

Abstract
The E5 oncoprotein is the major transforming protein of bovine papillomavirus type 1. This 44-residue transmembrane protein can interact with the platelet-derived growth factor receptor β, leading to ligand-independent activation and cell transformation. For productive interaction, E5 needs to dimerize via a C-terminal pair of cysteines, though a recent study suggested that its truncated transmembrane segment can dimerize on its own. To analyze the structure of the full protein in a membrane environment and elucidate the role of the Cys-Ser-Cys motif, we produced recombinantly the wild-type protein and four cysteine mutants. Comparison by circular dichroism in detergent micelles and lipid vesicular dispersion and by NMR in trifluoroethanol demonstrates that the absence of one or both cysteines does not influence the highly α-helical secondary structure, nor does it impair the ability of E5 to dimerize, observations that are further supported by sodium dodecylsulfate polyacrylamide gel electrophoresis. We also observed assemblies of higher order. Oriented circular dichroism in lipid bilayers shows that E5 is aligned as a transmembrane helix with a slight tilt angle, and that this membrane alignment is also independent of any cysteines. We conclude that the Cys-containing motif represents a disordered region of the protein that serves as an extra covalent connection for stabilization.
Introduction
Signaling cascades are usually triggered by extracellular ligands that bind to a receptor. Transmembrane tyrosine-kinase receptors are generally assumed to dimerize in this process and undergo autophosphorylation (1–5). A notable exception, where signaling is induced without the action of any ligand is the interaction of the oncoprotein E5 from bovine papillomavirus with the platelet-derived growth factor receptor β (PDGFR), which transforms epithelial cells and fibroblasts (6,7). The short E5 protein is itself present as a dimer in the membrane, assembled by two disulfide bridges and possible hydrophobic contacts (8,9). Here, we have investigated the role of these putative dimerization motifs within E5 from a structural point of view, to better understand the basis of its dimerization and interaction with PDGFR.
Papillomaviruses commonly cause hyperproliferating epithelial lesions and warts, and some of them also have transforming activity, e.g., cervical carcinomas can result from infection with human papillomavirus type 16 (10). Bovine papillomavirus type 1 (BPV) causes formation of fibropapillomas in infected cattle and sarcoids in horses. The primary transforming activity of BPV depends on the viral protein E5, a hydrophobic 44-amino-acid membrane protein, which has been found to be sufficient to transform rodent and human fibroblasts in tissue culture (11). The main target of E5 is the receptor tyrosine kinase PDGFR (6,12), but interactions with the epidermal growth factor receptor and the 16-kDa subunit of the vacuolar H+-ATPase have also been reported (13–16).
Several mutagenesis studies have shown that the two cysteines in the C-terminal luminal Cys37-Ser38-Cys39 (CSC) motif of E5 are important for induction of total DNA synthesis, host-cell transformation, and PDGFR binding (17–21). Double mutation of the two cysteines completely abolished the transforming activity of E5 and interfered with receptor interaction. The location of the cysteines appeared to be less important and compatible with a nonhelical conformation, as it was possible to move them from their original positions 37 and 39 to positions 34 and 42 or 36 and 40 and retain function, although when they were moved to positions 35 and 41 or 33 and 43, activation was lost (19). Furthermore, single-cysteine mutants were still able to transform cells and enhance DNA synthesis, albeit with decreased intensity (18,20). In conclusion, it was postulated that the cysteines are needed for dimerization of E5 before its interaction with PDGFR. In line with these observations, the addition of an extra leucine zipper dimerization motif to the C-terminus of E5 could partially overcome the destabilizing effect of a double-negative cysteine mutation (22). A study on the truncated transmembrane segment of E5, on the other hand, revealed that the hydrophobic helical part of the protein is able to dimerize on its own in vitro (9). Furthermore, recent results demonstrate that a segment lacking the luminal part of E5 specifically activates the PDGFR in vivo (23).
Despite extensive investigations in the past decades and the importance of E5 in virus-induced transformation, no high-resolution molecular structure of E5 is available. To our knowledge, there is only one polarized infrared (IR) spectroscopy study where synthetic E5 was shown to assume an α-helical secondary structure and a transmembrane orientation (8). Therefore, the role of the cysteines, which had been shown to be apparently essential for the transforming function of E5, needs to be reinvestigated. The conformational constraints imposed on the protein by one or more disulfide bridges are of particular interest, as there are numerous open questions concerning the secondary structure and bridging geometry in the CSC motif. For example, it might be expected that an α-helical fold would not support the formation of both intermolecular bridges at the same time for steric reasons, whereas a parallel or antiparallel β-strand would be compatible with this sequence. However, to our knowledge, there has been no discussion of these conformational aspects in the literature, nor has there been any attempt to distinguish experimentally between a parallel and a crossed-over arrangement of the two bridges.
To address these structural questions, we have designed several cysteine mutants of E5 (Fig. 1). Of these, the E5-CSC protein corresponds to the wild-type sequence, which can form two intermolecular disulfide bonds, whereas the E5-ASC, -CSA, and -ACA mutants can only form a single bridge, and E5-ASA is unable to undergo any covalent dimerization at all. Using circular dichroism (CD), oriented CD (OCD), and nuclear magnetic resonance (NMR) to compare the corresponding secondary structures and oligomerization behaviors in various membranous environments, we were thus able to address the conformation of the full E5 protein, with special attention to the impact of the disulfide bridges.
Figure 1.

Nomenclature and sequences of the native E5 (E5-CSC) and the four cysteine mutants used in our study. The gray box represents the putative transmembrane segment between Leu7 and Leu29. At the N-terminus, there is an additional glycine due to hydroxylamine cleavage. The amino acid substitutions between positions 37 and 39 of the different E5-mutants are highlighted.
