

Abstract
Adult stem cells represent a potential source for cell-based therapy of cancer. The present study evaluated the potential of bone marrow derived mesenchymal stem cells (MSC), genetically modified to express interferon (IFN)-α, for the treatment of lung metastasis in an immunocompetent mouse model of metastatic melanoma. A recombinant adeno-associated virus 6 vector (rAAV) encoding IFN-α was used to transduce mouse bone marrow-derived MSC ex vivo. Expression and bioactivity of the transgenic protein from rAAV-transduced MSC were confirmed prior to in vivo studies. A lung metastasis model of melanoma was developed by intravenous injection of B16F10 cells into 8-week-old C57BL/6 mice. Ten days later, MSC, transduced with rAAV-IFN-α or GFP were intravenously injected. One cohort of mice was sacrificed to determine the effects of the therapy at an earlier time point and another cohort was observed for long-term survival. Results indicated that systemic administration of MSC producing IFN-α reduced the growth of B16F10 melanoma cells and significantly prolonged survival. Immunohistochemistry analysis of the tumors from MSC-IFN-α treated animals indicated an increase in apoptosis and decrease in proliferation and blood vasculature. These data demonstrate the potential of adult MSC, constitutively producing IFN-α, to reduce the growth of lung metastasis in melanoma.
INTRODUCTION
Adult stem cells are currently being studied for tissue regeneration. One of the adult stem cells with proven plasticity is mesenchymal stem cell (MSC) of bone marrow origin. Originally known to differentiate into cells and tissues of mesodermal origin, MSC have been shown to differentiate into cells of ectodermal, endodermal and mesodermal origin under appropriate stimuli (1-3). In addition to their transdifferentiating nature, MSC constitute an ideal source for cell therapy. Some of the advantages of MSC as cell therapy vehicle are ease of isolation and ex vivo expansion, multi-lineage differentiation, lack of immune response and their ability to serve as cellular vehicles for the delivery of therapeutic proteins (4-7). Tumor microenvironment is known to provide preferential niche for MSC homing and survival (4). Primary and malignant tumor microenvironment is composed of tumor and normal cells including blood vessels, inflammatory cells and stromal fibroblasts. While stromal cells provide structural support for malignant cells and influence the growth and aggressiveness of tumors, the interaction resembles wound healing, which promotes engraftment of MSC to this region as in sites of injury (8-10). This provides an excellent opportunity to utilize MSC as therapeutic vehicles for the delivery of growth factors and cytokines for anti-tumor effects.
One of the cytokines with pleiotrophic anti-tumor property is type I interferon (IFN). Type I interferons (α and β) display a multitude of anti-tumor effects, including inhibition of cell proliferation, restriction of tumor angiogenesis, induction of apoptosis and activation of host defense against tumors (11,12). Previous studies have tested the potential of IFN-α as purified protein or using gene transfer approaches with viral and non-viral vectors in tumor models including melanoma (13-16).
Metastasis is the leading cause of mortality in melanoma as it is in most malignancies (17,18). Melanoma shows preferential metastasis to the brain, lung, liver, and skin (19,20). Cytokine-mediated tumor immunotherapy or tumor vaccination remains one of the most promising strategies for cancer gene therapy. To determine the potential of IFN-α-producing MSC in a therapy model of metastatic melanoma, the present study evaluated the anti-tumor activity of mouse MSC, transduced with a recombinant adeno-associated virus (rAAV) 6 expressing the murine IFN-α in a mouse melanoma lung metastasis model. Results indicated that IFN-α therapy using genetically modified MSC reduced the growth of B16F10 melanoma cells in the lungs and significantly prolonged survival. Collective evidence indicated that tumor apoptosis, inhibition of tumor cell proliferation and tumor vasculature as mechanisms responsible for the anti-tumor effects. These data demonstrate that rAAV-IFN-α transduced MSC therapy can be used to reduce the growth of metastatic melanoma.
MATERILAS AND METHODS
Cell lines
The human embryonal kidney cell line HEK293 and the mouse melanoma cell line B16F10 were purchased from the American Type Culture Collection (Manassas, VA). The HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml) and the B16F10 cells were maintained in DMEM with 4 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10%; fetal bovine serum and penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in a CO2 atmosphere.
Vector construction, packaging and purification
The open reading frame of mouse IFN-α was amplified by PCR from the plasmid pIFN-α1 (kind gift of Dr. Taniguchi, Japanese Foundation for Cancer Research, Japan) and cloned in a recombinant AAV plasmid pAAV-CA containing the CMV/chicken beta actin promoter. The amplified cDNA was verified by sequencing. Packaging of rAAV-IFN-α was performed by transient transfection in 293 cells with the helper plasmid pDP6, as described (21,22). Purification of rAAV was done by iodixanol density-gradient and heparin affinity chromatography as described (22). Particle titers of rAAV were determined by quantitative slot blot analysis and real-time PCR (23).
Western blot
rAAV-IFN-α was transduced at a multiplicity of infection (MOI) of 1000 and plasmid vector expressing IFN-α was transfected by lipofectamine 2000 to MSC in culture. After 48 hrs, mock-transduced and vector-transduced cells were lysed in lysis buffer (10 mM Tris-Cl, pH 7.4, 0.15 M NaCl, 5 mM EDTA, and 1% Triton X-100) containing 100 μg/ml protease inhibitors (Sigma-Aldrich; St. Louis, MO). Protein content of the lysates was determined using Bio-Rad Dc protein assay kit. Ten microgram of protein from each sample was separated in 10% SDS-PAGE and transblotted onto polyvinylidene fluoride (PVDF) membrane. The membranes were blocked in blocking buffer (5% powdered nonfat milk in PBS) containing 0.05% Tween-20. The blot was probed with a polyclonal rabbit anti-mouse IFN-α antibody at 1:500 dilution (PBL Biomedical Laboratories) followed by binding with an anti-rabbit secondary antibody at 1:1000 dilution (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Interferon-α protein band was detected with an enhanced chemiluminescence kit (ECL; Amersham Biosciences/GE Healthcare, Little Chalfont, UK).
