

Abstract
Anti-TB bioassay-directed fractionation led to the isolation of six carbazole alkaloids, as well as the γ-lactone derivative of oleic acid, from the CH2Cl2 extract of the stem bark of Micromelum hirsutum. The carbazoles include the new micromeline (2) and five known alkaloids: lansine (3), 3-methylcarbazole (4), methyl carbazole-3-carboxylate (5), 3-formylcarbazole (6), and 3-formyl-6-methoxycarbazole (7). Compound 1 was identified as the lactone derivative of oleic acid, (−)-Z-9-octadecene-4-olide, for which the trivial name micromolide (1) is suggested. It showed potent in vitro anti-TB activity against H37Rv (MIC: 1.5 μg/mL), a selectivity index (SI) of 63, and exhibited activity against the Erdman strain of M. tuberculosis in a J774 mouse macrophage model (EC90: 5.6 μg/mL). Thus, 1 appears worthy of further evaluation as a potential new anti-TB agent. Isolates 2, 3, 6 and 7 had anti-TB MIC values between 14.3 and 42.3 μg/mL, while compounds 4 and 5 were considered inactive (MIC > 128 μg/mL). Structure elucidation and identification were based on spectroscopic analysis, including MS, 1D/2D NMR, and a full 1H spin system analysis of 1.
Keywords: Micromelum hirsutum, Rutaceae, (−)-Z-9-octadecene-4-olide, micromeline, micromolide, carbazole alkaloids, anti-TB activity
Introduction
With approximately 3 million annual deaths in the 1990s [1], tuberculosis remains a leading cause of mortality worldwide. The current treatment of an infection, a cocktail of drugs including, for example, isoniazid, rifampin, ethambutol and pyrazinamide is prescribed for 2 months, followed by a continuation phase in which isoniazid and rifampin are taken for an additional 4 months. However, failure to successfully complete this treatment has frequently led to the emergence of multi-drug-resistant tuberculosis (MDR-TB) [1].
Both the cost of treating an MDR-TB patient and the treatment failure rate are much greater than for drug-sensitive TB. Therefore, there is an urgent need for new anti-mycobacterial drugs, particularly those effective against a persistent infection [1]. Our International Cooperative Biodiversity Group (ICBG) research project [2], involving the collaboration of institutions in Vietnam, Laos and USA, has as one of its objectives the discovery of anti-TB compounds from plants. Micromelum hirsutum Oliv. (Rutaceae) was found to be a promising lead against Mycobacterium tuberculosis in vitro. A review of the literature revealed no prior phytochemical reports on this plant. In the present study, anti-TB bioassay-directed fractionation of the CH2Cl2 extract of the stem bark of M. hirsutum led to the isolation of the γ-lactone derivative of oleic acid (1) beside six carbazole alkaloids. One of them, micromeline (2), was determined to be a new structure, whereas lansine (3), 3-methylcarbazole (4), methyl carbazole-3-carboxylate (5), 3-formylcarbazole (6), and 3-formyl-6-methoxycarbazole (7) were previously described. The lactone 1 was identified as the known compound, (−)-Z-9-octadecene-4-olide, and showed promising in vitro anti-TB activity with an MIC value of 1.5 μg/mL and a selectivity index (SI) of 63. Alkaloids 2,3,6 and 7 showed in vitro anti-TB activity with MIC values of 31.5,14.3,42.3 and 15.6 μg/mL, respectively, whereas alkaloids 4 and 5 were considered inactive (MIC > 128 μg/mL). The present paper reports the isolation, structure elucidation and/or dereplication, as well as the biological evaluation of these isolates.
Materials and Methods
General
Optical rotations were measured on a Perkin-Elmer model 241 polarimeter. IR spectra were run on a Jasco FT/IR-410 spectrometer, equipped with a Specac Silver Gate ATR system by applying a film on a germanium crystal. UV spectra were obtained with a Beckman DU-7 spectrometer. NMR spectra were recorded on Bruker DRX-500 MHz (11.7 Tesla) and DPX-300 MHz (7.0 Tesla) spectrometers. Chemical shifts (δ) were expressed in ppm with reference to the solvent signals. All NMR data were obtained by using standard pulse sequence supplied by the vendor. Column chromatography was carried out on silica gel (200 – 400 mesh, Natland International Corporation). Reversed-phase HPLC was carried out on a Waters 600E Delivery System pump, equipped with a Waters 996 photodiode detector, and a Phenomenex C18 column (250 × 50 mm, 10 μm, 120 Å), at a flow rate of 18 mL/min. Thin-layer chromatography was performed on Whatman glass-backed plates coated with 0.25 mm layers of silica gel 60. HRE-SI-MS were recorded on a Micromass QTOF-2 spectrometer. EI-MS were recorded on a JEOL GC mate II spectrometer.
