

Abstract
The majority of tumors arising in BRCA1 mutation carriers exhibit inactivation of p53, a key effector of cell death following DNA damage. Despite the loss of p53, BRCA1-deficient tumor cells exhibit increased sensitivity to cisplatin, and patients with BRCA1-associated ovarian carcinomas experience improved outcomes with platinum-based chemotherapy compared to sporadic cases. While it is known that chemosensitivity in BRCA1-associated cancers is associated with unrepaired DNA damage, the specific effector pathway mediating the cellular response to platinum-induced damage in these tumors is poorly understood. Here we demonstrate that the p53-related gene p73, encoding a pro-apoptotic protein which is linked to chemosensitivity in many settings, is upregulated through a novel epigenetic mechanism in both human and murine models of BRCA1-associated ovarian carcinoma. BRCA1-deficient ovarian carcinoma cells exhibit hypermethylation within a p73 regulatory region which includes the binding site for the p73 transcriptional repressor ZEB1, leading to abrogation of ZEB1 binding and increased expression of transactivating p73 isoforms (TAp73). Cisplatin chemotherapy induces TAp73 target genes specifically in BRCA1-deficient cells, and knockdown of TAp73 in these cells causes chemoresistance while having little or no effect on BRCA1-expressing tumor cells. In primary ovarian carcinomas, ZEB1 binding site methylation and TAp73 expression correlate with BRCA1 status and with clinical response. Together, these findings uncover a novel regulatory mechanism that supports the contribution of TAp73 as an important mediator of the response to platinum chemotherapy in a subset of ovarian carcinomas. TAp73 may represent a response predictor and potential therapeutic target for enhancing chemosensitivity in this disease.
Keywords: Ovarian Carcinoma, BRCA1, Cisplatin, TP73, Methylation, Chemosensitivity
Introduction
Ovarian cancers arising in the setting of germline mutation of the BRCA1 tumor suppressor gene exhibit particular clinical and molecular features, including predominantly serous histology, a high grade suggestive of a more aggressive malignancy, and distinct gene expression profiles (1, 2). Furthermore, these tumors exhibit mutational inactivation of p53 in up to 80% of cases (3). Inactivation of p53 is thought to promote tumor cell survival in the face of BRCA1-dependent defects in DNA repair (3). The unfavorable histological features and loss of functional p53 in BRCA1-associated ovarian cancers initially suggested that these tumors might be associated with chemoresistance and potentially a poor prognosis (1). Multiple studies, however, have confirmed that patients with BRCA1-associated ovarian carcinomas in fact experience a higher response rate and a more prolonged disease-free interval following platinum-based chemotherapy, as well as improved overall survival (3–5). In vitro studies of human ovarian cancer-derived cell lines have supported the view that loss of BRCA1 function is associated with selective chemosensitivity to platinum agents (6, 7). We and others have previously demonstrated that platinum sensitivity is associated with unrepaired DNA damage in BRCA1-deficient ovarian carcinoma cells (7, 8). Nevertheless, the specific effector pathway that mediates the response to this DNA damage in the absence of functional p53 has not been defined.
We have developed a murine model in which defined genetic events induce BRCA1-associated ovarian carcinomas that recapitulate the serous histology, the genetic instability, and the DNA damage sensitivity of human BRCA1-deficient ovarian carcinomas (8–10). We demonstrate here that BRCA1-deficient human and murine ovarian carcinoma cells exhibit selective up-regulation of transactivating isoforms of the p53-related gene p73 (TAp73), which is known to function as a mediator of the DNA damage response and chemosensitivity in many cellular contexts (11–14). We provide evidence that this TAp73 up-regulation occurs through a novel epigenetic mechanism, and that TAp73 serves as an important contributor to the platinum-induced DNA damage response and to chemosensitivity in ovarian carcinoma, both in vitro and in vivo. Our findings therefore define an epigenetic regulatory mechanism for p73 that plays a role in the clinical behavior of BRCA1-associated ovarian tumors.
Materials and Methods
Tissue culture and reagents
We (D. X., S. O.) generated the murine ovarian carcinoma cell lines (T1, T2, T3, TBR2, TBR5, TBR6). Western analysis was carried out to confirm the absence of whole-length BRCA1 proteins. These lines were maintained in DMEM/F12 containing 10% FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). The UWB1.289 human cell line was established (E. M. S.) as described (15); reconstituted BRCA1 expression was verified by QRT-PCR. Cells were maintained in 1:1 RPMI 1640/MEGM (Lonza) supplemented with 3% FBS. Chemosensitivity assays and antibody reagents are described in Supplementary Methods.
RNA analysis, lentiviral/retroviral production and chromatin immunoprecipitation (ChIP)
QRT-PCR analysis was performed as described (13). Specific forward and reverse primer sequences are provided in Table S1. High-titer amphotrophic retroviral and lentiviral stocks were generated by cotransfection with packaging vectors into 293T cells as described previously (13, 16). The targeted sequences are provided in Table S2. ChIP was carried out as described (16) using primers spanning different regions of p73 intron 1 (Table S3).
Bisulfite treatment and methylation analysis
For each cell line and tumor, 200–500ng of genomic DNA was used for bisulfite conversion using the EZ DNA Methylation-Gold Kit (Zymo Research Corp.). The 1.6 kb intronic region upstream of exon 2 of p73 was PCR amplified with primer sets designed using the program EpiDesignerBETA by Sequenom ® (Table S4). Following confirmation of desired product and quantification, methylation was quantitated in sequenced products by measurement of the height of the C and T peaks as described (17) using Chromas-Lite version 2.01 (Technelysiun Pty Ltd), and was expressed as C/(C+T) for each CpG site, and averaging the forward and reverse sequence results. Hypermethylation in clinical samples was defined as a value ≥ the methylated fraction in BRCA1-deficient UWB1.289 cells. Methylation status was validated in cell lines and in a representative subset of cases by HpaII/MspI digestion of unconverted DNA following PCR using primers shown in Table S4.
Patient sample acquisition and processing
Tumor specimens were obtained from the MGH Gynecologic Tissue Repository, from Cedars-Sinai Medical Center, and from the University of Washington. Use of tissues for this study was approved in each case by the respective Institutional Review Boards. Designation of tumors as BRCA1 wild-type or mutant resulted from clinical genetic testing. RNA/DNA was isolated from dry frozen tumor samples that had evidence of greater than 80% tumor volume with the GenElute Mammalian Total RNA and Genomic DNA Miniprep Kits following the manufacturer’s instructions (Sigma). For fixed specimens, microdissection or macrodissection of tissue sections was carried out following pathology review, and DNA was extracted using the RecoverALL Total Nucleic Acid Isolation Kit (Applied Biosystems), according to manufacturer’s instructions.
Additional methods are detailed in Supplemental Data.
Results
Cisplatin sensitivity of BRCA1-deficient ovarian carcinoma cells
To begin to explore pathways contributing to chemosensitivity in BRCA1-associated tumors, we first took advantage of our established murine model of BRCA1-deficient ovarian carcinoma (8). This model uses the RCAS avian retroviral system in order to infect primary ovarian surface epithelium (OSE) cells which express the TVA retroviral receptor transgene driven by the Keratin 5 (K5) promoter (Figure 1A) (18). To develop a model for BRCA1 loss of function we established K5-TVA mice harboring homozygous conditional p53lox/lox plus BRCA1lox/lox alleles. By infecting OSE cells derived from these mice with RCAS-Cre we showed that combined loss of p53 and BRCA1 along with one additional virus expressing the c-Myc oncogene was sufficient to induce tumors that exhibit many key features of human metastatic BRCA1-associated ovarian carcinoma (8). Three independent BRCA1-deficient tumor lines were derived from explants of these tumors (TBR2, TBR5, TBR6). As a control, we compared their phenotypic properties to three tumor lines (T1, T2, T3) resulting from p53 loss along with different combinations of any two of the oncogenes c-Myc, K-Ras, and AKT, which are required for ovarian tumorigenesis in the absence of BRCA1 deletion (Figure 1A) (9).
Figure 1. Loss of BRCA1 correlates with cisplatin sensitivity in murine and human ovarian carcinoma cells.

(A) Schema for murine ovarian carcinoma models. Primary ovarian epithelial cells of the indicated genotypes were targeted with RCAS retroviruses as shown, leading to tumor formation in vivo. See text for details. Each of the six cell lines listed at right was derived independently. (B) Increased cisplatin sensitivity is consistently observed in BRCA1-deficient (TBR) versus wild-type (T) ovarian carcinoma lines. Quantitative dose-response (MTT) assay was performed 72 hours post cisplatin treatment. (C) BRCA1 reconsitution induces cisplatin resistance in human ovarian carcinoma cells. BRCA1-deficient UWB1.289 cells were reconstituted with the control vector or BRCA1 followed by MTT assay at 72 hours. Error bars show SD for triplicate samples. (D) Apoptotic death induced by cisplatin (1μM, 72 hours) in BRCA1-deficient cells, shown by immunoblot for cleaved PARP-1; beta-tubulin (β-Tub) loading control.
In quantitative chemosensitivity assays, BRCA1-associated tumor lines consistently exhibited significantly increased sensitivity to cisplatin, but not to paclitaxel chemotherapy compared to BRCA1 wild-type tumor lines (Figure 1B and Figure S1) (8). The difference in platinum sensitivity was not related to the oncogenes used to transform these cells, since all three BRCA1 wild-type tumor cells exhibited significantly reduced sensitivity relative to BRCA1-deficient cells (Figure 1B), and the expression of either Akt or K-Ras in the BRCA1-deficient tumor cells did not significantly affect cisplatin sensitivity in these cells (not shown). As expected, chemosensitivity in BRCA1-deficient tumor cells was associated with substantial unrepaired DNA damage (8). Thus, this model recapitulates the BRCA1-associated chemosensitivity of human ovarian carcinomas.
In order to study cisplatin sensitivity in human ovarian carcinoma cells we tested UWB1.289 cells, which are derived from an ovarian cancer arising in a germline BRCA1 mutation-carrier and which lack expression of BRCA1 (15). These tumor cells are also deficient in p53 function due to an acquired somatic inactivating mutation (15). We compared cisplatin sensitivity in parental UWB1.289 cells stably reconstituted with either wild-type BRCA1 or the control vector. This reconstitution is physiologically relevant as it substantially restores defective checkpoint responses following ionizing radiation (15). BRCA1 reconstitution substantially abrogated platinum sensitivity in UWB1.289 cells as assessed in a quantitative dose-response curve (Figure 1C and Figure S1). Importantly, a clonogenic assay demonstrated the same result, confirming the increased platinum sensitivity of UWB1.289 cells relative to their isogenic BRCA1-expressing counterparts (Figure S1). We then sought to uncover the specific mechanisms involved in the response to cisplatin. We found that cisplatin induces apoptotic cell death in both murine and human BRCA1-deficient cells, as evidenced by cleavage of poly(ADP-ribose) polymerase-1 (PARP-1) on western analysis and by Annexin V staining on FACS analysis (Figure 1D and data not shown). Given that both the murine and human cells lack functional p53, our findings imply that cisplatin induces cell death in BRCA1-deficient cells at least in part through an apoptotic mechanism that is independent of p53.
Expression of p73 and its target genes correlate with cisplatin sensitivity in BRCA1-deficient cells
Following cisplatin treatment, we observed robust induction of the p53-regulated pro-apoptotic target genes Noxa, Puma, and p53AIP1 selectively in BRCA1-deficient murine and human cells, but little or no induction in their isogenic counterparts expressing wild-type BRCA1, even using cisplatin doses that cause substantial cell death in both populations (Figure 2A,B). These findings suggest a distinct cell death pathway in the absence of BRCA1. In particular, the p53-independent induction of these genes led us to consider that the related p53 family member p73 might contribute to the effects of cisplatin chemotherapy, given that p73 is known to be a direct activator of these particular target genes (19–21). Consistent with a role for p73 in the p53-independent DNA damage response in the specific context of BRCA1 deficiency, we first showed that lentiviral knockdown of BRCA1 led to induction of p73 following chemotherapy treatment of p53-deficient cells (Figure S2). We next examined p73 expression in wild-type and BRCA1-deficient murine ovarian carcinoma cells using an isoform-specific, quantitative reverse-transcription/real-time PCR (QRT-PCR) approach to distinguish between pro-apoptotic transactivating p73 isoforms (TAp73) and N-terminally truncated isoforms (ΔNp73). Remarkably, both murine and human BRCA1-deficient tumor lines showed substantially higher expression of TAp73 than any of the BRCA1-expressing lines (Figure 2C, D). In contrast, levels of ΔNp73 isoforms were comparable irrespective of BRCA1 status. Taken together, these data show that loss of BRCA1 function and platinum sensitivity correlate with both increased TAp73 and induction of TAp73 target genes in human and murine ovarian carcinoma.
Figure 2. Pro-apoptotic TAp73 and its target genes are selectively upregulated in BRCA1-deficient ovarian carcinoma cells.
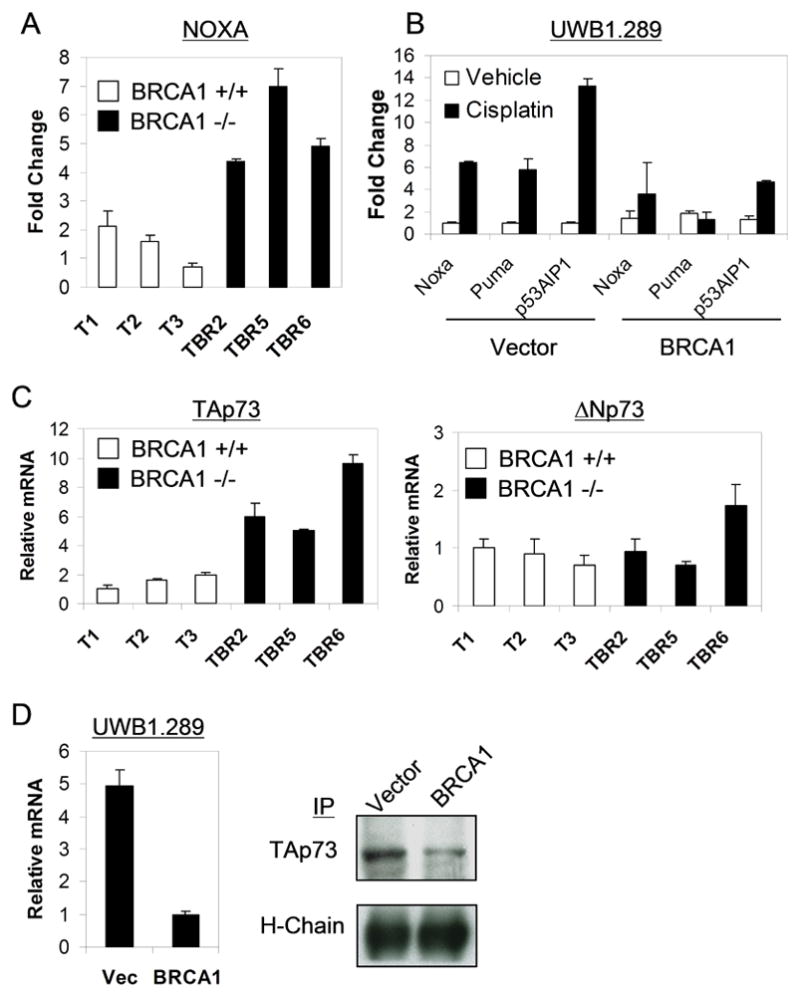
(A) Selective induction of Noxa in murine BRCA1-deficient (TBR) lines following cisplatin treatment (1μM, 48 hours), assessed by QRT-PCR. Error bars show SD for two independent experiments performed in triplicate. (B) Noxa, Puma, and p53AIP1 are selectively induced following cisplatin treatment (10μM, 72 hours) in UWB1.289 cells lacking BRCA1, as assessed by QRT-PCR. All values shown are relative to Vector/Vehicle cells. (C) Consistent upregulation of basal TAp73 but not ΔNp73 expression in BRCA1-deficient versus wild-type murine cells, as assessed by QRT-PCR. (D) Upregulation of TAp73 mRNA and protein is associated with BRCA1-deficiency in human ovarian carcinoma cells. Left, TAp73 QRT-PCR; right, IP/Immunoblot in UWB1.289 cells. Heavy chain (H-chain) is shown as a loading control.
TAp73 is required for cisplatin sensitivity in BRCA1-deficient ovarian carcinoma cells
We then sought to address directly whether TAp73 itself is an important mediator of the response to cisplatin in BRCA1-deficient cells in vitro and in vivo. We first ablated TAp73 expression using lentiviral RNA interference (RNAi) in both human and murine ovarian carcinoma cells, then quantitatively assayed cisplatin sensitivity. Consistently, knockdown of TAp73 induced significant cisplatin resistance in human BRCA1-deficient UWB1.289 cells, even though it did not affect proliferation or cell viability in the absence of cisplatin (Figure 3A and Figure S3). This chemoresistance was comparable to that observed in matched BRCA1-expressing UWB1.289 cells (Figure S3). TAp73 does not play a role in the BRCA1-expressing cells, which exhibit low levels of TAp73, as knockdown of TAp73 had little or no effect on chemosensitivity over a 5-log dose range of cisplatin (Figure 3A). Similarly, knockdown of TAp73 in all three murine BRCA1-deficient ovarian carcinoma lines using a distinct shRNA construct also induced substantial cisplatin resistance, while it showed no effect in BRCA1 wild-type cells at any dose (Figure 3B and Figure S3). Further supporting the specificity of these effects, TAp73 knockdown essentially abolished cisplatin-induced Noxa expression in the three murine BRCA1-deficient ovarian carcinoma lines (Figure 3C), while little or no effect was observed in BRCA1 wild-type cells (Figure 3C). The ability of TAp73 ablation to induce resistance to cisplatin was then confirmed in UWB1.289 cells using a clonogenic assay (Figure S3). Finally, retroviral overexpression of TAp73β was sufficient to induce significant cisplatin sensitivity in BRCA1-expressing cells (Figure S3). These experiments together argue that a TAp73-dependent transcriptional program is an important contributor to the chemosensitivity pathway in BRCA1-deficient ovarian carcinoma cells.
Figure 3. A TAp73-dependent transcriptional program is required for cisplatin sensitivity in ovarian carcinoma cells and tumors.

(A) Ablation of TAp73 consistently induces cisplatin resistance in BRCA1-deficient but not BRCA1-reconstituted UWB1.289 cells. Lentiviral transduction expressing a control (Vec) or TAp73-directed shRNA (TAp73si) was followed by MTT assay performed at 72 hours post cisplatin. Error bars represent SD for representative experiments performed in triplicate. (B) TAp73 is important for cisplatin sensitivity in murine BRCA1-deficient (TBR) but not BRCA1 wild-type (T) carcinoma cells. Lentiviral RNAi, cisplatin treatment and MTT assay as in (A). (C) TAp73-dependent Noxa induction by cisplatin is observed selectively in BRCA1-deficient lines. Cells described in (B) were treated with cisplatin (1μM, 72 hours) followed by RNA analysis using QRT-PCR. Error bars represent SD for two experiments performed in triplicate. (D) TAp73 is highly expressed in responsive (R; > 6 months recurrence-free survival) versus unresponsive (NR) tumors. QRT-PCR analysis from unselected primary tumors, expressed as Log2 TAp73 value. Note the mean TAp73 level is 10-fold higher (23.3) in responsive cases.
Given that a subset of sporadic ovarian cancers are thought to phenotypically resemble tumors arising in BRCA1/2 mutation carriers (22, 23), we hypothesized that some of these ovarian cancers would demonstrate increased expression of TAp73 in association with platinum sensitivity. To test this prediction we examined a series of unselected tumors from ovarian carcinoma patients treated with platinum-based chemotherapy following surgical resection. We first quantitated TAp73 expression by isoform-specific QRT-PCR in chemotherapy-responsive tumors (> 6 months recurrence-free survival, a standard clinical measure) versus unresponsive cases (24). The two groups of patients were otherwise well-matched for clinical characteristics including tumor histology, tumor grade and stage (Table S5). Remarkably, we observed a mean 10-fold higher level of TAp73 expression in responsive versus unresponsive tumors, a finding that was statistically significant (Figure 3D). Thus, TAp73 expression is associated with clinical platinum sensitivity, consistent with the data in our model systems demonstrating a direct role for p73 as a mediator of the cellular effects of cisplatin-induced DNA damage.
The ZEB1 transcriptional repressor binds the TAp73 locus and represses TAp73 levels selectively in BRCA1-expressing cells
We then sought to uncover the regulatory mechanism by which TAp73 was differentially expressed in BRCA1-deficient and -proficient human and murine ovarian carcinoma cells. Several mechanisms have been identified that may contribute to regulation of p73 expression in tumor cells, including 5′ promoter methylation (25), EGR-1 expression (26), and E2F1 dysregulation (27). We detected no significant methylation of the p73 promoter in any of our ovarian carcinoma cells, consistent with prior reports (Figure S4) (28). Furthermore, we did not find large differences in mRNA or protein levels of EGR-1 or of E2F family members by microarray analysis or immunoblot, respectively (data not shown).
An additional mechanism that is thought to contribute to endogenous p73 regulation involves the transcriptional repressor ZEB1 (also known as δEF1/zfhx1a). ZEB1 is a zinc finger and homeodomain-containing factor that binds to a regulatory region within the first intron of the TAp73 transcription unit and potently represses TAp73 mRNA expression (29, 30). However, we observed no consistent difference in ZEB1 expression levels between BRCA1-deficient and BRCA1-expressing cells (Figure S5). In order to test directly whether ZEB1 contributed to p73 regulation in these cells we inhibited endogenous ZEB1 using lentivirally expressed shRNA constructs (Figure 4A). Remarkably, knockdown of ZEB1 induced dramatic (5–15 fold) upregulation of TAp73 in all three ovarian lines expressing wild-type BRCA1, but had no effect on TAp73 expression in any of the three BRCA1-deficient lines (Figure 4A). Similarly, in human cells we observed high-level induction of TAp73 following ZEB1 knockdown in reconstituted UWB1.289 cells expressing wild-type BRCA1, but no induction in parental cells lacking BRCA1 function (Figure 4B). Thus, endogenous ZEB1 potently represses TAp73 only in cells expressing wild-type BRCA1. These results suggest that high levels of TAp73 observed in BRCA1-deficient cells may be due to loss of the repressive effect of ZEB1.
Figure 4. ZEB1-dependent repression of endogenous TAp73 is lost selectively in BRCA1-deficient ovarian carcinoma cells.

(A) Knockdown of endogenous ZEB1 (left) in wild-type (T) and BRCA1-deficient (TBR) murine ovarian carcinoma cells induces TAp73 de-repression (right) only in wild-type cells. QRT-PCR analysis 72 hours following infection with lentiviral control vector, non-specific (NS) shRNA, or ZEB1-directed shRNAs, normalized in each case to vector. Error bars show SD for two experiments in triplicate. (B) De-repression of TAp73 following endogenous ZEB1 knockdown in human BRCA1-expressing (BRCA1) but not BRCA1-deficient (Vector) ovarian carcinoma cells. Immunoblot 72 hours post lentiviral shRNA infection. (C, D) Specific binding by endogenous ZEB1 is absent in BRCA1-deficient murine (top) and human (bottom) ovarian carcinoma cells. Left, schematic diagram of the respective TAp73 genomic loci. Vertical dashes represent putative ZEB1 E-box motifs. Horizontal grey bars show fragments tested by ChIP; (*) indicates specific binding (> 100-fold over control antibody) detected by ChIP using a ZEB1-specific antibody, as shown at right. T1: wild-type BRCA1; TBR5: BRCA1-deficient murine cells. Vector: BRCA1-deficient; BRCA1: BRCA1-expresssing UWB1.289 cells. (#) Indicates no significant binding versus control antibody.
To pursue this hypothesis we next tested whether differential binding of ZEB1 to TAp73 regulatory sequences might explain a loss of ZEB1-mediated repression in BRCA1-deficient cells. ZEB repressors contain dual zinc fingers that bind to bipartite E-boxes (CACCT and CACCTG) whose orientation and spacing vary in different target genes (31, 32). In order to identify the endogenous ZEB1 binding site in murine cells we performed chromatin immunoprecipitation (ChIP) in ovarian carcinoma cells expressing wild-type BRCA1. We mapped robust and specific binding of ZEB1 to a 1kb region within the first intron of the TAp73 transcription unit that contains such a bipartite E-box motif (Figure 4C). Although significant binding of ZEB1 was consistently observed within this region in murine BRCA1 wild-type cells, in the isogenic BRCA1-deficient ovarian carcinoma cells ZEB1 binding was reduced to background levels (Figure 4C). Similarly, in human cells expressing wild-type BRCA1 we observed robust and specific binding of endogenous ZEB1 to a 550bp region containing bipartite E-box motifs (29, 30), yet virtually no specific binding was detected in isogenic BRCA1-deficient cells (Figure 4D). Taken together, these findings demonstrate that endogenous ZEB1 represses TAp73 expression in murine and human ovarian carcinoma cells expressing wild-type BRCA1, and that loss of BRCA1 function is associated with a loss of ZEB1 binding and its repressive effect on TAp73.
Methylation of the ZEB1 binding locus controls ZEB1 binding and TAp73 expression in human and murine cells
Since ZEB repressors are thought to bind directly to DNA (32), we asked whether chemical modification of DNA itself (e.g. methylation) might mediate differential binding of ZEB1 in cells expressing or lacking wild-type BRCA1. We first examined methylation of the ZEB1 binding region in these cells by digesting genomic DNA with either methylation sensitive or insensitive isoschizomeric restriction enzymes (HpaII or MspI, respectively), followed by PCR amplification. All three murine BRCA1 wild-type lines demonstrated hypomethylation of this locus, as assessed by the absence of a significant PCR product following HpaII digestion (Figure 5A). In contrast, in all BRCA1-deficient lines this locus was highly methylated and the DNA was therefore resistant to HpaII digestion. A remarkably similar pattern was observed upon analysis of the ZEB1 binding locus in human ovarian carcinoma cells: hypomethylation in BRCA1-reconstituted UWB1.289 cells, and hypermethylation in the BRCA1-deficient parental cells (Figure 5B).
Figure 5. BRCA1-associated differential methylation of the ZEB1 binding locus controls ZEB1 repressor binding and TAp73 expression.
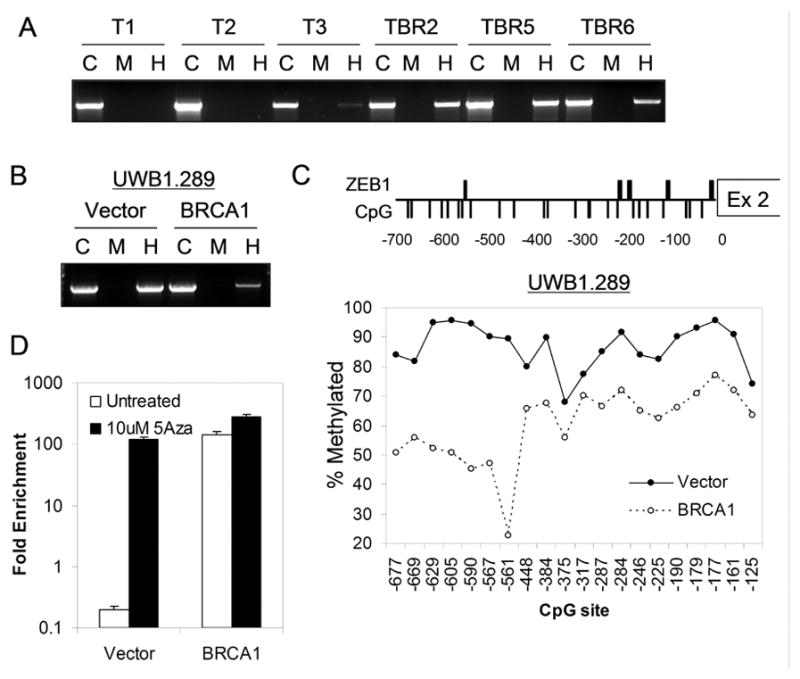
(A) Hypermethylation of the ZEB1 binding locus in murine BRCA1-deficient (TBR) versus wild-type (T) ovarian carcinoma cells. Genomic DNA was digested with MspI (M), HpaII (H), or undigested control (C) followed by PCR amplification of a 1.5kb fragment encompassing the ZEB1 binding locus. A ratio of H/C approaching 1:1 indicates hypermethylation. (B) Hypermethylation of BRCA1-deficient (Vector) versus BRCA1-reconstituted (BRCA1) human ovarian carcinoma cells. (C) Bisulfite sequencing showing consistent CpG hypermethylation across the ZEB1 binding locus in BRCA1-deficient human cells. Top, schematic showing putative ZEB binding motifs (thick bars) and CpG motifs (thin bars). Bottom, quantitation of methylation (see methods) for each CpG motif. The X-axis indicates positions of CpGs relative to exon 2. (D) Demethylation promotes ZEB1 binding in BRCA1-deficient cells. Fold-enrichment by ChIP 24 hours post 5-Aza treatment, using a ZEB1-specific versus control antibody, assayed by QPCR for fragment H5 (Figure 4D) in UWB1.289 cells.
To characterize the methylation of this locus in detail we performed bisulfite sequencing across the entire ZEB1 binding region identified by ChIP in UWB1.289 cells (Figure 5C). This region contains approximately 20 CpG dinucleotides, and we observed substantial hypomethylation of each CpG within this binding region in BRCA1-reconstituted cells compared to parental BRCA1-deficient cells (Figure 5C), in agreement with our results from MspI/HpaII digestion analysis in both murine and human cells. Based on these findings it is reasonable to hypothesize that hypermethylation may inhibit binding of the ZEB1 repressor, leading to a loss of ZEB1-mediated repression and a consequent increase in TAp73 levels.
In order to test this model directly we asked whether treatment with a 5-Azacytidine (5-Aza, a demethylating agent) would alter binding of ZEB1 to this locus. We observed more than 100-fold increase in ZEB1 binding in BRCA1-deficient cells following 5-Aza treatment compared to mock-treated cells, as assessed by quantitative ChIP (Figure 5D). Furthermore, 5-Aza treatment of BRCA1-expressing cells (which are already relatively hypomethylated) only increased ZEB1 binding by less than 2-fold (Figure 5D). Taken together, these data demonstrate that the binding of ZEB1 to the TAp73 regulatory region is controlled through an epigenetic mechanism, and that ZEB1 is bound and suppresses TAp73 expression in BRCA1-expressing but not BRCA1-deficient cells.
Correlation of ZEB1 binding site methylation and TAp73 expression with BRCA1 status and with clinical response in primary ovarian tumors
Our findings predict that ovarian cancers arising in patients with germline BRCA1 mutations will exhibit hypermethylation of the ZEB1 binding site within the p73 transcription unit compared to cancers arising in patients without such mutations. To test this prediction we obtained a series of such tumors that were well-matched in terms of both clinical stage and histological subtype (Table S6). Bisulfite sequencing within the ZEB1 binding region demonstrated higher methylation in BRCA1-associated tumors than in those expressing wild-type BRCA1 (Figure 6A). This finding was confirmed in a subset of cases by an independent method involving MspI/HpaII digestion of primary tumor DNA followed by PCR as shown in Figure 5 (data not shown). These findings support the association of BRCA1 deficiency with epigenetic regulation of the p73 locus in ovarian cancers in vivo.
Figure 6. ZEB1 binding site methylation correlates with BRCA1 status and with TAp73 levels in primary ovarian cancers.

(A) Increased methylation of residues within the ZEB binding site in primary tumors from BRCA1 mutation carriers (MUT, N=4) versus non-carriers (WT, N=8). Mean methylated fraction in each tumor is shown for CpG residues (−567, −561 and −542) which exhibit BRCA1-dependent methylation in vitro, assayed by bisulfite sequencing. (B) Reversion to wild-type BRCA1 expression in vivo is associated with hypomethylation of the ZEB1 binding site. Methylation-specific PCR was used to assay methylated (M) and unmethylated (U) resides as in (A), in the primary (BRCA1-deficient) and recurrent “revertant” (BRCA1-expressing) tumor. (C) TAp73 levels correlate with methylation of the ZEB1 binding region in primary ovarian carcinomas. Bisulphite sequencing of the entire ZEB binding region was performed in 25 primary tumors, and the mean fraction of methylated CpG residues was correlated with TAp73 expression assessed by QRT-PCR in the same tumor. P value by Fisher’s Exact Test. (D) Epigenetic regulation of ZEB-1 binding controls p73 expression. Tumors with BRCA1 pathway defects exhibit hypermethylation within p73 intron 1, preventing ZEB1 binding and leading to upregulation of TAp73, which in turn induces pro-apoptotic genes in response to cisplatin-induced DNA damage.
Recent reports have demonstrated that one mechanism by which both BRCA1 and BRCA2-associated ovarian cancers acquire resistance to chemotherapy in vivo is through re-expression of a functional protein (33–35). We therefore tested methylation of the ZEB1 binding site in patient-matched primary and recurrent tumors, in which the primary tumor contained only the mutant BRCA1 allele (185delAG) and exhibited no BRCA1 protein expression, while the recurrent, chemoresistant tumor exhibited expression of a wild-type sequence and detectable BRCA1 protein (35). We performed methylation-specific PCR at the ZEB1 binding locus within p73 (Figure 6B), after validating that this technique correlates with results obtained by both MspI/HpaII analysis and direct bisuphite sequencing (not shown). Remarkably, a substantial decrease in methylation was observed in the platinum-resistant, recurrent tumor relative to the hypermethylated primary tumor (Figure 6B). Thus, these results support our findings and provide in vivo evidence that functional BRCA1 status, which affects platinum chemosensitivity, is associated with epigenetic regulation of the pro-apoptotic TAp73 locus.
Finally, we tested the association of TAp73 expression with methylation of the ZEB1 binding locus in the entire cohort of unselected and BRCA1-associated tumors (Figures 3D, 6A). Thus, we performed bisulfite sequencing across the entire 500bp ZEB binding region, as well as QRT-PCR for TAp73. As predicted, those tumors exhibiting hypermethylation of this locus were more likely to express high levels of TAp73 (Figure 6C). In contrast, hypomethylated tumors almost exclusively expressed low levels of TAp73, comparable to the level expressed in BRCA1-reconstituted UWB1.289 cells. The pattern of methylation in the tumors recapitulated that seen in the UWB1.289 cell line (Figure 5C), in that differences in methylation were observed across all the CpG residues in region, rather than being restricted to a few residues (not shown). All together, these findings support the view that TAp73 is regulated through this epigenetic pathway and is an important mediator of the response to platinum-induced DNA damage in a subset of ovarian carcinomas (Figure 6D).
Discussion
Using murine and isogenic human ovarian carcinoma models, we demonstrate that BRCA1 is associated with a pathway for epigenetic regulation of TAp73, which we find to be an important contributor to chemosensitivity in BRCA1-deficient tumors. Previous studies have documented that TAp73 mediates cellular sensitivity to DNA damage in a variety of contexts (11, 12, 36, 37). Furthermore, we and others have demonstrated that TAp73 is activated specifically in the context of platinum chemotherapy (11, 13). We show here that TAp73 expression is substantially increased in BRCA1-deficient human and murine ovarian carcinoma cells. Treatment with cisplatin induces the pro-apoptotic transcriptional target genes of TAp73 selectively in these cells, and ablation of TAp73 promotes chemoresistance exclusively in BRCA1-deficient cells and blocks induction of these target genes. Together these findings strongly argue that a direct, TAp73-dependent effector pathway contributes to the chemotherapy response in these cells. Further support for this concept is provided by recent clinical studies that find a correlation between poor clinical outcome and increased expression of mRNA for non-transactivating p73 isoforms, which are thought to function as TAp73 antagonists, in ovarian and other cancers (38–40).
We provide substantial evidence that upregulation of TAp73 in BRCA1-deficient ovarian carcinoma cells is mediated through an epigenetic mechanism that controls DNA binding of the ZEB1 transcriptional repressor. The ability of DNA methylation to block binding by a transcriptional regulatory factor has been described in several other contexts (41). Regulation of a transcriptional repressor such as ZEB1 through this mechanism appears less common, however, as most described cases involve differential binding of transcriptional activators. It is also notable that methylation of the TAp73 proximal promoter was not correlated with methylation of the intronic ZEB1 binding region, consistent with prior reports that TAp73 promoter methylation is quite rare in ovarian carcinomas (28). Differential regulation of DNA methylation at these two loci is in keeping with emerging data from genome-wide studies that have uncovered distinct patterns of regulation in CpG-rich versus CpG poor regions (42). Thus, the proximal TAp73 promoter is a CpG-rich region that is bound by polycomb factors (43) and that exhibits a tumor-specific pattern of hypermethylation (44). In contrast, the intronic ZEB1 binding locus is a relatively CpG-poor region that may be subject to more dynamic changes in methylation status (42). While BRCA1 has not been previously associated with regulation of site-specific methylation, a putative BRCA1-regulated gene, GADD45, has been implicated in a demethylation pathway in zebrafish (45). However we did not find a difference in GADD45α or GADD45β expression between BRCA1-deficient and reconstituted ovarian carcinoma cells, and knockdown studies did not yield a GADD45-dependent difference in methylation at the ZEB1 binding locus (not shown). It will be of interest to determine whether the ability of BRCA1 to control site-specific methylation that we have uncovered is mechanistically related to its reported regulation of facultative heterochromatin formation on the X chromosome (46).
We show that TAp73 expression is correlated with the clinical response in unselected ovarian cancers, and we provide evidence that TAp73 is regulated in at least some of these tumors through the BRCA1-associated epigenetic mechanism. These observations are consistent with data demonstrating that chemosensitivity of a subset of sporadic ovarian carcinomas is associated with inactivation of a BRCA1/2-dependent pathway that also involves the Fanconi anemia proteins (22, 23, 47). Intriguingly, a recent report suggests that TAp73 is upregulated in cells from Fanconi anemia patients through a ZEB1-dependent epigenetic mechanism (48). An ongoing challenge for the field is to identify and classify the many mechanisms by which the BRCA1/2 pathway may be abrogated during ovarian epithelial tumorigenesis. Nevertheless, our findings do suggest that low levels of TAp73 may identify patients who will not benefit from standard platinum-based therapy, and who might therefore pursue other treatment options. Additionally, this work supports an emerging consensus that the TAp73 pathway might represent an attractive therapeutic target (14). Thus, future targeted therapeutics might be developed that trigger p73 expression and/or activation, thereby inducing apoptosis or sensitizing otherwise insensitive cells to DNA damaging agents.
Supplementary Material
Acknowledgments
We acknowledge Carolyn Koulouris, Petra Sergent, Darrell Borger and Susan Boisvert for technical and database support. This work was supported by the MGH ECOR Fund for Medical Discovery (CL); RO1 CA103924 (SO); the American Cancer Society Early Detection Professorship (SIOP-06-258-01-CCE) and the Milken Family Foundation (BYK); P50CA105009 and the Ovarian Cancer Research Fund (SO, BRR); the Ovarian Cancer Education and Awareness Network, and the Advanced Medical Research Foundation (BRR).
References
- 1.Lakhani SR, Manek S, Penault-Llorca F, et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin Cancer Res. 2004;10:2473–81. doi: 10.1158/1078-0432.ccr-1029-3. [DOI] [PubMed] [Google Scholar]
- 2.Jazaeri AA, Yee CJ, Sotiriou C, Brantley KR, Boyd J, Liu ET. Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. J Natl Cancer Inst. 2002;94:990–1000. doi: 10.1093/jnci/94.13.990. [DOI] [PubMed] [Google Scholar]
- 3.Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer. 2003;97:2187–95. doi: 10.1002/cncr.11310. [DOI] [PubMed] [Google Scholar]
- 4.Chetrit A, Hirsh-Yechezkel G, Ben-David Y, Lubin F, Friedman E, Sadetzki S. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J Clin Oncol. 2008;26:20–5. doi: 10.1200/JCO.2007.11.6905. [DOI] [PubMed] [Google Scholar]
- 5.Boyd J, Sonoda Y, Federici MG, et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. Jama. 2000;283:2260–5. doi: 10.1001/jama.283.17.2260. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy RD, Quinn JE, Mullan PB, Johnston PG, Harkin DP. The role of BRCA1 in the cellular response to chemotherapy. J Natl Cancer Inst. 2004;96:1659–68. doi: 10.1093/jnci/djh312. [DOI] [PubMed] [Google Scholar]
- 7.Zhou C, Huang P, Liu J. The carboxyl-terminal of BRCA1 is required for subnuclear assembly of RAD51 after treatment with cisplatin but not ionizing radiation in human breast and ovarian cancer cells. Biochem Biophys Res Commun. 2005;336:952–60. doi: 10.1016/j.bbrc.2005.08.197. [DOI] [PubMed] [Google Scholar]
- 8.Xing D, Orsulic S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer research. 2006;66:8949–53. doi: 10.1158/0008-5472.CAN-06-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell. 2002;1:53–62. doi: 10.1016/s1535-6108(01)00002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xing D, Orsulic S. A genetically defined mouse ovarian carcinoma model for the molecular characterization of pathway-targeted therapy and tumor resistance. Proc Natl Acad Sci U S A. 2005;102:6936–41. doi: 10.1073/pnas.0502256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gong JG, Costanzo A, Yang HQ, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–9. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 12.Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG., Jr Chemosensitivity linked to p73 function. Cancer Cell. 2003;3:403–10. doi: 10.1016/s1535-6108(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 13.Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest. 2007;117:1370–80. doi: 10.1172/JCI30866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lunghi P, Costanzo A, Mazzera L, Rizzoli V, Levrero M, Bonati A. The p53 family protein p73 provides new insights into cancer chemosensitivity and targeting. Clin Cancer Res. 2009;15:6495–502. doi: 10.1158/1078-0432.CCR-09-1229. [DOI] [PubMed] [Google Scholar]
- 15.DelloRusso C, Welcsh PL, Wang W, Garcia RL, King MC, Swisher EM. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol Cancer Res. 2007;5:35–45. doi: 10.1158/1541-7786.MCR-06-0234. [DOI] [PubMed] [Google Scholar]
- 16.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 17.Stirzaker C, Millar DS, Paul CL, et al. Extensive DNA methylation spanning the Rb promoter in retinoblastoma tumors. Cancer research. 1997;57:2229–37. [PubMed] [Google Scholar]
- 18.Orsulic S. An RCAS-TVA-based approach to designer mouse models. Mamm Genome. 2002;13:543–7. doi: 10.1007/s00335-002-4003-4. [DOI] [PubMed] [Google Scholar]
- 19.Flinterman M, Guelen L, Ezzati-Nik S, et al. E1A activates transcription of p73 and Noxa to induce apoptosis. J Biol Chem. 2005;280:5945–59. doi: 10.1074/jbc.M406661200. [DOI] [PubMed] [Google Scholar]
- 20.Melino G, Bernassola F, Ranalli M, et al. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;279:8076–83. doi: 10.1074/jbc.M307469200. [DOI] [PubMed] [Google Scholar]
- 21.Strano S, Monti O, Pediconi N, et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol Cell. 2005;18:447–59. doi: 10.1016/j.molcel.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 22.Kennedy RD, D’Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes & development. 2005;19:2925–40. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 23.Taniguchi T, Tischkowitz M, Ameziane N, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9:568–74. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 24.Fung-Kee-Fung M, Oliver T, Elit L, Oza A, Hirte HW, Bryson P. Optimal chemotherapy treatment for women with recurrent ovarian cancer. Curr Oncol. 2007;14:195–208. doi: 10.3747/co.2007.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corn PG, Kuerbitz SJ, van Noesel MM, et al. Transcriptional silencing of the p73 gene in acute lymphoblastic leukemia and Burkitt’s lymphoma is associated with 5′ CpG island methylation. Cancer research. 1999;59:3352–6. [PubMed] [Google Scholar]
- 26.Lee SW, Kim EJ, Um SJ. Transcriptional regulation of the p73 gene, a member of the p53 family, by early growth response-1 (Egr-1) Biochem Biophys Res Commun. 2007;362:1044–50. doi: 10.1016/j.bbrc.2007.08.128. [DOI] [PubMed] [Google Scholar]
- 27.Pediconi N, Ianari A, Costanzo A, et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol. 2003;5:552–8. doi: 10.1038/ncb998. [DOI] [PubMed] [Google Scholar]
- 28.Teodoridis JM, Hall J, Marsh S, et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer research. 2005;65:8961–7. doi: 10.1158/0008-5472.CAN-05-1187. [DOI] [PubMed] [Google Scholar]
- 29.Fontemaggi G, Gurtner A, Damalas A, et al. deltaEF1 repressor controls selectively p53 family members during differentiation. Oncogene. 2005;24:7273–80. doi: 10.1038/sj.onc.1208891. [DOI] [PubMed] [Google Scholar]
- 30.Fontemaggi G, Gurtner A, Strano S, et al. The transcriptional repressor ZEB regulates p73 expression at the crossroad between proliferation and differentiation. Mol Cell Biol. 2001;21:8461–70. doi: 10.1128/MCB.21.24.8461-8470.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 32.Remacle JE, Kraft H, Lerchner W, et al. New mode of DNA binding of multi-zinc finger transcription factors: deltaEF1 family members bind with two hands to two target sites. Embo J. 1999;18:5073–84. doi: 10.1093/emboj/18.18.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 34.Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer research. 2008;68:2581–6. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399:809–13. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 37.Yuan ZM, Shioya H, Ishiko T, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399:814–7. doi: 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- 38.Buhlmann S, Putzer BM. DNp73 a matter of cancer: mechanisms and clinical implications. Biochimica et biophysica acta. 2008;1785:207–16. doi: 10.1016/j.bbcan.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Concin N, Hofstetter G, Berger A, et al. Clinical relevance of dominant-negative p73 isoforms for responsiveness to chemotherapy and survival in ovarian cancer: evidence for a crucial p53-p73 cross-talk in vivo. Clin Cancer Res. 2005;11:8372–83. doi: 10.1158/1078-0432.CCR-05-0899. [DOI] [PubMed] [Google Scholar]
- 40.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371–86. [PubMed] [Google Scholar]
- 41.Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 42.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–70. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee TI, Jenner RG, Boyer LA, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–13. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16(Spec No 1):R50–9. doi: 10.1093/hmg/ddm018. [DOI] [PubMed] [Google Scholar]
- 45.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–12. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pageau GJ, Hall LL, Ganesan S, Livingston DM, Lawrence JB. The disappearing Barr body in breast and ovarian cancers. Nat Rev Cancer. 2007;7:628–33. doi: 10.1038/nrc2172. [DOI] [PubMed] [Google Scholar]
- 47.Weberpals JI, Clark-Knowles KV, Vanderhyden BC. Sporadic epithelial ovarian cancer: clinical relevance of BRCA1 inhibition in the DNA damage and repair pathway. J Clin Oncol. 2008;26:3259–67. doi: 10.1200/JCO.2007.11.3902. [DOI] [PubMed] [Google Scholar]
- 48.Pipaon C, Real PJ, Fernandez-Luna JL. Defective binding of transcriptional repressor ZEB via DNA methylation contributes to increased constitutive levels of p73 in Fanconi anemia cells. FEBS Lett. 2005;579:4610–4. doi: 10.1016/j.febslet.2005.07.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.