

Abstract
We describe a selective and a highly sensitive high-performance liquid chromatography-electrospray ionization-collision induced dissociation-tandem mass spectrometry (HPLC-ESI-CID-MS/MS) assay for the Aurora A kinase inhibitor MLN8237 in human plasma. The intraday precision based on the standard deviation of replicates of quality control samples ranged from 0.2 to 4% and with accuracy ranging from 96–102%. The interday precision ranged from 0.5 to 7% and the accuracy ranged from 93–105%. Stability studies showed that MLN8237 was stable both during the expected conditions for sample preparation and storage. The lower limit of quantification for MLN8237 was 5 ng/mL. The analytical method showed excellent sensitivity, precision, and accuracy. This method is robust and is being successfully employed in a Children’s Oncology Group Phase 1 Consortium study of MLN8237 in children with cancer.
Keywords: MLN8237, LC-ESI-CID-MS/MS, Plasma, Pediatric, Cancer
1. Introduction
The Aurora family of serine/threonine kinases are key regulators of mitosis and play a critical role in the regulation of chromosomal segregation and cytokinesis during mitotic progression. Over-expression of Aurora A kinase is associated with aneuoploidy and centrosome amplification resulting in transformation of normal cells. Thus, it is thought that Aurora A kinase is involved directly with oncogenesis. [1] Reduction of Aurora A using ribonucleic acid interference (RNAi) decreases Aurora A protein content and results in mitotic spindle defects, mitotic delay, and apoptosis in cancer cell lines. [2]
MLN8237 (sodium 4-{[9-chloro-7-(2-fluoro-6-methoxyphenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]amino}-2-methoxybenzoate) is a selective Aurora A kinase inhibitor that significantly increases event free survival in both neuroblastoma and acute lymphoblastic leukemia (ALL) xenograft models. [3] MLN8237 is currently being studied in pediatric clinical trials for the treatment of relapsed or refractory solid tumors and leukemia.
The objective of this investigation was to develop and validate a simple, selective and sensitive LC-ESI-CID-MS/MS method for the quantification of MLN8237 in human plasma to support an early phase pharmacokinetic study of MLN8237 in pediatric patients. Although previous assays for other Aurora kinase inhibitors have been reported, [4, 5] to our knowledge this is the first method developed for the detection of MLN8237 in human plasma.
2. Experimental
2.1 Reagant and chemicals
Sodium 4-{[9-chloro-7-(2-fluoro-6-methoxyphenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]amino}-2-methoxybenzoate (MLN8237) and MLN8237-D313C15N2 were provided by Millenium Pharmaceuticals (Cambridge, MA, USA). [figure 1] The different lots of drug-free (blank) human plasma were obtained from the blood bank at The Children’s Hospital of Philadelphia. HPLC-grade acetonitrile was purchased from Fisher-Scientific (Pittsburgh, PA, USA) and reagent-grade formic acid (~96%) was purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Deionized water was prepared using a Milli-Q water purifying system from Millipore Corp. (Bedford, MA, USA).
Figure 1.