Plant material
The initial stem bark sample (SV-0155) of Micromelum hirsutum (a tree of 10 m height with greenish-yellow flowers and aromatic leaves) was collected at the Cuc Phuong National Park, Vietnam (Bong, 20° 21′ N, 105° 35′ E, edge of valley forest) on March 19, 1999, and was documented by voucher herbarium specimens number Soejarto 10591. A voucher specimen was deposited each at the herbaria of Cuc Phuong National Park (CPNP), Vietnamese Academy of Science and Technology (formerly, National Center for Science and Technology) (HN; Hanoi), and the Field Museum (F; Chicago). A larger sample (SVA-0155, stem bark, 4.5 kg, documented by voucher specimens number Nguyen Manh Cuong 524) was re-collected from the same site at Cuc Phuong on November 2, 2001, for complete isolation work.
Extraction and isolation
The dried, milled stem bark (4.5 kg) of M. hirsutum was extracted with CH2Cl2 to yield 45.5 g extract, of which 32.1 g were chromatographed by vacuum liquid chromatography over a silica gel column (5.0 kg), which was developed by gradient elution with n-hexane/EtOAc/MeOH/H2O (H:E:M:W, 300 mL aliquots beginning with 100% H, increasing by 10 mL of the next polar solvent, until 100% MeOH, with three aqueous MeOH washes) to afford 9 fractions (F1 – F9) based on their TLC patterns (SiO2, H:E, 1: 1, an-isaldehyde reagent). Bioassay localized the anti-TB activity to fractions F2 (H:E 6: 4, 2.7 L VE) (5.0 g) and F3 (H:E 4: 6, 3.6 L VE) (7.8 g). Fraction F2 was chromatographed over a silica gel column (250 g) and eluted with mixtures of petroleum ether-acetone of increasing polarities (P:A, 500 mL aliquots from 100: 0 to 80: 20, increasing by 2% acetone, with 100% acetone wash) as mobile phase to afford 31 combined fractions (F10 – F40) based on their TLC patterns (SiO2, P:A, 8:2, 10% H2SO4 reagent). Fractions F15 (P:A, 9.2: 0.8, 300 mL VE) and F16 (P:A, 9.1:0.9, 600 mL VE) showed strong anti-TB activity with MICs of 1.96 and 3.72 μg/mL, respectively. F15 (347 mg) was further fractionated on a 16 g silica gel column. Elution was performed with a gradient of petroleum ether-CHCl3 (50 mL aliquots from P:C, 9: 1 increasing 10% CHCl3 to P:C 6:4) to yield compound 4 (2 mg, P:C, 9:1, 50 mL VE) and one fraction composed primarily of compound 1 (209 mg, P:C, 8:2, 50 mL, P:C, 7:3,100 mLVE) based on TLC patterns (C-18, MeOH:H2O,9.8:0.2,10% H2SO4 detection reagent). A portion (210 mg) of F16 (772 mg) was subjected to preparative HPLC separation on a Phenomenex C18 column and developed with MeOH-H2O (9:1), 18 mL/min; tR 79 – 83 min, 1422–1494 mL VE to obtain compound 1 (59 mg). Fraction 3 was chromatographed over a Sephadex LH-20 (250 g) column and isocratically eluted at 0.75 mL/min, in 25-mL fractions with CHCl3-MeOH (4:6) to afford 17 fractions (F41 – F57) based on TLC patterns (SiO2, P:A, 8:2,10% H2SO4 reagent). Fractions F53, F54 and F55 showed the same level of anti-TB activity (MIC: 16 μg/mL). Compound 3 (70 mg) was crystallized from F55 in acetone as yellow needles. Fractions F53 and F54 were pooled and chromatographed over a silica gel column (25 g), which was eluted with a petroleum ether-acetone gradient(500 mL aliquots from 100% P, increasing by 1 % acetone to P:A, 8: 2) to yield compounds 5 (18 mg, P:A, 9.5: 0.5, 60 mL), 7 (9 mg, P:A, 9.3: 0.7, 40 mL) and 2 (6 mg, P:A, 9: 1, 30 mL), in addition to fractions F69 – F75. Fraction 70 was repeatedly separated on silica gel columns using the same solvent system as above to yield pure compound 6 (14 mg, P:A, 9.4:0.6, 60 mL).
(−)-Z-9-Octadecene-4-olide [Micromolide] (1)
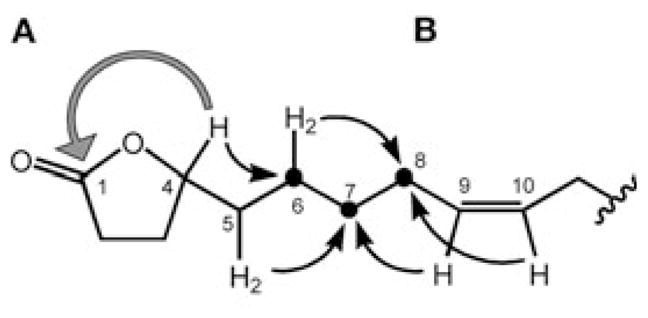
Colorless oil. : −19.8° (c 0.49, CHCl3); IR (film, Ge ATR): vmax = 2925, 2851,1773, 1163 cm−1; 1H-NMR (CDCl3, 300 MHz) and 13C-NMR (CDCl3, 75 MHz): see Table 1; EI-MS: m/z (rel. int.) = 280 ([M]+, 40), 220 (15), 195 (8), 181 (10), 167 (18), 153 (29), 137 (32), 136 (42), 123 (32), 109 (48), 95 (92), 85 (74), 81 (100), 67 (67).