Mouse MSC culture and vector transduction
C57BL/6 mice were purchased from the National Cancer Institute-Frederick Cancer Research Facility (Frederick, MD, USA). All animal protocols were performed following the guidelines of the Institutional Animal Care and Use Committee. To obtain bone marrow stromal cells, 6- to 8-week-old mice were sacrificed, bone marrow was flushed from the femur and tibia, and the marrow mononuclear cells were purified by ficoll gradient. Adherent bone marrow stromal cells were grown in α-MEM containing 10% FBS and supplemented with 10−9 M fibroblast growth factor (FGF)-2 to maintain the cells in pluripotent and undifferentiated state (16-18). Periodically, floating cells were removed and fresh medium replenished. Residual macrophages from the MSC culture were removed by IMAC using anti-mouse CD11b beads (BD Biosciences, San Diego, CA). After 14 days, the adherent stromal cells were split before attaining confluence to avoid possible onset of differentiation. The cells were routinely prepared and used for in vitro and in vivo studies as low passage cultures (passages 4–8). Undifferentiated MSC were transduced with 1000 MOI of rAAV6-IFN-α or rAAV6-GFP. Before transduction, the growth medium was removed and the cells washed once with serum-free Opti-MEM (Gibco-BRL, Gaithersburg, MD, USA). Virus infection was performed in 125 μl of Opti-MEM for 2 h at 37°C after which complete medium with FGF2 was added. One week later, cells were harvested by tryspinization and resuspended in PBS for injection. For transfection of MSC, cells were mock-transfected or transfected with a plasmid expressing IFN-α using LipofectAmin-2000 reagent (Invitrogen) as recommended by the manufacturer.
Cell proliferation assay
B16F10 cells were seeded in 96-well plates (Falcon 3072, Becton-Dickinson, Lincoln Park, NJ) at a density of 7.5×103 cells per well and incubated at 37°C for 24 hours. Supernatants from mock-transduced and rAAV6-IFN-α transduced MSC were added to the plate, and returned to the incubator for 3 days. MTT dye [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was added to each well for the last 4 hrs of treatment. The reaction was stopped with the addition of DMSO, and the optical density was determined at 570 nm in a multi-well plate reader (Bio-Rad, Model 3550, Mississauga, Canada). Background absorbance of the medium in the absence of cells was subtracted. All samples were assayed in triplicate, and the mean for each experiment was calculated. Results were expressed as a percentage of the control, which was considered as 100%.
In vivo studies
Eight-week-old female C57BL/6 mice were purchased from the National Cancer Institute-Frederick Cancer Research Facility (Frederick, MD). Handling of the animals was done in accordance with the guidelines of the Institutional Animal Care and Use Committee. Each group consisted of 10 mice and the experiment was repeated twice. For establishing lung metastasis, 5×104 B16F10 cells were injected by tail vein into each mouse. Ten days later, when tumors were established in the lungs, 5×105 MSC that were unmodified or transduced with rAAV6-IFN-α or rAAV6-GFP, or transfected with a plasmid expressing IFN-α were given by i.v. injection through tail vein in a volume of 200 μl. The MSC injections were given two times.
Cohorts of mice were sacrificed at different time points, tissues were harvested and 5-μm thick paraffin sections were made. Tissue sections were dewaxed in xylene, and then rehydrated in graded-alcohol. Endogenous peroxidase activity was blocked with 0·3% (v/v) hydrogen peroxide in methanol before washing the slides in water. Slides were incubated in citrate buffer (pH-6.0) for 20 minutes in a steamer for antigen retrieval for Ki67, anti-PARP-p85 and IFN-α. For Factor-VIII-related antigen, pepsin antigen retrieval was performed by incubating slides in 0.5 mg/ml pepsin A in pH 2.0 HCl (0.01N) for 15 minutes at 37 °C. Non-specific binding was blocked by incubating the slides in 5% (v/v) normal serum (depending on the animal in which the secondary antibody was raised). The primary antibodies were used in dilutions: anti-Ki67, 1:50 dilution (Abcam, Cambridge, MA); anti-PARP p85 fragment polyclonal antibody, 1:100 dilution (Promega Corp. Madison, WI); antibody for Factor-VIII related antigen, 1:40 dilution (Ventana Medical Systems, Inc. Tucson, AZ), and IFN-α antibody, 1:500 dilution (PBL Biomedical Laboratories, Piscataway, NJ). Following binding with primary antibodies, slides were incubated with respective secondary antibodies conjugated to horseradish peroxidase. The bound antibody was visualized using the peroxidase-based Vectastain Elite ABC Kit (Vector Laboratories Ltd, Peterborough, UK). The substrate reaction was stopped by washing the slides in running water. Finally, the slides were lightly counterstained in haematoxylin. To determine the proliferation and apoptotic indices, stained slides were examined under high power (x40). A minimum of 10 randomly chosen fields were counted to determine the total number of cells and that stained positive in each field. The percentage proliferation and apoptosis was calculated using the formula: (number of positively stained cells/total number of cells in a field) x 100. To determine microvessel density, the number of vessels was determined by counting 10 random fields of the highest vascular density in high-magnification (x40).
ELISA
Blood was collected from mice at different time points and sera were stored frozen in −80°C until use. To quantitate the amount of IFN-α protein, sera were used in triplicate in an ELISA, specific for mouse IFN-α (PBL Biomedical Laboratories; Piscataway, N.J.) according to the manufacturer’s protocol and analyzed at an absorbance of 450 nm. The minimum sensitivity of detection of the assay was 12.5 pg/ml.
Determination of MSC homing to lung
MSC homing to the tumor tissue in the lung in vivo was determined by using both labeling the cells with the fluorescent dye CM-DiI (Molecular Probes, Eugene, OR) and using MSC obtained from syngeneic, GFP transgeneic mice [C57BL/6-Tg(ACTbEGF)1Osb/J], prior to in vivo administration. In labeling studies, the dye was reconstituted at a concentration of 1 μg/μl in dimethyl sulfoxide (DMSO). Cultured MSC, transduced with rAAV-IFNα, were trypsinized, washed with PBS, and resuspended at a concentration of 106 cells/2 μg CM-DiI dye in 1 mL Dulbecco’s PBS. Cells were labeled by incubation at 37°C for 5 minutes followed by 15 minutes at 4°C in the dark. Unincorporated dye was then removed by centrifugation at 300 g for 5 minutes followed by 2 washes in PBS. Cells were resuspended in PBS and injected by tail vein into mice bearing B16F10 tumors in the lung. Twenty days later, mice were sacrificed and 5-μm thick paraffin sections were made of lungs. Visualization of labeled MSC was based on fluorescence of CM-Dil dye (Red) and expression of IFN-α was determined by using Alexa-Fluor 488 conjugated secondary antibody (Green) following incubation with the IFN-α primary antibody. Homing of MSC to the lungs using cells from GFP transgenic mice was detected by using a rabbit polyclone GFP antibody (Abcam, Cambridge, MA) at 1:500 dilution followed by binding with an Alexa-Fluor 594-conjugated donkey anti-rabbit secondary antibody at 1:1000 dilution. The stained slides were analyzed in a Leica DM 2RB fluorescence microscope. Quantitation of MSC homing in the tumor in lung was determined by counting 10 random fields of in high-power for both tumor cells and MSC (x40).
Analysis of immune effectors
Lungs of mice from different groups were disrupted using wire mesh screens, and erythrocytes were lysed by treatment with lysis buffered (8.29 g NH4Cl, 1.0 g KHCO3, and 0.037 g EDTA/liter). Mononuclear cells were centrifuged at 1500 rpm for 5 min. The cells were washed in PBS containing 2% (w/v) BSA and 0.2% (w/v) NaN3 and stained with FITC-conjugated anti-CD4 PE and anti-CD8 APC antibodies (BD PharMingen, San Diego, CA). The stained cells were subjected to FACS analysis to enumerate the percentage of positive cells. For infiltration of CD4+, CD8+ T cells and NK1.1 cells, frozen sections of the lung were acetone-fixed and blocked with 10% normal rabbit serum in PBS. Sections were then incubated with rat monoclonal antibody specific for mouse CD4, CD8 or NK1.1 (BD PharMingen, San Diego, CA), then incubated sequentially with a biotin-SP-conjugated goat anti-rat IgG secondary antibody (Jackson ImmunoResearch Laboratories, INC. West Grove, PA ) and horseradish peroxidase-conjugated avidin-biotin complex (Jackson ImmunoResearch Laboratories, INC. West Grove, PA ). Sections were developed with the peroxidase-based Vectastain Elite ABC Kit (Vector Laboratories Ltd, Peterborough, UK). The substrate reaction was stopped by washing the slides in running water. Finally, the slides were lightly counterstained in hematoxylin.
Statistical analysis
Data were compiled as mean ± standard error in quantitative experiments. For statistical analysis of differences between the groups, an unpaired Student’s t test was performed. P values <0.05 were considered to indicate significant difference between data sets.
RESULTS
High-level expression and bioactivity of rAAV encoding IFN-α1
The open read frame of murine IFN-α was amplified by PCR and cloned in a recombinant-AAV vector under CMV/chicken-β actin promoter (Figure 1A). High-level expression of the transgenic protein was confirmed by western blot analysis. MSC were mock-transduced or transduced with rAAV-IFNα1 or plasmid expressing IFNα and supernatants were harvested after 48 hours for western blot. Results, shown in Figure 1B indicating a strong band of 20 kD in rAAV-IFN-α transduction but not in the mock-transduced control established high-level expression of the transgenic protein.
Figure 1. Recombinant-AAV 6 expressing murine IFN-α.

An rAAV encoding human IFN-α under the control of CMV-chicken beta actin promoter (CBAp) was constructed using pSub201 vector (A). Expression of IFN-α in MSC was analyzed from the culture supernatant by Western blot following transduction with rAAV-IFN-α or plasmid expressing IFN-α (B).
To determine the bioactivity of rAAV produced IFN-α, transgene protein, MSC were either mock-infected or infected with rAAV-IFN-α at 1000 MOI. One week later, supernatants were harvested and concentrated. Interferon-α concentration was determined by ELISA and found to be 100 pg/ml of the supernatant, which amounts to approximately 5×10−4 pg/cell. For effects on cell proliferation, an MTT assay was performed in B16F10 cells. Results, shown in Figure 2 indicated a significant inhibition of tumor growth with 100 μl of supernatant from rAAV6-IFN-α transduced MSC, which was comparable to the effects observed with purified protein (p<0.001).
Figure 2. Effects of rAAV produced IFN-α on cell proliferation.
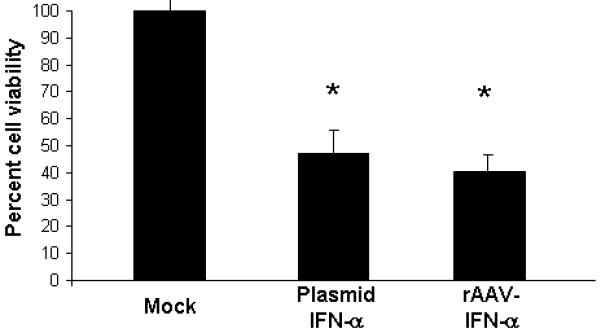
The bioactivity of rAAV produced IFN-α was determined in vitro in B16F10 cells by incubating the cells in conditioned media obtained from MSC cultures that were either mock-transduced or transduced with rAAV encoding IFN-α or transfected with a plasmid vector encoding IFN-α. The mean indicates average value from three independent experiments (*p<0.001 compared to mock).
rAAV-transduced MSC producing IFN-α significantly reduced the growth of B16F10 melanoma cells and prolonged survival
Groups of C57BL/6 mice (20 mice/group) were injected with 5×104 B16F10 cells by tail vain. Ten days later, mice were given no treatment or MSC (5×105) that was transduced with rAAV/IFN-α or rAAV/GFP. Cohorts of mice (n=10) from each group were sacrificed at day-20 to determine the effect of therapy and remaining mice (n=10) were monitored for long-term survival. Following treatment with MSC expressing IFN-α, there were significantly less metastatic colones in the lungs (p<0.001). This effect was found to be specific to the cytokine and not because of vector transduction or MSC since there was no significant therapeutic effect observed in mice treated with MSC transduced with rAAV expressing GFP (Figure 3A). MSC producing IFN-α was found to significantly increase the survival of mice. Whereas all animals in control group died by day 35, more than 70% of mice treated with MSC-IFNα survived beyond that time(p<0.01). Survival of 20% mice in this group was seen up to 80 days. There was no significant increase in survival of mice treated with MSC transduced with rAAV-GFP or MSC transfected with plasmid encoding IFN-α (Figure 3B).
