

Abstract
Carnitine acyltransferases catalyze the reversible exchange of acyl groups between coenzyme A (CoA) and carnitine. They have important roles in many cellular processes, especially the oxidation of long-chain fatty acids in the mitochondria for energy production, and are attractive targets for drug discovery against diabetes and obesity. To help define in molecular detail the catalytic mechanism of these enzymes, we report here the high resolution crystal structure of wild-type murine carnitine acetyltransferase (CrAT) in a ternary complex with its substrates acetyl-CoA and carnitine, and the structure of the S554A/M564G double mutant in a ternary complex with the substrates CoA and hexanoylcarnitine. Detailed analyses suggest these structures may be good mimics for the Michaelis complexes for the forward and reverse reactions of the enzyme, representing the first time that such complexes of CrAT have been studied in molecular detail. The structural information provides significant new insights into the catalytic mechanism of CrAT and possibly carnitine acyltransferases in general.
Carnitine acyltransferases catalyze the reversible exchange of acyl groups between carnitine and coenzyme A (CoA) (1-8). These enzymes are classified based on their preference for the chain length of the acyl groups. Carnitine acetyltransferase (CrAT) and carnitine octanoyltransferase (CrOT) prefer short- and medium-chain acyl groups as substrates, respectively. Carnitine palmitoyltransferases (CPTs) prefer long-chan acyl groups as substrates. Several different isoforms of CPTs have been identified in mammals, including CPT-Ia (liver isoform), CPT-Ib (muscle isoform), CPT-Ic (brain isoform), and CPT-II. The CPT-Is are integrally associated with the outer membrane of the mitochondria, whereas CPT-II is localized in the mitochondrial matrix and maybe loosely associated with the inner membrane of the mitochondria.
The CPTs catalyze the rate-limiting step in the β-oxidation of fatty acids in the mitochondria (4-9), which is the transport of fatty acids from the cytosol into the mitochondria. The CoA esters of fatty acids cannot cross the mitochondrial membranes. Instead, they must be converted to their carnitine esters, a reaction that is catalyzed by the CPT-Is, which can then be transported into the mitochondria. Once inside, the carnitine esters are converted back to the CoA esters through the action of CPT-II. Mutation and dysregulation of the CPTs are strongly linked to many serious, even fatal human diseases (4-6). CPT deficiencies are among the most common causes of inherited fatty acid oxidation disorders, and homozygous deletion of CPT-1a is lethal in mice (10). Inhibitors of CPT-Is may be efficacious for the treatment of type 2 diabetes (11,12), while agonists of these enzymes can stimulate fatty acid oxidation and may regulate body weight (13,14). CPT-I has also been proposed as a target for the treatment of heart failure (15).
The CrAT and CrOT enzymes may be important for the oxidation and transport of fatty acids from the peroxisomes to the mitochondria (4,5,7,8). CrAT may also have an important role in maintaining the CoA/acetyl-CoA balance in the cells (4,5,7,8).
The catalytic domains of carnitine acyltransferases contain about 600 amino acid residues, and share significant sequence identity (35% and higher). The CPT-Is contain an N-terminal extension of about 140 residues that are important for attachment to the mitochondrial membrane and other functions. We and others have reported the crystal structures of CrAT (16-21), CrOT (22), CPT-II (23,24), and the related enzyme choline acetyltransferase (25,26). The structures can be divided into two domains, N and C domains (Fig. 1), that share the same backbone fold with that of chloramphenicol acetyltransferase (CAT) and several other acetyltransferases (16,27).
Fig. 1.
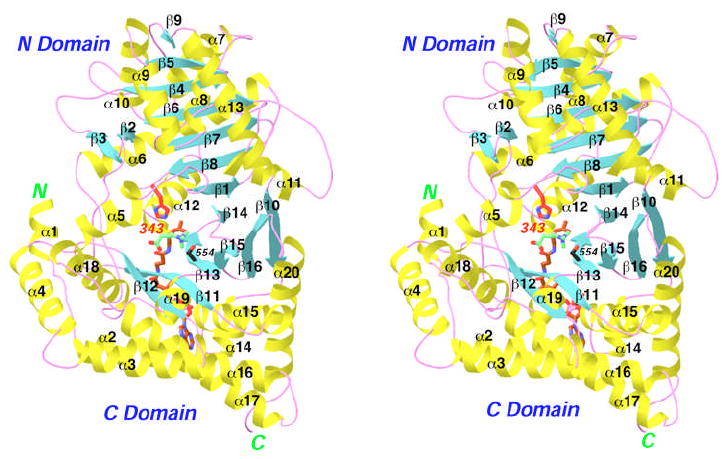
Structure of carnitine acetyltransferase (CrAT). Schematic drawing of the structure of wild-type mouse CrAT in a ternary complex with acetyl-CoA (in brown for carbon atoms) and carnitine (in green for carbon atoms). The catalytic His343 residue is shown in red, and Ser554 is shown in black for carbon atoms. Oxygen atoms are shown in red, nitrogen in blue, sulfur in yellow, and phosphorus in magenta. Produced with Ribbons (39).
The active site of the enzyme is located at the interface between the two domains, and the catalytic His residue is at the center of a tunnel that extends through the middle of the enzyme (Fig. 1) (21). Structures of the binary complexes with carnitine or CoA have defined the binding modes of these substrates, and suggested a catalytic mechanism in which the positive charge on the carnitine molecule helps to stabilize the oxyanion of the transition-state (substrate-assisted catalysis) (16). Additional stabilization of the oxyanion is provided by hydrogen-bonding to the side chain of a strictly conserved Ser residue (Ser554 in CrAT).
To further probe the catalytic mechanism, it is desirable to obtain structural information on the ternary complexes of this enzyme with its substrates or products. Alternatively, structures of the enzyme in complex with bisubstrate analogs could be used to provide insight on the catalysis, as has been carried out successfully for N-acetyltransferases (28,29). However, such compounds are not readily available for CrAT, and we have therefore developed protocols for trapping the ternary complexes of this enzyme. We report here the crystal structures of wild-type mouse CrAT in a ternary (Michaelis) complex with the substrates acetyl-CoA and carnitine, and in a ternary (dead-end) complex with CoA and carnitine, as well as the structure of the S554A/M564G double mutant in a ternary (Michaelis) complex with hexanoylcarnitine and CoA. The structures define in molecular detail the Michaelis complexes for both the forward and reverse reactions of the enzyme, and provide significant new insights into the catalytic mechanism of CrAT and possibly other carnitine acyltransferases as well.