Experimental Procedures
Construction of E5-CSC, E5-ASC, E5-CSA, E5-ACA, and E5-ASA
The E5 wild-type gene was synthesized and cloned into a pUC19 vector. The gene was recloned into a pMMHb vector (gift of Peter Kim, Howard Hughes Medical Institute, Chevy Chase, MD), which added an N-terminal trp-ΔLE-sequence (24) to direct the E5-trp-ΔLE-fusion protein into inclusion bodies. It also provided a His9-tag for affinity chromatography, and adjacent Asp and Gly residues for cleavage with hydroxylamine, which added an extra Gly at the N-terminus of the native E5 sequence. The mutants E5-ASA, E5-ACA, E5-CSA, and E5-ASC (Fig. 1) were generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene/Agilent Technologies, Waldbronn, Germany). All sequences were verified by DNA sequencing. The primer composition can be found in the Materials and Methods section in the Supporting Material.
Expression and purification of the E5 constructs
The plasmids were transfected into Escherichia coli strain BL21(DE3) or BL21(DE3) pLys (Novagen/Merck Chemicals, Darmstadt, Germany), and cells were grown to an optical density (OD600) of 0.6–0.8 in LB medium (standard composition) supplemented with 100 μg/ml ampicillin. In addition, 20 μg/ml chloramphenicol was added if expressed in E. coli BL21(DE3) pLysS. Expression was induced by addition of 0.2 mM isopropyl-β-D-thiogalactopyranoside, and cells were harvested after 6 h. For uniformly 15N-labeled proteins, the LB medium was replaced by M9 minimal medium supplemented with 0.5 g/L of 15(NH4)2SO4.
The cell pellet was lysed by sonication in 50 mM Tris-HCl, pH 8, 15% glycerol (v/v), 50 μg/ml lysozyme, and 1 mM NaN3 and centrifuged at 46,000 × g for 1 h. The cell debris was resuspended and sonicated in 50 mM Tris-HCl, pH 8, 1% deoxycholic acid (w/v), 1% IGEPAL-CA 630 (Sigma Aldrich, Hamburg, Germany) (v/v), 1 mM NaN3. After centrifugation (56,000 × g, 0.5 h), the resulting pellet was resuspended in 6 M guanidinium-HCl and sonicated again. Ten volumes of water were added to the suspension, and the insoluble material was collected by centrifugation at 30,000 × g for 1 h.
For cleavage of the trp-ΔLE-His9 tag, the pellet was resuspended in 6 M guanidinium HCl, 2 M hydroxylamine HCl, and 4.5 M LiOH, pH 9, and incubated for at least 6 h at 45°C under stirring. The digest was acidified with formic acid to pH 3 and filtered (0.22 μm). The products of the hydroxylamine cleavage were separated by high-performance liquid chromatography (HPLC) using a semipreparative (250 × 10 mm) reverse-phase C18 polymer column (Grace Silica, Düren, Germany). A focused gradient based on water/acetonitrile/i-propanol or 2-propanol solvent systems was employed. The identity and purity of the collected fractions were analyzed by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) (see Materials and Methods in the Supporting Material) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (Autoflex III, Bruker, Billerica, MA).
CD and OCD sample preparation
To reconstitute the E5 protein in detergent micelles or lipids for CD measurements, an appropriate amount of zwitterionic detergent or lipid was dissolved in pure 2,2,2-trifluoroethanol (TFE), whereas charged detergents like SDS were dissolved in TFE/water (50/50, v/v). 1-myristoyl-2-hydroxy-sn-glycero-3-phosphocholine (LMPC), 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (LPPC), and dodecylphosphocholine (DPC) were used as zwitterionic detergents; SDS and 1-palmitoyl-2-hydroxy-sn-glycero-3-(phospho-rac-(1-glycerol)) (LPPG) were used as anionic detergents; and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was used as a lipid. An aliquot of this detergent (or lipid) stock solution containing 2–30 mM detergent (or lipid) was added to an aliquot of the E5 stock solution in TFE (5–10 μmol protein). The solvents were evaporated by a stream of nitrogen and the samples were then lyophilized. To prevent oligomerization, samples were rehydrated with 200 μl acidified water (pH 3.3) and sonicated. The pH was adjusted to neutral with phosphate buffer. Lipidic samples of various protein/lipid ratios were incubated in an ultrasonic bath for 15 min at maximum power to generate small unilamellar vesicles, which were kept in the liquid crystalline state (∼30°C) until measurement.
CD spectroscopy
CD spectra of E5-CSC and the mutants were recorded in aqueous micelle solutions and vesicle suspensions as previously described (25). Spectra were recorded at 20°C for the micelles and at 30°C for the vesicles, i.e., above the phase-transition temperature of the corresponding lipids, using a water-thermostatted cell holder. Final CD spectra were averaged from three scans at a rate of 10 nm min−1, 8-s response time, and 1-nm bandwidth, and smoothed by the adaptive smoothing method, which is part of Spectra Analysis software (Jasco, Easton, MD). Secondary-structure analysis was performed using the CONTIN-LL (26,27) and CDSSTR (28) algorithms provided by the DICHROWEB server (28–30). The quality of the fit between the experimental and back-calculated spectra corresponding to the derived secondary structure was assessed from the normalized-root-mean-square deviation (NRMSD), with a value of <0.1 considered a good fit (30). For calculating the mean residue ellipticities used for secondary-structure estimation, the concentration of the E5 protein solutions was determined based on the absorbance of the protein at 280 nm (31). For secondary-structure deconvolution, we used the average concentration of E5, determined in all detergents used because the reconstitution efficiency, and thus the concentration, varied slightly from sample to sample.
OCD spectroscopy
Macroscopically oriented CD samples were prepared from the E5 vesicle suspensions used for the CD measurements by depositing a 50- to 100-μl aliquot of the sample (containing 0.2 mg lipid) on a quartz glass plate with a 20-mm diameter. In this way, we could generate thin lipid bilayer samples to avoid possible spectral artifacts caused by, e.g., linear dichroism (32). The water of the vesicle suspension was allowed to evaporate in a gentle stream of air until the sample appeared dry (a circular spot of ∼12-mm diameter). Samples were subsequently hydrated for 15 h at 30°C in a sample cell for OCD measurements manufactured in-house as previously described (33). A small volume of saturated K2SO4 salt solution (300–500 μl) was placed in the bottom of the cell to maintain 97% humidity. To reduce possible spectral artifacts caused by the linear dichroism arising from imperfections in the sample, strain in the quartz glass windows, or imperfect alignment of the windows (34), OCD spectra were recorded every 45.0° of rotation of the cell as an average of three scans using the same data acquisition parameters as in the normal CD measurements above (35). The eight spectra were subsequently averaged, and background spectra of lipid bilayers without protein were subtracted.
