

Abstract
Tolfenamic acid (TA) is a non-steroidal anti-inflammatory drug associated with anti-tumorigenic and pro-apoptotic properties in animal and in vitro models of cancer. However, the underlying cellular mechanisms by which TA exerts its effects are only partially understood. Activating transcription factor 3 (ATF3) is a member of the ATF/CREB subfamily of the basic region-leucine zipper family and has been known as a tumor suppressor in human colorectal cancer cells. The present study was performed to observe whether ATF3 mediates TA-induced apoptosis and to elucidate the molecular mechanism of ATF3 transcription induced by TA. TA treatment and ectopic expression of ATF3 increased apoptosis whereas knockdown of ATF3 resulted in significant repression of TA-activated apoptosis. The TA treatment also induced ATF3 promoter activity. Internal deletion and point mutation of the predicted ATF/C/EBP binding site in ATF3 promoter abolished luciferase activation by TA. Overexpression of ATF2 resulted in significant increase of ATF3 promoter activity, and electrophoretic mobility shift assay identified this region as a core sequence to which ATF2 binds. TA treatment resulted in an increase of ATF2 phosphorylation, which was followed by a subsequent increase of ATF3 transcription. Knockdown of ATF2 abolished TA-induced ATF3 expression. We further provide evidence that TA leads to increases of phospho-p38 MAPK, JNK, and ERK levels. Inhibition of these pathways using selective inhibitors and dominant negative constructs ameliorated TA-induced ATF3 expression and promoter activities. The current study demonstrates that TA stimulates ATF3 expression and subsequently induces apoptosis. These pathways are mediated through phosphorylation of ATF2, which is mediated by p38 MAPK, JNK, and ERK-dependent pathways.
Keywords: Tolfenamic acid, ATF2, ATF3, apoptosis, colorectal cancer
Introduction
Colorectal cancer is an important public health problem in the western world (Parkin et al., 2005) and the third leading cause of cancer-related death in the United States (Jemal et al., 2009). Chemoprevention by non-steroidal anti-inflammatory drugs (NSAIDs) has received much attention as a very attractive and promising strategy. A number of studies have demonstrated an inverse relationship between the consumption of NSAIDs and colorectal cancer (Gupta and Dubois, 2001), and thus molecular mechanisms by which NSAIDs affect chemopreventive activity in human colorectal cancer need to be elucidated.
Tolfenamic acid (TA) is a (N-(2-methyl-3-chlorophenyl)-anthranilic acid that shows a pharmacological profile characteristic of NSAIDs. TA has been broadly used for migraines and has shown fewer side effects than other NSAIDs in the upper gastro-intestine (Hansen, 1994). The first study of TA’s anti-cancer activity was performed in a pancreatic cancer model (Abdelrahim et al., 2006). TA treatment inhibited metastasis and tumorigenesis through suppression of vascular endothelial growth factor (VEGF) and its receptor (VEGFR1) (Abdelrahim et al., 2006; Abdelrahim, 2007). Using human colorectal cancer cells, our group observed that TA stimulates apoptosis and up-regulates the pro-apoptotic protein early growth response-1 (EGR-1), through epithelial-specific Ets-1 (ESE-1)-dependent transcriptional regulation (Lee et al., 2008). These data indicate that TA suppresses tumorigenesis through targeting various molecular mechanisms. Because the underlying cellular mechanisms by which TA exerts its effects are only partially understood, the molecular basis of apoptosis induction and the scope of its action in colorectal cancer need to be elucidated.
Activating transcription factor 3 (ATF3) is an ATF/CREB subfamily member, which is characterized by containing the basic region-leucine zipper (b-ZIP) DNA binding domain (Hai et al., 1988). ATF3 plays diverse biological roles and the expression of ATF3 is dramatically up-regulated in response to a variety of stress conditions in many different tissues (Hai and Hartman, 2001). ATF3 is rapidly induced in cells treated with growth-stimulating factors such as serum and growth factor (Iyer et al., 1999). ATF3 enhances DNA synthesis in hepatocytes (Allan et al., 2001) and involved in serum-induced cell proliferation as a target gene of c-myc (Tamura et al., 2005). However, ATF3 has a dichotomous role in cancer development and it is likely that pro- or anti-apoptotic mechanism of ATF3 is dependent on cell or tissue context (Miyazaki et al., 2009; Yin et al., 2008). The expression of ATF3 was repressed in human colorectal tumors compared to normal adjacent tissue (Bottone et al., 2004). Ectopic expression of ATF3 induced apoptosis (Yamaguchi et al., 2006) and suppressed growth of colorectal cancer cells (Fan et al., 2002) and Ras-stimulated tumorigenesis (Lu et al., 2006). In previous studies, our group reported that ATF3 was induced by treatment with anti-tumorigenic compounds including indole-3-carbinol (Lee et al., 2005), conjugated linoleic acid (Lee et al., 2006), epicatechin (Baek et al., 2004; Cho et al., 2007), berberine (Piyanuch et al., 2007), and phosphoinositide 3-kinase inhibitor (Yamaguchi et al., 2006).
ATF3 expression is influenced by various cell signaling and transcription factors. ATF3 is a key mediator of Krüppel-like factor 6 (KLF6)-induced apoptosis in prostate cancer cells (Huang et al., 2008) and mediates cyclooxygenase inhibitor-stimulated anti-invasive activity in human colorectal cancer (Bottone et al., 2005). ATF3 directly enhances p53 activation (Yan et al., 2005) and stabilize p53 protein (Yan and Boyd, 2006) as well as down-regulates cyclin D1 (Lu et al., 2006) and matrix metalloproteinase-2 expression (Chen and Wang, 2004). ATF3 also plays a crucial role in REIC/Dkk-3-induced apoptosis and down-regulation of inhibition of differentiation-1. (Kashiwakura et al., 2008). Overexpression of ATF3 prolonged the half-life of p73 by inhibiting its ubiquitination and thereby enhancing its transactivation and proapoptotic activities (Oh et al., 2008). ATF3 suppresses Toll-like receptor-mediated pathways (Whitmore et al., 2007) and directly represses nuclear factor erythroid-derived 2-related factor 2 (Nrf2) pathways (Brown et al., 2008). Thus, overexpression of ATF3 negatively regulates tumorigenic pathways through multiple mechanisms, although there are contradictory results in the literature.
In the present study, we investigated the transcriptional mechanism and biological significance of ATF3 expression in response to TA-activated apoptosis in human colorectal cancer model. Here, we report that ATF2 acts as a mediator in activation of the ATF3 gene by TA via MAPK-dependent pathways.
Materials and Methods
Materials
Human cancer cell lines (HCT-116, HT-29, LoVo, and SW480) and other cancer cell lines (A549, MCF-7, PC-3, and Spccy1) were purchased from American Type Culture Collection (Manassas, VA). Culture media were purchase from the followings; McCoy’s 5A (Bio Whittaker, Rockland ME), RPMI1640 (Mediatech, Herndon, VA), Ham’s F-12 (HyClone, Logan, UT), and DMEM (Invitrogen, Carlsbad, CA). Tolfenamic acid was purchased from Cayman Chemical (Ann Arbor, Michigan), and SB203580, U0126, PD98059, and SP600125 were purchased from Calbiochem (San Diego, CA). Antibodies for ATF3, ATF2, phospho-ERK, ERK, actin and siRNA for ATF3 and ATF2 were purchased from Santa Cruz (Santa Cruz, CA). Antibodies for PARP, phospho-JNK, JNK, phospho-p38MAPK, p38MAPK, and phospho-ATF2 (Thr71) were purchased from Cell Signaling (Beverly, MA). All chemicals were purchased from Fisher Scientific (Pittsburgh, PA), unless otherwise specified.
Cell culture
HCT-116 and HT-29 cells were maintained in McCoy’s 5A medium. SW480 cells were maintained in RPMI1640. LoVo, A549 and PC-3 cells were maintained in Ham’s F-12. MCF-7 and Spccy1 cells were maintained in DMEM. All culture media was supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 µg/mL streptomycin.
Promoters
Human ATF3 promoter constructs (pATF3-1850/+34 and pATF3-84/+34) were generously provided by Dr. S. Kitajima (Tokyo Medical and Dental University, Tokyo, Japan) and pATF3-1420/+34, pATF3-718/+34, and pATF3-514/+34 were produced from pATF3-1850/+34 using serial deletion (Cho et al., 2007). The internal deletion or point mutation constructs of the ATF3 promoter were constructed from wild type pATF3-84/+34 using the QuikChange II mutagenesis kit (Stratagene, La Jolla, CA) with indicated primers (Table 1).
Table 1.