Table 1.
1H and 13C-NMR spectral data of compound 1 (300/75 MHz, CDCl3, J in Hz)
| Position | Carbon | Proton | HMBC (→C) | ||
|---|---|---|---|---|---|
| δc[ppm] | δH[ppm] | Mult. | J [Hz] (→H) | ||
| 1 | 177.5 | ||||
| 2a + b | 29.1 | 2.528 | dd | 6.84 (3a), 9.51 (3b) | 1, 4 |
| 3a | 28.3 | 2.319 | ddt | 6.84 (2), −12.69 (3b), 6.62 (4) | 1, 4, 5 |
| b | 1.846 | ddt | 9.51 (2), −12.69 (3a), 5.60 (4) | 1, 4, 5 | |
| 4 | 81.2 | 4.484 | ddt | 6.62 (3a), 5.60 (3b), 6.84 (5a), 7.72 (5b) | 1, 5, 6 |
| 5a | 35.8 | 1.721 | dt | 6.84 (4), −11.98 (5b), 5.88 (6) | 6, 7 |
| b | 1.609 | ma | 6.00 (6), 7.72 (4), −11.98 (5a) | ||
| 6a + b | 25.1 | 1.408 | ma | 6.00 (5b), 5.88 (5a), 5.56 (7) | 7, 8 |
| 7a + b | 29.6 | 1.408 | ma | 5.56 (6), 5.50 (8), 0.45 (9)b | 6, 9 |
| 8a + b | 27.2 | 2.039 | dt | 5.50 (7), 6.81 (9) | 6, 10 |
| 9 | 130.7 | 5.372 | dtt | 0.45 (7)b, 6.81 (8), 10.94 (10) | 7, 11 |
| 10 | 129.4 | 5.328 | dtt | 10.94 (9), 6.71 (11), 0.49 (12)b | 8, 12 |
| 11a + b | 27.5 | 2.014 | dt | 6.71 (10) | 9 |
| 12a + b | 29.7 | 1.269 | ma | 0.49 (10)b | |
| 13a + b | 29.8 | 1.269 | ma | ||
| 14a + b | 29.9 | 1.269 | ma | ||
| 15a + b | 30.0 | 1.269 | ma | ||
| 16a + b | 32.2 | 1.269 | ma | 18 | |
| 17a + b | 22.9 | 1.269 | ma | 18 | |
| 18 | 14.4 | 0.881 | br t/m | 6.78 (17) | 16, 17 |
Overlapping peaks limited effective iteration, resulting in unresolved exact resonances and J values (see text).
An interesting observation is that, instead of the expected allylic4J9, 11 and 4J8, 10 couplings, W-type 4J7, 9 and 4J10, 12 long-range couplings are observed.
Micromeline (2)
Yellow microcrystalline powder. UV (acetone): λmax (log ε) = 316 (3.3), 329 (3.7) nm; IR (film): vmax = 3394, 3317, 2964, 2929, 2846, 1670, 1600, 1300, 1164, 1058, 812, 670 cm−1; 1H-NMR (acetone-d6, 300 MHz) and 13C-NMR (acetone-d6, 75 MHz), see Table 2; Negative HRESI-MS: m/z = 278.1183 [M − 1]− (calcd. for C18H16NO2: 278.1181).
Table 2.
1H- and 13C-NMRdata of compound 2 (300/75 MHz, acetone-d6, J in Hz)
| Position | Carbon | Proton | HMBC (→C) | ||
|---|---|---|---|---|---|
| δc[ppm] | δH[ppm] | Mult. (spinb) | J [Hz] (→H) | ||
| 1 | 111.9 | 7.596 | dd/mb(AMN) | 8.5 (2), 0.5 (4) | 3, 11 |
| 2 | 126.7 | 7.913 | dd (AMN) | 1.6 (4), 8.5 (1) | 4, 10, CHO |
| 3 | 129.5 | - | |||
| 4 | 127.4 | 8.648 | dd/mb(AMN) | 1.6 (2), 0.5 (1) | 2, 10, CHO |
| 5 | 122.8 | - | |||
| 6 | 149.5 | - | |||
| 7 | 116.7 | 7.114 | d | 8.5 (8) | 5, 6, 13 |
| 8 | 110.1 | 7.293 | d | 8.5 (7) | 6, 12 |
| 10 | 145.4 | - | |||
| 11 | 124.1 | - | |||
| 12 | 123.3a | - | |||
| 13 | 136.1 | - | |||
| 1′a + b | 26.3 | 4.023 | dqq (A2MX3Y3) | 6.5 (2′), 1.3 (5′), 0.4 (4′) | 6, 2′ |
| 2′ | 123.3a | 5.322 | tqq (A2MX3Y3) | 6.5 (1′), 1.2 (5′), 1.5 (4′) | |
| 3′ | 132.9 | ||||
| 4′ | 18.5 | 1.991 | tdq (A2MX3Y3) | 0.4 (1′), 1.5 (2′), 0.4 (5′) | 2′, 3′ |
| 5′ | 25.9 | 1.688 | tdq (A2MX3Y3) | 1.3 (1′), 1.2 (2′), 0.4 (4′) | 2′, 3′ |
| CHO | 191.9 | 10.054 | s | - | 3 |
| NH | 10.666 | br s | - | ||
| OH | 7.990 | br s | - | ||
Overlapping resonances, assigned to one CH and one C carbon by integration.
Multiplicities are given under nuclei first order assumptions, and are supplemented by the nomenclatures of the underlying spin system (spin).
Lansine (3)
Yellow needles. 13C-NMR (acetone-d6, 75 MHz): δ = 97.6 (C-1), 162.2 (C-2), 116.5 (C-3), 129.2 (C-4), 104.5 (C-5), 156.3 (C-6), 115.7 (C-7), 113.0 (C-8), 148.0 (C-10), 119.1 (C-11), 125.3(C-12), 136.8(C-13), 196.9(CHO), 56.6 (OCH3). (See Wu et al. [11] and Prakash et al. [12] for additional molecular data).
Anti-TB assay
Anti-TB activity of crude extracts, fractions and compounds against M. tuberculosis (H37Rv) was determined using the fluorometric microplate Alamar blue assay (MABA) as described previously [3]. Percent inhibition was defined as 1 – (test well fluorescence units/mean FU fluorescence units of triplicate wells containing only bacteria)x 100. The drug concentration effecting an inhibition of > 90% was considered as the MIC.
Cytotoxicity of compounds
Evaluation of the cytotoxic activity of compounds 1 – 7 in Vero cells (African green monkey kidney cells) was performed as described earlier [4] using the CellTiter 96 aqueous non-radioactive cell proliferation assay (Promega Corp., Madison, WI). The IC50 was defined as the reciprocal dilution resulting in 50% inhibition of the Vero cells.
Intracellular activity
The mouse macrophage cell line J774A.1 (ATCC TIB-67) was used to study the activity of samples against intracellular M. tuberculosis. Cells were cultured in Dulbecco’s modified Eagle’s medium (D-MEM) supplemented with 10% FBS, 1% glutamine, at 37°C and 5% CO2. The J774A.1 macrophage cell line was prepared using standard procedures (ATCC). Twenty mL of media (D-MEM) were dispensed into a 75 cm3 flask. Cells were detached with a scraper from the flask, centrifuged at 1000 rpm for 5 min, and re-suspended in 10 mL media. Cell number was counted using trypan blue and the cell suspension was adjusted to1–3×105 cells mL−1. The coverslips were placed in the 24-well plates (Fisher Scientific), and 1 mL cells were added to each well. Plates were incubated at 37 °C in a 5% CO2 incubator for 24 hours until confluent.
M. tuberculosis Erdman (ATCC 35 801) was diluted with the cell culture media to a final concentration of 1–3×105 CFU mL−1 and 500 μL were added to each well of another 24-well plate. The coverslips were transferred with the cells from the old plates to the new 24-well plates and infected for 2 – 3 hours at 37 °C in a CO2 incubator. Extracellular bacteria were removed by washing the coverslips with HBSS (Fisher Scientific). The coverslips were transferred to new 24-well plates and 1 mL of fresh media was added to each well. Cultures were incubated at 37 °C, 5 % CO2 for 7 days.
Cells were lysed using 0.25 % SDS (Fisher Scientific), and sonicated for 15 seconds. Pre-treatment samples (T0) were diluted (1: 10, 1: 100 and 1: 1000), and 0.1 mL of the undiluted suspension and the 3 dilutions were plated on 7H11 agar, and incubated for 3 weeks. Stock solutions of test samples were prepared in DMSO, diluted in D-MEM, and added to each well of new 24-well plates. Coverslip cultures were transferred to the plate and incubated for 7 days in the CO2 incubator.
After a week, cells from control and treated wells were lysed, and 3 dilutions were prepared. One hundred μL of dilutions were placed on 7H11 agar plates (T7). Colonies were counted after 16 – 20 days and compared with the control (T0). By comparing these data with CFU obtained from untreated control macrophages, the concentrations of the test compounds required to achieve a one log reduction (EC90) and a two log reduction (EC99) in viable M. tuberculosis were determined using CurveExpert 1.3 (Microsoft).
Results and Discussion
Anti-TB bioassay-directed fractionation of the CH2Cl2 extract of the stem bark of Micromelum hirsutum, screening against the virulent (H37Rv) strain of Mycobacterium tuberculosis, led to the isolation of six carbazole alkaloids (2 – 7) and theC18 γ-lactone 1.
Compound 1, was isolated as a colorless gum, : −19.8° (c, 0.49, CHCl3), and was identified as the previously described γ-lactone derivative of oleic acid ([5], [6], reporting 1H-NMR, IR and MS only). However, dereplication of 1 based on published spectroscopic/NMR data proved to be an impossible task due to inconsistencies and gaps in the published assignments, but also due to severe spectral overlap in both the 1H and 13C aliphatic regions. Therefore, and in order to substantiate the structure of this potent antimycobacterial agent, an ab initio structure elucidation was performed taking into account COSY, HMQC and HMBC correlations beside the 1D 1H- and 13C-NMR spectra. The results are compiled in Table 1. The molecular formula C18H32O2 of 1 was derived on the basis of MS results and 13C-NMR data. The 13C-NMR data additionally indicated that 1 contained one carbonyl (δ = 177.5) and two olefinic carbons (δ = 130.7, 129.4). The DEPT spectrum further indicated that it contained a methyl group (δ = 14.3) and an oxymethine carbon (δ = 81.2). Resonances of aliphatic methylene carbons (δ = 22.9 – 35.7) suggested the presence of a long aliphatic chain with one double bond. The carbonyl carbon chemical shift was consistent with the presence of a saturated γ-lactone ring, supported by the IR absorption band at 1773 cm−1 [8]. Further evidence for the γ-lactone partial structure came from the observation of an ion fragment (m/z = 85) in the EI-MS [5]. Critical to the establishment of the structure were (A) the unequivocal confirmation of the γ-lactone partial structure, (B) the location of the double bond in the aliphatic chain, and (C) the stereochemical assignment.
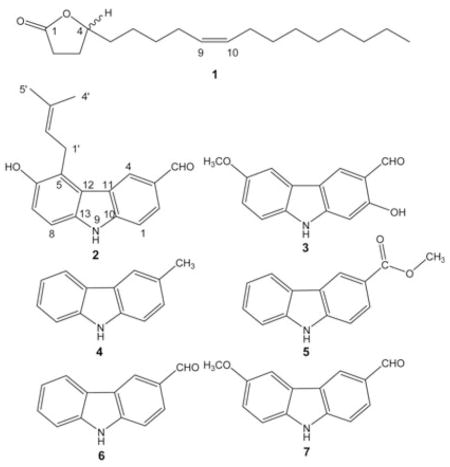
Regarding (A), several HMBC correlations (see Table 1) of the clearly resolved downfield 1H and 13C resonances led to the definitive assignment of a butanolide ring system. The lactone ring closure gave rise to a 3J correlation between H-4 and C-1. Severe spectral overlap made proof for the location of the double bond (B) a complicated task. Ultimately, it was possible through a combination of COSY, HMQC and HMBC data, processed with post-acquisition software. Fig. 1 shows the conclusive 3J couplings from the HMBC spectrum that were essential in unambiguously determining the location of the double bond at C-9/10. In contrast to the literature, the olefinic protons are not isochronous. This is due to the asymmetrical substitution of the double bond. Since the asymmetry is minimal, the difference in chemical shifts between carbons C-9 and C-10 is small (0.044 ppm), and, therefore, higher order effects can be seen. The dtt-like shaped multiplet shows a significant roofing effect as a result of these higher order interactions. Similar higher order effects were also observed in the two methylene protons (H2-8 and H2-11) immediately adjacent to the double bond. Spectral iteration and simulation, using the PERCH software package, replicated the peak characteristics and assigned the exact shift values and J values of the olefinic protons. The spectral overlap of the methylene, aliphatic resonances (H-12 to -17 and H-6 +-7) limited the achievable precision of the iteration process involved in a full-spin analysis of H-2-H-11. It can be estimated that at least 800 MHz spectra will be required to yield sufficient dispersion to allow successful iteration of H-2-H-11. These findings are in line with the fragmentation pattern observed in EI-MS (Fig. 2). The double bond was determined to be Z-configured on the basis of the 1H,1H-coupling constant (J < 12 Hz) and the 13C-NMR chemical shifts of the two adjacent carbons [9].
Fig. 1.
Conclusive proof for the structure of compound 1 with regards to (A) the γ-lactone arrangement and (B) the position of the double bond from the essential 3J HMBC correlations (see Table 1 for a complete NMR data set).
Fig. 2.
EI-MS fragmentation pattern of compound 1.
In order to determine the enantiomeric composition (C) of the natural isolate 1, a chiral LSR-NMR analysis according to the protocol recently established in our laboratory [7] was carried out using tris[3-(heptafluoropropylhydroxymethylene)-D-camphorato]-praseodymium(III), and concluded that the compound is enantiomerically pure. In order to assign the absolute stereochemistry of 1, we attempted to prepare its Mosher esters. However, after hydrolyzing the γ-lactone, the free alcohol spontaneously converted back into the butanolide during the following purification step. Thus, we are presently unable to report the absolute stereochemistry of the natural isolate 1. Compound 1 appears to be identical with previously described (−)-Z-9-octadecene-4-olide [5], [6], one of which has been reported to be enantiomerically pure [6]. However, because of inconsistencies in the published analytical data, an exact match with 1 cannot be shown. In order to address the stereochemical ambiguity remaining in both the literature [5], [6] and our work, and in following established drug discovery protocols, attempts are currently under way to synthesize 1, and to characterize and isolate minor side components/analogues that are likely to be contained in active fraction F16. Owing to the fact that 1 shows promising potential as a new anti-TB lead (see below), and since no trivial name has previously been suggested, the trivial name micromolide is proposed for 1.
