, Sycheva R, Golanov A. Molecular stratification of diagnostically challenging high-grade gliomas composed of small cells: the utility of fluorescence in situ hybridization. Clin Cancer Res. 2004;10(23):7820–7826. doi: 10.1158/1078-0432.CCR-04-0710. doi:10.1158/1078-0432.CCR-04-0710. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. doi:10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 3.Okamoto Y, Di Patre PL, Burkhard C, et al. Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol. 2004;108(1):49–56. doi: 10.1007/s00401-004-0861-z. [DOI] [PubMed] [Google Scholar]
- 4.Mischel PS, Nelson SF, Cloughesy TF. Molecular analysis of glioblastoma: pathway profiling and its implications for patient therapy. Cancer Biol Ther. 2003;2(3):242–247. doi: 10.4161/cbt.2.3.369. [DOI] [PubMed] [Google Scholar]
- 5.Czernicki T, Zegarska J, Paczek L, et al. Gene expression profile as a prognostic factor in high-grade gliomas. Int J Oncol. 2007;30(1):55–64. [PubMed] [Google Scholar]
- 6.El-Jawahri A, Patel D, Zhang M, Mladkova N, Chakravarti A. Biomarkers of clinical responsiveness in brain tumor patients: progress and potential. Mol Diagn Ther. 2008;12(4):199–208. doi: 10.1007/BF03256285. [DOI] [PubMed] [Google Scholar]
- 7.Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol. 2006;65(12):1181–1188. doi: 10.1097/01.jnen.0000248549.14962.b2. doi:10.1097/01.jnen.0000248549.14962.b2. [DOI] [PubMed] [Google Scholar]
- 8.Bouvier-Labit C, Chinot O, Ochi C, Gambarelli D, Dufour H, Figarella-Branger D. Prognostic significance of Ki67, p53 and epidermal growth factor receptor immunostaining in human glioblastomas. Neuropathol Appl Neurobiol. 1998;24(5):381–388. doi: 10.1046/j.1365-2990.1998.00137.x. doi:10.1046/j.1365-2990.1998.00137.x. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura M, Shimada K, Ishida E, Nakase H, Konishi N. Genetic analysis to complement histopathological diagnosis of brain tumors. Histol Histopathol. 2007;22(3):327–335. doi: 10.14670/HH-22.327. [DOI] [PubMed] [Google Scholar]
- 10.Homma T, Fukushima T, Vaccarella S, et al. Correlation among pathology, genotype, and patient outcomes in glioblastoma. J Neuropathol Exp Neurol. 2006;65(9):846–854. doi: 10.1097/01.jnen.0000235118.75182.94. doi:10.1097/01.jnen.0000235118.75182.94. [DOI] [PubMed] [Google Scholar]
- 11.Wager M, Menei P, Guilhot J, Levillain P, Michalak S, Bataille B, et al. Prognostic molecular markers with no impact on decision-making: the paradox of gliomas based on a prospective study. Br J Cancer. 2008;98(11):1830–1838. doi: 10.1038/sj.bjc.6604378. doi:10.1038/sj.bjc.6604378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Backlund LM, Nilsson BR, Liu L, Ichimura K, Collins VP. Mutations in Rb1 pathway-related genes are associated with poor prognosis in anaplastic astrocytomas. Br J Cancer. 2005;93(1):124–130. doi: 10.1038/sj.bjc.6602661. doi:10.1038/sj.bjc.6602661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murat A, Migliavacca E, Gorlia T, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26(18):3015–3024. doi: 10.1200/JCO.2007.15.7164. doi:10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- 14.Shai R, Shi T, Kremen TJ, et al. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene. 2003;22(31):4918–4923. doi: 10.1038/sj.onc.1206753. doi:10.1038/sj.onc.1206753. [DOI] [PubMed] [Google Scholar]
- 15.Kim S, Dougherty ER, Shmulevich I, et al. Identification of combination gene sets for glioma classification. Mol Cancer Ther. 2002;1(13):1229–1236. [PubMed] [Google Scholar]
- 16.Persson O, Krogh M, Saal LH, et al. Microarray analysis of gliomas reveals chromosomal position-associated gene expression patterns and identifies potential immunotherapy targets. J Neurooncol. 2007;85(1):11–24. doi: 10.1007/s11060-007-9383-6. doi:10.1007/s11060-007-9383-6. [DOI] [PubMed] [Google Scholar]
- 17.Shirahata M, Iwao-Koizumi K, Saito S, et al. Gene expression-based molecular diagnostic system for malignant gliomas is superior to histological diagnosis. Clin Cancer Res. 2007;13(24):7341–7356. doi: 10.1158/1078-0432.CCR-06-2789. doi:10.1158/1078-0432.CCR-06-2789. [DOI] [PubMed] [Google Scholar]
- 18.van den Boom J, Wolter M, Kuick R, et al. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163(3):1033–1043. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freije WA, Castro-Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64(18):6503–6510. doi: 10.1158/0008-5472.CAN-04-0452. doi:10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 20.Nutt CL, Mani DR, Betensky RA, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003;63(7):1602–1607. [PubMed] [Google Scholar]
- 21.Vital AL, Tabernero MD, Crespo I, et al. Intratumoral patterns of clonal evolution in gliomas. Neurogenetics. 2009;11(4):227–239. doi: 10.1007/s10048-009-0217-x. [DOI] [PubMed] [Google Scholar]
- 22.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification. WHO grading Astrocytic Tumours. Oligodendroglial Tumours, Chap. 08–67. IARC: Lyon; 2007. [Google Scholar]
- 23.Sayagues JM, Tabernero MD, Maillo A, Espinosa A, Rasillo A, Diaz P, et al. Intratumoral patterns of clonal evolution in meningiomas as defined by multicolor interphase fluorescence in situ hybridization (FISH): is there a relationship between histopathologically benign and atypical/anaplastic lesions? J Mol Diagn. 2004;6(4):316–325. doi: 10.1016/S1525-1578(10)60527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tabernero MD, Espinosa AB, Maillo A, Rebelo O, Vera JF, Sayagues JM, et al. Patient gender is associated with distinct patterns of chromosomal abnormalities and sex chromosome linked gene-expression profiles in meningiomas. Oncologist. 2007;12(10):1225–1236. doi: 10.1634/theoncologist.12-10-1225. doi:10.1634/theoncologist.12-10-1225. [DOI] [PubMed] [Google Scholar]
- 25.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. doi:10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 26.Breiman L. Random Forests. Mach Learn. 2001;45(1):5–32. doi:10.1023/A:1010933404324. [Google Scholar]
- 27.Reis RM, Konu-Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H. Genetic profile of gliosarcomas. Am J Pathol. 2000;156(2):425–432. doi: 10.1016/S0002-9440(10)64746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tena-Suck ML, Moreno-Jimenez S, Alonso M, Aguirre-Crux L, Sanchez A. Oligodendrogliomas in relation to astrocytes differentiation. Clinicopathologic and immunohistochemical study. Ann Diagn Pathol. 2008;12(5):313–321. doi: 10.1016/j.anndiagpath.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Jiang L, Huang CG, Lu YC, Luo C, Hu GH, Liu HM, et al. Expression of ubiquitin-conjugating enzyme E2C/UbcH10 in astrocytic tumors. Brain Res. 2008;1201:161–166. doi: 10.1016/j.brainres.2008.01.037. doi:10.1016/j.brainres.2008.01.037. [DOI] [PubMed] [Google Scholar]
- 30.Cabreiro F, Picot CR, Perichon M, Castel J, Friguet B, Petropoulos I. Overexpression of mitochondrial methionine sulfoxide reductase B2 protects leukemia cells from oxidative stress-induced cell death and protein damage. J Biol Chem. 2008;283(24):16673–16681. doi: 10.1074/jbc.M708580200. doi:10.1074/jbc.M708580200. [DOI] [PubMed] [Google Scholar]
- 31.Peng Y, Li CX, Chen F, Wang Z, Ligr M, Melamed J, et al. Stimulation of prostate cancer cellular proliferation and invasion by the androgen receptor co-activator ARA70. Am J Pathol. 2008;172(1):225–235. doi: 10.2353/ajpath.2008.070065. doi:10.2353/ajpath.2008.070065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136(3):473–484. doi: 10.1016/j.cell.2008.12.020. doi:10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 33.Chang JS, Yeh RF, Wiencke JK, Wiemels JL, Smirnov I, Pico AR, et al. Pathway analysis of single-nucleotide polymorphisms potentially associated with glioblastoma multiforme susceptibility using random forests. Cancer Epidemiol Biomarkers Prev. 2008;17(6):1368–1373. doi: 10.1158/1055-9965.EPI-07-2830. doi:10.1158/1055-9965.EPI-07-2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai ZC, Wei X, Shimizu T, Ramos E, Rohrbaugh M, Nikolaidis N, et al. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell. 2005;120(5):675–685. doi: 10.1016/j.cell.2004.12.036. doi:10.1016/j.cell.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 35.Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis. 2007;28(6):1133–1139. doi: 10.1093/carcin/bgm047. doi:10.1093/carcin/bgm047. [DOI] [PubMed] [Google Scholar]
- 36.Gudmundsson KO, Thorsteinsson L, Sigurjonsson OE, Keller JR, Olafsson K, Egeland T, et al. Gene expression analysis of hematopoietic progenitor cells identifies Dlg7 as a potential stem cell gene. Stem Cells. 2007;25(6):1498–1506. doi: 10.1634/stemcells.2005-0479. doi:10.1634/stemcells.2005-0479. [DOI] [PubMed] [Google Scholar]
- 37.Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther. 2006;5(5):1087–1098. doi: 10.1158/1535-7163.MCT-05-0375. doi:10.1158/1535-7163.MCT-05-0375. [DOI] [PubMed] [Google Scholar]
- 38.Arslantas A, Artan S, Oner U, Muslumanoglu MH, Ozdemir M, Durmaz R, et al. Genomic alterations in low-grade, anaplastic astrocytomas and glioblastomas. Pathol Oncol Res. 2007;13(1):39–46. doi: 10.1007/BF02893439. doi:10.1007/BF02893439. [DOI] [PubMed] [Google Scholar]
- 39.Dietschy T, Shevelev I, Stagljar I. The molecular role of the Rothmund-Thomson-, RAPADILINO- and Baller-Gerold-gene product, RECQL4: recent progress. Cell Mol Life Sci. 2007;64(7–8):796–802. doi: 10.1007/s00018-007-6468-5. doi:10.1007/s00018-007-6468-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6(1):e10. doi: 10.1371/journal.pmed.1000010. doi:10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gdynia G, Lehmann-Koch J, Sieber S, Tagscherer KE, Fassl A, Zentgraf H, et al. BLOC1S2 interacts with the HIPPI protein and sensitizes NCH89 glioblastoma cells to apoptosis. Apoptosis. 2008;13(3):437–447. doi: 10.1007/s10495-007-0176-3. doi:10.1007/s10495-007-0176-3. [DOI] [PubMed] [Google Scholar]
- 42.Jendrossek V, Kugler W, Erdlenbruch B, Eibl H, Lakomek M. Induction of differentiation and tetraploidy by long-term treatment of C6 rat glioma cells with erucylphosphocholine. Int J Oncol. 2001;19(4):673–680. doi: 10.3892/ijo.19.4.673. [DOI] [PubMed] [Google Scholar]
- 43.Chang Y, Berenson JR, Wang Z, Deuel TF. Dominant negative pleiotrophin induces tetraploidy and aneuploidy in U87MG human glioblastoma cells. Biochem Biophys Res Commun. 2006;351(2):336–339. doi: 10.1016/j.bbrc.2006.09.148. doi:10.1016/j.bbrc.2006.09.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmittling RJ, Archer GE, Mitchell DA, Heimberger A, Pegram C, Herndon JE, 2nd, et al. Detection of humoral response in patients with glioblastoma receiving EGFRvIII-KLH vaccines. J Immunol Methods. 2008;339(1):74–81. doi: 10.1016/j.jim.2008.08.004. doi:10.1016/j.jim.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Belda-Iniesta C, de Castro Carpeno J, Sereno M, Gonzalez-Baron M, Perona R. Epidermal growth factor receptor and glioblastoma multiforme: molecular basis for a new approach. Clin Transl Oncol. 2008;10(2):73–77. doi: 10.1007/s12094-008-0159-z. doi:10.1007/s12094-008-0159-z. [DOI] [PubMed] [Google Scholar]
- 46.Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci USA. 2009;106(8):2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martini M, Pallini R, Luongo G, Cenci T, Lucantoni C, Larocca LM. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int J Cancer. 2008;123(12):2955–2960. doi: 10.1002/ijc.23805. doi:10.1002/ijc.23805. [DOI] [PubMed] [Google Scholar]
- 48.Kishi Y, Okudaira S, Tanaka M, Hama K, Shida D, Kitayama J, et al. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J Biol Chem. 2006;281(25):17492–17500. doi: 10.1074/jbc.M601803200. doi:10.1074/jbc.M601803200. [DOI] [PubMed] [Google Scholar]
- 49.Hoelzinger DB, Nakada M, Demuth T, Rosensteel T, Reavie LB, Berens ME. Autotaxin: a secreted autocrine/paracrine factor that promotes glioma invasion. J Neurooncol. 2008;86(3):297–309. doi: 10.1007/s11060-007-9480-6. doi:10.1007/s11060-007-9480-6. [DOI] [PubMed] [Google Scholar]