

Abstract
Cys-loop receptors are molecular targets of general anesthetics, but the knowledge of anesthetic binding to these proteins remains limited. Here we investigate anesthetic binding to the bacterial Gloeobacter violaceus pentameric ligand-gated ion channel (GLIC), a structural homolog of cys-loop receptors, using an experimental and computational hybrid approach. Tryptophan fluorescence quenching experiments showed halothane and thiopental binding at three tryptophan-associated sites in the extracellular (EC) domain, transmembrane (TM) domain, and EC-TM interface of GLIC. An additional binding site at the EC-TM interface was predicted by docking analysis and validated by quenching experiments on the N200W GLIC mutant. The binding affinities (KD) of 2.3 ± 0.1 mM and 0.10 ± 0.01 mM were derived from the fluorescence quenching data of halothane and thiopental, respectively. Docking these anesthetics to the original GLIC crystal structure and the structures relaxed by molecular dynamics simulations revealed intrasubunit sites for most halothane binding and intersubunit sites for thiopental binding. Tryptophans were within reach of both intra- and intersubunit binding sites. Multiple molecular dynamics simulations on GLIC in the presence of halothane at different sites suggested that anesthetic binding at the EC-TM interface disrupted the critical interactions for channel gating, altered motion of the TM23 linker, and destabilized the open-channel conformation that can lead to inhibition of GLIC channel current. The study has not only provided insights into anesthetic binding in GLIC, but also demonstrated a successful fusion of experiments and computations for understanding anesthetic actions in complex proteins.
Introduction
Molecular mechanisms of general anesthesia remain unsolved. Although there is an ample amount of evidence for anesthetic modulation on functions of various ion channels in the central nervous system (1), the knowledge regarding where anesthetic sites of action are located and how anesthetics alter the functions of these channels is still limited. Cys-loop receptors, a superfamily of ligand-gated ion channels (LGICs), have been identified as potential anesthetic targets (1,2). Members of the LGIC family include glycine and GABA receptors with anion-selective channels and serotonin 5HT3 receptors and nicotinic acetylcholine receptors (nAChRs) with cation-selective channels. The receptors share considerable structural similarity. Each of them is assembled by five subunits, forming a homo- or heteropentameric ion channel, and each subunit consists of an extracellular (EC) domain, four transmembrane segments (TM1–TM4), and an intracellular domain. Binding of agonists to the EC domain activates the channel and produces ion current. The activities of LGICs could be altered by volatile and intravenous anesthetics. It is believed that anesthetics modulate the function of LGICs through direct binding to these receptors (1,2). Experimental proof of direct anesthetic binding to cys-loop receptors, however, has been presented only in a few examples (3–5). More investigations with atomic resolution are required for a comprehensive understanding of interactions between anesthetics and cys-loop receptors.
Gloeobacter violaceus pentameric ligand-gated ion channel (GLIC) is a bacterial homolog of open-channel nAChRs. Its x-ray crystal structures have been solved with resolutions of 2.9 Å (6) and 3.1 Å (7). A recent electrophysiology study validated the relevance of GLIC to anesthetic action (8). Similar to the findings in nAChRs, the channel current of GLIC could be inhibited by inhaled and intravenous general anesthetics. The low Hill number (0.3) implicated multiple binding sites for some anesthetics, but did not offer clues as to where the binding sites are and why binding induces channel inhibition. Collectively, the homologous nature of GLIC to nAChRs and the preceding experimental findings on GLIC provide several advantages for mapping anesthetic interaction sites in GLIC. The high-resolution structure of GLIC allows us to identify anesthetic binding sites with less ambiguity. The established knowledge of anesthetic inhibition on GLIC channel function defines the biological relevance of GLIC for exploring the molecular basis of anesthetic action. The availability of a relatively large quantity of GLIC through protein expression makes the investigation of anesthetic effects on structure and dynamics of GLIC more feasible.
The anesthetic binding sites have been exploited via various biophysical methods. NMR can sensitively detect the anesthetic binding sites in proteins with atomic resolution (9–15). In addition, NMR can also probe the structural and dynamical changes of proteins upon anesthetic binding (9–11,14). However, some technical challenges need to be overcome to acquire high-resolution information of anesthetic binding on integral protein complexes, such as GLIC. X-ray structures of anesthetic-protein complexes revealed anesthetic binding sites with atomic resolution, but the success has been limited, so far, in soluble proteins (16–19). Photoaffinity labeling identified the binding sites for halothane, [3H]azietomidate, and TDBzl-etomidate in nAChR (3,4,20) and [3H]azietomidate in the GABAA receptor (21). The application of this method for mapping binding sites is often limited due to a small number of anesthetics that are equipped with photolabeling probes (22). Tryptophan (TRP) fluorescence of proteins can be quenched effectively by anesthetic binding near the TRP residues of the proteins (23–25). Thus, steady-state fluorescence measurements are effective for mapping anesthetic binding sites and affinities (23–26). In the case of GLIC, steady-state fluorescence experiments are particularly suitable to search for anesthetic sites. Five intrinsic TRPs are located in three sites in the EC domain, transmembrane (TM) domain, and EC-TM interface of GLIC (Fig. 1). They can serve as natural probes for identifying anesthetic binding sites.
Figure 1.
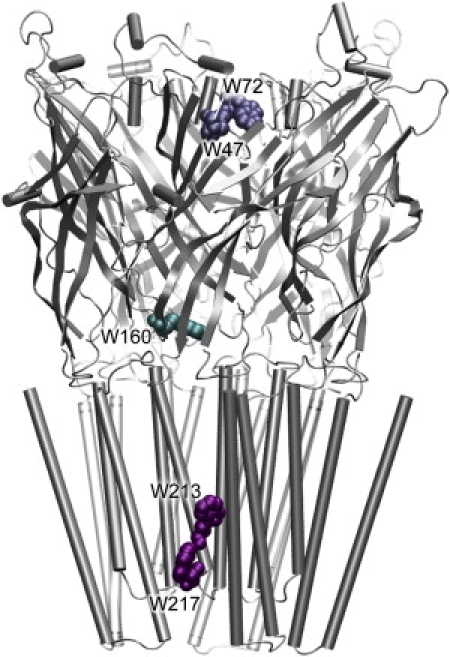
Three tryptophan-associated sites in the wild-type GLIC: W47 and W72 in the EC domain (termed as Site-TrpEC), W160 at the EC-TM interfacial region (termed as Site-TrpINT), and W213 and W217 in the TM domain (termed as Site-Trp™).
In this study, steady-state fluorescence quenching experiments were fused with computational predictions for exploring binding information of the volatile anesthetic halothane and intravenous anesthetic thiopental in GLIC. The fluorescence experiments suggested the binding of halothane and thiopental at four sites, including three intrinsic TRP-associated sites and one additional site at the EC-TM interface predicted by computer docking. The docking analysis provided not only predictions for potential anesthetic binding sites, but also details of anesthetic binding sites that the experiments could not reveal. The subsequent multiple molecular dynamics (MD) simulations of halothane binding to those experimentally predicted sites further explored the underlying cause for inhibition of GLIC channel current. Taken together, through experimental corroboration of computational prediction, our study offered what to our knowledge are the first insights into specific interactions between anesthetics and GLIC that facilitate the understanding of anesthetic modulation on functions of GLIC and homologous cys-loop receptors.
Materials and Methods
GLIC expression and purification
Wild-type GLIC plasmid was generously provided by Professor Raimund Dutzler's group of the University of Zürich, Zürich, Switzerland. The expression and purification of GLIC followed the previously published protocols (6,7). Briefly, GLIC was expressed in the Escherichia coli, extracted with n-dodecyl-β-D-maltoside (Anatrace, Affymetrix, Santa Clara, CA), and purified with the Ni-NTA column (GE Healthcare, Waukesha, WI) before and after HRV3C (GE Healthcare) digestion. The purified GLIC was stored at 4°C in 10 mM potassium phosphate (pH 8.0), 150 mM NaCl, and 0.025% n-dodecyl-β-D-maltoside. Before the fluorescence quenching experiments, the sample buffer was exchanged to 10 mM sodium acetate (pH ∼4) to ensure an open-channel conformation of GLIC (6,7). The final protein concentrations were determined using a DU800 UV/Vis Spectrometer (Beckman Coulter, Fullerton, CA).
Site-directed mutagenesis
Five tryptophan residues in a wild-type GLIC are situated at three different sites, as shown in Fig. 1. We generated three mutants to dissect the anesthetic binding information at each site. Each mutant contained tryptophan(s) at only one site; all other tryptophans were mutated to tyrosines. Thus, the mutant GLICEC has W47 and W72 in the extracellular domain, GLICINT has only W160 at the interface of extracellular and transmembrane domains, and GLIC™ has W213 and W217 in the transmembrane domain. N200 was predicted computationally as being involved in anesthetic binding. GLICN200W was made with replacing all natural tryptophans to tyrosines. All site-directed mutagenesis were done using the QuikChange Multi Site-Directed Mutagenesis Kit (Qiagen, Valencia, CA). The plasmid DNAs were collected and purified using the QIAGEN miniprep kit, and confirmed by sequencing. The mutants were expressed and purified using the same protocol as for wild-type GLIC.
Fluorescence quenching experiments
Anesthetic halothane or thiopental binding to GLIC was determined by steady-state intrinsic tryptophan fluorescence measurements on a Luminescence spectrometer LS50B (PerkinElmer, Waltham, MA). The tryptophan fluorescence was excited at 295 nm. The emission spectra were recorded in the ranges of 320–370 nm and 315–450 nm for halothane and thiopental quenched samples, respectively. The fluorescence peak maximums were ∼333–345 nm for the wild-type GLIC and four mutants. The excitation slit width was kept at 10 nm whereas the emission slit width was adjusted based on the fluorescence intensity of each sample. The recorded fluorescence spectra were the average of three or four scans. For each experiment, the sample premixed with an anesthetic was titrated to the same sample that was initially anesthetic free. The mixture was sealed in a quartz cell of 4-mm pathlength using a Teflon stopper. To prevent halothane evaporation, the premixed sample was stored in gas-tight glass vial and the fresh mixture was used at each halothane concentration. The fluorescence quenching experiments were also performed on GLIC using potassium bromide to evaluate quenching by Br− due to nonspecific binding or collision. The quenched fluorescence by anesthetics, Q, was normalized as
| (1) |
where F0 and F are the fluorescence intensity in the absence and presence of anesthetics, respectively. The maximum possible quenching (Qmax) at the infinite anesthetic concentration, the quenched fluorescence (Q) at anesthetics concentration ([A]), and the anesthetic binding affinity (KD) at the selected tryptophan residue follow the relationship (23,27)
| (2) |
Anesthetics docking in GLIC
Anesthetic sites in GLIC were predicted through flexible docking using the AUTODOCK program (version 3.0.05) (28) on the original crystal structure of GLIC (Protein DataBank (PDB): 3EAM) and snapshots of GLIC after MD simulations for 2 ns, 5 ns, and 10 ns. These relaxed GLIC structures may offer different docking results from the original crystal structure. Inhaled anesthetic halothane and intravenous anesthetic thiopental were investigated. Five-hundred independent docking runs were performed using a Lamarckian genetic algorithm. A grid spacing of 0.375 Å and grid size of 256 × 256 × 280 were utilized to span the entire protein structures. The maximum number of energy evaluations was set to 25,000,000. The docking energy is the average from the energies of the docked anesthetic at a same site. Docking occupancy represents the docking probability within 500 runs. The molecular surface (Connolly's surface) and volume of a putative anesthetics binding pocket were calculated using CASTp (29).
Molecular dynamics simulations and data analysis
MD simulations were performed on three systems:
-
1.
A control without anesthetics.
-
2.
Three halothane molecules near the TM23 loops in the crystal structure (termed as 3HAL-near-TM23).
-
3.
Four halothane molecules near TRP residues in the structure experienced a 5-ns MD simulation (termed as 4HAL-near-TRPs) (see Fig. S1 of the Supporting Material).
Halothane was chosen over other anesthetics because it has the optimized parameters required for MD simulations (30). Details on system preparation and MD simulations are provided in the Supporting Material. Briefly, deprotonations of acidic residues at pH = 4.6 were chosen randomly based on the Henderson-Hasselbach equation and the pKa calculations by Bocquet et al. (6). The x-ray structure of GLIC (PDB: 3EAM) was integrated with a fully hydrated and preequilibrated binary POPE-POPG (3:1) lipid mixture. Each simulation system was energy-minimized and followed by an equilibration simulation. MD simulations were performed using the NAMD2 package (31) with CHARMM27 force field with CMAP corrections (Ver. 31) (32). All three systems underwent unrestrained Nosé-Hoover constant pressure (P = 1 bar) and temperature (T = 310 K) (33,34) (NPT) simulation for >10 ns. An additional system of the mutant N200W GLIC was set up and equilibrated for 2 ns using similar protocols as described in the Supporting Material.
VMD (35) with home-developed scripts was used for data analysis and visualization. Pore-radius profiles were computed using the HOLE program (36). The system stability over the course of simulation was assessed using the Cα root mean-square deviation (RMSD) with respect to the crystal structure. The protein dynamics was evaluated from the root-mean-square fluctuation (RMSF).
Results and Discussion
Fluorescence quenching revealed multiple anesthetic binding sites in GLIC
Halothane and thiopental demonstrated high binding capabilities in GLIC. As shown in Fig. 2, both drugs quenched >80% of the intrinsic tryptophan fluorescence in a saturable manner. To prove that the observed quenching by halothane and thiopental resulted not from nonspecific collision, we performed the fluorescence quenching experiments on the same GLIC sample with KBr. The rationale for choosing Br− is based on the fact that the bromine atom of halothane and the negative charge of thiopental are considered as effective elements for quenching tryptophan fluorescence. Br− is highly soluble in GLIC samples and has nonspecific collision with GLIC. As expected, the Br− collision did not produce significant fluorescence quenching (<5%) on GLIC when Br− was in the same concentration range as halothane (Fig. 2 A) or even in a much higher concentration (data not shown). The saturable quenching pattern by halothane and thiopental was distinctly different from the insensitive quenching by Br−, suggesting that halothane and thiopental quenching was not due to nonspecific collisions.
Figure 2.

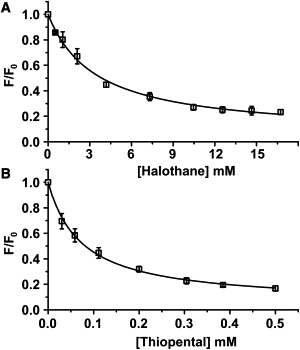
Fluorescence quenching of wild-type GLIC (square), GLICEC (circle), GLICINT (triangle), and GLIC™ (diamond) by various concentrations of (A) volatile anesthetic halothane and (B) intravenous anesthetic thiopental. (Solid lines) Nonlinear fitting of experimental data to Eq. 2. The experimental error bars (∼5%) are omitted for clarity. The maximum quenching Qmax and anesthetic disassociation constant KD from fittings are presented in Table 1. The quenching of wild-type GLIC by potassium bromide (pentagon) was included in panel A as reference.
The data fitting to Eq. 2 resulted in the dissociation constant KD of 0.10 ± 0.01 mM and the maximum quenching Qmax of 100 ± 2% for thiopental, and KD of 2.3 ± 0.1 mM and Qmax of 86 ± 1% for halothane. A higher binding affinity of thiopental than halothane in GLIC is consistent with their relative binding affinities in homologous cys-loop receptors and the difference of their clinical potencies (37,38). The experiments on individual GLIC mutants confirmed that both halothane and thiopental could quench all three TRP-associated sites (GLICEC, GLICINT, and GLIC™) up to saturation (Fig. 2). The KD and Qmax values of halothane and thiopental in GLICWT and the mutants are summarized in Table 1.
Table 1.
Results of GLIC fluorescence quenching by anesthetic halothane and thiopental
| GLIC | Halothane |
Thiopental |
||
|---|---|---|---|---|
| Qmax | KD (mM) | Qmax | KD (mM) | |
| GLICWT | 0.86 ± 0.01 | 2.3 ± 0.1 | 1.00 ± 0.02 | 0.10 ± 0.01 |
| GLICECD | 0.90 ± 0.02 | 1.3 ± 0.2 | 1.00 ± 0.05 | 0.11 ± 0.02 |
| GLICINT | 0.84 ± 0.02 | 1.7 ± 0.2 | 0.94 ± 0.02 | 0.067 ± 0.005 |
| GLICTMD | 0.85 ± 0.01 | 2.5 ± 0.1 | 1.00 ± 0.03 | 0.13 ± 0.01 |
| GLICN200W | 0.95 ± 0.03 | 3.4 ± 0.3 | 0.97 ± 0.01 | 0.067 ± 0.003 |
Values of apparent maximum quenching (Qmax) and anesthetic disassociation constant (KD) were derived from fitting the quenching data to Eq. 2 in the text.
Several notions can be derived from these data.
First, there are multiple anesthetic sites of similar binding affinities in GLIC. The finding implicates a possibility that multiple anesthetic sites may also exist in homologous proteins, such as cys-loop receptors. Indeed, our recent computational studies identified multiple halothane binding sites in neural nAChRs (39,40). Multiple residues of Torpedo californica nAChRs were also found photolabeled by [3H]azietomidate (3).
Secondly, because the inhaled anesthetic halothane and the intravenous anesthetic thiopental have quite different chemical properties and molecular sizes, it is unlikely that they occupy the same sites in GLIC. The same pattern of tryptophan quenching may not necessarily indicate the identical binding sites for halothane and thiopental.
How do those TRP-associated sites differ for halothane and thiopental?
Do halothane and thiopental have other interaction sites in GLIC besides the aforementioned three intrinsic TRP-associated sites?
How can anesthetic binding alter the function of GLIC?
The answers have been explored with anesthetic docking, additional experiments inspired by docking, and MD simulations.
An additional binding site at the EC-TM interface, predicted by docking and validated by fluorescence quenching
Docking thiopental and halothane to the x-ray structure of GLIC or its relaxed structures after MD simulations identified the binding sites that were compatible with fluorescence quenching. In addition, the docking predicted another major binding site consisting of N200 of TM1 and residues (E243, P247, and T249) of the TM23 loop in the adjacent subunit (termed as Site-near-TM23-loop; see Fig. S2). To verify this docking prediction, we mutated N200 to tryptophan and performed fluorescence quenching on GLICN200W. As shown in Fig. 3, both halothane and thiopental could quench the fluorescence of GLICN200W with binding affinities slightly weaker than or equivalent to those in three intrinsic tryptophan-associated sites. Compared to the intrinsic tryptophan-associated site in GLICINT, the Site-near-TM23-loop is closer to the TM domain at the EC-TM interface (Fig. 4 D). Nevertheless, the importance of the EC-TM interface for anesthetic binding is validated by this study.
Figure 3.
Fluorescence quenching of GLICN200W, a mutant of the anesthetic binding site predicted by computer docking, by (A) volatile anesthetic halothane and (B) intravenous anesthetics thiopental. The solid lines resulted from nonlinear fitting of the experimental data using Eq. 2. The error bars are the standard deviations of three experiments. The fitting results are included in Table 1.
Figure 4.
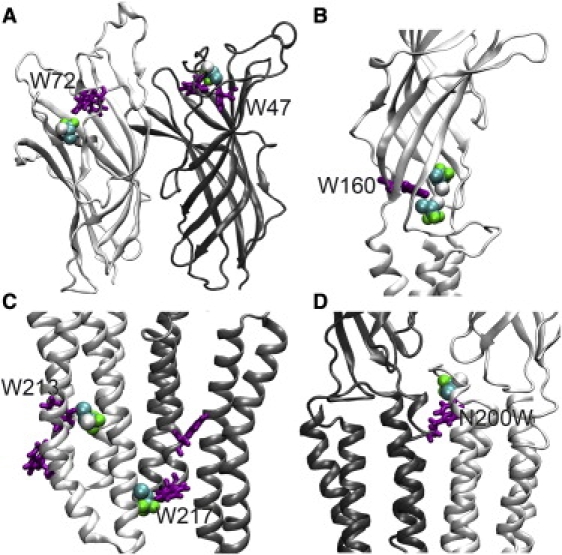
Halothane molecules (van der Waals format) at putative binding sites near TRP residues (licorice format) of GLIC or N200W of the mutant, suggested by halothane docking on the MD-relaxed protein structures. (A) Near W47 and W72 in the EC domains of GLIC; (B) near W160 at the EC-TM regions; (C) near W213 and W217 in the TM domains; and (D) near N200W at the EC-TM regions of the mutant. Note that most of halothane molecules are bound to the pocket within a subunit. Bromide or chloride atom of halothane interacts directly with the indole ring of TRP at most sites.
Halothane binding is more sensitive than thiopental to subtle changes in protein structure
The original crystal structure of GLIC and the structure snapshots after multiple nanosecond MD simulations yielded similar major binding sites for thiopental docking. Three intrinsic tryptophan-associated sites and the mutation site of N200W were identified for thiopental binding. Moreover, the docking also correctly predicted the site near W160 (termed Site-TrpINT) for the highest affinity of thiopental binding. Detailed docking results are summarized in Table S1.
In contrast, halothane docking seemed much more sensitive to subtle changes in GLIC structure. In the original crystal structure, the docking predicted the Site-near-TM23-loop and the site near D86 and D88 in the EC domain (termed as Site-near-EC-D86), but could not allocate the sites with intrinsic tryptophans. When halothane was docked to a relaxed structure, even a snapshot of GLIC after 2-ns simulation, those sites identified by our quenching experiments became apparent. Fig. 4 depicts representative halothane docking at these sites. The bromine or chlorine atom of halothane is oriented toward the indole ring of TRP in most sites that could yield effective fluorescence quenching.
The sensitivity of halothane docking to various subtle changes in GLIC structure may result from the intrinsically low binding affinity of halothane. The fact that both original x-ray and MD-relaxed structures of GLIC offered some, but not all, experimentally validated binding sites in halothane docking indicates the existence of the structural ensemble, in which GLIC structural variation is subtle enough to keep thiopental binding but sufficient to affect halothane binding. Alignment of the original x-ray structure and the structure after 5-ns MD simulations showed a small structural difference (Fig. S3). Relative to the original crystal structure, Cα RMSDs of a whole and an individual subunit structure of GLIC after 5-ns MD simulation were 2.0 Å and 1.2 Å, respectively. The MD relaxed structures showed comparatively looser intersubunit contacts and slightly expanded intrasubunits. Why did large size thiopental remain at the same binding sites whereas smaller halothane relocated to new sites when the GLIC structure changed from a rigid to a more relaxed form? It turned out that the binding pockets for halothane and thiopental were not entirely overlapped, even though both could quench the same tryptophan residues.
Different binding preference to intra- and intersubunit pockets
As shown in Fig. 4, halothane occupied predominately intrasubunit pockets and presumably quenched tryptophan fluorescence mainly through interaction within the subunit. In contrast, thiopental seemed to interact with tryptophans through the intersubunit pocket. Fig. 5 displays such an example, where the Site-TrpINT for thiopental was formed by residues of the F loop (or β8-β9) and β1-β2 loop from one subunit and K248 in the TM23 loop from the other subunit. Sulfur atom of thiopental interacts directly with the nearby indole ring of TRP. Although both halothane and thiopental were within reach of the highly conserved residues D122, D32, R192, and W160 in the GLIC (Fig. 4 B and Fig. 5), and W160 could be quenched by them in the fluorescence experiments, their binding pockets were not exactly overlapped. Smaller size halothane preferred the intrasubunit pocket for optimizing its interaction with surrounding resides. The intrasubunit pocket near W160 in the original crystal structure was too small (∼60 Å3) for halothane, but the pocket size expanded to a volume of 130∼300 Å3 through MD simulations and became large enough for halothane binding (Fig. S3). Thiopental is larger than halothane and could fit only into the intersubunit pocket. The larger surface area of thiopental made its interaction with protein more robust so that a 10% pocket volume change in the MD-relaxed structure did not change the docking results.
Figure 5.

Predicted binding for intravenous anesthetic thiopental near W160 at the pocket between two subunits colored in lime and orange. Docking was performed on a snapshot of GLIC after a 5-ns MD simulation in a fully hydrated POPG-POPE mixture. Only the docking with the lowest energy was shown. Thiopental is highlighted with mesh surfaces. Sulfur atom of thiopental is shown in a large yellow sphere. Oxygen, nitrogen, carbon, and hydrogen of anesthetics are shown in red, blue, tan, and white, respectively. Sulfur atom of thiopental interacts directly with the indole ring of TRP.
Possibilities for anesthetics to act as channel blockers
In addition to the aforementioned major sites with high occupancy and affinity, docking predicted possible binding inside the channel for intravenous anesthetic thiopental, but not for halothane. Thiopental could dock to a region between S226 and I240 inside the channel lumen with comparatively weaker binding affinity than the major binding sites (see Fig. 6). It is interesting to note that in the crystal structure (PDB: 3EAM), several detergent molecules are found inside the channel lumen at a region corresponding to the intravenous anesthetic docking sites revealed here. Although the validity of these docking predictions needs to be confirmed, these docking predictions are certainly valuable for designing future experiments.
Figure 6.
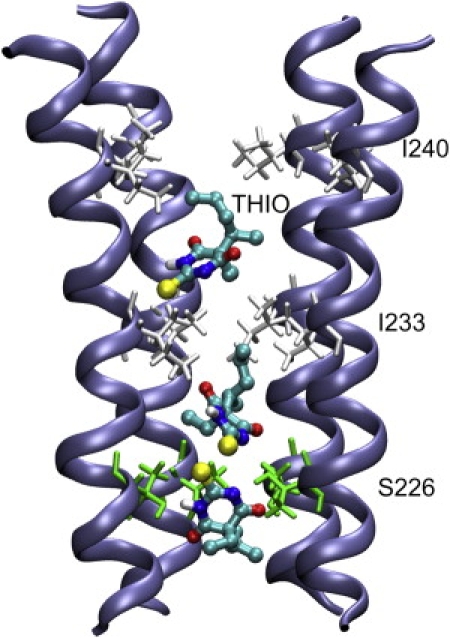
Putative binding of intravenous anesthetic thiopental inside the GLIC channel, predicted by docking on the MD relaxed GLIC structure. For clarity, only four TM2 subunits are shown. Residues S226, I233, and I240 are plotted in licorice format. Sulfur atom of thiopental is shown in a large sphere. Oxygen, nitrogen, carbon, and hydrogen of anesthitcs are colored differently. The docking energy of thiopental is −5.2 ± 0.3 Kcal/mol and the docking occupancy is 4%.
Halothane binding induced structural and dynamical consequences on GLIC
To determine how anesthetic binding affects GLIC dynamics that may impact GLIC function, we performed multiple MD simulations on several GLIC systems in the absence and presence of halothane (see Fig. S1). In all systems, GLIC pentameric structures remained stable and the maximum Cα RMSD from the initial crystal structure was only 2.4 ± 0.3 Å after 10-ns MD simulations. The backbone pore radius profiles of GLIC in all systems were similar to the one in the original crystal structure (see Fig. S4), indicating that halothane binding did not cause significant structural changes.
Over the course of >10-ns simulations, halothane resided stably in majority binding sites. The exceptions were found in the 4HAL-near-TRPs system, where two halothane molecules in the Site-TrpEC and Site-Trp™ moved away from their initial docked sites within a 1-ns MD simulation. This seemingly short residing time is probably long enough for fluorescence quenching, considering the picoseconds to nanoseconds lifetime of tryptophan fluorescence in protein (41,42).
The MD simulations demonstrated that halothane binding could affect the protein dynamics, especially at the EC/TM interface. Halothane binding to the Site-near-TM23-loop (see Fig. S1 A) decreased the RMSF of the TM23-loop and some regions in EC domains but significantly increased the RMSF of the β6-β7 loop (corresponding to the cys-loop in the nAChR), as demonstrated in Fig. 7. Both the TM23-loop and the cys-loop were suggested to play an important role in the channel gating for cys-loop receptors (43–45). Changes in their flexibility by halothane may impact on the function of GLIC. Moreover, halothane was found to disrupt interactions between critical residues of GLIC. In the 4HAL-near-TRPs system, halothane near W160 perturbed the salt bridges between D32 and R192 (see Fig. 8), increased the probability of breaking D32-R192 salt bridges by >40%, and increased residue fluctuation near the binding sites by >20%. Homologous salt-bridges in nAChR, i.e., α1-E45 and α1-R209 nAChR, were suggested to be particularly important for channel gating and functions (44). Removing these salt-bridges by mutations dramatically reduced the open channel stability in the ligand-gated ion channels (45–47). Thus, halothane binding near W160 may inhibit the channel current through decreasing open channel stability. Because thiopental binding to the W160 site was also confirmed, it is very likely that thiopental inhibits GLIC function via a similar mechanism.
Figure 7.
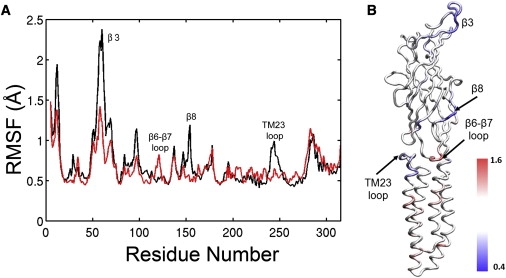
(A) Comparison of the subunit root-mean-squared-fluctuation (RMSF) of GLIC in the absence (black) and presence (red) of halothane near N200 over the last 1-ns simulation. (B) The ratio of RMSFs in the presence and absence of halothane is color-coded onto a subunit structure of GLIC. The regions having values significantly smaller (i.e., reducing RMSF) or greater (i.e., increasing RMSF) than one are colored in blue or red, respectively. The regions in white color had no significant change in RMSF.
Figure 8.
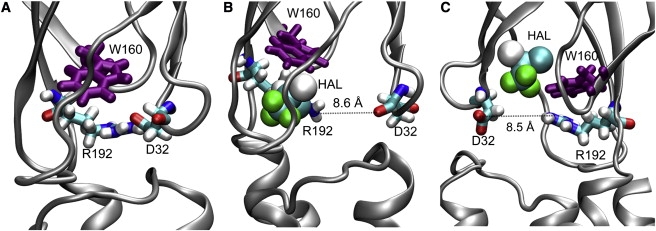
(A) Salt bridge between D32 and R192 in the control system of GLIC. The salt bridge was perturbed by halothane binding near W160. (B) Halothane interacts with R192, and (C) halothane interacts with D32. Halothane molecules are shown in van der Waals format and protein residues are presented in licorice format.
Conclusions
Several important conclusions have emerged in this experimental and computational hybrid study.
First, multiple anesthetic binding sites of similar binding affinities exist in open-channel GLIC. Previous investigations of nAChRs also found multiple anesthetic interaction sites in both open- and closed-channel structures (3,4,26,40,48). It is possible that this is a common property shared by all cys-loop receptors.
Secondly, smaller size volatile anesthetics, such as halothane, prefer the binding sites within a subunit, whereas larger size intravenous anesthetics, such as thiopental, prefer the binding sites between two subunits. Sometimes the intra- and intersubunit binding pockets could partially overlap, as evidenced in our finding that tryptophans in GLIC could be quenched by both halothane and thiopental.
Thirdly, anesthetic binding at the EC-TM interface of GLIC destabilized the open-channel conformation, which could be the major cause for inhibition of the channel current (8).
Finally, it seems premature to conclude that intravenous anesthetics can block the GLIC channel solely based on the docking results. However, such a prediction is worth testing in future experiments, considering that indeed there are several detergent molecules inside the GLIC channel in one of the original crystal structures (6).
Acknowledgments
The authors thank Drs. Tommy Tillman, Dan Willenbring, and Murali Jayaraman for their helpful discussion and Dr. Willenbring for the ab initio calculations of thiopental structures used for docking. The authors also thank Dr. Ricarda J. C. Hilf for her helpful discussion for GLIC expression and purification.
This research was supported in part by the National Science Foundation through TeraGrid resources provided by the Pittsburgh Supercomputing Center. TeraGrid systems are hosted by Indiana University, Laboratory of Neuro Imaging, National Center for Atmospheric Research, National Center for Supercomputing Applications, National Institute for Computational Sciences, Oak Ridge National Laboratory, Pittsburgh Supercomputing Center, Purdue University, San Diego Supercomputer Center, Texas Advanced Computing Center, and University of California, Argonne National Laboratory. This research was also supported by grants from the National Institutes of Health (Nos. R01GM066358, R01GM056257, and R37GM049202).
Supporting Material
References
- 1.Franks N.P., Lieb W.R. Molecular and cellular mechanisms of general anesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 2.Hemmings H.C., Jr., Akabas M.H., Harrison N.L. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol. Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Chiara D.C., Hong F.H., Cohen J.B. Time-resolved photolabeling of the nicotinic acetylcholine receptor by [3H]azietomidate, an open-state inhibitor. Mol. Pharmacol. 2009;75:1084–1095. doi: 10.1124/mol.108.054353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiara D.C., Dangott L.J., Cohen J.B. Identification of nicotinic acetylcholine receptor amino acids photolabeled by the volatile anesthetic halothane. Biochemistry. 2003;42:13457–13467. doi: 10.1021/bi0351561. [DOI] [PubMed] [Google Scholar]
- 5.Eckenhoff R.G. An inhalational anesthetic binding domain in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 1996;93:2807–2810. doi: 10.1073/pnas.93.7.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bocquet N., Nury H., Corringer P.J. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 7.Hilf R.J.C., Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–118. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- 8.Weng Y., Yang L., Sonner J.M. Anesthetic sensitivity of the Gloeobacter violaceus proton-gated ion channel. Anesth. Analg. 2010;110:59–63. doi: 10.1213/ANE.0b013e3181c4bc69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bondarenko V., Yushmanov V.E., Tang P. NMR study of general anesthetic interaction with nAChR β2 subunit. Biophys. J. 2008;94:1681–1688. doi: 10.1529/biophysj.107.116772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canlas C.G., Cui T., Tang P. Anesthetic modulation of protein dynamics: insight from an NMR study. J. Phys. Chem. B. 2008;112:14312–14318. doi: 10.1021/jp805952w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui T., Bondarenko V., Tang P. Four-α-helix bundle with designed anesthetic binding pockets. Part II. Halothane effects on structure and dynamics. Biophys. J. 2008;94:4464–4472. doi: 10.1529/biophysj.107.117853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang P., Hu J., Xu Y. Distinctly different interactions of anesthetic and nonimmobilizer with transmembrane channel peptides. Biophys. J. 1999;77:739–746. doi: 10.1016/S0006-3495(99)76928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang P., Eckenhoff R.G., Xu Y. General anesthetic binding to gramicidin A: the structural requirements. Biophys. J. 2000;78:1804–1809. doi: 10.1016/S0006-3495(00)76730-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang P., Simplaceanu V., Xu Y. Structural consequences of anesthetic and nonimmobilizer interaction with gramicidin A channels. Biophys. J. 1999;76:2346–2350. doi: 10.1016/S0006-3495(99)77391-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Y., Seto T., Firestone L. NMR study of volatile anesthetic binding to nicotinic acetylcholine receptors. Biophys. J. 2000;78:746–751. doi: 10.1016/S0006-3495(00)76632-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franks N.P., Jenkins A., Brick P. Structural basis for the inhibition of firefly luciferase by a general anesthetic. Biophys. J. 1998;75:2205–2211. doi: 10.1016/S0006-3495(98)77664-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharya A.A., Curry S., Franks N.P. Binding of the general anesthetics propofol and halothane to human serum albumin. High resolution crystal structures. J. Biol. Chem. 2000;275:38731–38738. doi: 10.1074/jbc.M005460200. [DOI] [PubMed] [Google Scholar]
- 18.Liu R., Loll P.J., Eckenhoff R.G. Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 2005;19:567–576. doi: 10.1096/fj.04-3171com. [DOI] [PubMed] [Google Scholar]
- 19.Vedula L.S., Brannigan G., Loll P.J. A unitary anesthetic binding site at high resolution. J. Biol. Chem. 2009;284:24176–24184. doi: 10.1074/jbc.M109.017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nirthanan S., Garcia G., 3rd, Cohen J.B. Identification of binding sites in the nicotinic acetylcholine receptor for TDBzl-etomidate, a photoreactive positive allosteric effector. J. Biol. Chem. 2008;283:22051–22062. doi: 10.1074/jbc.M801332200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li G.D., Chiara D.C., Cohen J.B. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckenhoff R.G., Xi J., Dailey W.P. Analgesia. Humana Press; Totowa, NJ: 2010. Inhalational anesthetic photolabeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson J.S., Eckenhoff R.G., Dutton P.L. Binding of halothane to serum albumin demonstrated using tryptophan fluorescence. Anesthesiology. 1995;83:316–324. doi: 10.1097/00000542-199508000-00012. [DOI] [PubMed] [Google Scholar]
- 24.Solt K., Johansson J.S. Binding of the active metabolite of chloral hydrate, 2,2,2-trichloroethanol, to serum albumin demonstrated using tryptophan fluorescence quenching. Pharmacology. 2002;64:152–159. doi: 10.1159/000056165. [DOI] [PubMed] [Google Scholar]
- 25.Johansson J.S., Manderson G.A., Eckenhoff R.G. Binding of the volatile general anesthetics halothane and isoflurane to a mammalian β-barrel protein. FEBS J. 2005;272:573–581. doi: 10.1111/j.1742-4658.2004.04500.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu J., Strzalka J., Blasie J.K. Mechanism of interaction between the general anesthetic halothane and a model ion channel protein. II. Fluorescence and vibrational spectroscopy using a cyanophenylalanine probe. Biophys. J. 2009;96:4176–4187. doi: 10.1016/j.bpj.2009.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solt K., Johansson J.S., