

Summary
Cell cycle checkpoints are implemented to safeguard genome, avoiding the accumulation of genetic errors1–2. Checkpoint loss results in genomic instability and contributes to the evolution of cancer. Among G1-, S-, G2- and M-phase checkpoints, genetic studies indicate the essence of an intact S phase checkpoint in maintaining genome integrity3–4. Although the basic framework of S phase checkpoint in higher eukaryotes has been outlined, the mechanistic details remain to be elucidated. Human chromosome 11 band q23 translocations disrupting the MLL/HRX/ALL-1 gene lead to poor prognostic leukemias5–9. Here we assign MLL as a novel effector in the mammalian S phase checkpoint network and identify checkpoint dysfunction as an underlying mechanism of MLL leukemias. MLL is phosphorylated at serine 516 by ATR in response to genotoxic stress in S phase, which disrupts its interaction with and thereby degradation by the SCFSkp2 E3 ligase, leading to its accumulation. Stabilized MLL protein accumulates on chromatin, methylates histone H3K4 at late replication origins, and inhibits the loading of CDC45 to delay DNA replication. Cells deficient in MLL exhibited radioresistant DNA synthesis (RDS) and chromatid-type genomic abnormalities, indicative of S phase checkpoint dysfunction. Reconstitution of MLL−/− mouse embryonic fibroblasts (MEFs) with wild-type but not S516A or ΔSET mutant MLL rescues the S phase checkpoint defects. Moreover, murine myeloid progenitor cells (MPCs) carrying an MLL-CBP knock-in allele that mimics human t(11;16) leukemia exhibit a severe RDS phenotype. MLL-fusions function as dominant negative mutants that abrogate the ATR-mediated phosphorylation/stabilization of wild-type MLL upon DNA damage and thus compromise the S phase checkpoint. Altogether, our study identifies MLL as a key constituent of the mammalian DNA damage response pathway and deregulation of the S phase checkpoint incurred by MLL translocations likely contributes to the pathogenesis of human MLL leukemias.
Leukemogenic MLL translocations fuse the common MLL N-terminal 1,400 aa in frame with more than 60 partners8. The MLL gene encodes a 500 kD precursor MLL500 which is processed by Taspase110 to generate mature, heterodimerized MLLN320/C180. MLL participates in embryogenesis, cell fate, cell cycle and stem cell function7,11–14, in part by methylating histone H3 lysine 4 (H3K4) through its C-terminal SET domain15. Although the importance of HOX gene deregulation in the pathogenesis of MLL leukemias has been extensively investigated5–8, physiological MLL-fusion knock-in mouse models indicate that HOX gene aberrations alone are insufficient to initiate MLL leukemias7,16.
MLL participates in the cell cycle control12,17–19 and exhibits a biphasic expression with peaks at G1/S and G2/M transitions12. This unique, two peaks are conferred by proteasome-mediated degradation–SCFSkp2 and APCCdc20 degrade MLL at S and M phases, respectively12. Why MLL needs to be degraded in S and M phases is unclear. The observation that over-expression of MLL impedes S phase progression12 raises a testable thesis that MLL may accumulate in S phase upon DNA damage to delay DNA replication for repair. Indeed, tested DNA perturbation agents, including aphidocolin, hydroxyurea (HU), ultraviolet light (UV), etoposide, and γ-ionizing irradiation (γ-IR), induced the MLL protein expression, (Fig. 1a and Supplementary Fig. 1a, b). The MLL protein was induced upon DNA damage in S but not G1 or M phases through a transcription-independent mechanism (Fig. 1b and Supplementary Fig. 1c).
Figure 1. MLL accumulates in S phase upon DNA insults and MLL dysfunction results in S phase checkpoint defects.

a, 293T cells were treated with aphidocolin (10 µM), hydroxyurea (1 mM), UV (20 J/m2), etoposide (25 µM), or γ-IR (5 Gy) and then analyzed by anti-MLL immunoblots. b, Synchronized 293T cells were subjected to γ-IR. c, Metaphase spread of MEFs after mitomycin C treatment. Arrowheads indicate chromosomal errors. d, Control- and MLL- knockdown 293T cells (left panel) and wild-type and MLL−/− MEFs (right panel) were subjected to RDS assays. e, MPCs were subjected to RDS assays. Data shown in c–e are mean ± s.d. of three independent experiments.