Materials and Methods
Mutagenesis, protein expression and purification
Residues 30–626 of wild-type mouse CrAT was sub-cloned into the pET28a vector (Novagen) and over-expressed in E. coli (16). The expression construct contains an N-terminal hexa-histidine tag. The S554A/M564G double mutant was created with the QuikChange kit (Stratagene) and verified by sequencing.
The wild-type and mutant proteins were purified following the same protocol, with nickel-agarose, anion exchange, and gel filtration chromatography. The protein was concentrated to 32 mg/ml in a solution containing 20 mM Tris (pH 8.5) and 200 mM NaCl, flash-frozen in liquid nitrogen in the presence of 5% (v/v) glycerol, and stored at −80°C.
Protein Crystallization
Crystals of wild-type CrAT in complex with carnitine and acetyl-CoA were obtained at 4°C by the sitting-drop vapor diffusion method. The reservoir solution contained 100 mM Tris (pH 8.5), 100 mM NaCl, 14% (w/v) PEG 3350, and 2.3 mM carnitine, and the protein was at 17 mg/ml concentration. The crystals were soaked in a solution containing 100 mM Bis-Tris (pH 6.5), 150 mM NaCl, 20% (w/v) PEG 3350, 2.5 mM carnitine, 5 mM acetyl-CoA, and 25% (v/v) ethylene glycol for 3 minutes before being flash-frozen in liquid nitrogen for data collection at 100 K. They belong to space group C2, with cell parameters of a = 163.9 Å, b = 89.2 Å, c = 122.6 Å, and β = 128.9°. There are two CrAT molecules in the crystallographic asymmetric unit.
Using this soaking protocol, crystals of wild-type CrAT in complex with carnitine and CoA have also been obtained, the acetyl group being hydrolyzed during the soaking experiment.
Crystals of the S554A/M564G mutant in complex with hexanoylcarnitine and CoA were obtained at 4°C by the sitting-drop vapor diffusion method. The reservoir solution contained 100 mM Tris (pH 8.5), 18% (w/v) PEG 3350, and 2.3 mM carnitine, and the protein was at 10 mg/ml concentration. The crystals were soaked in a solution containing 100 mM Bis-Tris (pH 6.5), 150 mM NaCl, 18% (w/v) PEG 3350, 2.5 mM carnitine, 5 mM hexanoyl-CoA, and 25% (v/v) ethylene glycol for 3 minutes before being flash-frozen for data collection. They belong to space group C2 with cell parameters of a = 164.1 Å, b = 89.1 Å, c = 122.8 Å, and β = 129.0°. These mutant crystals are isomorphous to the ternary complexes of wild-type CrAT described above, with two CrAT molecules in the crystallographic asymmetric unit.
Data collection, structure determination and refinement
X-ray diffraction data were collected on an ADSC Quantum-4 CCD at the X4A beamline of the National Synchrotron Light Source (NSLS). The diffraction images were processed and scaled with the HKL package (30).
These ternary complex crystals have cell parameters that are similar to those of the free enzyme and binary complexes of wild-type CrAT (16), with about 5 Å difference in the a axis and 3 Å in the c axis. The initial structure was determined by the molecular replacement method, with the program COMO (31), using the structure of the free enzyme as the search model. The structure refinement was carried out with the program CNS (32). Manual adjustment of the atomic model against the electron density was performed with the program O (33). The crystallographic information are summarized in Table 1.
Table 1.
Summary of crystallographic information
| Enzyme | Wild-type | Wild-type | S554A/M564G |
|---|---|---|---|
| Substrate | Carnitine + acetyl-CoA | Carnitine + CoA | Hexanoylcarnitine + CoA |
| Resolution range (Å) | 30-2.2 | 30-1.9 | 30-2.1 |
| Number of observations | 195,424 | 204,773 | 225,110 |
| Rmerge (%)a | 4.0 (9.8) | 7.4 (40.6) | 4.5 (10.8) |
| I/σI | 24.6 (7.8) | 10.8 (1.9) | 22.5 (6.4) |
| Redundancy | 3.0 (2.4) | 1.9 (1.8) | 3.0 (2.1) |
| Number of reflections | 65,954 | 101,205 | 75,758 |
| Completeness (%) | 95 (80) | 92 (77) | 94 (80) |
| R factor (%)b | 17.0 (19.4) | 19.3 (31.0) | 17.7 (21.1) |
| Free R factor (%) | 22.3 (24.9) | 23.5 (32.7) | 21.9 (25.5) |
| Average B for protein atoms (Å2) | 24 | 22 | 21 |
| Average B for ligands (Å2) | 22 | 22 | 33 |
| Residues in most favored region of the Ramachandran plot (%) | 90.4 | 91.7 | 90.7 |
| Residue in disallowed region of Ramachandran plot | Ile 116 | Ile 116 | Ile 116 |
| rmsd in bond lengths (Å) | 0.006 | 0.006 | 0.006 |
| rmsd in bond angles (°) | 1.1 | 1.2 | 1.1 |
| Protein Data Bank entry code | 2H3P | 2H3U | 2H3W |
. The numbers in parenthesis are for the highest resolution shell.
Kinetic studies
The kinetic parameters of the wild-type and mutant CrAT were determined using an endpoint fluorometric assay (34). The reaction buffer contained 40 mM Hepes (pH 7.8), 1.5 mM EDTA, and 3-500 μM of acetyl-CoA or hexanoyl-CoA. Each reaction contained 10-200 ng of wild-type enzyme or the M564G mutant, or 1-4 μg of the S554A or S554A/M564G mutant, in a volume of 600 μl. The reactions were initiated by the addition of carnitine (1.5 mM final concentration), allowed to progress for 10 min. at room temperature, and then stopped by heat treatment at 70 °C. The free CoA product was reacted with 7-fluorobenz-2-oxa-1,3-diazole-4-sulfonamide (ABD-F, Molecular Probes) by incubation at 42 °C for 40 min., and the fluorescence was recorded at 535 nm (excitation wavelength 405 nm) using a 96-well plate reader (200 μl solution per well; Perkin Elmer). Control reactions in the absence of carnitine were carried out to remove the background. The kinetic data were fitted to the Michaelis-Menten equation (Table 2).