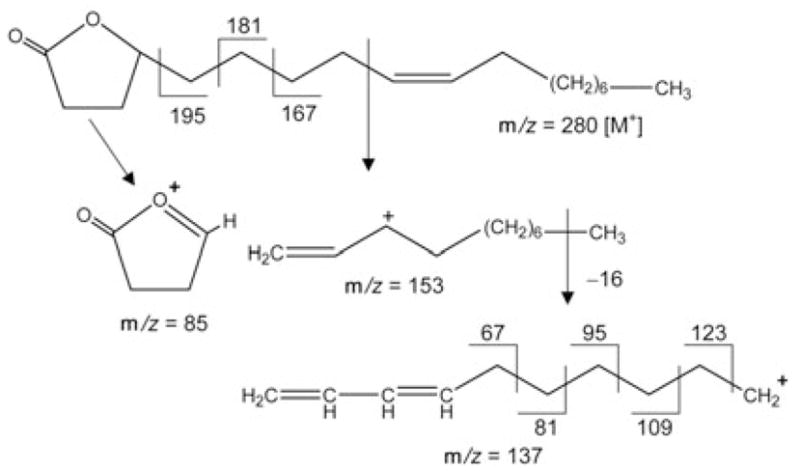
The carbazole alkaloid 2 was obtained as yellow crystals, and the molecular formula determined as C18H17NO2 by negative HRESI-MS at m/z = 278.1183 [M − 1]− (calcd.: 278.1181). Based on the analysis of its IR, 1H-NMRand 13C-NMR spectra, 2 was recognized as a carbazole alkaloid [10], [11]. The IR spectrum showed the presence of NH (3317 cm−1), OH (3394 cm−1) and conjugated carbonyl (1670 cm−1) functions, which were also observed in the 1H- and 13C-NMR spectra [δ = 10.666 (br s, NH), 10.054 (s, CHO), 7.990 (br s, OH) and δ = 191.9 (aldehyde CH)]. It was shown to be an analogue of 3-formylcarbazole (6) by comparison with published NMR data [10]. As in 6, the aldehyde group in 2 is attached to C-3 due to the presence of signals due to an AMN spin system centered at δ 8. = 648, 7.913 and 7.596, respectively (Table 2). The presence of HMBC long-range correlations between the carbon signal at δ = 191.9 (CHO) and the proton signals at δ = 8.648 (H-4) and 7.913 (H-2) supported this finding. However, in contrast to the ABMN spin-spin system of 6, 2 only showed a set of AB-type signals at δ = 7.114 (H-7) and 7.293 (H-8) with a shared coupling of 8.5 Hz, suggesting the presence of two additional substituents in the second benzene ring attached to either C-5 and C-6, or C-7 and C-8. One of the substituents was determined to be a 3-methylbut-2-enyl (prenyl) group based on in-depth analyses of the COSY, HMQC and HMBC correlations, and due to the fact that the set of 1H-NMR signals showed the complex J pattern (A2MX3Y3, see Table 2), which perfectly resembled the fingerprint of the prenyl groups of prenylated chalcones and flavanones recently isolated in our laboratory [16]. The site of linkage of the prenyl residue was determined to be C-5 due to the presence of NOE correlations of the proton resonating at δ = 8.648 (H-4) with the protons at δ = 4.023 (H2–1′) and 1.991 (H-4′) (Fig. 3). The second substituent was determined to be a hydroxy group based on the 1H- and 13C-NMR data [δH = 7.990 (br s, OH) and δC = 149.5]. It could be placed to C-6 due to the presence of HMBC correlations between the methylene signals of the prenyl residue at δH = 4.023 (H2–1′)and the hydroxylated C-6 carbon δC = 149.5. Thus, the structure of compound 2 was determined to be 6-hydroxy-5-(3-methylbut-2-enyl)-9H-carbazole-3-carbaldehyde, a new carbazole alkaloid named micromeline.
Fig. 3.
Selected NOESY correlations establishing the site of prenylation in 2.
Compounds 3 – 7 were shown to be the known carbazole alkaloids lansine (3) [11], [12], 3-methylcarbazole (4) [13], [14], methyl carbazole-3-carboxylate (5) [10], 3-formylcarbazole (6) [10] and 3-formyl-6-methoxycarbazole (7) [10] by comparison of their NMR data and physical properties to those reported in the literature. Since no 13C-NMR data of 3 have been published in the literature, they are presented in the Materials and Methods section of the current report.