The S phase checkpoint senses DNA damage, activates ATM/ATR, inhibits the firing of late replication origins, and enlists repair machineries. “Chromatid-type” genomic errors, accrued during S phase, include quadriradials, triradials, and chromatid gaps and breaks20. Metaphase spread analysis demonstrated a higher incidence of chromatid-type errors in mitomycin C treated MLL−/− than wild-type cells (Fig. 1c). Cells with compromised S phase checkpoints exhibit radioresistant DNA synthesis (RDS)3. Knockdown of MLL in 293T cells or genetic deletion of MLL in MEFs resulted in RDS (Fig. 1d), confirming a critical role of wild-type MLL in the mammalian S phase checkpoint. To explore whether MLL-fusions incur S phase checkpoint defects, we generated myeloid precursor cells (MPCs) from MLL+/ex7(stop)CBP mice that carry a knock-in inducible MLLex7-CBP allele (Supplementary Fig. 2)21. MLL+/ex7(stop)CBP MPCs retained only one copy of wild-type MLL and consequently exhibited a partial RDS phenotype (Fig. 1e). Remarkably, a severe RDS phenotype was observed in MLL+/ex7-CBP MPCs (Fig. 1e). These data suggest that MLL-CBP functions as a dominant negative mutant that actively compromises the S phase checkpoint, contributing to the acquisition of additional chromosomal translocations observed in MLL+/ex7-CBP leukemias21. Furthermore, expression of MLL-AF4 or MLL-AF9 in Jurkat T cells resulted in an RDS phenotype despite the presence of two wild-type MLL alleles (Supplementary Fig. 3). Consistently, expression of MLL-ENL in progenitor cells increased chromosomal abnormalities upon etoposide treatment22.
MLL is normally degraded in S phase by SCFSkp2 which directly binds to the N-terminal 1,400 aa of MLL12. It is conceivable that signal transduction triggered by DNA damage disrupts the MLL-Skp2 interaction and thereby induces MLL, which was indeed observed (Fig. 2a). As the DNA damage response network relays signals mainly through phosphorylation, we examined whether inhibition of proximal kinases including ATM, ATR and DNA-PKcs prohibited the DNA damage-induced MLL accumulation. LY294002 and Wortmannin abolished the MLL accumulation upon DNA damage (Fig. 2b). To specify key kinase(s) required for such signaling, we employed MEFs with deletion of ATM, ATR or DNA-PKcs23–24. Deficiency in ATR greatly reduced the accumulation of MLL upon DNA damage (Fig. 2c and Supplementary Fig.4), identifying ATR as the principal kinase for the MLL induction.
Figure 2. ATR signaling prevents the SCFSkp2-mediated degradation of MLL.

a, 293T cells were transfected with the FLAG-MLL expressing construct for 2 days, treated with 1 mM HU for the indicated times, and then subjected to anti-FLAG co-immunoprecipitation assays. Immunoprecipitated FLAG-MLL and co-immunoprecipitated Skp2 were determined by anti-FLAG and anti-Skp2 immunoblots, respectively. b, 293T cells synchronized at mid-S phase were treated with HU in the presence of LY294002 (50 µM) or Wortmannin (10 µM) and then subjected to anti-MLLC180 immunoblots. c, Genetic deletion of ATR prevented the DNA damage-induced accumulation of MLL. MEFs were treated with γ-IR (5 Gy) or HU and then subjected to anti-MLL immunoblots.
As DNA damage signals disrupt the MLL-Skp2 interaction, ATR might directly or indirectly phosphorylate MLL and/or Skp2, leading to their dissociation. Bioinformatics analysis (scansite.mit.edu) identified a candidate ATM/ATR site at conserved serine 516 (LPISQSP) of MLL (Fig. 3a). In contrast to the disrupted interaction between wild-type MLL and Skp2 upon HU treatment, a comparable interaction was detected between MLL(S516A) and Skp2 irrespective of DNA insults (Fig. 3b), suggesting that S516 phosphorylation dissociates MLL from Skp2. The S516 of MLL became phosphorylated after HU treatment (Fig. 3b), correlating with diminished MLL-Skp2 interaction. To assess if ATR can directly phosphorylate MLL, we performed in vitro kinase assays using affinity-purified ATR and recombinant MLL proteins. As ATR needs to be fully activated by TopBP1 or Claspin25, purified ATR only weakly phosphorylated wild-type but not S516A MLL protein (Fig. 3c). Once activated by TopBP1, ATR effectively phosphorylated MLL, detected by both anti-phospho-ATM/ATR substrate and anti-phospho-MLL(S516) antibodies (Fig. 3c). To assess the functional significance of S516 phosphorylation, MLL−/− MEFs reconstituted with wild-type or S516A human MLL (Supplementary Fig. 5) were subjected to RDS and metasphase spread assays (Fig. 3d, e). Unlike wild-type MLL, S516A MLL failed to fully rescue the RDS defects and chromatid-type errors of MLL−/− MEFs (Fig. 3d, e). Although S516A MLL is defective in the S phase checkpoint, its interaction with Menin and WDR5 and targeting to the promoters of HoxA9 and Meis1 remain intact as wild-type MLL (Supplementary Fig. 6).