Figure 3. In vivo experiment.
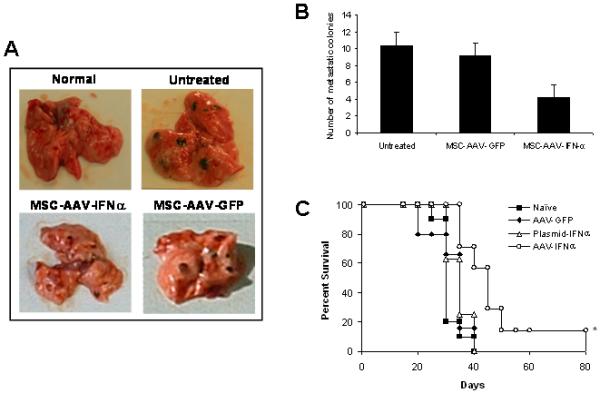
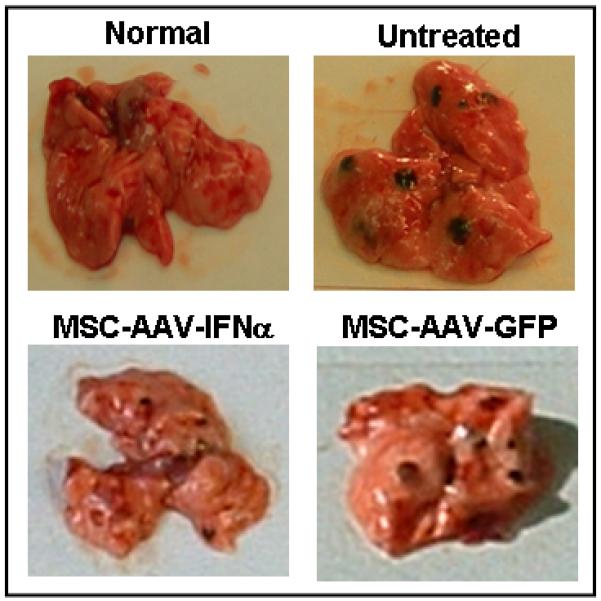
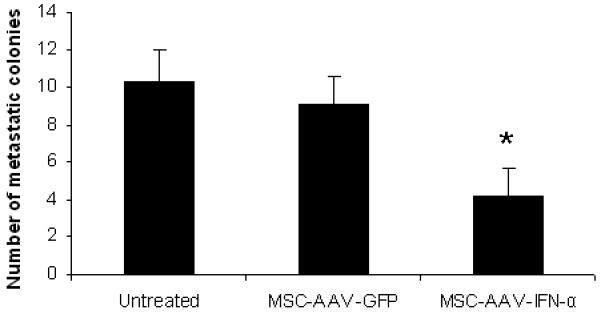
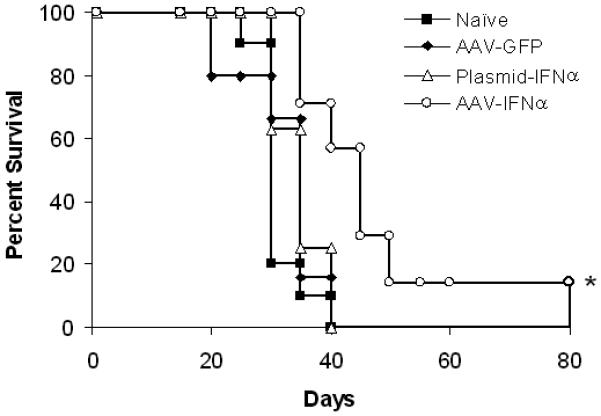
Cohorts of C57BL/6 mice were injected with 5×104 B16F10 cells by tail vain. Ten days later, mice were given no treatment or MSC (5×105) that was transduced with rAAV-IFN-α or GFP, or transfected with a plasmid encoding IFN-α. Cohorts of mice from each group (n=10) were sacrificed at day 20 to determine the effect of therapy and remaining mice (n=10) were allowed for long-term survival. The in vivo experiment was repeated twice. Images represent lungs from indicated treatment group on day 20 (Figure 3A) and the number of metastatic colonies (*p<0.001; Figure 3B). Survival index is given in Figure 3C (*p<0.01).
MSC homing to tumors in the lung and expressing IFN-α
In order to track homing of transplanted MSC to tumors in the lungs, rAAV-IFN-α transduced MSC were labeled using the cell tracker dye CM-DiI (Red) or isolated MSC form GFP transgenic mice. Labeled MSC or GFP positive MSC were intravenously administered to mice bearing B16F10 tumors in the lungs. Twenty days after the administration of both MSC, the animals were sacrificed and tissues harvested. Microscopic and immunohistochemistry analysis of lung sections provided evidence for homing of MSC to the tumor site in the lungs as shown in Figure 4A. Expression of IFN-α in MSC that homed to lungs was also confirmed by staining with an IFN-α antibody. The quantification data showed approximately 3.6 labeled-MSC and 3.2 GFP-positive MSC per 1000 tumor cells in the lung (Figure 4C).
Figure 4. Immunofluorescence analysis of MSC homing to tumors in the lung, and systemic IFN-α level following therapy.
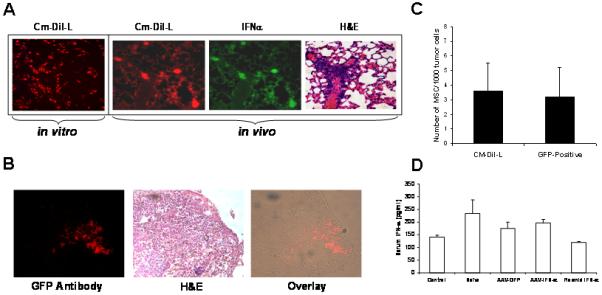


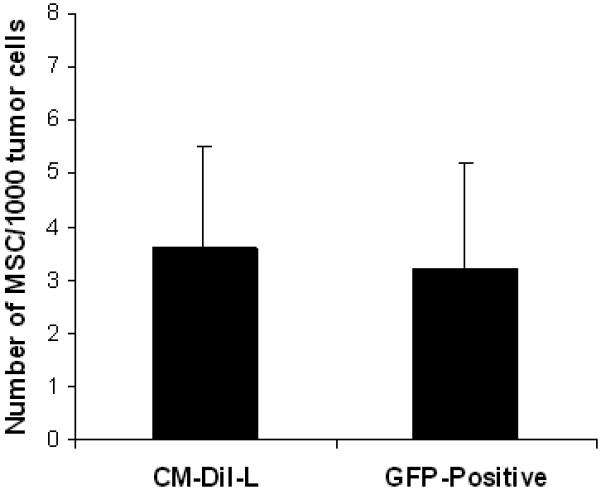
MSC were labeled with the cell tracker dye CM-DiI (Red) prior to in vivo administration. C57BL/6 mice bearing B16F10 tumors (n=3) in the lungs were injected with 5×105 MSC transduced with rAAV-IFN-α and labeled with CM-DiI. Twenty days later, mice were sacrificed and lungs harvested, and paraffin sections made. The sections were stained with IFN-α antibody (green). Tumor-homed MSC were identified by CM-DiI staining (Red) (Figure 4A). Similarly, 5×105 MSC, isolated from GFP transgenic mice were systemicall injected into C57BL/6 mice bearing B16F10 tumors in the lungs (n=3). Twenty days later, mice were sacrificed and lungs harvested, and paraffin sections made. MSC homing to tumor site was confirmed by staining with anti-GFP antibody followed fluorescence secondary antibody (Red). H&E staining indicates areas of the tumor in the lungs (Figure 4B) Quantitation of the number of MSC to tumor cells is given in Figure 4C. An ELISA for IFN-α levels was performed in triplicate with pooled serum samples from mice in different groups, obtained on day-20 following MSC administration (Figure 4D).
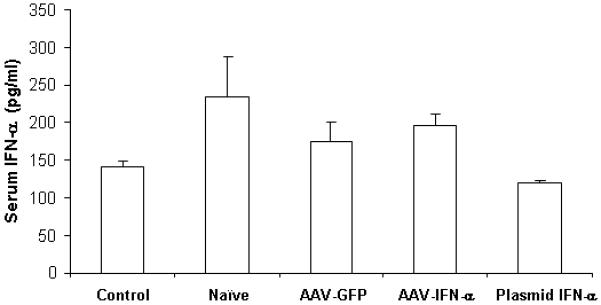
Genetically modified MSC therapy does not increase systemic levels of IFN-α
To determine if IFN-α expressing MSC produced systemically higher concentrations of the cytokine, an ELISA was performed using serum samples collected at different times following therapy. Results indicated no significant difference in systemic interferon-α level after MSC therapy among different groups at different time points (Figure 4B).
Effects of IFN-α producing MSC on tumor cell proliferation, apoptosis and neovasculature
Results of immunohistochemistry and the quantitative analysis indicated a significant decrease in cell proliferation (p<0.0002) and tumor vasculature (p<0.0001) and an increase in apoptosis (p<0.0005) following treatment with MSC producing IFN-α compared to untreated animals. No such difference was seen in tumors of mice treated with MSC expressing GFP suggesting once again that the effects were specific to IFN-α (Figure 5A-C).
Figure 5. Immunochemistry of lungs with B16F10 tumors for apoptosis, cell proliferation and neovasculature.
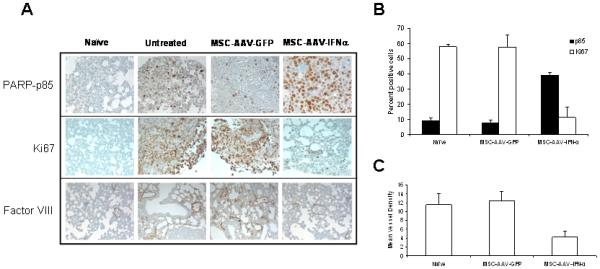
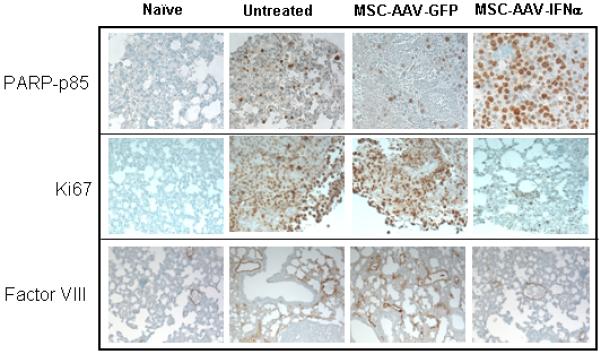
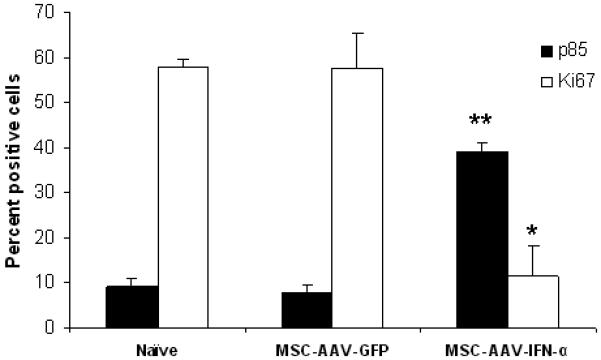
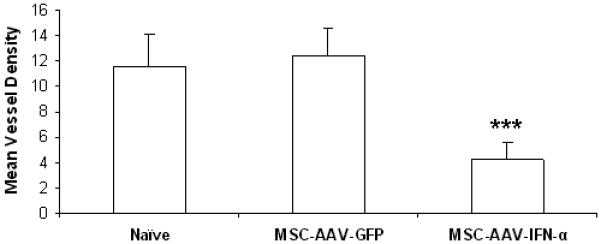
For immunochemical analysis of tumor cell proliferation, apoptosis and microvessel density, lungs were paraffin-embedded and sectioned. Staining was performed with anti-PARP-p85 fragment polyclonal antibody for apoptosis, anti-Ki67 antibody for proliferation, and Factor-VIII antibody for endothelial cells. Slides were minimally counterstained with haematoxylin (Figure 5A). Quantitative analysis of proliferation and apoptotic indices were calculated by counting positive cells in 10 random fields at x40 (*p<0.0002; **p<0.0005; Figure 5B). Blood vessel counts were determined by counting the number of vessels in 10 randomly chosen areas of Factor-III stained sections in x40 (***p<0.0001; Figure 5C).
Anti-tumor effects of MSC transfected with a plasmid vector expressing IFN-α
We have also compared in parallel if MSC transfected with a plasmid vector expressing IFN-α would have similar or better anti-tumor effects. Results of the MTT proliferation assay indicated a comparable growth inhibition to that of rAAV transduced MSC (Figure 2). However, results of the in vivo study showed that treatment with MSC, transfected with the plasmid expressing IFN-α, provided only modest increase in survival compared to mice in the rAAV-IFN-α group (p>0.05; Figure 3).
Analysis of immune effectors
We have examined the infiltration of CD8+ and CD4+ T and NK1.1 cells in the lung after injection of MSCs-IFN-α, MSCs-GFP, MSC-plasmid-α, compared with tumor only and normal control by immunohistochemistry (figure 6A). Results showed no significant differences between different groups in these three cell populations. Results of immunohistochemistry also corroborated analysis of immune effectors by flow cytometry. There was no significant difference in CD8 or CD4 T cell population between IFN-α treated and control groups (p>0.05; Figure 6B).
Figure 6. Determination of immune effectors in the lung following IFN-α therapy.
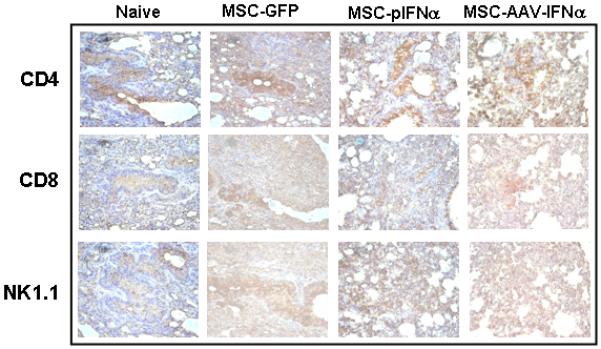
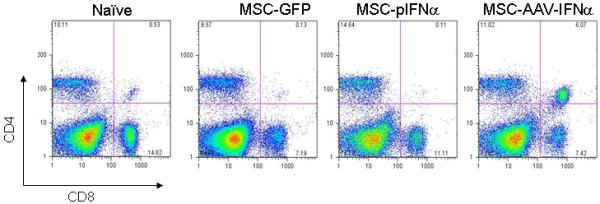
MSC, transduced with rAAV-IFN-α or rAAV-GFP or transfected with a plasmid encoding IFN-α were injected intravenously into C57BL/6 mice with lung metastases of B16F10 cells (n=3 in each group). Twenty days later, lung was harvested; frozen sections were made from half of the tissue for immunohistochemistry. Staining was performed with rat monoclonal antibody specific for mouse CD4, CD8 or NK1.1. Slides were minimally counterstained with haematoxylin (Figure 6A). Mononuclear cells from tumor tissue were isolated and stained with FITC-conjugated anti-CD4 and PE-conjugated anti-CD8 antibodies and analyzed by flow cytometry (Figure 6B).
DISCUSSION
The choice of IFN-α over other cytokines for anti-tumor effects on melanoma cells is based on its pleiotrophic effect that includes enhancing antigen presentation by dendritic cells, upregulation of co-stimulatory molecules, promoting differentiation of lymphoid cells and stimulating the activation of effector cells including cytotoxic macrophages, NK cells, LAK cells, and CTLs, induction of apoptosis, and affecting endothelial cells by inhibiting angiogenesis (24). Studies have shown that allogeneic MSC can be detected after intravenous infusion in cartilage, lung, bone marrow and spleen for up to 6 months. Moreover, autologous ex vivo expanded MSC have been infused into patients without adverse effects (25). Several phase I studies with allogeneic MSC revealed no specific cellular immune response against the transplanted cells, explaining prolonged survival of MSC in patients (26-28). With the intent of using MSC as the delivery vehicles for therapeutic transgenes, several studies have reported efficient ex vivo transduction of MSC with different viral vectors such as adenoviral and retroviral vectors (29-32). Moreover, treatment of human melanoma with human MSC expressing IFN-β in a xenogeneic mouse model led to a pronounced tumor reduction (4).
Whereas systemically higher amounts of IFN-α may prove useful in immunomodulatory effects, other properties including induction of apoptosis and anti-angiogenesis may have undesired consequences in tissues other than tumor cells. Several side effects of high-dose IFN-α therapy have been reported in preclinical studies and clinical trials including neuropsychiatric, hematologic and hepatic nature (24,33,34). Most of these studies have been conducted with high doses of purified IFN-α, which resulted in such adverse effects. In contrast, the use of MSC to express IFN-α specifically at the sites of tumor growth without significantly elevated amounts in systemic circulation would greatly decrease the undesired side effects of the cytokine. Results of the present study, indicating absence of an increase in systemic IFN-α levels yet promoting anti-tumor activity through induction of apoptosis and anti-angiogenesis suggests the potential of utilizing MSC as cellular vehicles. Despite a significant reduction in the tumor growth and increase in survival index, mice eventually succumbed to tumor indicating that this therapy may not be sufficient as a primary or stand-alone therapy. Considering the complex nature of tumor growth and refractoriness of tumors for any given therapy, it remains possible that certain population of tumor cells may develop resistance to IFN-α. Thus, a combined approach with other therapies would augment the effects of MSC-based cell therapy using IFN-α. Studies have reported the adjuvant effects of IFN-α to endostatin, thalidomide and radiation therapies (35-37). One of the promising new therapies for melanoma is immunotherapy using melanoma-specific antigens. Clinical immunotherapy studies on melanoma patients have indicated mixed outcome. While some patients have shown remarkable reduction of the tumor and evidence of robust melanoma-specific CTL response, others have shown only modest responses (38). However, in the present study, the systematic levels of IFN-α did not show a significant increase following the MSC-AAV-IFN-α therapy despite significant anti-tumor effects suggesting that that local effects of MSC-produced IFN-α in the tumor microenvironment. Further, absence of of a significant infiltration of CD8+ T cells in the tumor suggests that the anti-tumor effect was more by induction of tumor cells and tumor endothelial cell apoptosis. It remains to be seen whether the absence of an increase in CD8+ T cells is due to the absence of high systemic levels of IFN-α observed in the studies and whether this will also correlate to dendritic cell function in the draining lymph nodes. It remains possible that by increasing the amount of IFN-α production from the cell-based therapy, improved anti-tumor effects may be achieved including activation of the host immune system and dendritic cell function. However, caution must be exercised while using high numbers of MSC while delivering them by systemic route. A major concern in tumor immunotherapy that limits the potential of this approach is inefficient immune effectors produced by the tumor vaccines. Thus, the strategy described here to deliver IFN-α using MSC at the tumor site should overcome limitations of immunotherapy as an adjuvant therapy. When the B16 melanoma cells were tested for SDF-1 and HGF levels by RT-PCR, results indicated that neither SDF-1 nor HGF was overexpressed (data not shown). Previous studies by adenoviral gene transfer in B16 melanoma cells have used the vector to overexpress SDF-1 to achieve dendritic cell function (41). Thus, it may be unlikely to use CXCR4 and c-met antagonists to inhibit chemotaxis of MSC to the lungs.
A point of consideration of this therapy is the longevity of MSC in tumor microenvironment and their lineage differentiation. An advantage of using MSC for ex vivo therapy is their immunoprivileged nature. Although MSC express MHC class I, the absence of CD40, CD80 and CD86, required for providing co-stimulatory signals necessary for antigen presentation make them attractive for repeat administration. Further, because rAAV do not encode any vector structural proteins and in ex vivo strategy the capsid proteins are degraded much before in vivo infusion, genetically modified MSC, using rAAV, will have great utility in multiple applications if needed. Prior to clinical translation of this approach, it will be important to determine whether constitutive expression of IFN-α in MSC would induce lineage differentiation. Overall, the present study indicates the potential of genetically modified MSC expressing IFN-α for metastatic melanoma.
ACKNOWLEDGEMENTS
Financial support from the National Institutes of Health grants R01CA98817, R01AR50251 and the U.S. Army Department of Defense grants BC044440 and PC050949 is gratefully acknowledged.
REFERENCES
- 1.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 2.Friedenstein AJP, Petrokova KV. Osteogenesis in transplants of bone marrow cells. Journal of Embryological Experimental Morphology. 1966;16:381–390. [PubMed] [Google Scholar]
- 3.Barry FP, Murphy JM. Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004;36:568–84. doi: 10.1016/j.biocel.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Studeny M, Marini Frank C, Champlin RE, et al. Bone Marrow-derived Mesenchymal Stem Cells as Vehicles for Interferon-β; Delivery into Tumors. Cancer Research. 2002;62:3603–08. [PubMed] [Google Scholar]
- 5.Kassem M, Kristiansen M, Abdallah BM. Mesenchymal stem cells: cell biology and potential use in therapy. Basic and Clinical Pharmacology and Toxicology. 2004;95:209–214. doi: 10.1111/j.1742-7843.2004.pto950502.x. [DOI] [PubMed] [Google Scholar]
- 6.Pereboeva L, Curiel DT. Cellular vehicles for cancer gene therapy: current status and future potential. BioDrugs. 2004;18:361–385. doi: 10.2165/00063030-200418060-00003. [DOI] [PubMed] [Google Scholar]
- 7.Nakamizo A, Marini F, Amano T, et al. Human bone marrow–derived mesenchymal stem cells in the treatment of gliomas. Cancer Research. 2005;65:3307–3318. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 8.Studeny M, Marini FC, Dembinski JL, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. Journal of the National Cancer Institute. 2004;96:1593–603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 9.Bissell MJ, Radisky D. Putting tumors in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ben-Baruch A. Host microenvironment in breast cancer development: inflammatory cells, cytokines and chemokines in breast cancer progression: reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003;5:31–6. doi: 10.1186/bcr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annu Rev Biochem. 1987;56:727–77. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 12.Borden EC. Interferons: pleiotropic cellular modulators. Clin Immunol Immunopathol. 1992;62:S18–24. doi: 10.1016/0090-1229(92)90037-o. [DOI] [PubMed] [Google Scholar]
- 13.Mecchia M, Matarrese P, Malorni W, et al. Type I consensus interferon (CIFN) gene transfer into human melanoma cells up-regulates p53 and enhances cisplatin-induced apoptosis: implications for new therapeutic strategies with IFN-alpha. Gene Ther. 2000;7:167–179. doi: 10.1038/sj.gt.3301059. [DOI] [PubMed] [Google Scholar]
- 14.Heller LC, Ingram SF, Lucas ML, et al. Effect of electrically mediated intratumor and intramuscular delivery of a plasmid encoding IFN alpha on visible B16 mouse melanomas. Technol Cancer Res Treat. 2002;1:205–209. doi: 10.1177/153303460200100305. [DOI] [PubMed] [Google Scholar]
- 15.Papageorgiou A, Kamat A, Benedict WF, et al. Combination therapy with IFN-alpha plus bortezomib induces apoptosis and inhibits angiogenesis in human bladder cancer cells. Mol Cancer Ther. 2006;5:3032–3041. doi: 10.1158/1535-7163.MCT-05-0474. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Y, Tibensky I, Schmidt J, et al. Interferon-alpha in combination with chemotherapy has potent antiangiogenic properties in an orthotopic mouse model for pancreatic adenocarcinoma. J Immunother. 2008;31:28–33. doi: 10.1097/CJI.0b013e318157c682. [DOI] [PubMed] [Google Scholar]
- 17.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 18.Miller AJ, Mihm MC., Jr. Melanoma. N Engl J Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 19.Garrison M, Nathanson L. Prognosis and staging in melanoma. Semin Oncol. 1996;23:725–733. [PubMed] [Google Scholar]
- 20.Avril MF, Charpentier P, Margulis A, Guillaume JC. Regression of primary melanoma with metastases. Cancer. 1992;69:1377–1381. doi: 10.1002/1097-0142(19920315)69:6<1377::aid-cncr2820690613>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 21.Grimm D, Kern A, Rittner K, et al. Novel tools for production and purification of recombinant adeno associated virus vectors. Hum Gene Ther. 1998;9:2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- 22.Zolotukhin S, Byrne BJ, Mason E, et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 23.Ponnazhagan S, Mahendra G, Kumar S, et al. Adeno-associated virus 2-mediated antiangiogenic cancer gene therapy: long-term efficacy of a vector encoding angiostatin and endostatin over vectors encoding a single factor. Cancer Res. 2004;64:1781–1787. doi: 10.1158/0008-5472.can-03-1786. [DOI] [PubMed] [Google Scholar]
- 24.Lens M. Cutaneous melanoma: interferon alpha adjuvant therapy for patients at high risk for recurrent disease. Dermatol Ther. 2006;19:9–18. doi: 10.1111/j.1529-8019.2005.00051.x. [DOI] [PubMed] [Google Scholar]
- 25.Koc ON, Gerson SL, Cooper BW, et al. Rapid hematopoietic recovery after coinfusion of tautologies-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast patients receiving high-dose chemotherapy. J Clin Oncol. 2000;18:307–316. doi: 10.1200/JCO.2000.18.2.307. [DOI] [PubMed] [Google Scholar]
- 26.Koc ON, Day J, Nieder M, et al. Allogenic mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler syndrome (MPS-IH) Bone Marrow Transplant. 2002;30:215–222. doi: 10.1038/sj.bmt.1703650. [DOI] [PubMed] [Google Scholar]
- 27.Lazarus HM, Koc ON, Devine SM, et al. Cotransplantation of HLA-identical sibling culture-expanded mesenchymal stem cell and hematopoietic stem cells in hematologic malignancy patients. Biol Blood Marrow Transplant. 2005;11:389–398. doi: 10.1016/j.bbmt.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Ryan JM, Barry FP, Murphy JM, et al. Mesenchymal stem cells avoid allogeneic rejection. J inflamm. 2005;2:8–19. doi: 10.1186/1476-9255-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conget PA, Minguell JJ. Adenoviral-mediated gene transfer into ex vivo expanded human bone marrow mesenchymal progenitor cells. Exp Hematol. 2000;28:382–390. doi: 10.1016/s0301-472x(00)00134-x. [DOI] [PubMed] [Google Scholar]
- 30.Tsuda H, Wada T, Ito Y, et al. Efficient BMP2 gene transfer and bone formation of mesenchymal stem cells by a fiber-mutant adenoviral vector. Mol Ther. 2003;7:354–365. doi: 10.1016/s1525-0016(02)00062-x. [DOI] [PubMed] [Google Scholar]
- 31.Ding LM, Saylors R, Munshi NC. The stromal cell as a vehicle for ex vivo gene transfer. Blood. 1995;86:1625–1625. [Google Scholar]
- 32.Majumdar MK, Jaiswal N, Mackay AM, et al. High efficiency gene transfer into mesenchymal stem cells (MSCs) and maintenance of transgene expression with differentiation. Blood. 1997;90:4617–4617. [Google Scholar]
- 33.Sleijfer S, Bannink M, Van Gool AR, et al. Side effects of interferon-alpha therapy. Pharm World Sci. 2005;27:423–431. doi: 10.1007/s11096-005-1319-7. [DOI] [PubMed] [Google Scholar]
- 34.Van Gool AR, Kruit WH, Engels FK, et al. Neuropsychiatric side effects of interferon-alfa therapy. Pharm World Sci. 2003;25:11–20. doi: 10.1023/a:1022449613907. [DOI] [PubMed] [Google Scholar]
- 35.Moschos SJ, Odoux C, Land SR, et al. Endostatin plus interferon-alpha2b therapy for metastatic melanoma: a novel combination of antiangiogenic and immunomodulatory agents. Melanoma Res. 2007;17:193–200. doi: 10.1097/CMR.0b013e3281ad91a3. [DOI] [PubMed] [Google Scholar]
- 36.Hutchins LF, Moon J, Clark JI, et al. Evaluation of interferon alpha-2B and thalidomide in patients with disseminated malignant melanoma, phase 2, SWOG 0026. Cancer. 2007;110:2269–2275. doi: 10.1002/cncr.23035. [DOI] [PubMed] [Google Scholar]
- 37.Conill C, Jorcano S, Domingo-Domènech J, et al. Toxicity of combined treatment of adjuvant irradiation and interferon alpha2b in high-risk melanoma patients. Melanoma Res. 2007;17:304–309. doi: 10.1097/CMR.0b013e3282c3a6ed. [DOI] [PubMed] [Google Scholar]
- 38.Terando AM, Faries MB, Morton DL. Vaccine therapy for melanoma: current status and future directions. Vaccine. 2007;25:S2, B4–16. doi: 10.1016/j.vaccine.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 39.Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–3506. doi: 10.1182/blood-2007-02-069716. [DOI] [PubMed] [Google Scholar]
- 40.Selmani Z, Naji A, Zidi I, et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells. 2008;26:212–222. doi: 10.1634/stemcells.2007-0554. [DOI] [PubMed] [Google Scholar]
- 41.Fushimi T, O’Connor TP, Crystal R. Adenoviral gene transfer of stromal cell-derived factor-1 to murine tumors induces the accumulation of dendritic cells and suppresses tumor growth. Cancer Res. 2006;66:3513–3522. doi: 10.1158/0008-5472.CAN-05-1493. [DOI] [PubMed] [Google Scholar]