Table 2.
Kinetic parameters of wild-type and mutant CrAT
| With acetyl-CoA as the substratea | |||
| kcat (s−1) | Km (μM) | kcat/Km (M-1 s-1) (×106) | |
| Wild-type | 97.6 ± 3.1 | 21.0 ± 2.0 | 4.7 ± 0.3 |
| S554A | 4.0 ± 0.4 | 92.1 ± 21.6 | 0.043 ± 0.007 |
| M564G | 28.4 ± 1.5 | 74.6 ± 10.9 | 0.38 ± 0.04 |
| S554A/M564G | 1.4 ± 0.1 | 59.6 ± 7.5 | 0.024 ± 0.002 |
| H343A | NAb | NA | NA |
| H343E | 15.4 ± 0.8 | 32.4 ± 4.4 | 0.48 ± 0.04 |
| With hexanoyl-CoA as the substratea | |||
| kcat (s−1) | Km (μM) | kcat/Km (M-1 s-1) (×106) | |
| Wild-type | 30.4 ± 0.8 | 103.4 ± 6.7 | 0.29 ± 0.01 |
| S554A | 1.42 ± 0.04 | 41.5 ± 3.1 | 0.034 ± 0.002 |
| M564G | 204.7 ± 8.2 | 22.4 ± 3.5 | 9.1 ± 1.2 |
| S554A/M564G | 8.2 ± 0.5 | 111.8 ± 15.9 | 0.073 ± 0.007 |
| H343A | NA | NA | NA |
| H343E | NA | NA | NA |
Carnitine is present at saturating concentrations (1.5 mM). Each assay was repeated two to three times to ensure reproducibility.
NA: No appreciable activity with up to 50 μg protein in the reaction.
Atomic Coordinates
The atomic coordinates have been deposited in the Protein Data Bank with the accession codes 2H3P, 2H3U, 2H3W.
Results and Discussion
Strategies to obtain the ternary complexes of CrAT
Our structures of the CoA and carnitine binary complexes of wild-type CrAT defined the binding modes of these two substrates on their own (16). We have also determined the crystal structure of the F565A mutant in a ternary (dead-end) complex with carnitine and CoA (17). However, to fully understand the catalysis by this enzyme, the structure of the ternary (Michaelis) complex with carnitine and acetyl-CoA (or acetylcarnitine and CoA) is needed. Our earlier attempts at soaking wild-type CrAT crystals with acetyl-CoA showed that the acetyl group is quickly hydrolyzed, even in the crystalline state (16). This suggested that the catalytic activity of the enzyme must be reduced before there is any possibility of capturing the structure of the ternary (Michaelis) complex.
Previous studies have indicated that His343 is the general base in the catalysis by CrAT (4,5). Crystal structures show that this side chain is held in an unusual conformation, with a hydrogen-bond between the side chain Nδ2 atom and the main chain carbonyl oxygen (16). A similar conformation for the catalytic His residue is observed in the related enzymes chloramphenicol acetyltransferase (27) and dihydrolipoyl transacetylase (35), and this conformation may enhance the reactivity of the His residue as a general base.
As a first attempt to reduce the activity of the enzyme, we mutated the catalytic His343 residue of CrAT to Ala or Glu. Our kinetic studies showed that the H343A mutant is inactive towards both acetyl-CoA and hexanoyl-CoA substrates, even when 50 μg of the enzyme were used in the assay (Table 2). In comparison, only 10 ng of the wild-type enzyme is needed to obtain robust activity. This suggests that the H343A mutation caused a >5,000-fold reduction in the activity of CrAT, consistent with the crucial role of this residue in catalysis. On the other hand, the H343E mutant has only a 10-fold loss in kcat/Km towards the acetyl-CoA substrate (Table 2), suggesting that the carboxylic side chain of the Glu residue can partially function as the general base for catalysis. The H343E mutant however does not show appreciable activity towards the hexanoyl-CoA substrate (Table 2). The presence of the negative charge may disfavor substrates with longer aliphatic chains.
Unfortunately, we could not observe any electron density for the CoA molecule in the active site region for either mutant (data not shown), indicating that it was mostly disordered in these mutants. Therefore, it appears that the His343 side chain may also play a role in stabilizing the conformation of the CoA molecule.
Based on our structural analysis, the Ser554 residue is part of the oxyanion hole and helps to stabilize the tetrahedral transition-state intermediate of the catalysis (16). Earlier kinetic studies on CrOT showed that mutation of this Ser residue to Ala produced a 10-fold decrease in the kcat while having little impact on the Km for carnitine (36). Therefore, we produced the S554A mutant of CrAT for our attempts to obtain the ternary (Michaelis) complexes. Our kinetic studies indicated a 25-fold reduction in the kcat and a 4-fold increase in the Km for acetyl-CoA of this mutant (Table 2), suggesting that the S554A mutation may have a somewhat larger impact on the catalysis by CrAT than CrOT. We also produced the S554A/M564G double mutant, as the latter mutation increases the activity of CrAT towards medium-chain fatty acids (such as hexanoyl-CoA) (17,37). This double mutant could then allow us to determine the binding modes of medium-chain acyl groups. Our kinetic data on this double mutant as well as the M564G single mutant (Table 2) are fully consistent with kinetic data reported earlier (17,37).
While mutagenesis is a powerful way to reduce the catalytic activity of CrAT, there could be concerns in the resulting structures as to the impact of the mutations. Therefore, we also developed a soaking protocol that would allow us to capture the ternary (Michaelis) complex for the wild-type enzyme, by sampling the pH of the soaking solution and the soaking time. The optimal pH of CrAT is 7.4, and most of the earlier soaking experiments were carried out at this pH. By lowering the pH of the soaking solution, we should be able to reduce the catalytic activity of the enzyme, as it can be expected that the His343 catalytic residue becomes a weaker base at lower pH, especially if it becomes protonated. We have varied the pH of the soaking solution from 5.5 to 8.5, together with using short soaking times (1 to 5 min.), to obtain the simultaneous binding mode of carnitine and acetyl-CoA to the wild-type enzyme. It should be noted that the actual pH of the system could also change during the cryo-freezing procedure. Structural comparisons with the binary complexes, which were obtained near the optimal pH of the enzyme, show that the reduction in the pH did not cause a significant change in the active site of the enzyme (see below).