Primer sequence for internal deletion or point mutation clones of ATF3 promoters
| Internal deletion |
Forward | Reverse |
|---|---|---|
| ΔIL-6 | 5’-gtaagcttgcaacacggagtaaacgaccgc-3’ | 5’-actccgtgttgcaagcttacttagatcgca-3’ |
| ΔDTF-1 | 5’-gcctgggactggagtaaacgac-3’ | 5’-ggcggcgcggtcgtttactccagtcccagg-3’ |
| ΔGCN-4 | 5’-gcctgggactggcaacacgacgaccgcgcc-3’ | 5’-caggctggcggcgcggtcgtcgtgttgcc-3’ |
| ΔSp1 | 5’-gagtaaacgaagcctgagggctataaaagg-3’ | 5’-ccctcaggcttcgtttactccgtgttgcca-3’ |
| ΔYi | 5’-gagtaaacgaccgcgccgccctataaaagg-3’ | 5’-gttgcatcaccccttttatagggcggcgcgg-3’ |
| ΔGATA | 5’-gaccgcgccgccagcctgagaaggggtgatg-3’ | 5’-gagagcgttgcatcaccccttctcaggctgg-3’ |
| ΔATF/C/EBP | 5’-cctgagggctataaaaggggcgctctccaag-3’ | 5’-cgactgtggcttggagagcgccccttttatag-3’ |
| ΔCBFA-1 | 5’-gtgatgcaacgctctccaagagtcgcacgc-3’ | 5’-gcgcctggctgcgtgcgactcttggagagc-3’ |
| Point mutation | Forward | Reverse |
| mut-22/-19/-18 | 5’-ctataaaaggggtaatataacgctctccaag-3’ | 5’-cttggagagcgttatattaccccttttatag-3’ |
| mut-22/-19 | 5’-ctataaaaggggtaatacaacgctctccaag-3’ | 5’-cttggagagcgttgtattaccccttttatag-3’ |
| mut-19/-18 | 5’-ctataaaaggggtgatataacgctctccaag-3’ | 5’-cttggagagcgttatatcaccccttttatag-3’ |
Expression vectors
Full-length C/EBPα, C/EBPδ, CHOP, GATA5, and NF-IL3 cDNAs were amplified from human lung cDNA (Clontech, Mountain View, CA) using ReadyMix Taq polymerase (Sigma, St. Louis, MO) with indicated primers (Table 2). PCR was performed for 30 cycles at 94°C for 1 min, 55°C for 1 min, and 72°C for 2 min. PCR product of C/EBPα, C/EBPδ, CHOP, GATA5, and NF-IL3 were sub-cloned into pcDNA3.1/V5/His TOPO vector (Invitrogen, Carlsbad, CA) to generate the V5-His-tagged clones. For C/EBPβ expression vector, pOTB7 vector containing full length C/EBPβ cDNA (Open Biosystems, Huntsville, AL) were digested using EcoRI and Xho I and sub-cloned into pcDNA3.1 (Invitrogen) to generate pcDNA3.1/C/EBPβ. The pCG-ATF3 expression construct was generously provided by Dr. T. Hai (Ohio State University, Columbus, OH). The ATF2 expression vector (pEBG2T-GST-ATF2-6His) and CREB expression vector were kindly provided by Dr. Philip Cohen (University of Dundee, Scotland, UK) and Dr. Joo-Heon Yoon (Yonsei University, Seoul, Korea), respectively. The wild type and dominant negative construct of ERK2 were kindly provided by Dr. Melanie Cobb (University of Texas Southwestern, Dallas, TX). The wild type and dominant negative construct of p38 MAPK was previously described (Hao et al., 2007).
Table 2.
Primer sequence for cloning of expression vectors
| Gene | Forward | Reverse |
|---|---|---|
| C/EBPα | 5’-tgccgggagaactctaactc-3’ | 5’-caccggaatctcctagtcctg-3’ |
| C/EBPδ | 5’-aggtgacagcctcgcttg-3’ | 5’-gtatgggtcgttgctgagtctct-3’ |
| CHOP | 5’-agactgatccaactgcagag-3’ | 5’-tgcttggtgcagattcacc-3’ |
| GATA5 | 5’-ccctgcccgctggtcaagaccacg-3’ | 5’-gactcagtgggtggtctgttcca-3’ |
| NF-IL3 | 5’-ttgttctcctacacacatagatagggtaa-3’ | 5’-tcagcataatacaaaatggactgc-3’ |
Caspase 3/7 enzyme activity
Enzyme activity of capsapse 3/7 was analyzed using Apo-ONE Homogeneous Caspase-Glo 3/7 Assay kit (Promega, Madison, WI) according to the manufacturer’s protocol. The cells were harvested with RIPA buffer containing protease inhibitors, and the same volume of caspase-Glo 3/7 reagent was added to the cell lysates (50 µg protein) in 96-well plates and incubated at room temperature in the dark for 1h. The luminescence was measured using a FLX800 microplate reader (BioTek, Winooski, VT).
Transient transfections
Transient transfections were performed using Lipofectamine (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction. HCT-116 cells were plated in 12-well plates at the concentration of 2×105cells/well. The next day, plasmid mixtures containing 0.5 µg of ATF3 promoter linked to luciferase and 0.05 µg of pRL-null vector were transfected for 5 h and rescued with fresh media for 24 h. The transfected cells were exposed to DMSO or 30 µM of TA for 24 h. The cells were harvested in 1X luciferase lysis buffer, and luciferase activity was normalized to the pRL-null luciferase activity using a dual luciferase assay kit (Promega, Madison, WI). For the co-transfection experiment, 0.25 µg of ATF3 promoter and 0.25 µg of expression vectors were co-transfected with 0.05 µg of pRL-null vector as described above.
Isolation of RNA and semi-quantitative RTPCR
Total RNA was prepared using a RNA isolation kit (Eppendorf, Hamburg, Germany). Total RNA (1 µg) was reverse-transcribed with the iScript cDNA kit (BioRad, Hercules, CA) according to the manufacturer’s instruction. PCR was carried out using ReadyMix Taq polymerase (Sigma) with primers for human ATF3 and GAPDH as follows: ATF3: forward 5’-gtttgaggattttgctaacctgac-3’, and reverse 5’-agctgcaatcttatttctttctcgt-3’; GAPDH: forward 5’- gggctgcttttaactctggt-3’, and reverse 5’- tggcaggtttttctagacgg-3’.
Western analysis
Cells were washed with PBS and cell lysates were isolated in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (1 mM PMSF, 5 µg/ml aprotinin, 5 µg/ml leupeptin) and phosphatase inhibitors (1 mM Na3VO4, 1 mM NaF) and centrifuged at 10,000×g for 5 min at 4°C. Protein concentration was determined by the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL) using bovine serum albumin (BSA) as the standard. The proteins were separated on SDS-PAGE and transferred to nitrocellulose membranes (Osmonics, Minnetonka, MN). The membranes were incubated with a specific primary antiserum in TBS containing 0.05% Tween 20 (TSB-T) and 5% nonfat dry milk at 4°C overnight. After three washes with TBS-T, the blots were incubated with peroxidase-conjugated IgG for 1 h at room temperature, visualized using ECL (Amersham Biosciences, Piscataway, NJ) and quantified by Scion Image Software (Scion Corp., Frederick, MD).
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared by the manufacturer’s protocols (Active Motif, Carlsbad, CA). Oligonucleotide probes were end-labeled with biotin using the following sequences: 5'-aggggtgatgcaacgctctcaggggtgatgcaacgctctc-3'. Nuclear protein (5 µg) was incubated with biotin-labeled oligonucleotide probes (100 nM) and 1X binding buffer (Promega, Madison, WI) at room temperature for 20 min. For competition assay, nuclear extracts were preincubated with the unlabeled oligonucleotide (10x or 100x) for 10 min. For supershift assay, nuclear extracts were preincubated with antibodies for phosphor-ATF2 or ATF2 for 10 min prior to binding reactions. DNA-protein complexes were resolved by 5% nondenaturing polyacrylamide gel and developed using the protocol of LightShift Chemiluminescent EMSA kit (Pierce, Rockford, IL).