NMR spectroscopy
For NMR experiments, 1 mg of protein powder was dissolved in TFE and sonicated for at least 5 min. Undissolved particles were removed by centrifugation to get a clear solution and D2O (20% (v/v)) was added. For chemical-shift calibration, and to compare relative signal intensities, 0.2 mM 2,2-dimethyl-2-silpentane-5-sulfonic acid was added. 15N-HSQC spectra were acquired at 37°C on a Bruker Avance II 600 spectrometer equipped with a broadband triple resonance probe, with 128 scans/increment and a total of 128 increments in the indirect dimension. Data were processed and analyzed with TOPSPIN software (Bruker-BioSpin, Rheinstetten, Germany).
Results
Production of the wild-type E5 protein and its four cysteine mutants
E5 and its mutants (Fig. 1) were expressed in E. coli as fusion proteins to a trp-ΔLE sequence, which directs the protein into inclusion bodies. As these turned out to contain virtually pure fusion protein, no further affinity chromatography step was required. The trp-ΔLE-His9 tag was cleaved off with hydroxylamine, which added a Gly to the N-terminus of all constructs. HPLC was used to separate the mixture of protein monomers and dimers, as illustrated in Fig. 2 A for representative mutants and redox conditions. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis of the collected fractions showed that the peak at the lower retention time contained monomeric E5 (5.2 kDa), whereas the other fraction at higher retention time was primarily composed of dimers (10.4 kDa). This latter fraction could be converted to the monomer by addition of the reducing agent tris-(2-carboxyethyl)-phosphine (TCEP). The cysteine-free E5-ASA mutant eluted as a single fraction at the earlier retention time, corroborating that this peak corresponds to the monomer (Fig. 2 A).
Figure 2.
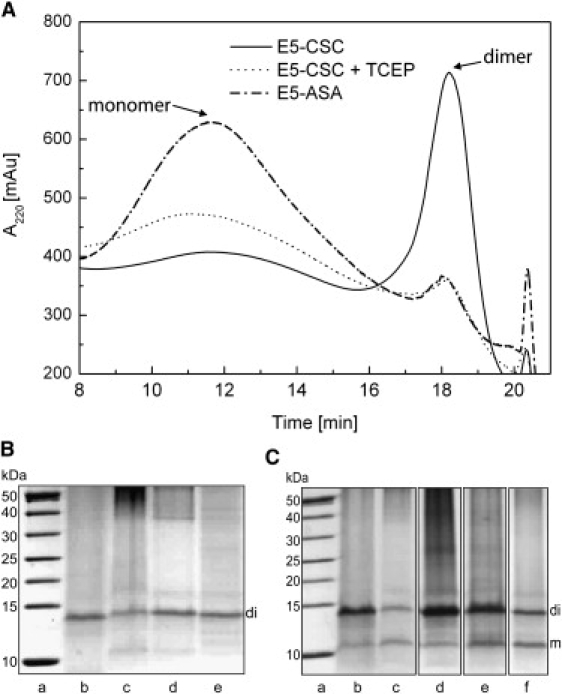
(A) Chromatographic analysis of E5-CSC and the cysteine-free mutant E5-ASA. In the HPLC trace, the wild-type protein eluted as two distinct fractions dominated by monomers (retention time of 12 min) or dimers (retention time of 18 min). The molecules in the dimer fraction could be converted into momomers by reduction of the disulfide bridges with TCEP. E5-ASA consisted mostly of monomers, as expected. (B and C) SDS-PAGE of the dimeric (B) and monomeric (C) fractions under nonreducing conditions. Lanes represent a molecular weight marker (a), E5-CSC (b), E5-ASC (c), E5-CSA (d), E5-ACA (e), and E5-ASA (f).
SDS-PAGE analysis (under reducing and nonreducing conditions) showed one band, at apparent 14 kDa, for the dimer fractions (Fig. 2 B) and two bands, at apparent 11 kDa and 14 kDa, for the monomer fractions (Fig. 2 C). These bands correspond to monomeric and dimeric E5, respectively, as this hydrophobic protein is known to migrate at higher apparent masses on SDS-PAGE. Despite the fact that E5-ASA is not able to form a covalent dimer, this construct also showed a dimer band. Further bands at higher molecular masses were also observed, indicating the presence of higher oligomers under all SDS-PAGE conditions.
Only the HPLC purified dimer fractions of the Cys-containing E5 proteins were used for further structural analysis. For comparison in the case of E5-ASA, the fraction eluting at the retention time of the monomer was used.
CD spectroscopy
In pure TFE, all E5 variants showed a pronounced α-helical conformation (Fig. S1 A in the Supporting Material). The highly congruent CD spectra indicated no differences in helicity between the wild-type protein and any of its mutants. Estimation of the helical fraction from the observed mean residue molar ellipticity (MRE) at 220 nm (36,37) resulted in an α-helical content of ∼70%, which corresponds to ∼31 residues (out of 45 total) adopting this conformation.
A more relevant membranelike environment was generated by reconstitution in zwitterionic (LMPC, LPPC, and DPC) and anionic detergents (SDS and LPPG), and in uncharged liposomes (DMPC/LMPC). Fig. 3 A depicts representative CD spectra of E5 and its mutants in LPPC micelles. Again, all variants showed a typical α-helical spectrum, but the MRE values decreased by ∼10–15% compared to TFE values. The same picture was obtained also in DPC, LMPC, LPPG, and SDS (Fig. S1, B–E). It is interesting that monomeric E5-ASA showed the same secondary structure as dimeric E5 wild-type and single-cysteine mutants. Addition of TCEP to reduce the disulfide bridge(s) had no effect on the secondary structure of either the wild-type E5 protein or its mutants (Fig. S2), in agreement with the results for E5-ASA.
Figure 3.
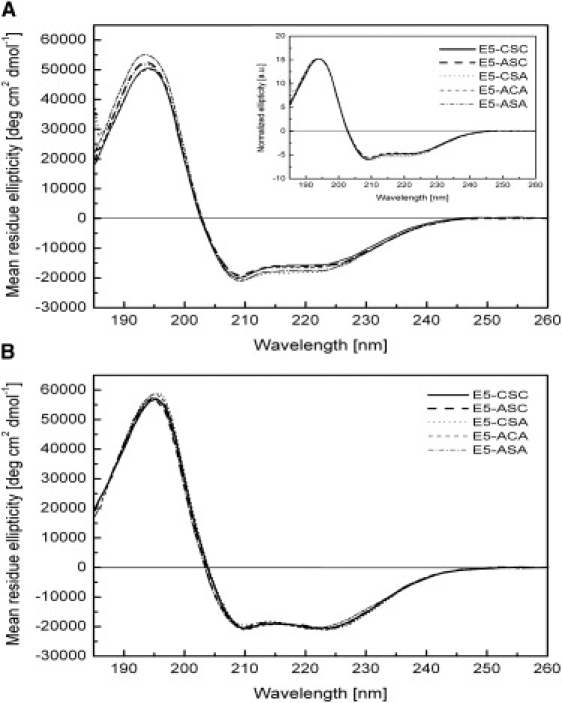
CD spectra of E5-CSC and mutants in detergent micelles and liposomes. (A) Aqueous solution of 10 mM LPPC micelles at 20°C, showing a predominantly α-helical conformation of the proteins. (Inset) Measured CD spectra normalized to the maximum ellipticity at 194 nm. (B) DMPC/LMPC vesicle suspensions (pH 3) with a protein/lipid ratio of 1:300 (mol/mol) measured above the lipid phase transition at 30°C.
In zwitterionic DMPC/LMPC liposomes, the negative signal intensity at 222 nm was increased relative to that at 208 nm (Fig. 3 B). This is interesting, as the ratio between the minima has been interpreted previously as reflecting the coiled-coil structure of two parallel oriented helices (38), though this interpretation is not generally accepted and secondary-structure prediction programs (see below) do not take it into account.
Secondary-structure estimation with the algorithm CONTIN-LL (26,27) or the CDSSTR algorithm (Table S1 and Table S2) showed that, within error, all mutants have the same amount of α-helical secondary structure as the wild-type. The overall helicity of the proteins ranged from ∼75% in LPPC and DPC to ∼82% in LMPC, LPPG, and SDS micelles and DMPC/LMPC liposomes. The slight differences most likely result from small errors in the concentration determination (Materials and Methods in the Supporting Material). In general, CONTIN-LL yielded a ∼25% higher total helical fraction (regular and distorted helices) compared to single-wavelength analysis at 220 nm (fH in Table S1). However, the latter values nicely matched the prediction of CONTIN-LL for the regular helix fractions, αR, which—depending on the detergent or lipid—were in the range 51–56%.
Influence of pH and temperature
We noticed that the pH used during reconstitution of the protein into detergents or lipids had a dramatic influence on the shape and intensity of the CD spectrum. The highest apparent secondary-structure contents were obtained when the proteins were initially kept under acidic conditions during reconstitution and the pH adjusted only afterward to neutral. To maintain well-defined conditions, we dialyzed the HPLC fraction against pure H2O at neutral pH and lyophilized afterward. The pH in these samples could be easily adjusted to the desired value by rehydration with phosphate buffer or water.
However, samples prepared from these stock solutions at neutral pH yielded turbid solutions. Fig. 4 shows representative CD spectra of E5-CSC in DPC micelles reconstituted under such conditions. These CD spectra at pH 7 were still reminiscent of α-helices, but the peak at 208 nm was significantly decreased in intensity, whereas the one at 197 nm was slightly red-shifted and the crossover point moved from 203 to 206 nm. An estimation of the secondary-structure content was not possible with any of the available programs. Not only did the turbidity in these samples suggest protein aggregation, but in some preparations the intensity at short wavelengths was severely reduced due to absorption flattening or differential light-scattering effects, which is a typical sign of aggregation (39). The corresponding ultraviolet (UV) spectra exhibit a distinct baseline offset and strong exponential increase of apparent absorption at shorter wavelengths due to scattering (Fig. S4). It is interesting that all samples became clear when the pH was adjusted to acidic conditions (pH 3), resulting in CD spectra with a characteristic α-helical lineshape and appropriate intensities. The UV spectra of the acidified CD samples showed the artifact-free absorption spectrum of peptide chromophores with a maximum around 190 nm and minimal baseline offset.
Figure 4.
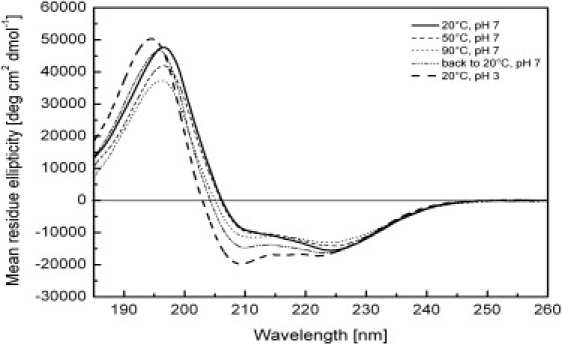
CD spectra showing the pH and temperature dependence of E5-CSC, acquired in 10 mM DPC micellar solution. By heating to 90°C and cooling back to 20°C, the aggregated protein at pH 7 could be converted into almost the same regular helical structure as the acidified sample maintained at pH 3.
Aggregation could also be largely reversed if the samples were heated up to 90°C and cooled back to room temperature. The corresponding spectra of aggregated E5-CSC in DPC micelles are shown in Fig. 4 (and the same for E5-ACA and E5-ASA in DPC in Fig. S3). Only relatively small spectral changes occurred up to 50°C, and up to 90°C a predominantly helical spectrum was observed. An unexpected finding was the more intense α-helical spectrum when the sample was cooled back to 20°C. This indicated disassembly of the aggregates. Disassembly under the conditions chosen was not complete, however, as the refolded spectrum does not fully correspond to the α-helical spectrum obtained at 20°C and pH 3.
Oriented CD
To characterize the alignment of the different E5 proteins in the lipid bilayer we used OCD, which is a powerful tool for the study of α-helical segments of membrane-bound proteins in their native lipid environment. To discriminate between a transmembrane orientation and a surface alignment of well-folded α-helical segments, the most informative feature is the presence or absence of a negative band around 208 nm (25). Each E5 protein was reconstituted at a protein/lipid ratio of ∼1:300 in DMPC/LMPC bilayers and spread onto the OCD quartz window to give a macroscopically oriented membrane sample. To verify that well-oriented bilayers were formed, solid-state 31P-NMR spectra were measured from samples prepared in the same way (Fig. S6 and Materials and Methods in the Supporting Material).
All E5 variants had a mostly α-helical conformation in the lipid bilayer (Fig. 5). Despite the standardized protocol for sample reconstitution, OCD spectra are less reproducible than CD spectra in solution, probably due to slight differences in drying the vesicle suspensions. To account for these potential errors, spectra of two to six separate OCD samples were averaged for each E5 variant. Normalization to the intensity at 225 nm (Fig. 5, inset) showed that any remaining differences mainly occurred below 200 nm, probably because of residual linear dichroic effects. For preparation of the E5 wild-type and mutant OCD samples, only small unilamellar vesicle dispersions were used, because these are less likely to cause absorption flattening (39). The representative UV spectra of E5 wild-type, E5-ACA, and E5-ASA (Fig. S5) are free of absorption flattening and scattering artifacts. Since all variants possessed a distinct residual negative intensity of the 208-nm band, a perfectly upright transmembrane orientation of E5 could be ruled out. The ratio of the negative band at 208 nm to that at 225 nm, being close to 0.7, could be attributed to one of two scenarios: 1), the observed spectrum is the sum of two populations arising from parallel and perpendicularly aligned helices, or 2), the E5 transmembrane segment assumes a uniform but slightly tilted alignment in the lipid bilayer. An argument in favor of the second scenario is the distinct hydrophobicity of the protein, which favors a transmembrane alignment. We thus estimated the alignment of the E5 helix to be tilted by an angle of ∼26–30° with respect to the membrane normal. This angle was calculated from the helix orientational order parameter, which was obtained from the CD difference spectra of the protein aligned in planar oriented bilayers and that in lipid vesicles (the isotropic case) (40,41).
Figure 5.
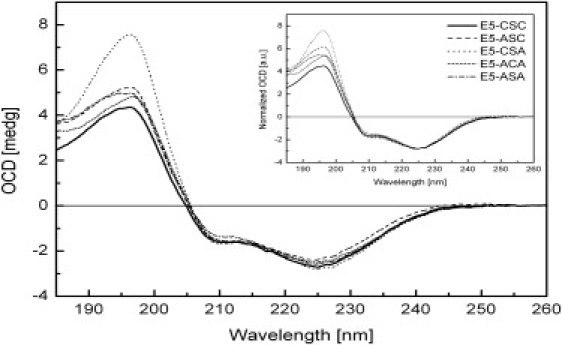
OCD spectra of E5-CSC and mutants in oriented lipid bilayers of DMPC/LMPC, acquired at 30°C and 97% relative humidity. (Inset) The spectra are normalized to the same intensity (225 nm) to illustrate the similarity in the lineshapes.
NMR spectroscopy
We tried to reconstitute E5 in DPC or SDS micelles, but despite a clear appearance of the solution, the resulting spectra suffered from severe exchange broadening. This could not be mended by using either low or high peptide concentrations (a range of 50 μM to 1 mM was tested) or by changing buffer, sample temperature, or detergent concentration. On the other hand, the spectrum quality improved dramatically in 80% TFE at 37°C (Fig. 6, A–D) and the resonances were well dispersed. TFE is known to realize internal hydrogen bond potential and may favor additional structuring that is not stably present in other solvents. We do not think, however, that this is detrimental to our aim, because we sought to understand whether the C-terminal part is capable at all of forming secondary structure, and whether the disulfide bridges influence this. Our CD measurements show that the helicity of E5 is increased in TFE, but we are strongly convinced that the organic solvent at best induces secondary structure (which in more nativelike conditions is eventually less stable) but does not impose structuring that opposes the native conformation.
Figure 6.
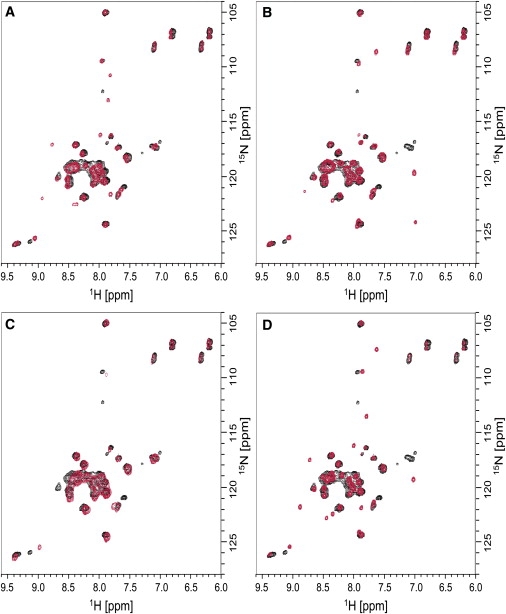
Superposition of the 1H15N-HSQC spectra of wild-type E5-CSC (black) and mutants (red) E5-ASC (A), E5-CSA (B), E5-ACA (C), and E5-ASA (D) in 80% TFE.
Superposition of the mutant spectra with the wild-type demonstrated that the presence and relative position of the disulfide bond had only minor effects on the overall protein structure, because the majority of the peaks remained unaffected. Although the observed differences might reflect some subtle changes in the arrangement of the C-terminal part of E5, it is more likely that they are attributable to the local effects arising from the substitution of the cysteines, in accordance with the results obtained from CD and OCD. Thus, the overall similarity of the NMR spectra confirmed that the three-dimensional structure of E5 remains unaffected by the presence of two, one, or no disulfide bridges.
Discussion
Dimerization of E5 is considered to be an essential step for productive interaction with the PDGFR. Early studies had demonstrated the functional importance of the two conserved cysteines at position 37 and 39, and it was commonly assumed that E5 homodimerizes via two intermolecular disulfide bridges (17–21). A recent study on the truncated transmembrane segment, however, reported that this part of E5 per se is able to dimerize in vitro (9). This finding is further confirmed by our results, which indicate that the cysteine-free mutant of E5 forms stable dimers in SDS-PAGE and shows no structural differences from the wild-type or the single-cysteine mutants. Given the previous mutagenesis experiments, we were surprised to find in our experiments that the presence of the disulfide bridge(s) had an influence on neither the secondary structure nor the thermal stability of E5. Both E5-CSC and the single-cysteine mutants E5-ASC, E5-CSA, and E5-ACA, which had been designed to examine the influence of the position of the disulfide bridge, displayed virtually identical structural features in all CD and NMR experiments. Even the cysteine-free mutant E5-ASA, which is incapable of covalently dimerizing and had been designed to represent a monomeric form of E5, was indistinguishable from the wild-type. OCD in lipid bilayers and NMR analysis in TFE corroborated the absence of fundamental structural differences between the various mutants and the wild-type protein, either in solution or in a proper membrane environment.
Prediction programs (SOSUI (42) or TMHMM server (43)) define the region from Leu7 to Leu29 as a transmembrane helix with a typical length of 23 residues (Fig. 1). From an inspection of the E5 sequence, however, the total hydrophobic region extends from Leu4 to at least Trp32, with Asn3 being an ideal N-capping residue for a helix. This extended length fits well with the total α-helical content estimated by deconvolution of the CD spectra (for TFE, 31 amino acids; in the comparatively thin DMPC bilayer, 25 residues). A caveat for these CD-derived values is, however, that current secondary-structure prediction programs use a CD reference data set that contains mainly globular soluble proteins, which may not be fully adequate for membrane proteins (44). By single-wavelength deconvolution, an α-helical content of ∼50–60% (23–27 residues) can be estimated (ƒH in Table S1), which would correspond approximately to the predicted transmembrane part and also fit to the regular helix fraction, αR. An additional fraction of ∼25% (9–11 residues) distorted helix (αD) has emerged from this analysis, which most reasonably reflects distorted helical stretches directly adjacent to both ends of the transmembrane segment beyond the lipid bilayer (Fig. 7). The higher α-helicity induced by TFE indeed suggests that the α-helix can extend beyond the transmembrane part.
Figure 7.
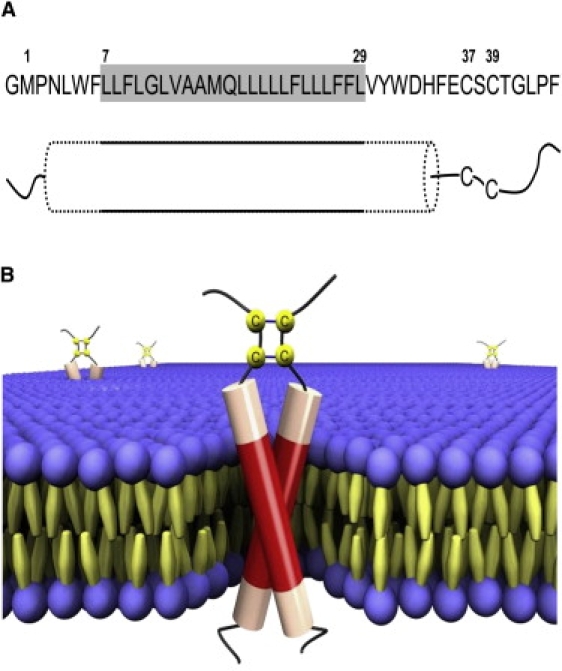
(A) Proposed model of the membrane-inserted structure of E5. The gray box represents the putative transmembrane segment that forms the regular helix fraction (solid outline), which is flanked by distorted helical regions (dotted outline) that extend beyond the lipid bilayer. The cysteines are located in the adjacent unstructured stretch near the C-terminus. (B) Three-dimensional scheme of the E5 dimer in a lipid bilayer based on the model in A. The regular helix parts are shown as red and the distorted helical regions as pale red in the cylinders, and the unstructured parts are shown as black lines. The cysteines are arranged to allow parallel disulfide bridges between Cys37-Cys37 and Cys39-Cys39. The sketch was made using POV-Ray (Persistence of Vision Raytracer).
In a previous study on the wild-type E5 protein in DMPC bilayers, an even longer, continuous helix of 39 residues was postulated, encompassing the CSC motif (8). That model structure, however, would place the two cysteines (position 37 and 39) onto opposite faces of the helix, which is sterically difficult to reconcile with the formation of two simultaneous intermolecular disulfide bridges in a dimer. Therefore, we interpret our observed values of 50% regular and up to 80% total helical content in terms of a structural model in which parts of the extramembranous regions of E5 do not form a continuous α-helix. The results of our mutation studies confirm that this is indeed the case, as the cysteines must lie outside the helical region.
Support for this structural model comes from the results reported by DiMaio and co-workers. By fusing a leucine zipper dimerization motif N-terminally to the truncated (Cys-free) transmembrane fragment of E5, they imposed different arrangements on the monomers within a dimer and sampled different interfaces in this way (22). Only one of their fusion proteins was as efficient as the wild-type in transformation, whereas the activity of the others dropped considerably. Hence they could show that only one specific interfacial arrangement leads to functional activity of E5. In such a particular, fixed dimer arrangement, the cysteines are obviously not able to simultaneously form a disulfide bridge to the other monomer if arranged as an α-helix. Moreover, we found that shifting the position of the cysteine bridge within the motifs CSA/ACA/ASC (in an α-helix this would result in a side-chain rotation of ∼100°) had no influence on the secondary and tertiary structure of E5, nor on its interaction with the lipid bilayer. Thus, it is highly likely that the cysteine-containing part is unstructured. Furthermore, we can conclude that the Cys-containing stretch of E5 is most likely arranged in a parallel fashion in the dimer, allowing bridges to form between Cys37-Cys37 and Cys39-Cys39 (rather than being crossed over between Cys37 and Cys39), because the E5-ASC and E5-CSA mutants can dimerize just as readily as the wild-type E5 and do not show significant differences in the NMR spectra (Fig. 6).