Structure of CrAT in a ternary (Michaelis) complex with carnitine and acetyl-CoA
The crystal structure of wild-type murine CrAT in a ternary complex with carnitine and acetyl-CoA has been determined at 2.2 Å resolution (Fig. 1). The atomic model contains residues 30-625 of the enzyme, with excellent agreement to the crystallographic data and expected geometric parameters (Table 1). The majority of the residues (90%) are in the most favored region of the Ramachandran plot, while residue Ile116 is the only one in the disallowed region (Table 1). This residue has clearly defined electron density, and is also in the disallowed region in the structures of the free enzyme and the binary complexes.
To obtain this structure, wild-type murine CrAT was first co-crystallized with carnitine, and then soaked in a solution containing 5 mM acetyl-CoA at pH 6.5 for 3 min. There are two molecules of CrAT in the crystallographic asymmetric unit. Clear electron density for acetyl-CoA (Fig. 2A) and carnitine was observed in the active site of one of these molecules, while the other active site contained carnitine and CoA. This suggests that the hydrolysis of the acetyl-CoA substrate can still occur at the lowered pH, and the enzyme is still catalytically active in the crystalline environment. Using this protocol, we also successfully obtained the crystal structure of the S554A mutant of CrAT in a ternary complex with acetyl-CoA and carnitine (data not shown). From our experiments, it appears that pH 6.5 is the optimal soaking condition for observing the electron density of acyl groups in the active site of CrAT.
Fig. 2.
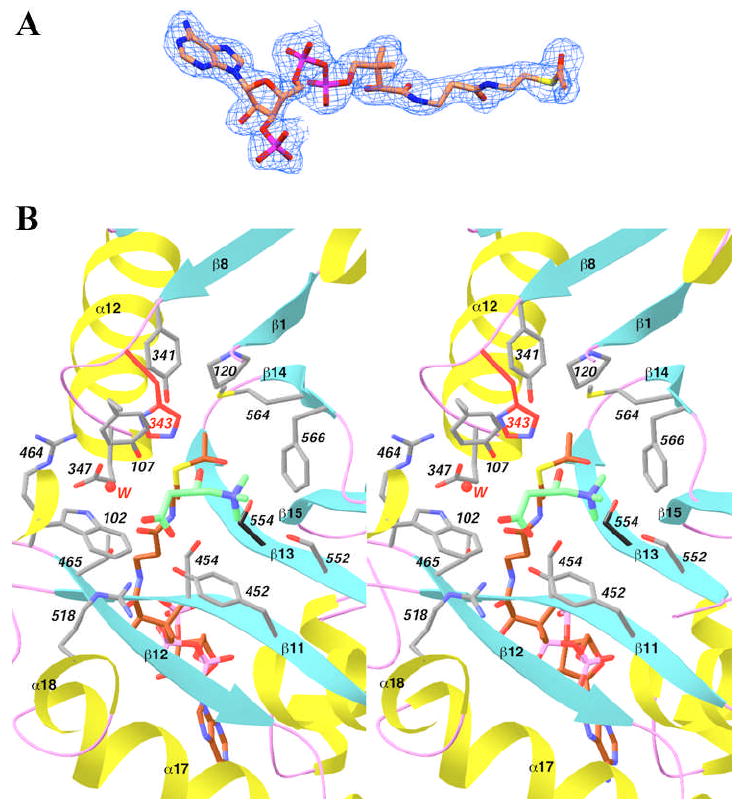
Structure of wild-type CrAT in a ternary complex with acetyl-CoA and carnitine. (A). Final 2Fo−Fc electron density map for acetyl-CoA at 2.2 Å resolution, contoured at 1σ. Produced with Setor (40). (B). Stereo drawing showing the active site of CrAT in the ternary complex with acetyl-CoA (in brown) and carnitine (in green). The side chain of His343 is shown in red, and Ser554 in black. Residues 104-117 have been omitted for clarity (except for the side chain of Tyr107). A water molecule involved in carnitine binding is shown as a sphere in red. Produced with Ribbons (39).
Even with the optimized soaking conditions, more than 20 crystals were examined to obtain the structure of the ternary complex with carnitine and acetyl-CoA. The other crystals contain either the (dead-end) ternary complex of carnitine and CoA (see next section), or they have weak electron density for the CoA or acetyl-CoA molecule in the active site.
The reaction catalyzed by CrAT is readily reversible, and the equilibrium constant of the reaction in solution is close to one (4,5). Therefore, one might expect that a mixture of reactants and products will be present in our crystals, but we observed only the reactants in the structure. This might be related to the fact that our experiments are performed in a crystalline environment. There are two molecules of CrAT in these crystals. We routinely see only CoA and carnitine in one of these molecules (dead-end complex), while the other CrAT molecule could contain carnitine and acetyl-CoA (but we have not observed acetylcarnitine in our studies so far). It might be possible that acetyl-CoA was observed in this molecule because the hydrolysis reaction has not occurred to a significant extent. An alternative explanation is that the crystalline environment may have affected the catalytic activity (and possibly the reaction equilibrium) of the enzyme. At the same time, we do not see any recognizable structural differences near the active site region between the two molecules.
The overall structure of the ternary complex with carnitine and acetyl-CoA is highly similar to that of the free enzyme, the binary complex with carnitine or CoA, as well as the ternary complex with carnitine and CoA. The rms distance between any pair of these structures is about 0.3 Å, for roughly 590 equivalent Cα atoms. The largest difference among these structures is generally located near residues 368-374 (α12-β9 loop, Fig. 1), in the loop connecting the N and C domains. Residues in this loop have very weak electron density, and the structural differences are therefore due to flexibility of these residues. In the active site region of the enzyme, only small conformational differences are observed among these structures (with the exception of the Glu347 residue, see below). Therefore, CrAT appears to be a lock-and-key enzyme, and does not undergo significant conformational changes upon substrate binding.
As was observed with the binary complexes (16), the acetyl-CoA and carnitine substrates are bound in the active site tunnel of CrAT (Fig. 3A), located on opposite sides of the His343 catalytic base in this tunnel (Fig. 1). The carboxylate group of carnitine is involved in an intricate network of hydrogen-bonding and ion-pair interactions, while the trimethylammonium group is located in a mostly hydrophobic binding site (Fig. 2B) (16). The acetyl group of the acetyl-CoA molecule is bound such that its carbonyl oxygen atom is pointed towards the Ser554 side chain (Fig. 2B), within hydrogen-bonding distance (2.9 Å). The methyl group is pointed towards the acyl-group binding site of these enzymes, with Met564 at the bottom of this pocket (Fig. 2B) (16,17,22,37).
Fig. 3.
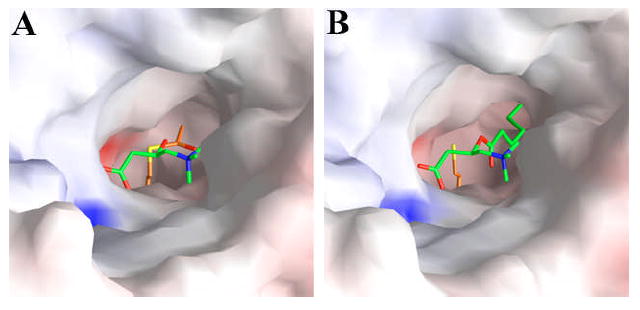
Molecular surface of the active site region of CrAT. (A). Molecular surface of the active site region of wild-type CrAT, in the ternary complex with carnitine and acetyl-CoA. (B). Molecular surface of the active site region of the S554A/M564G double mutant of CrAT, in the ternary complex with hexanoylcarnitine and CoA. Produced with Grasp (41).
Detailed structural analysis of this ternary complex suggests that it may be a good mimic of the expected Michaelis complex for CrAT. In the complex, the hydroxyl group of carnitine is hydrogen-bonded to the side chain of His343 (Fig. 2B), with a distance of 2.7 Å. This should facilitate the deprotonation of this hydroxyl group by the His343 catalytic base. At the same time, this hydroxyl group is located within 3 Å of the carbonyl carbon of the acetyl group in acetyl-CoA, and the line linking these two atoms is almost perpendicular to the plane of the acetyl group (Fig. 4A). As a result, the hydroxyl group of carnitine appears to be located in the correct position to initiate the nucleophilic attack, after being deprotonated by His343, on the carbonyl carbon of acetyl-CoA. Therefore, our structure of the ternary complex of CrAT with acetyl-CoA and carnitine may correspond to that of the Michaelis complex of the enzyme in the forward reaction, converting acetyl-CoA and carnitine to CoA and acetylcarnitine.
Fig. 4.
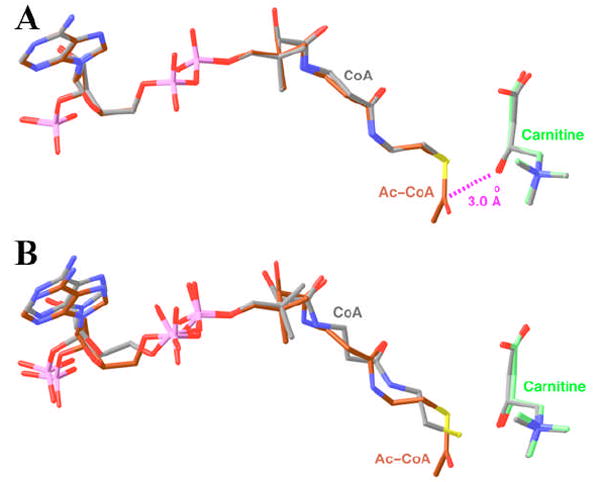
Comparison of the binding modes of CoA and carnitine in ternary and binary complexes. (A). Overlay of the binding modes of acetyl-CoA (in brown) and carnitine (in green) in one ternary complex with that of CoA and carnitine (in gray) in another ternary complex. (B). Overlay of the binding modes of acetyl-CoA (in brown) and carnitine (in green) in the ternary complex with that of CoA and carnitine (in gray) in their respective binary complexes (16). Produced with Ribbons (39).
Structure of CrAT in a ternary (dead-end) complex with carnitine and CoA
The crystal structure of wild-type murine CrAT in a ternary (dead-end) complex with carnitine and CoA has been determined at 1.9 Å resolution (Table 1). Although the crystal was soaked with acetyl-CoA, it appears to have been hydrolyzed completely in both CrAT molecules in the asymmetric unit, and the acetate product has left the active site region of the enzyme (or it is disordered as we did not observe any electron density for this product).
Besides the two ternary complexes in this crystal, several other structures of the dead-end complex have also been determined, including one of the CrAT molecules in the crystal described in the previous section and the ternary complexes of the F565A mutant that we described earlier (17). The binding modes of CoA and carnitine in these ternary (dead-end) complexes are highly similar to each other, as well as to that of acetyl-CoA and carnitine in the ternary (Michaelis) complex (Fig. 4A). The carnitine and the CoA portion of these molecules are located at essentially the same position.
The binding mode of carnitine in the ternary complex confirms our observations earlier from the binary complex (16) (Fig. 4B). In comparison, the binding mode of CoA in the binary complex (16) shows significant differences to that in the ternary complex (Fig. 4B). Structural differences can be recognized along the entire length of the (acetyl-)CoA molecule, and the largest difference is at the sulfur atom of CoA, with a distance of 1.2 Å between the two binding modes (Fig. 4B). However, it is not clear whether our structural observations represent a genuine difference in the binding mode of CoA in the binary and the ternary complexes, as the structure of the binary complex was determined using a crystal that had suffered significant damage during the soaking process (16).