Statistical analysis
Statistical analysis was performed with Student’s unpaired t test, with statistical significance set at *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Results
TA induced ATF3 expression in human colorectal cancer and other cancer cells
A number of studies indicate that NSAIDs prevent or suppress tumor development in human colorectal cancer (Gupta and Dubois, 2001). To observe which NSAIDs induce ATF3 expression, we treated HCT-116 cells for 24 h with 30 µM of various NSAIDs: conventional (diclofenac, ibuprofen, aspirin, tolfenamic acid, naproxen) and COX-2 selective (SC-236, DFU, celecoxib) or COX-1 selective (SC-560). As a result, TA and celecoxib increased ATF3 expression (Fig. 1A). Sulindac sulfide also increased ATF3 expression in these cells (data not shown). The level of ATF3 protein dramatically rose in the cells treated with 20 µM TA for 24 h, and ATF3 mRNA increased in a dose-dependent manner (Fig. 1B). We also tested ATF3 expression in other colorectal or types of cancer cells (Fig. 1C and D, ). The increased ATF3 expression was observed in HT-29, LoVo and SW480 cells in different concentrations, indicating that ATF3 induction by TA is observed in other colorectal cancer cells. TA also induced ATF3 expression in lung (A549) and prostate (PC-3) cancer cells, but not in breast (MCF-7) and head and neck (Spccy1) cancer cells. It is notable that basal expression of ATF3 is very high in Spccy1 cells.
Figure 1. Induction of ATF3 expression in tolfenamic acid (TA)-treated cancer cells.
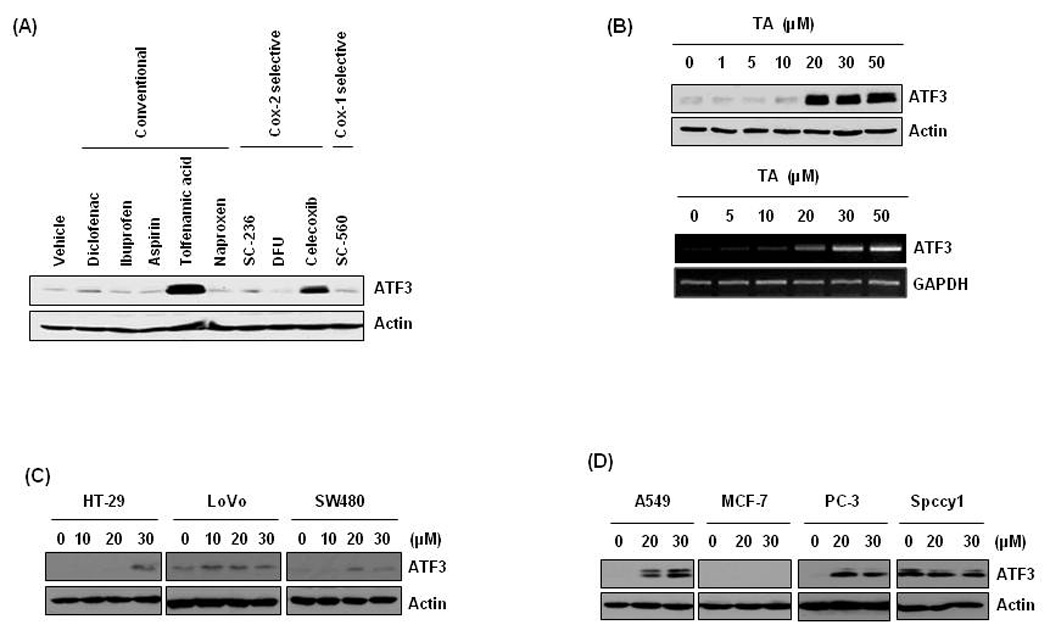
(A) HCT-116 cells were treated with various NSAIDs for 24 h at 30 µM. Total cell lysates were harvested and subsequently Western blot analysis was performed for ATF3 and actin. (B) The cells were treated with 0, 1, 5, 10, 20, 30, and 50 µM of TA for 24 h and Western blot was performed for ATF3 and actin (upper panel). The cells were treated with 0, 5, 10, 20, 30, and 50 µM of TA for 24 h and semiquantatitive RTPCR was performed for ATF3 and GAPDH (lower panel). (C) Other human colorectal cancer cells (HT-29, LoVo and SW480) were treated with 0, 10, 20 and 30 µM of TA for 24 h. (D) Lung (A549), breast (MCF-7), prostate (PC-3), and head and neck (Spccy1) cancer cells were treated with 0, 20, and 30 µM of TA for 24 h.
ATF3 mediated TA-induced apoptosis
Previously, we reported that human colorectal cancer cells treated with TA increased apoptosis in dose-dependent manner (Lee et al., 2008). To see whether ATF3 expression affects apoptosis, enzyme activity of caspase 3/7 was measured in empty- (CONT) or ATF3 expression vector (pCG-ATF3)-transfected cells. As shown in Fig. 2A, overexpression of ATF3 increased caspase 3/7 activity, indicating that ATF3 is one of pivotal genes in the pathway of apoptosis. Next, to investigate whether TA-induced apoptosis is associated with ATF3 expression, the cells were transfected with ATF3 siRNA, and PARP cleavage (Fig. 2B) and capsapase activity (Fig. 2C) were measured. As a result, knockdown of ATF3 significantly suppressed PARP cleavage and enzyme activity, suggesting that ATF3 at least partially plays a significant role in apoptosis induced by TA.
Figure 2. Role of ATF3 expression in TA-induced apoptosis.
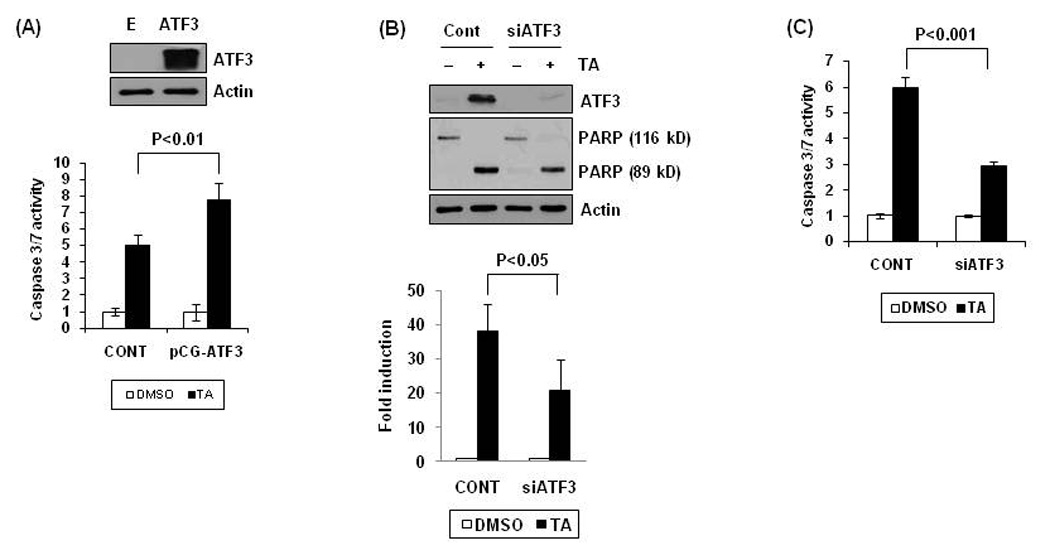
(A) pCG-ATF3 expression vector was transfected into cells using Lipofectamine and then the cells were treated with 30 µM of TA for 24 h. Caspase 3/7 enzyme activity was measured as described in Materials and Methods. The graph represents of three independent experiments. (B) HCT-116 cells were transfected with control siRNA (100 nM) or ATF3 siRNA (100 nM) for 24 h using a TransIT-TKO transfection reagent and then treated with 30 µM of TA for 24 h. Western blot analysis was performed for ATF3, PARP, and actin (upper panel). Data from three independent experiments were densitometrically analysed using Scion Image (Scion Corporation, Frederick, MD) and cleaved PARP versus actin was quantified and expressed as fold induction (Lower panel). (C) After transfection with control or ATF3 siRNA as described in (B), caspase 3/7 enzyme activity was measured. The data represent mean ± SD from three independent experiments.
Effect of TA on ATF3 gene promoter activity
To investigate whether TA affects transcriptional regulation of the ATF3 gene, promoter activity was measured using five serial deletion constructs (pATF3-1850/+34, pATF3-1420/+34, pATF3-718/+34, pATF3-514/+34 and pATF3-84/+34) (Cai et al., 2000). The promoters were transfected into HCT-116 cells and treated with 30 µM of TA for 24 h. As shown in Fig. 3A, TA treatment resulted in a dramatic increase of promoter activity, and fold inductions of luciferase activities were 7.7, 6.1, 6.2, 6.9, and 4.8 in pATF3-1850/+34, pATF3-1420/+34, pATF3-718/+34, pATF3-514/+34 and pATF3-84/+34-transfected cells, respectively, whereas fold induction of luciferase in empty vector-transfected cells was 1.4. These data indicate that the −84 and +34 region of promoter is responsible for TA’s effects. To identify potential regulatory cis-acting elements that mediate the stimulatory effects of TA, the TFSEARCH Program was used to search for regions of conserved transcription factor binding site within the −84 and +34 region. The ATF3 gene promoter (pATF3-84/+34) contained multiple potential transcription factor-binding sites including IL-6, DTF-1, GCN-4, Sp1, Yi, GATA, ATF/C/EBP, and CBFA-1 (Fig. 3B, upper panel). To confirm the responsible site for the transactivation of ATF3 gene by TA, we constructed eight deletion clones lacking each binding site. HCT-116 cells were transfected with deletion constructs and then exposed to DMSO or TA for 24 h, and subsequently luciferase activity measured. As shown in Fig. 3B (lower panel), transfection of wild type pATF3-84/+34 promoter increased luciferase activity by 5.2-fold whereas transfection of promoter lacking the ATF/C/EBP binding site increased luciferase activity by 1.2-fold, which is the same with basal induction by TA found in cells transfected with empty vector (pGL3-Basic). Internal deletion of IL-6, Sp1, Yi, and CBFA-1 binding sites slightly increased luciferase activity induced by TA. Other deletion constructs did not block TA-induced luciferase activities.
Figure 3. ATF3 promoter assay in TA-treated HCT-116 cells.
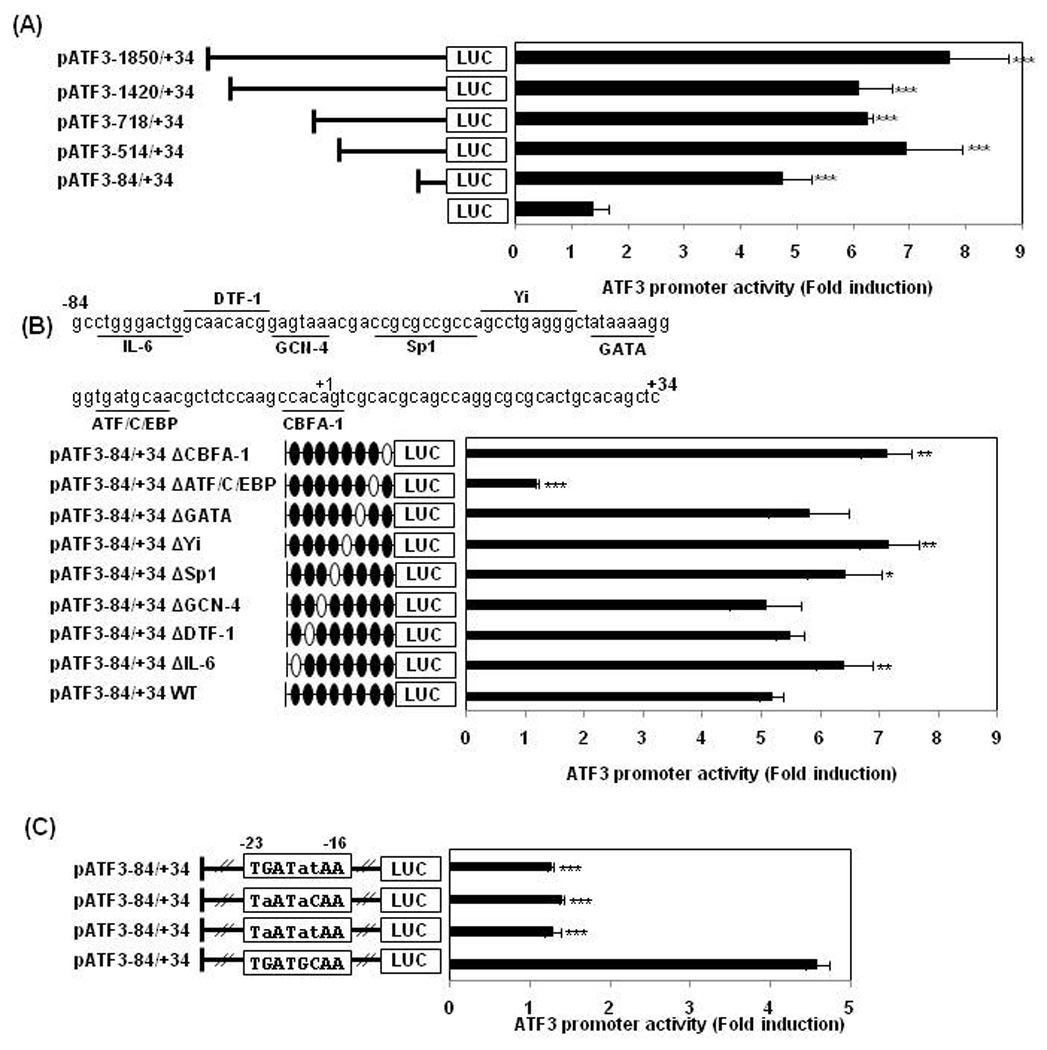
(A) Five deletion ATF3 promoter constructs (0.5 µg) were co-transfected with pRL-null vector (0.05 µg) into HCT-116 cells. The cells were treated with DMSO or 30 µM of TA for 24 h and luciferase activity was measured. Fold induction refers to ratio of luciferase activity of TA-treated cells compared to DMSO-treated cells. ***, P < 0.001 versus empty (pGL3 basic) vector-transfected cells. The data represent mean ± SD from three independent experiments. (B) Upper panel: the putative transcription binding sites within the −84 to +34 region in the ATF3 promoter. The underlines represent the binding sites of the indicated transcription factors which is used for construction of internal deletion clones. Lower panel: each internal deletion clone of pATF3-84/+34 (0.5 µg) was co-transfected with pRL-null vector (0.05 µg) into the cells and then treated with 30 µM of TA for 24 h. The open ovals in the promoter represent the binding site that is internally deleted. *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus wild type (WT)-transfected cells. The data represent mean ± SD from three independent experiments. (C) The pATF3-84/+34 construct was point-mutated as described in Materials and Methods. Each mutant clone (0.5 µg) was co-transfected with pRL-null vector (0.05 µg) into the cells and then treated with 30 µM of TA for 24 h. ***, P < 0.001 versus wild type (WT)-transfected cells. The data represent mean ± SD from three independent experiments.
To obtain further evidence that the ATF/C/EBP binding site is responsible for activation of ATF3 transcription by TA, we constructed three point mutation clones replacing two or three nucleotides within the ATF/C/EBP binding site as described in Fig. 3C. Wild type pATF3-84/+34 resulted in 4.7-fold induction of luciferase activity. However, all clones having point mutations in the ATF/C/EBP binding site completely blocked induction of luciferase activity by TA, which is comparable with empty vector-transfected cells. These results indicate that the region spanning −23 and −16 in the promoter of ATF3 plays an essential role in mediating the effect of TA on ATF3 transactivation.
ATF2 mediated ATF3 promoter activation by TA
ATF and C/EBP proteins have highly conserved DNA binding domain and share their binding sites. The sequence of the C/EBP binding oligonucleotide (TGATGCAA) closely resembles the consensus sequence of CREB (TCACGTCA) and ATF2/3 (TGACGT(C/A)(G/A)). To investigate which transcription factor is able to affect ATF3 transcription, we measured ATF3 promoter activity after co-transfecting expression vectors including CREB, ATF2, ATF3, C/EBPα, C/EBPδ, C/EBPβ, CHOP (C/EBPζ), GATA5, and NF-IL3. These proteins are able to bind to TGATGCAA site at some extend. Expression of transfected vectors was confirmed by Western blot analysis (data not shown). As shown in Fig. 4A, ATF2 expression caused a dramatic increase of TA-induced luciferase activity, compared with empty vector-transfected cells. Interestingly, expression of CREB, ATF3, C/EBPs and GATA5 significantly decreased TA-induced ATF3 promoter activation, implying that these transcription factors may act as suppressors of the ATF3 gene. ATF2-induced activation of ATF3 promoter activity was completely abolished in the presence of a construct lacking the ATF/C/EBP (−23 to −16), suggesting that ATF2 is necessary for activation of the ATF3 promoter induced by TA (Fig. 4B).
Figure 4. A role of ATF2 in TA-induced ATF3 transactivation.
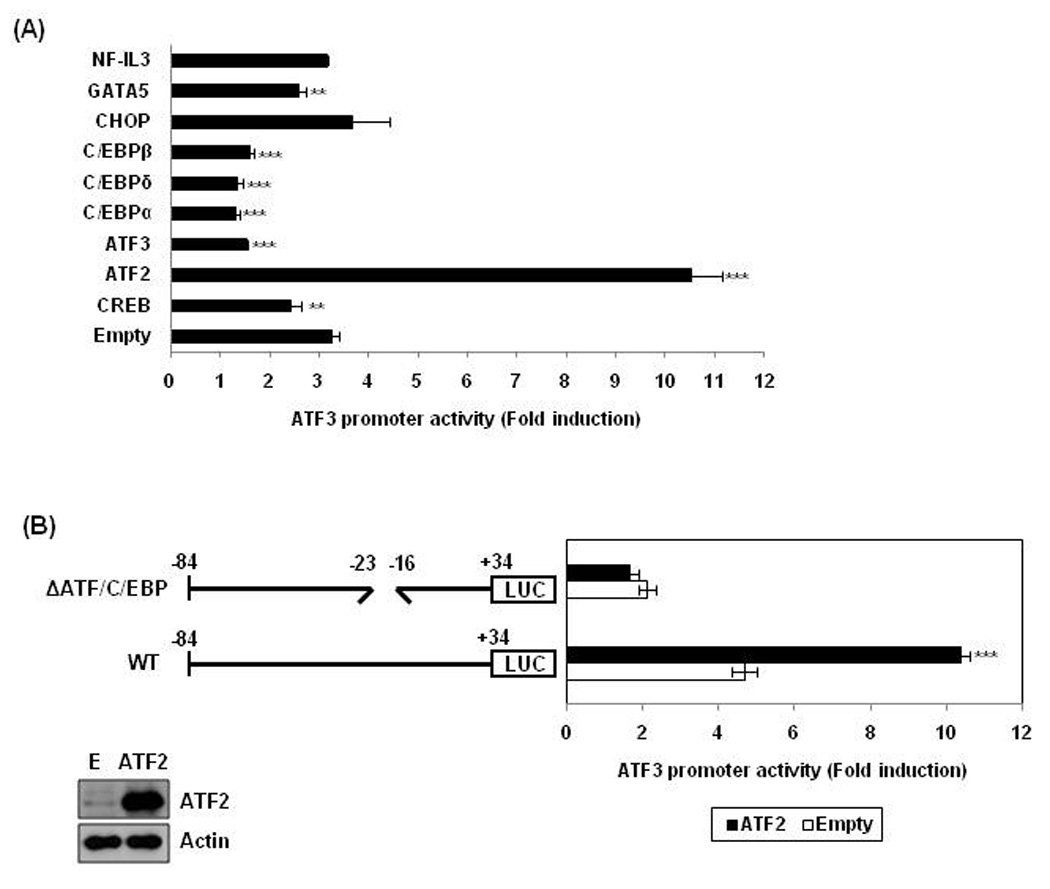
(A) pATF3-84/+34 (0.5 µg) and pRL-null vector (0.05 µg) were co-transfected into cells with indicated expression vector and then treated with 30 µM of TA for 24 h. **, P < 0.01; ***, P < 0.001 versus empty vector-transfected cells. The data represent mean ± SD from three independent experiments. (B) HCT-116 cells were co-transfected with wild type pATF3-84/+34 or internal deletion clone lacking the ATF/C/EBP binding site (pATF3-84/+34Δ−23/−16) in the presence of empty (E) or ATF2 expression vector. Then, the cells were treated with 30 µM of TA for 24 h. The results are presented as the means ± S.D. of three independent transfections. The overexpression of ATF2 was confirmed by Western analysis using ATF2 antibody.
ATF2 bound to the putative ATF/C/EBP binding site in the ATF3 promoter region
To examine whether transactivation of the ATF3 gene promoter by the ATF/C/EBP binding site is direct or indirect, we performed electrophoretic mobility shift assays (EMSA) using oligonucleotide probes derived from the ATF3 promoter (−23 to −16). As shown in Fig. 5A, TA caused an induction of formation of the DNA-protein complex. Preincubation of nuclear extracts with 10X or 100X excess unlabeled oligonucleotide abolished the binding activity (Fig. 5B, Lane 3,4) suggesting that the binding protein is sequence specific. To identify the binding protein, supershift assay was performed using antibody against ATF2 and phospho-ATF2. Preincubation of nuclear extracts with a phospho-ATF2-specific antibody resulted in reduction of the protein-DNA complexes, whereas antibody for IgG and ATF2 did not affect the DNA-protein complex, suggesting that the binding protein is phospho-ATF2 (Fig. 5C).
Figure 5. Binding of ATF2 to the ATF3 promoter.
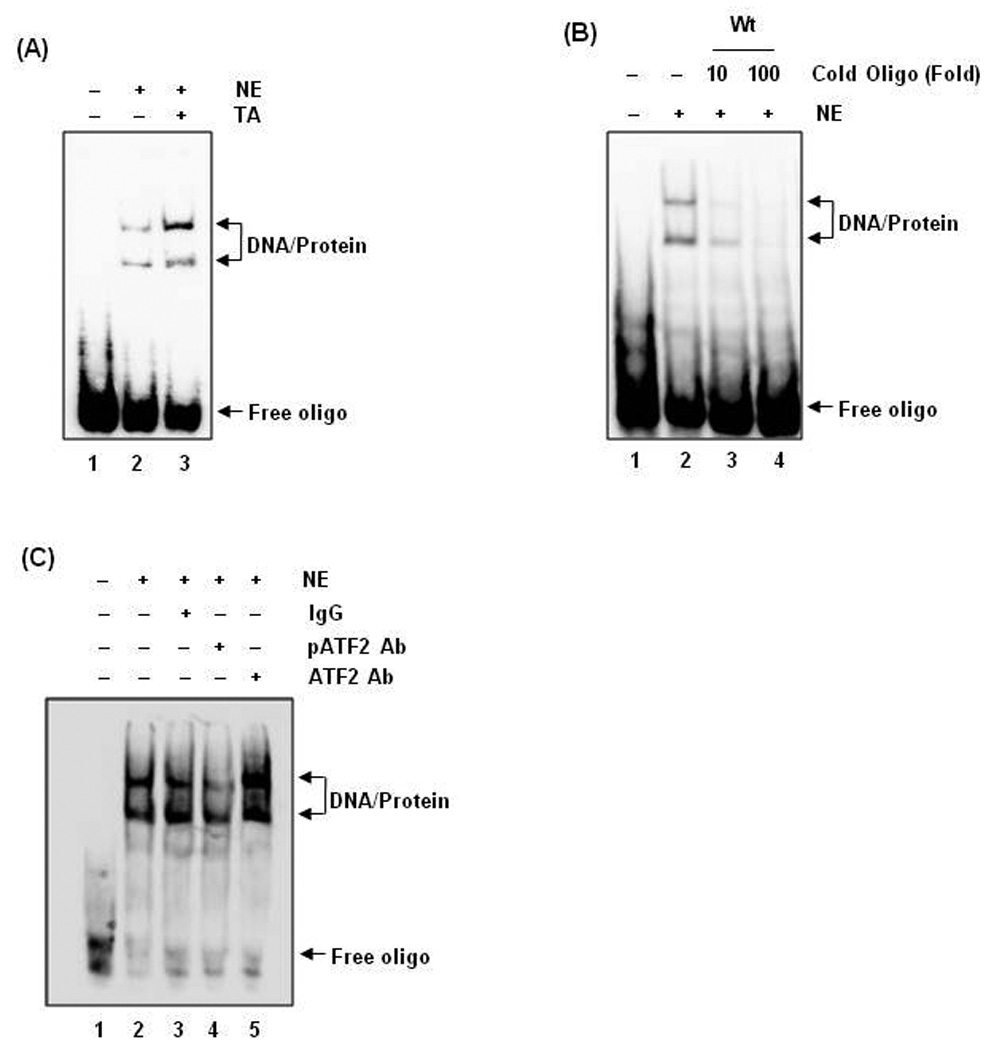
(A) Nuclear extract protein (5 µg) were prepared and incubated with biotin-labeled oligonucleotide containing ATF/C/EBP binding sites (−23/−16) as described in Materials and Methods. Oligonucleotide probes contained the following sequences: 5'- aggggtgatgcaacgctctcaggggtgatgcaacgctctc-3'. Specific protein-DNA complexes are indicated by arrows. (B) Nuclear extract protein from TA-treated cells was preincubated with a 10- or 100-fold excess of unlabeled oligonucleotide for 10 min and then incubated with biotin-labeled oligonucleotide and 1X binding buffer (Promega) at room temperature for 20 min. (C) Nuclear extract protein was preincubated with specific antibodies against IgG, phospho-ATF2, or ATF2 for 10 min and then incubated with labeled oligonucleotide and 1X binding buffer (Promega) at room temperature for 20 min. DNA-protein complexes were resolved by 5% nondenaturing polyacrylamide gel and developed using the protocol of LightShift Chemiluminescent EMSA kit (Pierce).
TA induced phosphorylation of ATF2, and knockdown of ATF2 decreased TA-induced ATF3 expression
Phosphorylation of ATF2 has been shown to be crucial to its ability to bind to cognate DNA sequences and activate gene transcription. Thus, we determined whether TA affects phosphorylation of ATF2. As shown in Fig. 6A, phospho-ATF2 started to increase at 15 min and reached a maximum at 30 min after TA treatment. The induction of ATF2 phosphorylation was followed by high induction of ATF3 expression 2 h after TA treatment. ATF3 mRNA levels began to increase at 1 h and showed time-dependent increase (Fig. 6B). To observe the role of ATF2 in TA-induced ATF3 expression, the cells were transfected with control or ATF2 siRNA and ATF3 expression measured. As shown in Fig. 6C, knockdown of ATF2 significantly ameliorated ATF3 expression by TA, confirming that ATF2 mediates TA-induced ATF3 expression.
Figure 6. Increase of ATF2 phosphorylation by TA treatment.
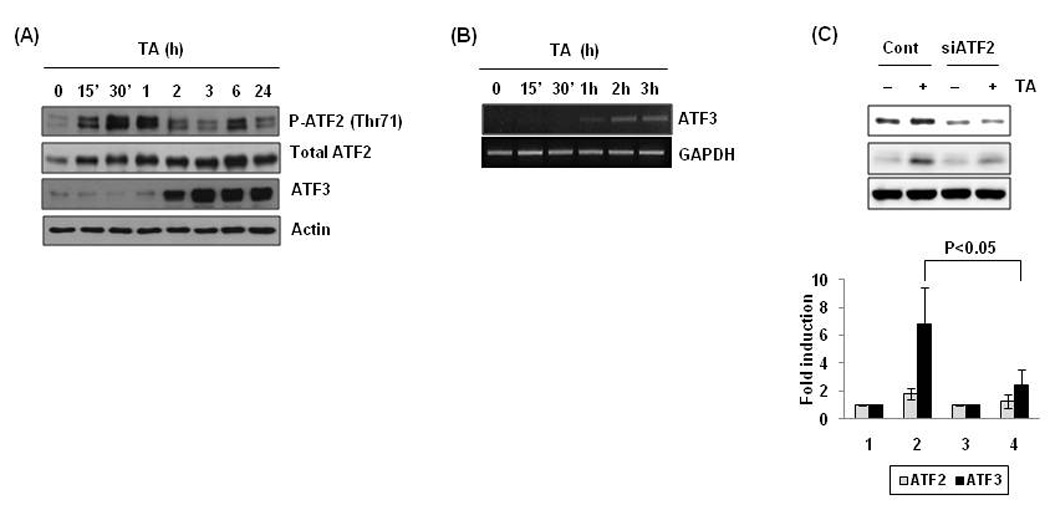
(A) Cells were treated with 30 µM of TA for the indicated times, and Western blot was performed for p-ATF2, ATF2, ATF3, and actin. (B) The cells were treated with 30 µM of TA for the time indicated and semi-quantative RTPCR was performed for ATF3 and GAPDH. (C) The cells were transfected with control siRNA (100 nM) or ATF2 siRNA (100 nM) for 24 h using a TransIT-TKO transfection reagent and then treated with 30 µM of TA for 2 h. Western blot analysis was performed for ATF2, ATF3, and actin (upper panel). Data from three independent experiments were densitometrically analysed using Scion Image (Scion Corporation, Frederick, MD) and ATF3 versus actin was quantified and expressed as fold induction (Lower panel).
Phosphorylation of ATF2 and ATF3 expression was mediated by MAPK pathways
MAPK signaling is an important pathway affecting ATF2 phosphorylation (Ouwens et al., 2002). So, we examined whether TA treatment affects MAPK pathways. As shown in Fig. 7A, TA enhanced phosphorylation of ERK, JNK, and p38MAPK proteins. To investigate whether these kinases affect TA-induced ATF3 expression, HCT-116 cells were pre-treated with selective inhibitors for p38MAPK, ERK, and JNK and treated with DMSO and 30 µM of TA. As shown in Fig 7B, C, and D, in the presence of inhibitors against p38MAPK (SB203580), ERK (U0126 and PD98059), or JNK kinase (SP600125), TA-induced phosphorylation of ATF2 and expression of ATF3 was inhibited. To confirm the ERK and p38 MAPK dependency of ATF3 expression, we transfected wild type or dominant negative construct of ERK2 and p38 MAPK, and compared TA-induced ATF3 expression. As shown in Fig. 7E, TA-activated ATF3 expression was blocked in the presence of dominant negative kinases of ERK2 and p38. In addition, pre-treatment with inhibitors of ERK, p38MAPK and JNK suppressed ATF3 promoter activity (Fig. 7F). Overall, three major MAPK pathways implicated in TA-induced phosphorylation of ATF2.
Figure 7. Upstream kinase for ATF2 phosphorylation and ATF3 expression.
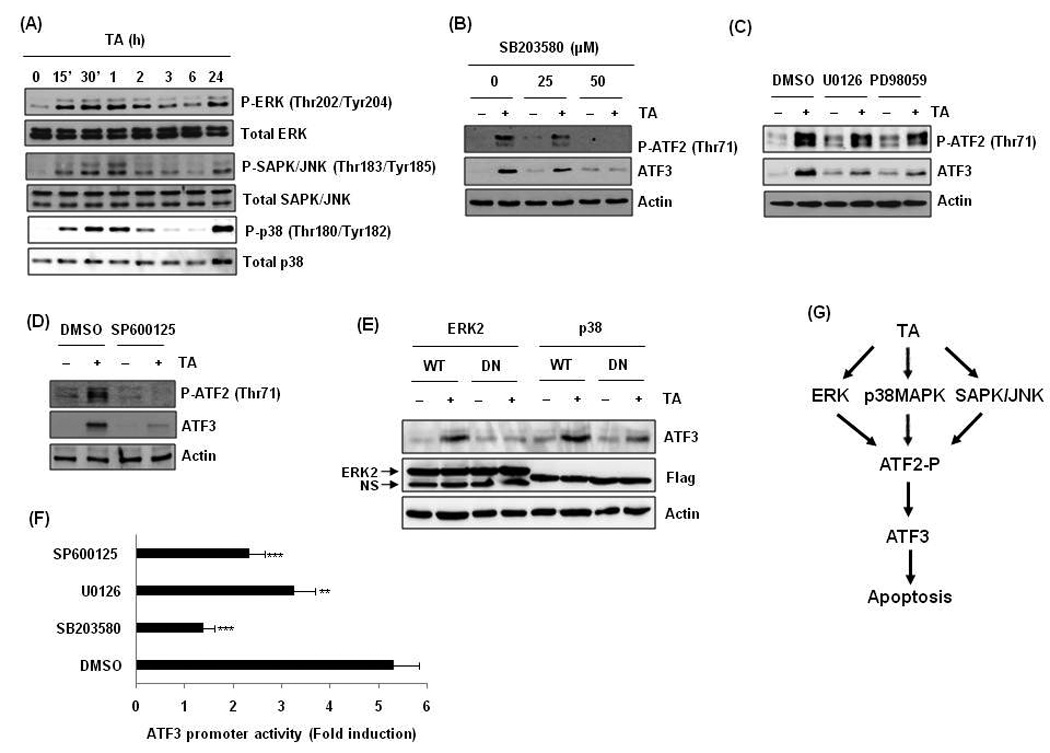
(A) Cells were treated with 30 µM of TA for the indicated times, and Western blot was performed for p-ERK, ERK, p-SAPK/JNK, SAPK/JNK, p-p38, and p38. (B) Cells were pre-treated with indicated concentrations of SB203585 and then treated with 30 µM of TA for 2 h. (C) Cells were pre-treated with U0126 (10 µM) or PD98059 (40 µM) and then treated with 30 µM of TA for 2 h. (D) Cells were pre-treated with SP600125 (30 µM) and then treated with 30 µM of TA for 2 h. (E) Cells were transfected with wild type (WT) and dominant negative (DN) mutant construct of ERK2 or p38 kinase and then treated with 30 µM of TA for 2 h. (F) Cells were transfected with pATF3-84/+34 and pRL-null vector and pre-treated with indicated concentrations (SB203580, 50 µM; U0126, 10 µM; SP600125, 30 µM) of inhibitors for 30 min and then treated with 30 µM of TA for 24 h. **, P < 0.01; ***, P < 0.001 versus DMSO pretreated cells. (G) Proposed mechanism that TA activates ATF3 transcription and apoptosis in human colorectal cancer cells.
Discussion
The anti-tumorigenic effects of TA in recent studies have raised an important question regarding the underlying molecular mechanisms. Here, we demonstrate direct evidence that TA enhances apoptosis of human colorectal cancer cells through activation of ATF3 gene transcription via ATF2 phosphorylation in human colorectal cancer cells.
ATF3 markedly accelerated TA-induced apoptosis and knockdown of ATF3 expression ameliorate TA-induced apoptosis (Fig. 2). These data indicate significant relevance of ATF3 gene expression to TA-mediated cancer suppression and a contributory role of ATF3 in caspase 3 and 7-mediated pathways.
Internal deletion and point mutation of the ATF/C/EBP binding site (−23/−16) in the upstream regulatory region of the ATF3 promoter completely blocked the ability of TA to stimulate promoter activity, suggesting that proteins bound to this site mediate the ability of TA to stimulate ATF3 promoter activity. EMSA data showed that phospho-ATF2 is present in complexes bound to the ATF/C/EBP binding site in the ATF3 promoter and this binding is increased by TA. Furthermore, overexpression of ATF2 activates the ability of TA to stimulate gene expression via ATF3 transactivation. On the contrary, knockdown of ATF2 ameliorated TA-induced ATF3 expression. This study indicates that ATF2 directly regulates ATF3 gene expression induced by TA.
ATF2, another member of the ATF/CREB family of basic region-leucine zipper protein, plays an important role in the cellular stress response (Maekawa et al., 1989). ATF2 expression has been associated with maintenance of cancer cell phenotype. ATF2 directly targets genes such as c-jun, cyclins, TNFα, TGFβ, and DNA polymerase b, which are known to play important roles in the stress response, immune response, cell growth and differentiation (Bhoumik and Ronai, 2008). However, other studies demonstrate anti-proliferative or apoptotic role of ATF2. Heterozygous Atf-2 mutant mice are highly prone to mammary tumors (Maekawa et al., 2007), and ATF2 plays significant role in hypoxia-induced apoptosis and suppresses development of mammary tumors (Maekawa et al., 2007). Suppression of mammary tumors by ATF2 is associated with activation of tumor suppressor genes Maspin and GADD45a (Maekawa et al., 2008). In keratinocytes, the loss of ATF2 promotes the tumor formation, suggesting a tumor suppressor role of ATF2 in skin (Bhoumik et al., 2008). Genetic variants of the ATF-2 gene were also detected in 5 of the 46 (10.6%) lung cancers (Woo et al., 2002). Further, constitutively nuclear-localized ATF2 suppressed ionizing radiation-induced prostate cancer progression (Deng et al., 2008). So far, the biological effect of ATF2 expression has not been studied in colorectal cancer models. Although ATF2 expression did not affect apoptosis directly in human colorectal cancer cells (data not shown), it is likely that phosphorylation of ATF2 contributes to enhancement of apoptosis via activation of their target genes such as ATF3. Likewise, activation of signal transduction pathways by TA treatment may result in the phosphorylation of ATF2 and regulation of its activity as suggested previously. In fact, the promising anticancer agent 3,3’-diindolylmethane (DIM) activates both JNK and p38 pathways, resulting in ATF2 phosphorylation (Xue et al., 2005). Thus, selection of the target gene, apoptotic or antiapoptotic, depends on the type of stress signal and the presence of other binding partners.
Interestingly, protein level of ATF2 was abruptly increased at an early time point after TA treatment (Fig. 6A). It is likely that the increase of total protein of ATF2 is associated with its phosphorylation because phosphorylation of ATF2 protects ATF2 from ubiquitination and subsequent degradation (Fuchs et al., 2000).
This study also indicated that TA treatment not only caused activation of p38MAPK and JNK, but also increased phosphorylation of ERK. Although we did not examine the changes of ATF2 Thr69 in this study, it has been shown that p38MAPK phosphorylates ATF2 on Thr69 and Thr71 residues. The ability of ATF2 to bind to cognate DNA sequence and activate its target gene depends on the phosphorylation of ATF2. The current data show that phosphorylation of ATF2 by TA is positively regulated by the p38MAPK, ERK, and JNK pathways, which subsequently modulate transcriptional activity of ATF3. This highlights a new role for the kinase pathways in the control of cell growth and apoptosis initiated by TA.
One interesting finding of this study is that overexpression of ATF3, all C/EBPs, CREB and GATA5 suppressed TA-induced ATF3 transactivation (Fig. 4A). This is consistent with data that ATF3 auto-represses itself transcriptionally through the same ATF binding site we indicated (Wolfgang et al., 2000). Although it is unclear how those transcription factors repress ATF3 transactivation, we may speculate that activation of ATF2 by phosphorylation sequesters inhibitory promoter activity by other bZIP proteins.
In conclusion, the current study provides information on the molecular mechanism of anti-tumorigenic activity by TA. MAPK pathways influence TA-induced phosphorylation of ATF2 and subsequently activate ATF3 expression. The resulting ATF3 activation induces apoptosis in human colon cancer cells (Fig. 7G).
Acknowledgements
We thank Dr. T. Hai (Ohio State University) and Dr. S. Kitajima (Tokyo Medical and Dental University, Tokyo, Japan) for providing the pCG-ATF3 construct and ATF3 promoter, respectively. We thank Dr. Philip Cohen (University of Dundee, Scotland, UK) and Dr. Joo-Heon Yoon (Yonsei University, Seoul, Korea) for providing pEBG2T-GST-ATF2-6His and CREB expression vector, respectively. We thank Dr. Melanie Cobb (University of Texas Southwestern Medical Center, Dallas, TX) for providing wild type and dominant negative ERK2 expression vector. We also thank Misty Bailey for her critical reading of manuscript. This work was supported by grants from the American Cancer Society (CNE-111611), National Institutes of Health (R01CA108975), and the University of Tennessee, Center of Excellence in Livestock Diseases and Human Health to SJB.
References
- Abdelrahim M, Baker CH, Abbruzzese JL, Safe S. Tolfenamic acid and pancreatic cancer growth, angiogenesis, and Sp protein degradation. J Natl Cancer Inst. 2006;98:855–868. doi: 10.1093/jnci/djj232. [DOI] [PubMed] [Google Scholar]
- Abdelrahim MB, Abbruzzese CH, Sheikh-Hamad JL, Liu D, Cho SD, Yoon K, Safe S. Regulation of vascular endothelial growth factor receptor-1 expression by specificity proteins 1, 3, and 4 in pancreatic cancer cells. Cancer Res. 2007;67(7):3286–3294. doi: 10.1158/0008-5472.CAN-06-3831. [DOI] [PubMed] [Google Scholar]
- Allan AL, Albanese C, Pestell RG, LaMarre J. Activating transcription factor 3 induces DNA synthesis and expression of cyclin D1 in hepatocytes. J Biol Chem. 2001;276:27272–27280. doi: 10.1074/jbc.M103196200. [DOI] [PubMed] [Google Scholar]
- Baek SJ, Kim JS, Jackson FR, Eling TE, McEntee MF, Lee SH. Epicatechin gallate-induced expression of NAG-1 is associated with growth inhibition and apoptosis in colon cancer cells. Carcinogenesis. 2004;25:2425–2432. doi: 10.1093/carcin/bgh255. [DOI] [PubMed] [Google Scholar]
- Bhoumik A, Fichtman B, Derossi C, Breitwieser W, Kluger HM, Davis S, et al. Suppressor role of activating transcription factor 2 (ATF2) in skin cancer. Proc Natl Acad Sci U S A. 2008;105:1674–1679. doi: 10.1073/pnas.0706057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoumik A, Ronai Z. ATF2: a transcription factor that elicits oncogenic or tumor suppressor activities. Cell Cycle. 2008;7:2341–2345. doi: 10.4161/cc.6388. [DOI] [PubMed] [Google Scholar]
- Bottone FG, Jr, Martinez JM, Alston-Mills B, Eling TE. Gene modulation by Cox-1 and Cox-2 specific inhibitors in human colorectal carcinoma cancer cells. Carcinogenesis. 2004;25:349–357. doi: 10.1093/carcin/bgh016. [DOI] [PubMed] [Google Scholar]
- Bottone FG, Jr, Moon Y, Kim JS, Alston-Mills B, Ishibashi M, Eling TE. The anti-invasive activity of cyclooxygenase inhibitors is regulated by the transcription factor ATF3 (activating transcription factor 3) Mol Cancer Ther. 2005;4:693–703. doi: 10.1158/1535-7163.MCT-04-0337. [DOI] [PubMed] [Google Scholar]
- Brown SL, Sekhar KR, Rachakonda G, Sasi S, Freeman ML. Activating transcription factor 3 is a novel repressor of the nuclear factor erythroid-derived 2-related factor 2 (Nrf2)-regulated stress pathway. Cancer Res. 2008;68:364–368. doi: 10.1158/0008-5472.CAN-07-2170. [DOI] [PubMed] [Google Scholar]
- Cai Y, Zhang C, Nawa T, Aso T, Tanaka M, Oshiro S, et al. Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH(2)-terminal kinase and promoter response element. Blood. 2000;96:2140–2148. [PubMed] [Google Scholar]
- Chen HH, Wang DL. Nitric oxide inhibits matrix metalloproteinase-2 expression via the induction of activating transcription factor 3 in endothelial cells. Mol Pharmacol. 2004;65:1130–1140. doi: 10.1124/mol.65.5.1130. [DOI] [PubMed] [Google Scholar]
- Cho KN, Sukhthankar M, Lee SH, Yoon JH, Baek SJ. Green tea catechin (−)-epicatechin gallate induces tumour suppressor protein ATF3 via EGR-1 activation. Eur J Cancer. 2007;43:2404–2412. doi: 10.1016/j.ejca.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ, et al. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res. 2008;68:9663–9670. doi: 10.1158/0008-5472.CAN-08-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Jin S, Amundson SA, Tong T, Fan W, Zhao H, et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene. 2002;21:7488–7496. doi: 10.1038/sj.onc.1205896. [DOI] [PubMed] [Google Scholar]
- Fuchs SY, Tappin I, Ronai Z. Stability of the ATF2 transcription factor is regulated by phosphorylation and dephosphorylation. J Biol Chem. 2000;275:12560–12564. doi: 10.1074/jbc.275.17.12560. [DOI] [PubMed] [Google Scholar]
- Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- Hai TW, Liu F, Allegretto EA, Karin M, Green MR. A family of immunologically related transcription factors that includes multiple forms of ATF and AP-1. Genes Dev. 1988;2:1216–1226. doi: 10.1101/gad.2.10.1216. [DOI] [PubMed] [Google Scholar]
- Hansen PE. Tolfenamic acid in acute and prophylactic treatment of migraine: a review. Pharmacol Toxicol. 1994;75(Suppl 2):81–82. doi: 10.1111/j.1600-0773.1994.tb02005.x. [DOI] [PubMed] [Google Scholar]
- Hao F, Tan M, Xu X, Han J, Miller DD, Tigyi G, et al. Lysophosphatidic acid induces prostate cancer PC3 cell migration via activation of LPA(1), p42 and p38alpha. Biochim Biophys Acta. 2007;1771:883–892. doi: 10.1016/j.bbalip.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Li X, Guo B. KLF6 induces apoptosis in prostate cancer cells through up-regulation of ATF3. J Biol Chem. 2008;283:29795–29801. doi: 10.1074/jbc.M802515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JC, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- Kashiwakura Y, Ochiai K, Watanabe M, Abarzua F, Sakaguchi M, Takaoka M, et al. Down-regulation of inhibition of differentiation-1 via activation of activating transcription factor 3 and Smad regulates REIC/Dickkopf-3-induced apoptosis. Cancer Res. 2008;68:8333–8341. doi: 10.1158/0008-5472.CAN-08-0080. [DOI] [PubMed] [Google Scholar]
- Lee SH, Bahn JH, Choi CK, Whitlock NC, English AE, Safe S, et al. ESE-1/EGR-1 pathway plays a role in tolfenamic acid-induced apoptosis in colorectal cancer cells. Mol Cancer Ther. 2008;7:3739–3750. doi: 10.1158/1535-7163.MCT-08-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Kim JS, Yamaguchi K, Eling TE, Baek SJ. Indole-3-carbinol and 3,3'-diindolylmethane induce expression of NAG-1 in a p53-independent manner. Biochem Biophys Res Commun. 2005;328:63–69. doi: 10.1016/j.bbrc.2004.12.138. [DOI] [PubMed] [Google Scholar]
- Lee SH, Yamaguchi K, Kim JS, Eling TE, Safe S, Park Y, et al. Conjugated linoleic acid stimulates an anti-tumorigenic protein NAG-1 in an isomer specific manner. Carcinogenesis. 2006;27:972–981. doi: 10.1093/carcin/bgi268. [DOI] [PubMed] [Google Scholar]
- Lu D, Wolfgang CD, Hai T. Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem. 2006;281:10473–10481. doi: 10.1074/jbc.M509278200. [DOI] [PubMed] [Google Scholar]
- Maekawa T, Sakura H, Kanei-Ishii C, Sudo T, Yoshimura T, Fujisawa J, et al. Leucine zipper structure of the protein CRE-BP1 binding to the cyclic AMP response element in brain. EMBO J. 1989;8:2023–2028. doi: 10.1002/j.1460-2075.1989.tb03610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, Sano Y, Shinagawa T, Rahman Z, Sakuma T, Nomura S, et al. ATF-2 controls transcription of Maspin and GADD45 alpha genes independently from p53 to suppress mammary tumors. Oncogene. 2008;27:1045–1054. doi: 10.1038/sj.onc.1210727. [DOI] [PubMed] [Google Scholar]
- Maekawa T, Shinagawa T, Sano Y, Sakuma T, Nomura S, Nagasaki K, et al. Reduced levels of ATF-2 predispose mice to mammary tumors. Mol Cell Biol. 2007;27:1730–1744. doi: 10.1128/MCB.01579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Inoue S, Yamada K, Watanabe M, Liu Q, Watanabe T, et al. Differential usage of alternate promoters of the human stress response gene ATF3 in stress response and cancer cells. Nucleic Acids Res. 2009;37:1438–1451. doi: 10.1093/nar/gkn1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh YK, Lee HJ, Jeong MH, Rhee M, Mo JW, Song EH, et al. Role of activating transcription factor 3 on TAp73 stability and apoptosis in paclitaxel-treated cervical cancer cells. Mol Cancer Res. 2008;6:1232–1249. doi: 10.1158/1541-7786.MCR-07-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouwens DM, de Ruiter ND, van der Zon GC, Carter AP, Schouten J, van der Burgt C, et al. Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras-MEK-ERK pathway and of Thr69 through RalGDS-Src-p38. EMBO J. 2002;21:3782–3793. doi: 10.1093/emboj/cdf361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- Piyanuch R, Sukhthankar M, Wandee G, Baek SJ. Berberine, a natural isoquinoline alkaloid, induces NAG-1 and ATF3 expression in human colorectal cancer cells. Cancer Lett. 2007;258:230–240. doi: 10.1016/j.canlet.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Hua B, Adachi S, Guney I, Kawauchi J, Morioka M, et al. Stress response gene ATF3 is a target of c-myc in serum-induced cell proliferation. Embo J. 2005;24:2590–2601. doi: 10.1038/sj.emboj.7600742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore MM, Iparraguirre A, Kubelka L, Weninger W, Hai T, Williams BR. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. 2007;179:3622–3630. doi: 10.4049/jimmunol.179.6.3622. [DOI] [PubMed] [Google Scholar]
- Wolfgang CD, Liang G, Okamoto Y, Allen AE, Hai T. Transcriptional autorepression of the stress-inducible gene ATF3. J Biol Chem. 2000;275:16865–16870. doi: 10.1074/jbc.M909637199. [DOI] [PubMed] [Google Scholar]
- Woo IS, Kohno T, Inoue K, Ishii S, Yokota J. Infrequent mutations of the activating transcription factor-2 gene in human lung cancer, neuroblastoma and breast cancer. Int J Oncol. 2002;20:527–531. [PubMed] [Google Scholar]
- Xue L, Firestone GL, Bjeldanes LF. DIM stimulates IFNgamma gene expression in human breast cancer cells via the specific activation of JNK and p38 pathways. Oncogene. 2005;24:2343–2353. doi: 10.1038/sj.onc.1208434. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Lee SH, Kim JS, Wimalasena J, Kitajima S, Baek SJ. Activating transcription factor 3 and early growth response 1 are the novel targets of LY294002 in a phosphatidylinositol 3-kinase-independent pathway. Cancer Res. 2006;66:2376–2384. doi: 10.1158/0008-5472.CAN-05-1987. [DOI] [PubMed] [Google Scholar]
- Yan C, Boyd DD. ATF3 regulates the stability of p53: a link to cancer. Cell Cycle. 2006;5:926–929. doi: 10.4161/cc.5.9.2714. [DOI] [PubMed] [Google Scholar]
- Yan C, Lu D, Hai T, Boyd DD. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. Embo J. 2005;24:2425–2435. doi: 10.1038/sj.emboj.7600712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Dewille JW, Hai T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene. 2008;27:2118–2127. doi: 10.1038/sj.onc.1210861. [DOI] [PubMed] [Google Scholar]