The crude CH2Cl2 extract, fractions and isolates of M. hirsutum were tested against M. tuberculosis H37Rv in the MABA assay system (Table 3). All isolates were also tested for cytotoxicity toward Vero cells (derived from African green monkey kidney) in the MTS assay system to determine the selective index value (SI, SI = IC50 Vero cell/MIC TB). Rifampin was used as a positive control in the assay. The CH2Cl2 extract of M. hirsutum had an MIC of 12.5 μg/mL. Compound 1 showed promising in vitro anti-TB activity with an MIC of 1.5 μg/mL and a selectivity index (SI) of 63 (Table 3). This level of selectivity compares favorably with some of the less potent anti-TB agents such as ethambutol, and is well above the value of 10 that is required to move into advanced testing in a major on-going TB drug discovery program [17]. Compound 1 was therefore further evaluated in the J774 mouse macrophage cell line infected with M. tuberculosis Erdman, a strain more virulent than H37Rv (Fig. 4). The concentration of 5.6 μg/mL required to achieve a level of growth inhibition of 90% relative to control cultures, while higher than that required for the same level of inhibition in axenic medium (MIC = 1.5 μg/mL), is still well below the IC50 value of 1 for J774 cells, suggesting that growth inhibition of M. tuberculosis is not secondary to host cell toxicity. A three-fold higher concentration of 1 was required to achieve an additional log10 reduction in cfu (EC99); this was consistent with that observed for rifampin and streptomycin (Table 3). This compound appears worthy of further evaluation in in vivo models of tuberculosis infection in mice as well as to serve as a template for an analoging program.
Table 3.
Anti-tuberculosis and cytotoxic activities, and resulting selectivity indices (SI) of Micromelum constituents 1 – 7
| Material/compound | MIC[μg/ml]a | Cytotoxicity IC50 [μg/ml]b | SIc | Macrophage assayd (μg/ml) | ||
|---|---|---|---|---|---|---|
| VERO | J774 | EC90 | EC99 | EC99 | ||
| CHCl3 extract | 12.5 | |||||
| 1 | 1.5 ± 0.4 | 95 | 40.71 | 63 | 5.6 | 18.9 |
| 2 | 31.5 ± 0.2 | > 102 | >3 | |||
| 3 | 14.3 ± 0.9 | > 102 | >7 | |||
| 4 | > 128 | > 102 | ||||
| 5 | > 128 | > 102 | ||||
| 6 | 42.3 ± 0.5 | 101 | ||||
| 7 | 15.6 ± 0.2 | > 102 | >7 | |||
| Rifampine | 0.040 ± 0.017 | 100 | 2500 | 0.05 | 0.18 | |
| Streptomycin | 0.1 | 0.3 | ||||
Minimum inhibitory concentration (MIC90) values against M. tuberculosis (H37Rv) in the MABA assay.
Medium inhibitory concentration (IC50) on Vero cells.
Selectivity Index (SI) calculated by the quotient of the VERO IC50 and the MIC90.
Concentration effecting a reduction of M. tuberculosis cfu of 90% (EC90) or 99% relative to untreated control.
Positive control; negative control was solvent.
Fig. 4.
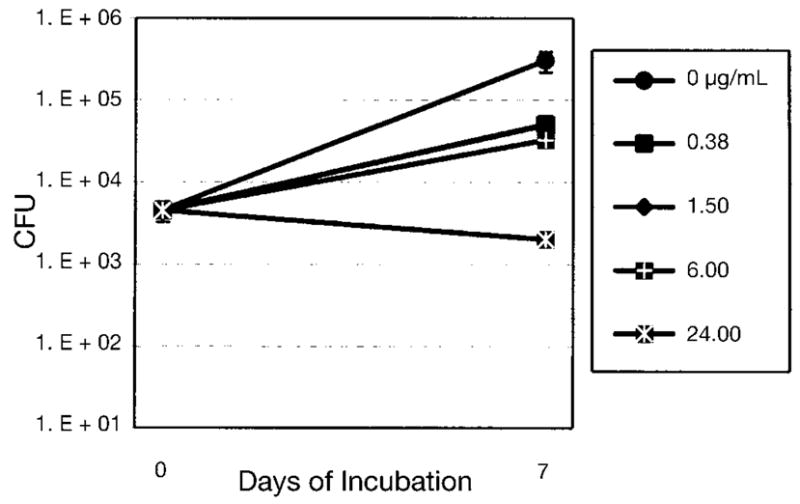
Evaluation of 1 in the J774 mouse macrophage cell line infected with M. tuberculosis Erdman, a strain somewhat more virulent than H37Rv.
The carbazoles 2, 3, 6 and 7 had MICs ranging from 14.3 to 42.3 μg/mL. Compounds 4 and 5 are inactive. Inhibitory activity of carbazole alkaloids against M. tuberculosis was previously reported against the H37Ra rather than the H37Rv strain [15] for methyl carbazole-3-carboxylate (5) and 3-formylcarbazole (6) with MIC values of 50 and 100 μg/ml, respectively. Although the number of studied carbazole alkaloids evaluated is limited to six compounds, preliminary conclusions about anti-TB structure-activity relationships can be drawn as follows. Carbazole alkaloids possessing an aldehyde group at the C3 position (2, 3, 6 and 7) exhibit significantly greater activity than those lacking this functionality (4 and 5). The presence of a methoxy group in 3 and 7 renders compounds at least twice as active as their free-phenolic counterparts 2 and 6. Further carbazole alkaloids will have to be biologically evaluated, before definitive structure-activity correlations can be established.
Acknowledgments
The present research project was supported by NIH Grant 1 UO1-TW01015-01, administered by the Fogarty International Center as part of an International Cooperative Biodiversity Groups (ICBG) program, through funds from NIH, NSF, and Foreign Agricultural Service of the USDA. Permit for the collection and export of plant material for this study was granted by the Ministry of Agriculture and Rural Development, Hanoi, Vietnam, through a letter dated September 15,1998, Ref. No. 3551/BNN/KHCN, and from the Cuc Phuong National Park, through a letter dated September 16,1998. The authors are grateful to the Research Resources Center, University of Illinois at Chicago, for supporting the Bruker DRX 500 MHz used in this study, and for the acquisition of the MS data. GFP thanks Dr. D. Lankin, UIC, for helpful discussions.
References
- 1.Smith CV, Sharma V, Sacchettini JC. TB drug discovery: addressing issues of persistence and resistance. Tuberculosis. 2004;84:45–55. doi: 10.1016/j.tube.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 2.Soejarto DD, Gyllenhaal C, Regalado JC, Pezzuto JM, Fong HHS, Tan GT, et al. Studies on biodiversity of Vietnam and Laos: The UIC-based ICBG program. Pharm Bio. 1999;37 (Supplement):100–13. [Google Scholar]
- 3.Collins LS, Franzblau SG. Microplate Alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Ch. 1997;41:1004–9. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantrell CL, Lu T, Fronczek FR, Fischer NH, Adams LB, Franzblau SG. Antimycobacterial cycloartanes from Borrichia frutescens. J Nat Prod. 1996;59:1131–6. doi: 10.1021/np960551w. [DOI] [PubMed] [Google Scholar]
- 5.Bohlmann F, Abraham WR. Neue prenylfavanone aus Helichrysum hypocephalum. Phytochemistry. 1979;18:1851–3. [Google Scholar]
- 6.Cosse AA, Bartelt RJ, James DG, Petroski RJ. Identification of a female-specific, antennally active volatile compound of the currant steam girdler. J Chem Ecol. 2001;27:1841–53. doi: 10.1023/a:1010412826373. [DOI] [PubMed] [Google Scholar]
- 7.Jaki B, Franzblau SG, Pauli GF. An NMR method towards routine chiral determination of natural products. Phytochemical Analysis. 2004 doi: 10.1002/pca.760. in press. [DOI] [PubMed] [Google Scholar]
- 8.Jakupovic J, Zdero C, Grenz M, Tsichritzis F, Lehmann L, Hashemi-Nejad SM, Bohlmann F. Twenty-one acylphloroglucinol derivatives and further constituents from south African Helichrysum species. Phytochemistry. 1989;28:1119–31. [Google Scholar]
- 9.Gunstone FD. High-resolution 13C NMR spectroscopy of lipids. The Oily Press; Dundee: 1993. p. 35. [Google Scholar]
- 10.Li WS, McChesney JD, El-Feraly FS. Carbazole alkaloids from Clausena lansium. Phytochemistry. 1991;30:343–6. [Google Scholar]
- 11.Wu TS, Huang SC, Wu PL, Teng CM. Carbazole alkaloids from Clausena excavata and their biological activity. Phytochemistry. 1996;43:133–40. doi: 10.1016/0031-9422(96)00212-9. [DOI] [PubMed] [Google Scholar]
- 12.Prakash D, Raj K, Kapil RS, Popli SP. Chemical constituents of Clausena lansium: part 1 - structure of lansamide-I and lansine. Indian J Chem. 1980;19B:1075–6. [Google Scholar]
- 13.Chakrabarty M, Batabyal A. Indolisation of cyclohexanone phenylhydrazones using phosphorous trichloride. Indian J Chem. 1992;31B:199–201. [Google Scholar]
- 14.Chakraborty DP, Das KC, Basak SP. New syntheses of isomeric methyl carbazoles. J Indian Chem Soc. 1968;45:84–6. [Google Scholar]
- 15.Sunthitikawinsakul A, Kongkathip N, Kongkathip B, Phonnakhu S, Daly JW, Spande TF, Nimit Y, Rochanaruangraj S. Coumarins and carbazoles from Clausena excavata exhibited antimycobacterial and anti-fungal activities. Planta Medica. 2003;69:155–7. doi: 10.1055/s-2003-37716. [DOI] [PubMed] [Google Scholar]
- 16.Chadwick LR. Ph.D. Dissertation: Estrogens and congeners from spent hops. University of Illinois at Chicago: Department of Medicinal Chemistry and Pharmacognosy; Chicago: 2004. [Google Scholar]
- 17.Orme I. Search for new drugs for treatment of tuberculosis. Antimicrob Agents Ch. 2001;45:1943–6. doi: 10.1128/AAC.45.7.1943-1946.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pauli GF. qNMR - A versatile concept for the validation of natural product reference compounds. Phytochemical Analysis. 2001;12:28–42. doi: 10.1002/1099-1565(200101/02)12:1<28::AID-PCA549>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]