Structure of the S554A/M564G mutant in a ternary (Michaelis) complex with hexanoylcarnitine and CoA
The crystal structure of the S554A/M564G mutant of murine CrAT in a ternary complex with hexanoylcarnitine and CoA has been determined at 2.1 Å resolution (Table 1). The rms distance between this structure and those of the free enzyme and the other complexes is 0.3 Å, suggesting that there are no overall structural differences among them. Although the crystal was grown in the presence of carnitine and then soaked with hexanoyl-CoA at pH 6.5, the acyl-transfer reaction appears to have proceeded to completion in the crystalline state, such that hexanoylcarnitine (Fig. 5A) and CoA (Fig. 5B) are observed in the active sites of both CrAT molecules in the crystallographic asymmetric unit. The observed electron density is inconsistent with the presence of hexanoyl-CoA in the crystal (Fig. 5B). As discussed earlier for the wild-type enzyme, we do not know the exact reason why only the products (hexanoylcarnitine and CoA) are observed in our structure. It could be possible that the S554A/M564G double mutation, and the crystalline environment, may have affected the equilibrium of the reaction.
Fig. 5.
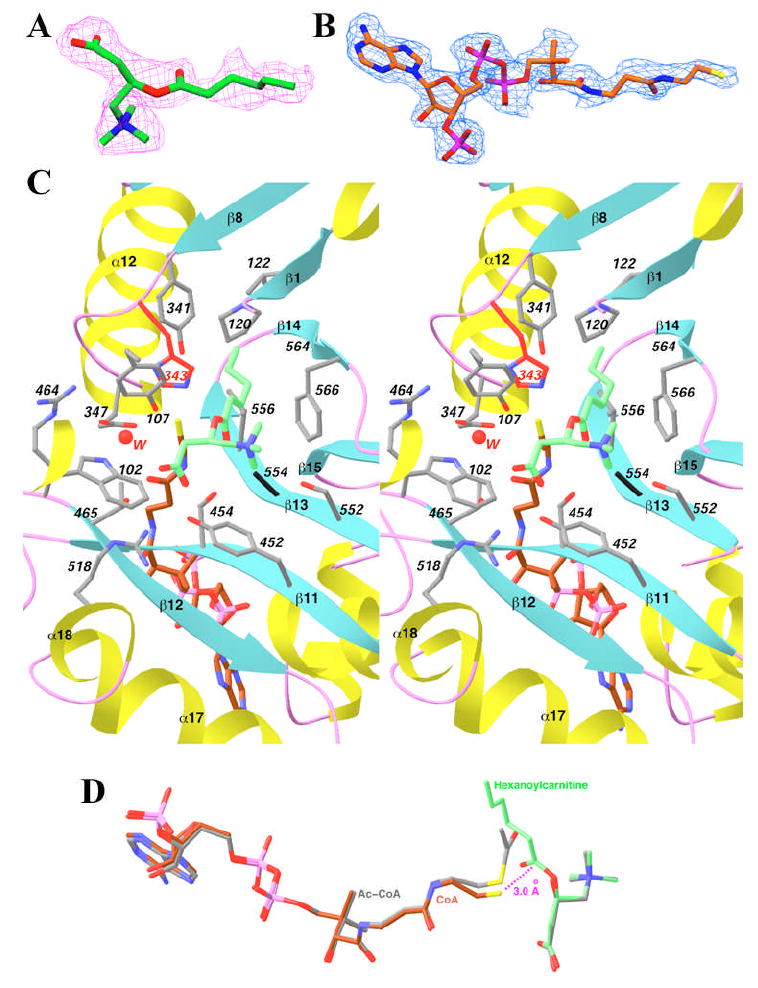
Structure of the S554A/M564G mutant in a ternary complex with hexanoylcarnitine and CoA. (A). Final 2Fo−Fc electron density map for hexanoylcarnitine at 2.1 Å resolution, contoured at 1σ. (B). Final 2Fo−Fc electron density map for CoA at 2.1 Å resolution, contoured at 1σ. Produced with Setor (40). (C). Stereo drawing showing the active site of the S554A/M564G mutant in the ternary complex with hexanoylcarnitine (in green) and CoA (in brown). The side chain of His343 is shown in red, and Ala554 in black. Residues 104-117 have been omitted for clarity (except for the side chain of Tyr107). A water molecule involved in carnitine binding is shown as a sphere in red. (D). Overlay of the binding modes of hexanoylcarnitine (in green) and CoA (in brown) in one ternary complex with that of acetyl-CoA and carnitine (in gray) in the other ternary complex. Produced with Ribbons (39).
The hexanoyl group of the substrate/product is positioned in the acyl group binding site of the enzyme, enlarged due to the absence of the Met564 side chain (Figs. 3B, 5C) (17,37). This pocket is located between strands β1, β8, β13, β14 and helix α12 (Fig. 5C) (16), and several hydrophobic side chains from these secondary structure elements (such as Pro120, Val122, Val352 and Val556) line the sides of this pocket. The hexanoyl group is not fully extended in the binding site (Fig. 5C), which is different from the mostly extended conformation of octanoylcarnitine bound to CrOT that we reported earlier (22). The position of the carnitine portion is essentially the same as that of carnitine itself in other binary and ternary complexes (Fig. 5D), suggesting that acylation does not affect the bound position of this compound.
The bound position of CoA in this ternary complex is similar to that of CoA or acetyl-CoA in the other ternary complexes (Fig. 5D). Only the thiol group of CoA shows distinct differences in its position in this complex, with a distance of about 0.6 Å to that in the other ternary complexes (Fig. 5D). The thiol group is within 3 Å of the carbonyl carbon of hexanoylcarnitine, and it is placed at the correct position to initiate the nucleophilic attack on this carbon atom (Fig. 5D). Therefore, this ternary complex may be a good mimic for the Michaelis complex for the reverse reaction catalyzed by CrAT, transfer of the acyl group from acylcarnitine to CoA.
The side chain of Glu347 in this ternary complex assumes a different conformation as compared to that in the other ternary complexes (compare Fig. 5C with 2B). In the ternary complex with hexanoylcarnitine and CoA, the conformation of Glu347 is similar to that seen in the free enzyme of CrAT and the binary complex with carnitine (16), having a mono-dentate interaction with Arg464 (Fig. 5C). In all the other ternary complexes as well as the binary complex with CoA, the Glu347 side chain assumes a different rotamer, where it makes a bi-dentate ion-pair interaction with the side chain of Arg464 (Fig. 2B). In this conformation, the Glu347 side chain is still capable of hydrogen-bonding to the water molecule that mediates the binding of the carboxylate group of carnitine (Fig. 2B). It remains to be seen whether this conformational switching in the side chain of Glu347 has any functional importance. This residue is a Glu only in the CrAT family of enzymes, whereas it is an Asp in CrOT and the CPTs. In the crystal structure of CrOT, this Asp side chain is involved in binding the carnitine substrate, and it also shows bi-dentate interactions with the Arg residue that is equivalent to Arg464 of CrAT (22).
Implications for the catalytic mechanism of CrAT
Our structural information on the two ternary (Michaelis) complexes provides significant new insights into the catalytic mechanism of CrAT (Fig. 6). The His343 side chain is located in the center of the active site, and the structural information is fully consistent with the important catalytic role of this residue. For the forward reaction, conversion of acetyl-CoA and carnitine to CoA and acetylcarnitine, the hydroxyl group of carnitine is deprotonated by the His343 residue and attacks the carbonyl carbon of acetyl-CoA to initiate the reaction. Our structure model suggests that the resulting oxyanion would be about 5 Å from the positive charge on the trimethylammonium group of carnitine (Fig. 6), which should allow favorably electrostatic interactions that helps to stabilize the transition-state. The oxyanion is also within hydrogen-bonding distance of the side chain of Ser554 (Fig. 6). The positively-charged trimethylammonium group should also reduce the pKa of the hydroxyl group of carnitine, facilitating its deprotonation by the His343 catalytic base.
Fig. 6.
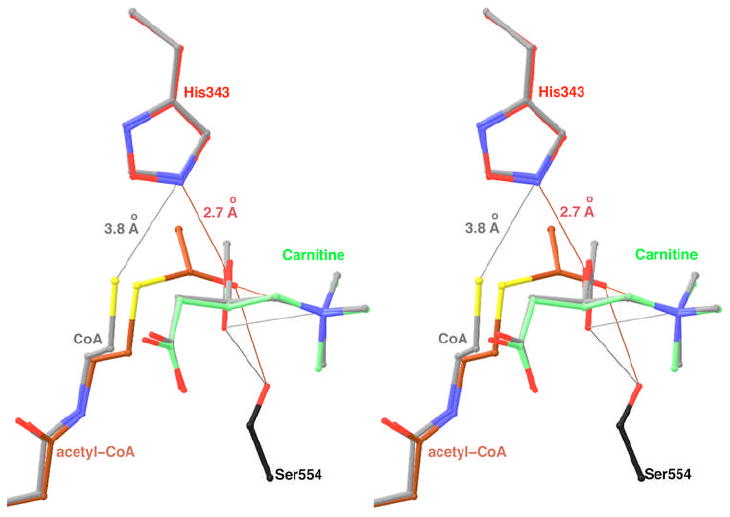
Models for the Michaelis complex of CrAT for the forward and reverse reactions. The acetyl-CoA and carnitine substrates for the forward reaction are shown in brown and green, respectively. The substrates for the reverse reaction are shown in gray. The conformation of acetylcarnitine is modeled from that of hexanoylcarnitine. The side chain of His343 is shown in red and gray for the forward and reverse reactions, respectively. The Ser554 side chain is shown in black. The thin lines indicate some of the interactions in the complexes. Produced with Ribbons (39).
For the reverse direction, conversion of CoA and acetylcarnitine to acetyl-CoA and carnitine, the thiol group of CoA is too far (3.8 Å) from the His343 side chain to be in direct hydrogen-bonding interactions (Fig. 6). However, as the thiol group has a much lower pKa than the hydroxyl group and can be deprotonated more easily, direct interactions with the catalytic base may not be necessary. In fact, the separation between the Cys and His residues in the catalytic dyad of caspases (about 5 Å) is also significantly longer than hydrogen-bonding distances (38). Therefore, our structural information suggests that the CoA substrate may not be directly hydrogen-bonded to the His343 residue at the start of the reverse reaction. The oxyanion is expected to be located within 3.5 Å of the trimethylammonium group of carnitine (Fig. 6), and this favorable electrostatic interaction is the dominant force that stabilizes the transition-state of the reaction (16).
The locations of the acetyl groups of acetyl-CoA and acetylcarnitine are different in the two ternary complexes (Fig. 6). In fact, the planes of the carbonyl group are almost perpendicular in the two complexes. While it may be possible that the actual positions of these groups in the genuine Michaelis complex are different to some extent from what we observed here in the ternary complexes, the observed positions do place the acetyl groups at roughly the correct position for nucleophilic attacks by the other substrate (Fig. 6). Therefore, our model suggests that the acetyl group may move by about 2 Å after the acyl-transfer reaction.
In summary, we have determined the crystal structures of CrAT in ternary complexes with acetyl-CoA and carnitine, or hexanoylcarnitine and CoA. The former may be a good mimic of the Michaelis complex for the forward reaction of the enzyme, whereas the latter may be a good mimic of the Michaelis complex for the reverse reaction. Detailed structural analyses on these complexes have provided significant new insights into the catalytic mechanism of CrAT and possibly carnitine acyltransferases in general.
Acknowledgments
We thank Randy Abramowitz and John Schwanof for setting up the X4A beamline at NSLS; Reka Letso, Andras Bauer and Brent Stockwell for help with the fluorescence plate reader. This research is supported in part by a grant (DK67238 to LT) from the National Institutes of Health.
References
- 1.Bieber LL. Ann Rev Biochem. 1988;57:261–283. doi: 10.1146/annurev.bi.57.070188.001401. [DOI] [PubMed] [Google Scholar]
- 2.Foster DW. Ann NY Acad Sci. 2004;1033:1–16. doi: 10.1196/annals.1320.001. [DOI] [PubMed] [Google Scholar]
- 3.Colucci WJ, Gandour RD. Bioorg Chem. 1988;16:307–334. [Google Scholar]
- 4.McGarry JD, Brown NF. Eur J Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- 5.Ramsay RR, Gandour RD, van der Leij FR. Biochim Biophys Acta. 2001;1546:21–43. doi: 10.1016/s0167-4838(01)00147-9. [DOI] [PubMed] [Google Scholar]
- 6.Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Mol Aspects Med. 2004;25:495–520. doi: 10.1016/j.mam.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Steiber A, Kerner J, Hoppel CL. Mol Aspects Med. 2004;25:455–473. doi: 10.1016/j.mam.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Ramsay RR, Zammit VA. Mol Aspects Med. 2004;25:475–493. doi: 10.1016/j.mam.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Kerner J, Hoppel C. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/s1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- 10.Nyman LR, Cox KB, Hoppel CL, Kerner J, Barnoski BL, Hamm DA, Tian L, Schoeb TR, Wood PA. Mol Gen Metab. 2005;86:179–187. doi: 10.1016/j.ymgme.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 11.Anderson RC. Curr Pharm Des. 1998;4:1–16. [PubMed] [Google Scholar]
- 12.Lenhard JM, Gottschalk WK. Advanced Drug Delivery Reviews. 2002;54:1199–1212. doi: 10.1016/s0169-409x(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 13.Thupari JN, Landree LE, Ronnett GV, Kuhajda FP. Proc Natl Acad Sci USA. 2002;99:9498–9502. doi: 10.1073/pnas.132128899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronnett GV, Kim E-K, Landree LE, Tu Y. Physiol Behav. 2005;85:25–35. doi: 10.1016/j.physbeh.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Mengi SA, Dhalla NS. Am J Cardiovasc Drugs. 2004;4:201–209. doi: 10.2165/00129784-200404040-00001. [DOI] [PubMed] [Google Scholar]
- 16.Jogl G, Tong L. Cell. 2003;112:113–122. doi: 10.1016/s0092-8674(02)01228-x. [DOI] [PubMed] [Google Scholar]
- 17.Hsiao Y-S, Jogl G, Tong L. J Biol Chem. 2004;279:31584–31589. doi: 10.1074/jbc.M403484200. [DOI] [PubMed] [Google Scholar]
- 18.Wu D, Govindasamy L, Lian W, Gu Y, Kukar T, Agbandje-McKenna M, McKenna R. J Biol Chem. 2003;278:13159–13165. doi: 10.1074/jbc.M212356200. [DOI] [PubMed] [Google Scholar]
- 19.Ramsay RR, Naismith JH. Trends Biochem Sci. 2003;28:343–346. doi: 10.1016/S0968-0004(03)00137-3. [DOI] [PubMed] [Google Scholar]
- 20.Govindasamy L, Kukar T, Lian W, Pedersen B, Gu Y, Agbandje-McKenna M, Jin S, McKenna R, Wu D. J Struct Biol. 2004;146:416–424. doi: 10.1016/j.jsb.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Jogl G, Hsiao Y-S, Tong L. Ann NY Acad Sci. 2004;1033:17–29. doi: 10.1196/annals.1320.002. [DOI] [PubMed] [Google Scholar]
- 22.Jogl G, Hsiao Y-S, Tong L. J Biol Chem. 2005;280:738–744. doi: 10.1074/jbc.M409894200. [DOI] [PubMed] [Google Scholar]
- 23.Rufer AC, Thoma R, Benz J, Stihle M, Gsell B, de Roo E, Banner DW, Mueller F, Chomienne O, Hennig M. Structure. 2006;14:713–723. doi: 10.1016/j.str.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 24.Hsiao Y-S, Jogl G, Esser V, Tong L. Biochem Biophys Res Commun. 2006;346:974–980. doi: 10.1016/j.bbrc.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai Y, Cronin CN, Engel AG, Ohno K, Hersh LB, Rodgers DW. EMBO J. 2004;23:2047–2058. doi: 10.1038/sj.emboj.7600221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Govindasamy L, Pedersen B, Lian W, Kukar T, Gu Y, Jin S, Agbandje-McKenna M, Wu D, McKenna R. J Struct Biol. 2004;148:226–235. doi: 10.1016/j.jsb.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Leslie AGW, Moody PC, Shaw WV. Proc Natl Acad Sci USA. 1988;85:4133–4137. doi: 10.1073/pnas.85.12.4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheibner KA, De Angelis J, Burley SK, Cole PA. J Biol Chem. 2002;277:18118–18126. doi: 10.1074/jbc.M200595200. [DOI] [PubMed] [Google Scholar]
- 29.Hickman AB, Namboodiri MA, Klein DC, Dyda F. Cell. 1999;97:361–369. doi: 10.1016/s0092-8674(00)80745-x. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Jogl G, Tao X, Xu Y, Tong L. Acta Cryst. 2001;D57:1127–1134. doi: 10.1107/s0907444901006783. [DOI] [PubMed] [Google Scholar]
- 32.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Hassett RP, Crockett EL. Anal Biochem. 2000;287:176–179. doi: 10.1006/abio.2000.4799. [DOI] [PubMed] [Google Scholar]
- 35.Mattevi A, Obmolova G, Schulze E, Kalk KH, Westphal AH, de Kok A, Hol WGJ. Science. 1992;255:1544–1550. doi: 10.1126/science.1549782. [DOI] [PubMed] [Google Scholar]
- 36.Cronin CN. Biochem Biophys Res Commun. 1997;238:784–789. doi: 10.1006/bbrc.1997.7390. [DOI] [PubMed] [Google Scholar]
- 37.Cordente AG, Lopez-Vinas E, Vazquez MI, Swiegers JH, Pretorius IS, Gomez-Puertas P, Hegardt FG, Asins G, Serra D. J Biol Chem. 2004;279:33899–33908. doi: 10.1074/jbc.M402685200. [DOI] [PubMed] [Google Scholar]
- 38.Watt W, Koeplinger KA, Mildner AM, Heinrikson RL, Tomasselli AG, Watenpaugh KD. Structure. 1999;7:1135–1143. doi: 10.1016/s0969-2126(99)80180-4. [DOI] [PubMed] [Google Scholar]
- 39.Carson M. J Mol Graphics. 1987;5:103–106. [Google Scholar]
- 40.Evans SV. J Mol Graphics. 1993;11:134–138. doi: 10.1016/0263-7855(93)87009-t. [DOI] [PubMed] [Google Scholar]
- 41.Nicholls A, Sharp KA, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]