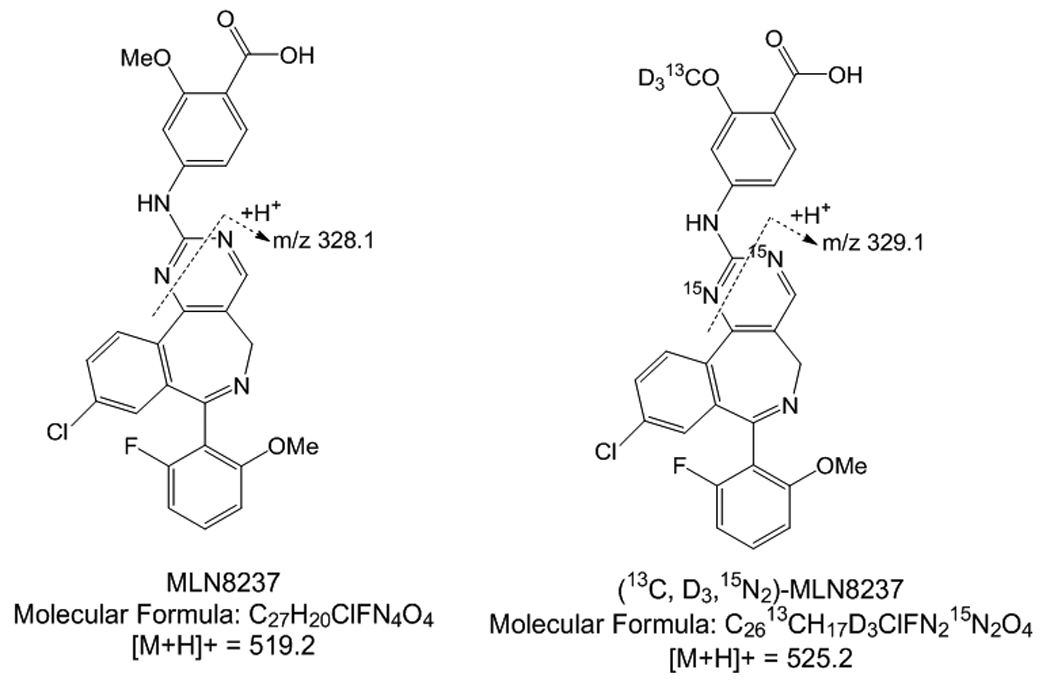
MLN8237 and MLN8237 Internal Standard molecular structure
2.2 Liquid chromatography
The Shimadzu HPLC system consisted of two LC-20AD delivery pumps, a DGU-20A5 Shimadzu vacuum degasser, a SIL-20AC Shimadzu autosampler and a CBM-20A system controller (Shimadzu Scientific Instruments; Columbia, MD, USA). HPLC separations were performed on a Thermo Scientific Hypersil Gold C18 analytical column (50 mm × 2.1 mm, 3.5 μm 120 A). Mobile phase A consisted of water with 0.1% formic acid and mobile phase B consisted of acetonitrile with 0.1% formic acid. The linear gradient was as follows: 0.00–0.25 minutes mobile phase A 80%, mobile phase B 20%; 0.25–2.25 minutes mobile phase A 10%, mobile phase B 90%; 2.26–4 minutes mobile phase A 80%, mobile phase B 20%. The flow rate was 0.25 ml/min and 25 μL of the sample was injected for each analysis. The column and autosampler were maintained at room temperature and 10° C, respectively. An electronic valve actuator with a Rheodyne selector valve was used to divert LC flow to waste, at the first 1 minute, when no data acquisition was taking place.
2.3 Mass spectrometry analysis
Samples were analyzed with an ABI/Sciex 4000 triple quadropole mass spectrometer equipped with Turbo Ionspray. Software for controlling this equipment, acquiring and processing data was Analyst version 1.4.1 software (MDS Sciex; Toronto, Canada). Nitrogen was used as the nebulizer, auxillary, collision and curtain gases. Analytes were detected by low energy CID-MS/MS (positive ion mode) using multiple reaction monitoring (MRM) with a dwell time of 100 ms. For the initial determination of the precursor and product of ion spectra a solution of 1 μg/mL MLN8237 or internal standard in mobile phase was infused directly into the ion sources with a Harvard Apparatus syringe pump at a flow rate of 10 μL/min. The most intense precursor-to-fragment transitions using positive turbo spray were: MLN8237, m/z 519.22/328.10; MLN8237IS-525.24/329.10.
The conditions for ionization of MLN8237 and internal standard were optimized using individual standard solutions, each at 500 ng/mL, which were infused by a syringe pump through a Tee device at a flow rate of 10 μL/min into the stream of mobile phase eluting from the LC column through a mixing Tee and then into the turbo spray source, to mimic the LC-ESI-CID-MS/MS conditions. The main working parameters of the mass spectrometer were: collision activate dissociation (CAD) gas, medium; curtain gas, 35; Gas 1 (nebulizer gas) 50; Gas 2 (heater gas) 50; source temperature 600 °C. The optimized declustering potential (DP), entrance potential (EP), collision energy (CE), collision cell exit potential (CXP) were set at 101, 10, 49, 4 for MLN8237; 111, 10, 45, 8 for MLN8237IS.
2.4 Preparation of standards and quality control (QC) samples
Two independent MLN8237 stock solutions were prepared: standard solutions were prepared from one stock solution and QC samples were prepared from the other. The primary stock solutions of MLN8237 were prepared by dissolving MLN8237 in 1% formic acid in acectonitrile producing a concentration of 1 mg/mL and were stored at −20 °C. The primary stock solution was diluted in acetonitrile to prepare intermediate stock solutions of 100 μg/mL. Working solutions of MLN8237 were freshly prepared by appropriately diluting the respective stock solutions with plasma. Seven standards containing MLN8237 concentrations of 5, 20, 50, 200, 500, 2000 and 5000 ng/mL were prepared by adding the appropriate volumes of working solution into 1.5 mL eppendorf tubes containing plasma. Four QC concentrations were prepared in the same manner by adding appropriate volumes of working solution to obtain concentrations of 8, 40, 400 and 4000 ng/mL, representing low, medium and high QCs. The internal standard stock solution was prepared by dissolving 1 mg MLN8237-D313C15N2 per mL of 0.1% formic acid in acetonitrile and diluting into acetonitrile to a concentration of 10 μg/mL. Internal standard working solution was prepared by diluting the internal standard stock solution with 0.1% formic acid in acetonitrile into a single working solution with a final concentration of 10 ng/mL. Amber glass vials for storing stock solution were used.
2.5 Sample preparation
To 100 μL of human plasma sample, 500 μL of acetonitrile containing 10 ng/mL of internal standard was added in a 96 well plate. The sample was vortex-mixed for 10 minutes and centrifuged at 4,000 rpm for 20 minutes. A volume of 25 μL of the supernatant was injected into the LC-MS/MS system.
2.6 Method validation
Method validation and documentation were performed according to guidelines set by the United States Food and Drug Administration (FDA) for bioanalytical method validation. [6] This method was validated in terms of linearity, specificity, low limit of quantitation (LLOQ), recovery, intra- and inter-day accuracy and precision, and stability of the analyte during sample storage and processing procedures. Each analytical run included a double blank sample (without internal standard), a blank sample (with internal standard), seven standard concentrations for calibration, and replicate sets of QC samples: low (LQC) 8 ng/mL, medium QCs (MQC) 40 ng/mL, 400 ng/mL, and high QC (HQC) 4000 ng/mL.
2.6.1 Linearity and sensitivity
For the evaluation of the linearity of the standard calibration curve, the analyses of MLN8237 in plasma samples were performed on three independent days using fresh preparations. The calibrations curves were prepared over a linear range of 5–5000 ng/ml at seven concentrations of 5, 20, 50, 200, 500, 2000 and 5000 ng/mL. Each calibration curves consisted of a double blank sample, a blank sample, a plasma blank sample and seven calibrator concentrations. Another double blank sample was analyzed immediately following the highest concentration standard in each run to monitor for carry-over of MLN8237 or the internal standard.
The calibration curve was developed using the following criteria: (1) the mean value should be within ±15% of the theoretical value, except at the LLOQ, where it should not deviate by more than ±20%; (2) the precision around the mean value should not exceed a 15% coefficient of variation (CV), except for LLOQ, where it should not exceed a 20% CV. (3) at least 75% of the non-zero standards of each nominal concentration should meet the above criteria; (4) the correlation coefficient (r) should be greater than or equal to 0.98.
Each calibration curve was constructed by plotting the analyte to internal standard peak area ratio (y) against the analyte concentrations (x). The calibration curves were fitted using a least-square linear regression model y=ax+b, weighted by 1/x2 using the Analyst ® software. The resulting a, b and c parameters were used to determine back-calculated concentrations, which were then statistically evaluated.
2.6.2 Specificity
The specificity was defined as non-interference at retention times of MLN8237 from the endogenous plasma components and no cross-interference between MLN8237 and internal standard using the proposed extraction procedure and LC-ESI-CID-MS/MS conditions. Six different lots of blank (MLN8237 free plasma) were evaluated with and without internal standard to assess the specificity of the method.
2.6.3 Accuracy and precision
The intra- and inter-assay precisions were determined using the CV (%), and the intra- and inter-assay accuracies were expressed as the percent difference between the measured concentration and the nominal concentration. The % accuracy of the method was expressed by the formula: % accuracy = (measured concentration)/(nominal concentration) × 100.
Intra-assay precision and accuracy were calculated using replicate (n=5) determinations for each concentration of the spiked plasma sample during a single analytical run. Inter-assay precision and accuracy were calculated using replicate (n=5) determination of each concentration made on three separate days.
2.6.4 Recovery (extraction efficiency) and matrix effect
The extraction efficiency of MLN8237 was determined by analyzing three replicates of MLN8237 plasma samples at four QC concentrations of 8, 40, 400 and 4000 ng/mL, respectively. Recovery was calculated by comparing the peak areas of MLN8237 added into blank plasma and extracted using the protein precipitation procedure with those obtained from MLN8237 spiked directly into post-protein precipitation solvent at the four QC concentrations. The matrix effect was measured by comparing the peak response of the post-extracted spiked sample with those of the pure standards containing equivalent amounts of the MLN8237 prepared in mobile phases.
2.6.5 Stability study
The stability of MLN8237 in human plasma was assessed by analyzing replicates QC samples at concentrations of 8, 40, 400 and 4000 ng/mL, during the sample and storage procedure. For all stability studies, freshly prepared and stability testing QC samples were evaluated by using a freshly prepared standard curve for the measurement. The short term stability was assessed after exposure of the plasma samples to room temperature for 4 hours. The free/thaw stability was determined after three freeze/thaw cycles (room temperature to −20 °C). The concentrations obtained from all stability studies were compared to freshly prepared QC samples, and the percentage concentration deviation was calculated. The analytes were considered stable in human plasma when the concentration difference was less than 15% between the freshly prepared samples and the stability testing samples.
3. Results
3.1 Linearity and sensitivity
The method was validated at the above criteria and found to be linear from the concentrations of 5 to 5000 ng/mL. A representative calibration curve for MLN8237 is shown in Figure 2. The correlation co-efficient from inter-day analysis was found to be greater than 0.98 in all cases. The LLOQ was 5 ng/mL, demonstrating a %CV of less than 5% (precision) and an accuracy of greater than 95%, with a signal-to-noise (S/N) ratio of greater than 10. A representative chromatogram of double blank, LLOQ and the upper limit of quantification samples (ULOQ) and internal standard are shown in Figure 3. No carry over of peaks were observed at the retention time or ion channels of MLN8237 or IS.
Figure 2.