Figure 3. Phosphorylation of MLL at serine 516 by ATR disrupts its interaction with Skp2 and is required for the integrity of S phase checkpoint.
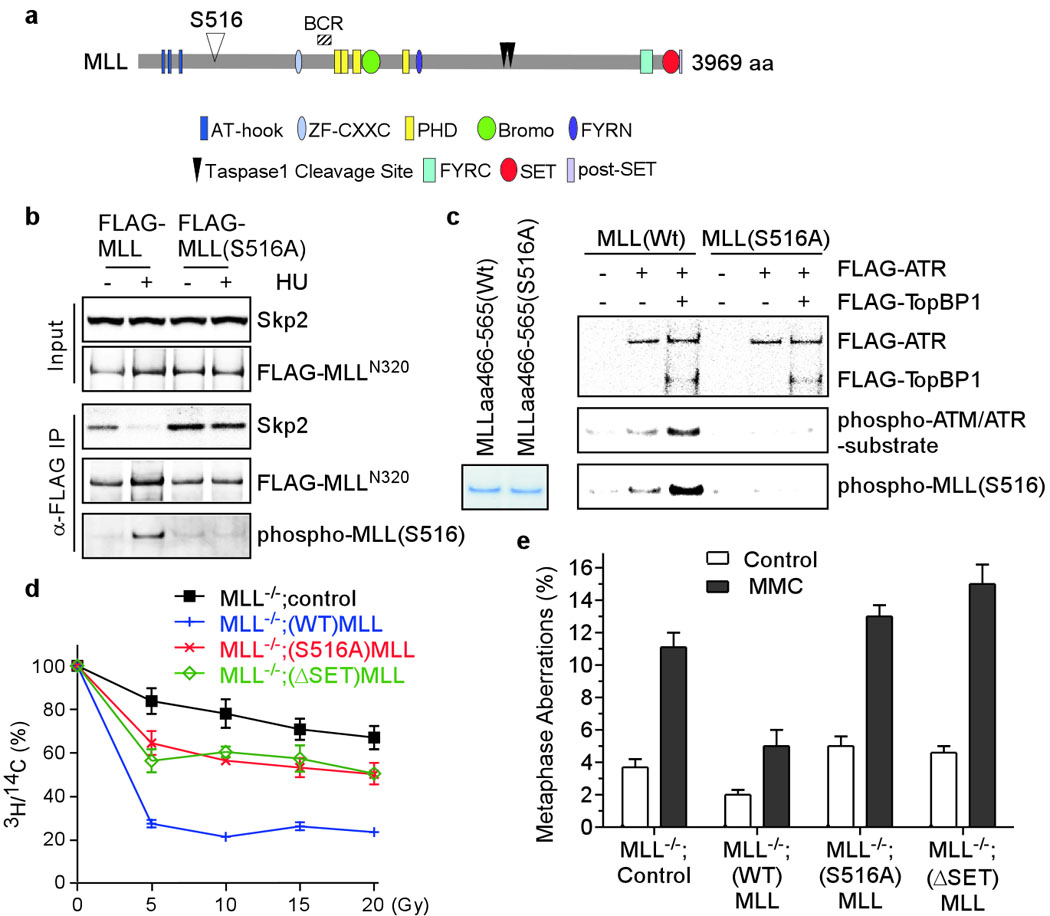
a, A diagram of MLL domains. b, 293T cells were transfected with FLAG-MLL for 2 days, treated with 1 mM HU for 1 hour, and then subjected to anti-FLAG co-immunoprecipitation assays. Results were analyzed by immunoblots. c, Recombinant MLL (aa 466–565) was incubated with FLAG-ATR ± FLAG-TopBP1 for in vitro kinase reactions. Results were assessed by anti-phospho-ATM/ATR substrate and anti-phospho-MLL(S516) Western blots. d, FRT+;MLL−/− MEFs (Supplementary Fig. 5) reconstituted with wild-type, S516A or ΔSET human MLL were subjected to RDS assays. e, MEFs described in d were subjected to metaphase spread analysis. Data shown in d and e are mean ± s.d. of three independent experiments.
To investigate the mechanism(s) by which MLL engages S phase checkpoint, we determined if MLL deficiency affects the upstream signal transduction upon DNA damage. Like wild-type MEFs, MLL−/− cells were competent in the formation of γH2AX foci (Supplementary Fig. 7a). The autophosphorylation of ATM, the S139 phosphorylation of H2AX, the ATM-mediated activating phosphorylation of Chk2, and the ATR-mediated activating phosphorylation of Chk1 were not affected by the MLL deficiency (Supplementary Fig. 7b, c). Furthermore, the phosphorylation of SMC1 by ATM/ATR, the degradation of CDC25A signaled by Chk kinases, and the Y15 phosphorylation of CDK2 were also not altered in MLL deficient cells (Supplementary Fig. 7d).
The key effector step at the initiation of DNA replication is the loading of CDC45 onto the pre-replication complex (pre-RC) which consists of ORC and the MCM2-7 complex26. The chromatin association of CDC45 correlates well with DNA synthesis, and thus marks the firing of replication origins26. Supporting the role of MLL in S phase checkpoint, an aberrant chromatin association of CDC45 was observed in MLL deficient cells upon DNA insults, whereas the chromatin association of MCM2 was not altered (Fig. 4a and Supplementary Fig. 8). The S phase checkpoint commenced at ATM/ATR ultimately inhibits CDC45 loading, in part through inactivating CDK2 and DDK3,3,27–28, which was not affected by the MLL deficiency (Supplementary Figs. 7d and 9). Furthermore, the MLL-mediated inhibition of chromatin association of CDC45 was transcription-independent (Supplementary Fig. 10). ChIP (chromatin immunoprecipitation) assays on the β-globin origin, a well characterized late replication origin in 293T cells29, demonstrated that MLL accumulated and methylated H3K4 at the β-globin origin upon DNA damage, resulting in a decreased CDC45 occupancy (Fig. 4b). These data suggest that the histone methyl transferase (HMT) activity of MLL may be required for the execution of S phase checkpoint, which is corroborated by the inability of ΔSET MLL mutant to fully correct the RDS defects and chromatid-type errors of MLL−/− MEFs (Fig. 3d, e). In fact, histone H3 directly interacted with CDC45 and this interaction was greatly compromised when H3K4 was trimethylated (Fig. 4c, d). Data presented thus far support a model in which stabilized MLL accumulates on chromatin to methylate H3K4 at late replication origins upon S phase checkpoint activation, which inhibits CDC45 loading and thereby delays DNA replication (Fig. 4h). Although MLL likely methylates H3K4 at all late replication origins upon DNA damage, genome-wide studies are required to conclude such a mechanism.
Figure 4. Upon DNA damage MLL accumulates on chromatin to methylate H3K4, resulting in diminished CDC45 loading.

a, 293T cells were synchronized in S phase, treated with 25 µM etoposide, fractionated, and subjected to immunoblots using the indicated antibodies. TCE, total cell extracts. b, ChIP assays were performed using the indicated antibodies after etoposide treatment. Immunoprecipitated β-globin replication origin was amplified by PCR. c. Anti-CDC45 immunoprecipitates from 293T nuclear extracts were assessed by anti-histone H3 and anti-H3K4me3 immunoblots. d, The indicated biotin-conjugated histone H3 tails (aa1–21, 2µg) were incubated with nuclear extracts or recombinant GST-CDC45, and then subjected to pull down assays using strepavidin beads. The precipitated H3 peptides were visualized by Coomassie blue stain, whereas co-precipitated CDC45 was detected by anti-CDC45 immunoblots. e, The indicated Jurkat cells were treated with 25 µM etoposide for 90 minutes, fractionated, and then subjected to immunoblots using the indicated antibodies. Of note, MLLC180 denotes endogenous wild-type MLL. f, ChIP assays were performed using the indicated antibodies on Jurkat cells after etoposide treatment. Immunoprecipitated β-globin replication origin was amplified by PCR. g, 293T cells transfected with FLAG-MLL ± FLAG-MLL-AF9 were treated with HU for 90 minutes and then subjected to co-immunoprecipitation assays using the indicated antibodies. h, Model depicts how MLL and MLL-fusions affect the S checkpoint response.
We next investigated how MLL-fusions function as dominant negative mutants in the S phase checkpoint. Of note, all MLL-fusions have lost their C-terminal SET domain (Fig. 3a) and tested MLL-fusions were resistant to the Skp2-mediated degradation due to impaired interaction12. Accordingly, MLL-AF4 and MLL-AF9 were not further stabilized upon DNA damage despite the increased S516 phosphorylation (Supplementary Fig. 11). Chromatin association assays revealed an aberrant loading of CDC45 upon DNA damage in Jurkat T cells that stably express MLL-AF4 or MLL-AF9 (Fig. 4e). While MLL-AF4 and MLL-AF9 stably bound to chromatin, wild-type MLL (MLLC180) failed to accumulate on chromatin in the presence of MLL-Fusions (Fig. 4e). Consequently, ChIP assays demonstrated a stable association of FLAG-MLL-fusions, an ablated accumulation of wild-type MLL (MLLC180), a failed induction of H3K4me3, and an aberrant loading of CDC45 on the late replication origin upon genotoxic stress (Fig. 4f). Altogether, these data suggest that MLL-fusions function as dominant negative mutants by preventing the stabilization of wild-type MLL upon DNA insults. As the ATR-mediated phosphorylation and dissociation of wild-type MLL from Skp2 constitutes the initiating step of MLL-mediated S phase checkpoint response, MLL-fusions might prevent the stabilization of wild-type MLL upon DNA damage by abrogating the S516 phosphorylation of wild-type MLL and thus preserving the interaction between wild-type MLL and Skp2. Since ATR associates with chromatin and becomes active upon DNA insults25, the pre-occupancy of MLL-fusions on chromatin would prohibit the access of wild-type MLL to the activated ATR. Co-expression of MLL-AF9 with wild-type MLL abrogated the S516 phosphorylation of wild-type MLL but not MLL-AF9 upon DNA insults, leading to a constitutive interaction/degradation of wild-type MLL by Skp2 (Fig. 4g). In summary, MLL-fusions stably associate with chromatin and prevent the stabilization/targeting of wild-type MLL to the late replication origin upon DNA damage, which abolish the trimethylation of H3K4 and result in an aberrant loading of CDC45 and dysfunction of S phase checkpoints (Fig. 4e–h and Supplementary Fig. 12). The discovery of MLL in executing S phase checkpoint provides new mechanistic insights concerning not only normal cell biology but also the pathology underlying MLL leukemias.
METHODS SUMMARY
Human embryonic kidney 293T cells, a cell line commonly utilized in assessing the S phase checkpoint response30, was synchronized as previously described12. Both shRNA-mediated knockdown and qRT-PCR of MLL have been described12. Genetically defined MEFs including ATRf/+, ATRf/−, ATRf/+;ATM−/−, ATRf/−;ATM−/−, DNA-PKcs−/−, and DNA-PKcs+/+ MEFs have been described23–24. The MLLex7(stop)CBP mice and the generation of MPCs have been described20–21. The modified Flp-In system is illustrated in Supplementary Fig. 5.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
METHODS
Plasmid constructions, antibodies, and Western blots
The N-terminal FLAG-tagged full-length human MLL cDNA was inserted into the pCI-neo vector (Promega). The S516A MLL mutant was generated using the Quickchange site-directed mutagenesis kit (Stratagene). The cDNA of wild-type and mutant MLL was inserted into pcDNA5-FRT to generate constructs utilized in the Flp-In system (Invitrogen). creERT2 was inserted into the retroviral vector MSCV Puro (Clontech). pBJ5.1-FLAG-ATR and pFLAG-TopBP1 were provided by Drs. Stuart Schreiber and Weei-Chin Lin31, respectively. Transfection was performed according to the manufacture’s protocol using Lipofectamine 2000 (Invitrogen). The anti-N-terminus MLL (anti-MLLN320) and anti-C-terminus MLL (anti-MLLC180) antibodies have been described12,32. Polyclonal anti-phosphoMLL(S516) antibody was raised against EVHPPLPI(p)SQSPENE (Antagene). Commercially available antibodies against Skp2 (Santa Cruz Biotechnology), CDC45 (Santa Cruz Biotechnology), MCM2 (BD Transduction Laboratories), β-Actin (Sigma), FLAG (Sigma), phospho-(Ser/Thr) ATM/ATR substrate antibody (Cell Signaling Technology), Chk1 (Cell Signaling Technology), phospho-Chk1(Ser317) (Cell Signaling Technology), Chk2 (Cell Signaling Technology), phospho-Chk2(Thr68) (Cell Signaling Technology), phospho-Histone H2AX(Ser139) (Abcam), phospho-ATM(Ser1981) (Abcam), Menin (Abcam), WDR5 (Abcam), phosphor-Histone H1(Thr146) (Abcam), Histone H1 (Abcam), Histone H3 (Abcam), Histone H3K4me3 (Abcam), phospho-MCM2(Ser53) (Abcam), SMC1 (Abcam), phospho-SMC1(Ser966) (Abcam), Cdc25A (Abcam), CDK2 (Abcam), and phospho-CDK2(Y15) (Abcam) were purchased from individual venders for Western blot analyses. Antibodies were detected using the enhanced chemiluminescence method (Western Lightning, PerkinElmer). Western blot signals were acquired with the LAS-3000 Imaging system (FujiFilm) and then analyzed by ImageGauge software (FujiFilm) as previously described33.
Cell culture and synchronization
293T, NIH3T3, HeLa and hTERT-BJ1 (Invitrogen) cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS), nonessential amino acids, L-glutamine (Invitrogen). MLL−/−, ATRf/+, ATRf/−, ATRf/+;ATM−/−, ATRf/−;ATM−/−, DNA-PKcs−/−, and DNA-PKcs+/+ MEFs have been described23–24,34. To generate ATR−/− MEFs, ATRf/− MEFs were first stably transduced with creERT2 and then treated with 0.5 µM 4-hydroxytamoxifen to delete ATR. Resulted ATR−/− MEFs were confirmed by genomic PCR and immediately subjected to experimentations. Human embryonic kidney 293T cells are commonly used to assess S phase checkpoint defects30. Cell synchronization procedures have been described12 and confirmed by FACS analyses. In brief, to synchronize in G1 phase, 293T cells were treated with 200 µM mimosine for 16 hours. To enrich in S phase, 293T cells were synchronized by double-thymidine block and released for 3 hours. To synchronize cells in M phase, 293T cells were treated by nocodazole for 18 hours.
Generation of myeloid precursor cells (MPCs)
The MLL-CBP knock-in allele (MLLex7-CBP) was designed to recapitulate human t(11;16) MLL-CBP myeloid leukemias21. The MLLex7(stop)CBP allele was engineered by inserting a floxed “stop cassette” proximal to the knock-in CBP fusion that could be removed upon the expression of cre recombinase, generating the MLLex7-CBP allele (Supplementary Fig. 2b). The generation of MPCs has been described20. Mice were sacrificed by CO2 asphyxiation and bone marrow was harvested from 6 MLL+/ex7(stop)CBPand 6 age-, sex-matched, wild-type littermate mice (Supplementary Fig. 2)21. MPCs were isolated by lineage depletion (Lin: CD3, Gr-1, B220, ter119) followed by positive selection of Sca-1+ cells using magnetic beads (Miltenyi). These cells were co-cultured with irradiated (30 Gy) NIH 3T3 hph-HOX11 retrovirus producer cells in infection medium (IMDM, 20% FBS, 100 U/ml penicillin-streptomycin, 2 mM glutamine, 10 ng/ml IL-3, 20 ng/ml stem cell factor, 10 ng/ml GMCSF, and 2 ng/ml GCSF) for 3 days. Cells grew in suspension were expanded in IMDM supplemented with 20% FBS, 100 U/ml penicillin-streptomycin, 2 mM glutamine, and 10% WEHI conditioned medium as the source of IL-3. To generate MLL+/ex7-CBP MPCs, MLL+/ex7(stop)CBP MPCs were first retrovirally transduced with creERT2 by spin inoculation, selected with puromycin, and then treated with 0.5 µM 4-OHT for 2 days to delete the “stop cassette”, which was then confirmed by genomic PCR as described21.
Reconstitute MLL−/− MEFs using the Flp-In system
MLL−/− MEFs were engineered to carry one FRT cassette (Supplementary Fig. 5a). These FRT+;MLL−/− MEFs were co-transfected with pcDNA-FRT vectors expressing wild-type, S516A, or ΔSET human MLL plus pOG44 which encodes the Flp recombinase and subjected to hygromycin B selection. Recombination was verified by genomic DNA PCR and the protein expression was determined by Western blot analyses.
Generate Jurkat MLL-AF4 and Jurkat MLL-AF9 cells using the Flp-In system
The Flp-In™ Jurkat cells (Invitrogen) were co-transfected with pcDNA-FRT MLL-AF4 or MLL-AF9 expressing vectors plus pOG44 which encodes the Flp recombinase and subjected to hygromycin B selection. Recombination was verified by genomic DNA PCR.
Radio-resistant DNA synthesis assays
The procedures of RDS have been described35. In brief, cells were first labeled with 20 nCi/ml 14C-thymidine for 24 h to monitor the rate of proliferation of individual cell lines under normal growth condition. These cells were then irradiated with indicated dose of γ-IR. Forty-five minutes post-IR, culture medium was replaced with 2.5 µCi/ml 3H-thymidine containing medium. Thirty minutes after 3H-thymidine labeling, cells were then lysed in 0.25 M NaOH. The radioactivity was measured using an LS 6500 Liquid Scintillation Counter and the [3H]/[14C] ratio was calculated as described35.
shRNA-mediated knockdown, qRT–PCR, and immunofluorescence assays
Both shRNA-mediated knockdown and qRT-PCR of MLL have been described12. MEFs grown on LabTek II chamber slides (Nunc) were treated with 25 µM etoposide for 15 minutes, washed twice with PBS, fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked with 3% BSA in PBS for 60 min. Fixed cells were then incubated with anti-γH2AX (Abcam) antibody for 1 hour, washed twice with PBS, and incubated with Alexa488-conjugated anti-mouse (Molecular Probes) antibody for an additional 30 minutes. Microscopy was performed using an Olympus IX51 microscope attached to a Spotcam (Diagnostic). Images were captured and analyzed by SPOTcam program as described36.
Metaphase spread, chromatin association, and ChIP assays
Metaphase spread assays have been described37. In brief, MEFs of the indicated genotypes were treated with 20 ng/ml mitomycin C for 36 hours before subjected to metaphase spread analysis. Chromatin-enriched fractions were purified as described38. ChIP assays were performed using the Magna ChIP A Kit (Millipore) according to the manufacture’s protocol. One µg of pre-immune rabbit IgG, anti-CDC45, or anti-MLLC180 antibody was utilized for each ChIP reaction. Precipitated DNA was amplified using β-globin origin, HoxA9 and Meis1 specific primers29,17 and the PCR products were analyzed by agarose gel electrophoresis (2%) and visualized with ethidium bromide under UV.
In vitro kinase assays
293T cells were transfected with pcDNA3-FLAG-ATR or pFLAG-TopBP1 using Lipofectamine 2000 (Invitrogen). Twenty-four hours post transfection, cells were treated with 20 µM of etoposide for 1 hour and then lysed in RIPA buffer. FLAG-tagged protein was immunoprecipitated with anti-FLAG M2 agarose (Sigma) and eluted with 3×FLAG peptide. His-tagged wild-type and S516A MLL aa 466–565 fragments were purified using TALON beads (Clontech) and eluted with Imidazole according to the manufacturer’s protocol. In vitro kinase assays were performed by incubating 0.5 µg of recombinant MLL fragments with purified ATR plus/minus TopBP1 in kinase buffer that contains 10 µM ATP at 30°C for 30 minutes. Proteins were separated by SDS-PAGE and phosphorylation was visualized by indicated phospho-specific antibodies.
Peptide pull-down assays
For the preparation of nuclear extracts, 293T nuclei were resuspended in RIPA buffer, sonicated, and precleared for 1 hour with streptavidin beads (GE Healthcare). Individual biotin conjugated histone H3 tails (aa 1–21, 2µg) (Millipore) were incubated with either nuclear extracts or 1µg of recombinant GST-CDC45 in 300 µl of binding buffer (50 mM Tris-HCl, pH 7.5, NaCl 150 mM, Triton X-100 0.1%), precipitated using streptavidin beads, resolved in NuPAGE, and subjected to Western or Coomassie blue stain analyses.
BrUTP incorporation assays
BrUTP incorporation assay was performed using Lipofectamine 2000 (Invitrogen). Briefly, Lipofectamine 2000 was mixed with 10 mM BrUTP (Sigma) and incubated for 15 min at room temperature before transfection. 293T cells were washed twice with OPTI-MEM (Invitrogen), and 100 µl of the transfection mix was added to each well of a chamber slide. After incubation for 2 hours, the cells were fixed with 4% paraformaldehyde and used for immunostaining with BrdU antibody (clone Bu-33 Sigma).
Supplementary Material
Acknowledgements
We thank Drs. Jean Y. Wang and Zhongsheng You for insightful discussions during the inception and the completion of this study, respectively. H.L. is supported by the Scholar award of the American Society of Hematology. The MLL+/ex7(stop)CBP mice were kindly provided by Drs. Scott Armstrong and late Stanley Korsmeyer. This study is supported by CA119008, Scholar award of the American Society of Hematology, and Scholar award of the American Cancer Society to J.J.-D.H. and CA129537/CA123232 to T.K.P.
Footnotes
Author Contributions
H.L. designed and performed the experiments; T.K.P. designed some experiments; S.T., R.K., and T.D.W. performed some experiments; E.J.B. generated essential tools; and E.H.C. and J.J.H. designed the experiments and supervised the project.
Author information
Reprints and permission information is available at www.nature.com/reprints.
References
- 1.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 2.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 3.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 4.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 5.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nature reviews. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Perales S, Cano F, Lobato MN, Rabbitts TH. MLL gene fusions in human leukaemias: in vivo modelling to recapitulate these primary tumourigenic events. International journal of hematology. 2008;87:3–9. doi: 10.1007/s12185-007-0001-3. [DOI] [PubMed] [Google Scholar]
- 7.Liu H, Cheng EH, Hsieh JJ. MLL fusions: pathways to leukemia. Cancer biology & therapy. 2009;8:1204–1211. doi: 10.4161/cbt.8.13.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer C, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23:1490–1499. doi: 10.1038/leu.2009.33. [DOI] [PubMed] [Google Scholar]
- 9.Liedtke M, Cleary ML. Therapeutic targeting of MLL. Blood. 2009;113:6061–6068. doi: 10.1182/blood-2008-12-197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsieh JJ, Cheng EH, Korsmeyer SJ. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell. 2003;115:293–303. doi: 10.1016/s0092-8674(03)00816-x. [DOI] [PubMed] [Google Scholar]
- 11.Jude CD, et al. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu H, Cheng EH, Hsieh JJ. Bimodal degradation of MLL by SCFSkp2 and APCCdc20 assures cell cycle execution: a critical regulatory circuit lost in leukemogenic MLL fusions. Genes Dev. 2007;21:2385–2398. doi: 10.1101/gad.1574507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeda S, et al. Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev. 2006;20:2397–2409. doi: 10.1101/gad.1449406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 15.Milne TA, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 16.Kumar AR, et al. Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood. 2004;103:1823–1828. doi: 10.1182/blood-2003-07-2582. [DOI] [PubMed] [Google Scholar]
- 17.Milne TA, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia ZB, et al. The MLL fusion gene, MLL-AF4, regulates cyclin-dependent kinase inhibitor CDKN1B (p27kip1) expression. Proc Natl Acad Sci U S A. 2005;102:14028–14033. doi: 10.1073/pnas.0506464102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell. 2007;27:107–119. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 20.Zinkel SS, et al. A role for proapoptotic BID in the DNA-damage response. Cell. 2005;122:579–591. doi: 10.1016/j.cell.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, et al. Conditional MLL-CBP targets GMP and models therapy-related myeloproliferative disease. Embo J. 2005;24:368–381. doi: 10.1038/sj.emboj.7600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eguchi M, et al. MLL chimeric protein activation renders cells vulnerable to chromosomal damage: an explanation for the very short latency of infant leukemia. Genes Chromosomes Cancer. 2006;45:754–760. doi: 10.1002/gcc.20338. [DOI] [PubMed] [Google Scholar]
- 23.Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–628. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taccioli GE, et al. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity. 1998;9:355–366. doi: 10.1016/s1074-7613(00)80618-4. [DOI] [PubMed] [Google Scholar]
- 25.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- 27.Santocanale C, Diffley JF. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–618. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- 28.Sheu YJ, Stillman B. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol Cell. 2006;24:101–113. doi: 10.1016/j.molcel.2006.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goren A, Tabib A, Hecht M, Cedar H. DNA replication timing of the human beta-globin domain is controlled by histone modification at the origin. Genes Dev. 2008;22:1319–1324. doi: 10.1101/gad.468308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim DS, et al. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 31.Liu K, Paik JC, Wang B, Lin FT, Lin WC. Regulation of TopBP1 oligomerization by Akt/PKB for cell survival. Embo J. 2006;25:4795–4807. doi: 10.1038/sj.emboj.7601355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsieh JJ, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23:186–194. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim H, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 34.Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci U S A. 1998;95:10632–10636. doi: 10.1073/pnas.95.18.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theunissen JW, Petrini JH. Methods for studying the cellular response to DNA damage: influence of the Mre11 complex on chromosome metabolism. Methods Enzymol. 2006;409:251–284. doi: 10.1016/S0076-6879(05)09015-4. [DOI] [PubMed] [Google Scholar]
- 36.Tu HC, et al. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc Natl Acad Sci U S A. 2009;106:1093–1098. doi: 10.1073/pnas.0808173106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta A, et al. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol Cell Biol. 2008;28:397–409. doi: 10.1128/MCB.01045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.