The existence of several different types of dimerization motifs, namely two covalent disulfide bridges and a putative hydrophobic leucine zipper (8) in such a small protein is remarkable. Although we can only speculate in the light of previous functional studies, it is possible that the cysteines may confer additional stabilization to the dimer within the cellular environment, or that they may be important for initiation of folding.
One remarkable observation is that all E5 variants were prone to aggregation, depending on the sample conditions, and that acidification or heating above 90°C reversed this self-assembly. Since this tendency was independent of the presence of any disulfide bonds, the formation of higher covalent oligomers through the second cysteine bridge can be ruled out. Such an oligomerization had not been noticed for the truncated transmembrane part of E5 (9), which ended with His34 but contained three additional N-terminal and one C-terminal Lys to increase solubility. Similar pH- and temperature-dependent effects had been noted in the literature for designer-made helical peptides. It was shown by electron microscopy that they form higher-order associates of coiled coils (45,46). In addition to His34, two acidic residues, Asp33 and Glu36, are located at the juxta-membrane C-terminal position. At pH 7.0, ionization of these residues could lead to a change in the net charge from +1 (at pH 3) down to −2 (depending on the protonation state of His). Moreover, if we assume that this part of the sequence is still included in the α-helix, the two acidic side chains not only would be adjacent to one another but would increase the helix dipole moment considerably at neutral pH. Maybe this effect prevents proper reconstitution into the detergents or lipids, as we observed, so that an alternative pathway to form coiled-coil aggregates becomes energetically more favorable. Below pH 4, only one positive charge (of the histidine) remains, which diminishes the helix dipole moment and eventually allows better interaction with the membrane.
Acknowledgments
The authors thank Dr. Ralf Heinzmann for his help in preparing the scheme of the E5 structure using the POV-Ray program.
Supporting Material
References
- 1.Yarden Y., Schlessinger J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry. 1987;26:1443–1451. doi: 10.1021/bi00379a035. [DOI] [PubMed] [Google Scholar]
- 2.Ullrich A., Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 3.Yarden Y., Schlessinger J. Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry. 1987;26:1434–1442. doi: 10.1021/bi00379a034. [DOI] [PubMed] [Google Scholar]
- 4.Jorissen R.N., Walker F., Burgess A.W. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp. Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 5.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 6.Petti L., Nilson L.A., DiMaio D. Activation of the platelet-derived growth factor receptor by the bovine papillomavirus E5 transforming protein. EMBO J. 1991;10:845–855. doi: 10.1002/j.1460-2075.1991.tb08017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlegel R., Wade-Glass M., Yang Y.C. The E5 transforming gene of bovine papillomavirus encodes a small, hydrophobic polypeptide. Science. 1986;233:464–467. doi: 10.1126/science.3014660. [DOI] [PubMed] [Google Scholar]
- 8.Surti T., Klein O., Smith S.O. Structural models of the bovine papillomavirus E5 protein. Proteins. 1998;33:601–612. [PubMed] [Google Scholar]
- 9.Oates J., Hicks M., Dixon A.M. In vitro dimerization of the bovine papillomavirus E5 protein transmembrane domain. Biochemistry. 2008;47:8985–8992. doi: 10.1021/bi8006252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Villiers E.M., Wagner D., zur Hausen H. Human papillomavirus infections in women with and without abnormal cervical cytology. Lancet. 1987;2:703–706. doi: 10.1016/s0140-6736(87)91072-5. [DOI] [PubMed] [Google Scholar]
- 11.Burkhardt A., DiMaio D., Schlegel R. Genetic and biochemical definition of the bovine papillomavirus E5 transforming protein. EMBO J. 1987;6:2381–2385. doi: 10.1002/j.1460-2075.1987.tb02515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petti L., DiMaio D. Specific interaction between the bovine papillomavirus E5 transforming protein and the β receptor for platelet-derived growth factor in stably transformed and acutely transfected cells. J. Virol. 1994;68:3582–3592. doi: 10.1128/jvi.68.6.3582-3592.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin P., Vass W.C., Velu T.J. The bovine papillomavirus E5 transforming protein can stimulate the transforming activity of EGF and CSF-1 receptors. Cell. 1989;59:21–32. doi: 10.1016/0092-8674(89)90866-0. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein D.J., Finbow M.E., Schlegel R. Bovine papillomavirus E5 oncoprotein binds to the 16K component of vacuolar H+-ATPases. Nature. 1991;352:347–349. doi: 10.1038/352347a0. [DOI] [PubMed] [Google Scholar]
- 15.Goldstein D.J., Kulke R., Schlegel R. A glutamine residue in the membrane-associating domain of the bovine papillomavirus type 1 E5 oncoprotein mediates its binding to a transmembrane component of the vacuolar H+-ATPase. J. Virol. 1992;66:405–413. doi: 10.1128/jvi.66.1.405-413.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein D.J., Andresson T., Schlegel R. The BPV-1 E5 protein, the 16 kDa membrane pore-forming protein and the PDGF receptor exist in a complex that is dependent on hydrophobic transmembrane interactions. EMBO J. 1992;11:4851–4859. doi: 10.1002/j.1460-2075.1992.tb05591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horwitz B.H., Burkhardt A.L., DiMaio D. 44-amino-acid E5 transforming protein of bovine papillomavirus requires a hydrophobic core and specific carboxyl-terminal amino acids. Mol. Cell. Biol. 1988;8:4071–4078. doi: 10.1128/mcb.8.10.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rawls J.A., Loewenstein P.M., Green M. Mutational analysis of bovine papillomavirus type 1 E5 peptide domains involved in induction of cellular DNA synthesis. J. Virol. 1989;63:4962–4964. doi: 10.1128/jvi.63.11.4962-4964.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer A.N., Xu Y.F., Donoghue D.J. Cellular transformation by a transmembrane peptide: structural requirements for the bovine papillomavirus E5 oncoprotein. Proc. Natl. Acad. Sci. USA. 1994;91:4634–4638. doi: 10.1073/pnas.91.11.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Settleman J., Fazeli A., DiMaio D. Genetic evidence that acute morphologic transformation, induction of cellular DNA synthesis, and focus formation are mediated by a single activity of the bovine papillomavirus E5 protein. Mol. Cell. Biol. 1989;9:5563–5572. doi: 10.1128/mcb.9.12.5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilson L.A., Gottlieb R.L., DiMaio D. Mutational analysis of the interaction between the bovine papillomavirus E5 transforming protein and the endogenous β receptor for platelet-derived growth factor in mouse C127 cells. J. Virol. 1995;69:5869–5874. doi: 10.1128/jvi.69.9.5869-5874.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mattoon D., Gupta K., DiMaio D. Identification of the transmembrane dimer interface of the bovine papillomavirus E5 protein. Oncogene. 2001;20:3824–3834. doi: 10.1038/sj.onc.1204523. [DOI] [PubMed] [Google Scholar]
- 23.Talbert-Slagle K., Marlatt S., Dimaio D. Artificial transmembrane oncoproteins smaller than the bovine papillomavirus E5 protein redefine sequence requirements for activation of the platelet-derived growth factor β receptor. J. Virol. 2009;83:9773–9785. doi: 10.1128/JVI.00946-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miozzari G.F., Yanofsky C. Translation of the leader region of the Escherichia coli tryptophan operon. J. Bacteriol. 1978;133:1457–1466. doi: 10.1128/jb.133.3.1457-1466.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lange C., Müller S.D., Ulrich A.S. Structure analysis of the protein translocating channel TatA in membranes using a multi-construct approach. Biochim. Biophys. Acta. 2007;1768:2627–2634. doi: 10.1016/j.bbamem.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 26.Provencher S.W., Glöckner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 27.van Stokkum I.H., Spoelder H.J., Groen F.C. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990;191:110–118. doi: 10.1016/0003-2697(90)90396-q. [DOI] [PubMed] [Google Scholar]
- 28.Sreerama N., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 29.Lobley A., Whitmore L., Wallace B.A. DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics. 2002;18:211–212. doi: 10.1093/bioinformatics/18.1.211. [DOI] [PubMed] [Google Scholar]
- 30.Whitmore L., Wallace B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32(Web Server issue):W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pace C.N., Vajdos F., Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson N.J., Cassim J.Y. Evidence for an α II-type helical conformation for bacteriorhodopsin in the purple membrane. Biochemistry. 1989;28:2134–2139. [Google Scholar]
- 33.Bürck J., Roth S., Ulrich A.S. Conformation and membrane orientation of amphiphilic helical peptides by oriented circular dichroism. Biophys. J. 2008;95:3872–3881. doi: 10.1529/biophysj.108.136085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Y., Huang H.W., Olah G.A. Method of oriented circular dichroism. Biophys. J. 1990;57:797–806. doi: 10.1016/S0006-3495(90)82599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen F.Y., Lee M.T., Huang H.W. Sigmoidal concentration dependence of antimicrobial peptide activities: a case study on alamethicin. Biophys. J. 2002;82:908–914. doi: 10.1016/S0006-3495(02)75452-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J.T., Wu C.S., Martinez H.M. Calculation of protein conformation from circular dichroism. Methods Enzymol. 1986;130:208–269. doi: 10.1016/0076-6879(86)30013-2. [DOI] [PubMed] [Google Scholar]
- 37.Bulheller B.M., Rodger A., Hirst J.D. Circular and linear dichroism of proteins. Phys. Chem. Chem. Phys. 2007;9:2020–2035. doi: 10.1039/b615870f. [DOI] [PubMed] [Google Scholar]
- 38.Monera O.D., Zhou N.E., Hodges R.S. Comparison of antiparallel and parallel two-stranded α-helical coiled-coils. Design, synthesis, and characterization. J. Biol. Chem. 1993;268:19218–19227. [PubMed] [Google Scholar]
- 39.Mao D., Wallace B.A. Differential light scattering and absorption flattening optical effects are minimal in the circular dichroism spectra of small unilamellar vesicles. Biochemistry. 1984;23:2667–2673. doi: 10.1021/bi00307a020. [DOI] [PubMed] [Google Scholar]
- 40.Vogel H. Comparison of the conformation and orientation of alamethicin and melittin in lipid membranes. Biochemistry. 1987;26:4562–4572. doi: 10.1021/bi00388a060. [DOI] [PubMed] [Google Scholar]
- 41.Clayton A.H., Sawyer W.H. Oriented circular dichroism of a class A amphipathic helix in aligned phospholipid multilayers. Biochim. Biophys. Acta. 2000;1467:124–130. doi: 10.1016/s0005-2736(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 42.Hirokawa T., Boon-Chieng S., Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 43.Krogh A., Larsson B., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 44.Wallace B.A., Lees J.G., Janes R.W. Analyses of circular dichroism spectra of membrane proteins. Protein Sci. 2003;12:875–884. doi: 10.1110/ps.0229603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pagel K., Wagner S.C., Koksch B. Random coils, β-sheet ribbons, and α-helical fibers: one peptide adopting three different secondary structures at will. J. Am. Chem. Soc. 2006;128:2196–2197. doi: 10.1021/ja057450h. [DOI] [PubMed] [Google Scholar]
- 46.Potekhin S.A., Melnik T.N., Kajava A.V. De novo design of fibrils made of short α-helical coiled coil peptides. Chem. Biol. 2001;8:1025–1032. doi: 10.1016/s1074-5521(01)00073-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.