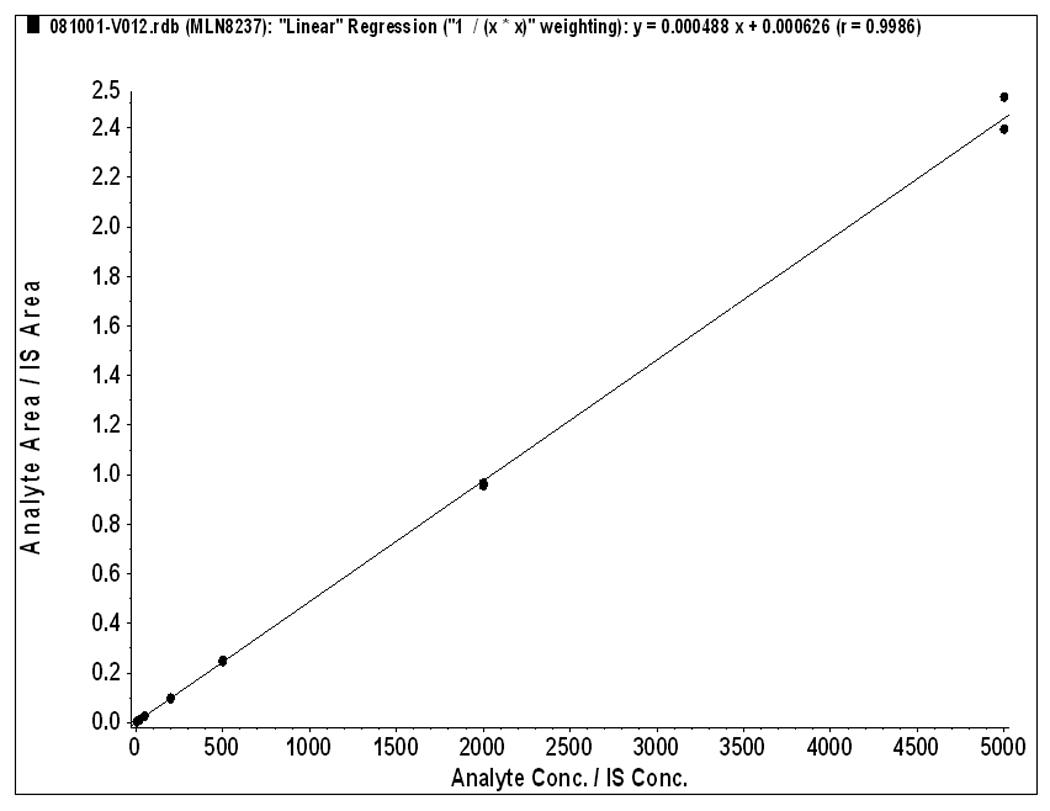
Calibration Curve for MLN8237 in human plasma
Figure 3.
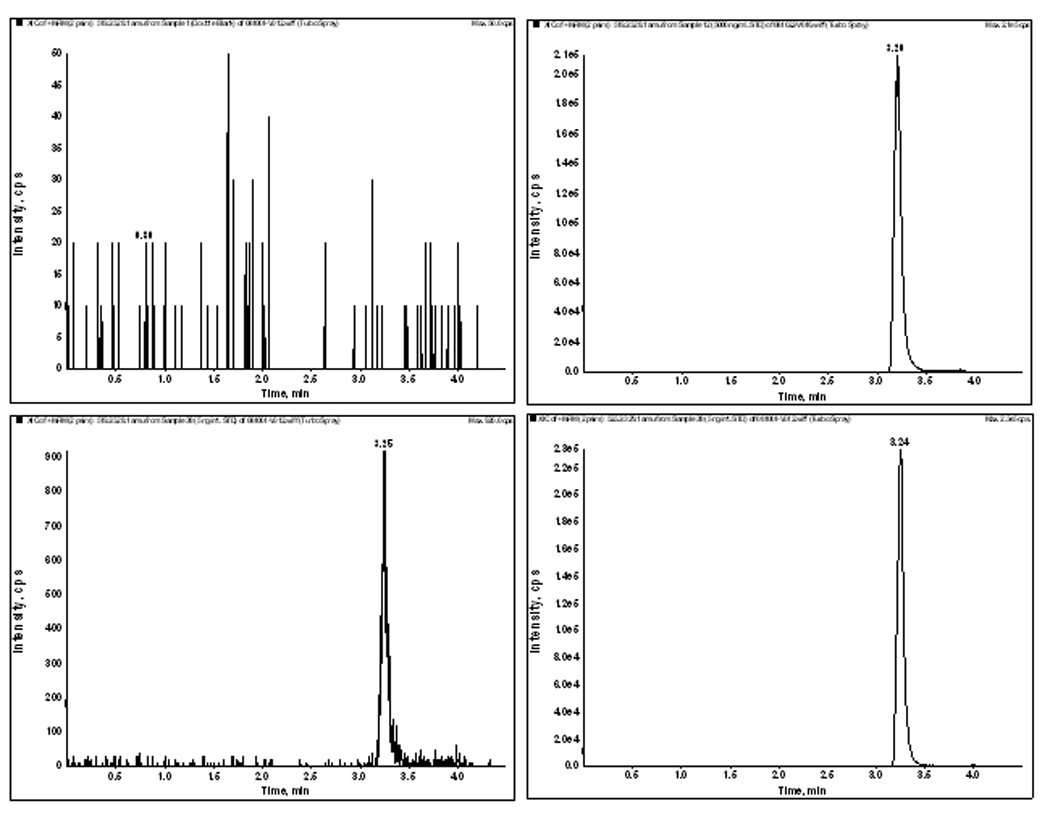
- Double Blank
- LLOQ 5 ng/mL (MRM: 519.22/328.10)
- ULOQ 5000 ng/mL (MRM: 519.22/328.10)
- Internal Standard 5ng/mL (MRM: 525.24/329.10)
3.2. Precision and Accuracy
At the seven calibration standards, the inter-day precision ranged from 0.5–7% and the accuracy ranged from 93–105% (Table 1). All subsequent validation results are depicted in Table 2. The intraday precision ranged from 0.2–4% with the accuracy ranging from 96–102%. The inter-day precision ranged from 1–3.5% with the accuracy ranging from 101–103%. These data confirm that the present method has a satisfactory accuracy, precision and reproducibility for the quantification of MLN8237 throughout a wide dynamic range.
Table 1.
Accuracy and precision of MLN8237 calibration standards in human plasma
| Nominal concentration (ng/mL) |
Mean | CV (%) | Accuracy (%) |
|---|---|---|---|
| 5 | 4.96 | 0.89 | 99.1 |
| 20 | 21.2 | 6.26 | 106 |
| 50 | 46.6 | 6.66 | 93.3 |
| 200 | 201 | 0.77 | 100.7 |
| 500 | 482 | 3.67 | 96.3 |
| 2000 | 2103 | 5.09 | 105.2 |
| 5000 | 4953 | 0.91 | 96.1 |
Table 2.
Summary of validation outcomes for MLN8237 in human plasma
| Intraday (n=5) |
Interday (n=3 days) |
Recovery (n=5) |
Matrix Effect (n=5) |
Stability RT 4 h |
Stability, 3 cycles freeze (−80°C)/thaw |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal conc. (ng/mL) |
Mean | CV (%) |
Accuracy (%) |
Mean | CV (%) |
Accuracy (%) |
Recovery (%) |
SD | CV | Matrix Effect (%) |
SD | CV (%) |
Mean | CV (%) |
Accuracy (%) |
Mean | CV (%) |
Accuracy (%) |
| 8 | 7.7 | 4 | 96 | 8.2 | 3 | 103 | 97.8 | 2.8 | 8.3 | 107 | 21 | 3.8 | 9.6 | 21 | 119 | 8.7 | 9 | 109 |
| 40 | 40 | 0.5 | 99.5 | 41 | 1.8 | 102 | 91.2 | 2.6 | 11 | 81 | 5.8 | 1.9 | 44.7 | 12 | 111 | 45.1 | 13 | 112 |
| 400 | 407 | 2 | 102 | 407 | 3.1 | 102 | 101 | 0.9 | 4.1 | 93 | 1.9 | 5.4 | 449 | 12 | 112 | 468 | 17 | 116 |
| 4000 | 3989 | 0.3 | 99.7 | 4131 | 3.3 | 103 | 101 | 4.1 | 0.2 | 105 | 1.6 | 0.8 | 4336 | 8.4 | 108 | 4661 | 17 | 116 |
3.3. Recovery
At 8, 40, 400 and 4000 ng/mL QC concentrations the percent extraction recoveries (mean ± % SD) after five replicates were 97 ± 27, 91 ± 2.6, 101 ± 0.9, and 100 ± 4, respectively (Table 2). Data indicated that the extraction efficiency for MLN8237 using protein precipitation was satisfactory and was not concentration dependent.
3.4. Assay specificity and ionization suppression (matrix effect)
Matrix effect can affect the reproducibility from the analyte or the internal standard of the assay. The matrix effect, or intensity of ion suppression or enhancement is caused by co-eluting matrix components. The matrix effect of MLN8237 and IS were calculated using the following formula: % matrix effect = (A/B) × 100%. A represents the corresponding peak areas of the analytes in spiked plasma post-precipitation and B represents peak responses of the pure standards prepared in mobile phases. A value of >100% indicated ionization enhancement, and a value of <100% indicated ionization suppression. The matrix effect was tested on the four QC concentrations and three individual lots of blank plasma were evaluated. As shown in Table 2, only minor differences were observed between the pure standards and the post-extracted spiked samples, illustrating that the HPLC separation conditions had little effect by background signal of plasma after simple protein precipitation clean up step.
3.5. Analyte stability
The stability of MLN8237 was investigated to cover expected conditions during sample preparation and storage for all samples, which include stability data from freeze/thaw and bench top, or short-term, stability. The precision for freeze/thaw samples ranged from 9–16% and the accuracy ranged from 109–116%. The results indicated that the analyte was stable in plasma for three cycles when stored at −80 °C and thawed to room temperature. The precision for bench-top stability ranged from 8–20% and the accuracy ranged from 108–119% (table 2). This indicated reliable stability under the experimental conditions of the analytical runs.
4. Discussion
We developed a MLN8237 assay that has a simple and rapid sample preparation and a selective and sensitive LC-ESI-CID-MS/MS method capable of analyzing small volumes of plasma samples, needed for use in pediatric patients. The LC-ESI-CID-MS/MS assay reported here accurately and precisely quantitates MLN8237 concentration in 100 μL plasma specimens with a limit of quantification of 5 ng/mL. The intra-day and inter-day coefficients of variation were less than 4 and 5% respectively at all concentrations tested. With linearity up to 5000 ng/mL, the assay is well suited for use in pediatric pharmacokinetic studies. We currently are utilizing this assay to support a Children’s Oncology Group Phase 1 Consortium study of MLN8237.
Acknowledgments
Supported in part by grants to the Children’s Oncology Group from the National Cancer Institute (U01 CA97452) and Millenium Pharmaceuticals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gautschi O, et al. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14(6):1639–1648. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 2.Oslob JD. Discovery of a potent and selective aurora kinase inhibitor. Bioorg Med Chem Lett. 2008;18(17):4880–4884. doi: 10.1016/j.bmcl.2008.07.073. [DOI] [PubMed] [Google Scholar]
- 3.Maris JM, et al. Initial testing of the aurora kinase a inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatr Blood Cancer. 2010 doi: 10.1002/pbc.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steeghs N, et al. Phase I pharmacokinetic and pharmacodynamic study of the aurora kinase inhibitor danusertib in patients with advanced or metastatic solid tumors. J Clin Oncol. 2009;27(30):5094–5101. doi: 10.1200/JCO.2008.21.6655. [DOI] [PubMed] [Google Scholar]
- 5.Traynor AM, et al. Phase I dose escalation study of MK-0457, a novel Aurora kinase inhibitor, in adult patients with advanced solid tumors. Cancer Chemother Pharmacol. 2010 doi: 10.1007/s00280-010-1318-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.FDA. Guidance for Industry: Bioanalytical Methods Validation. 2001 Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf.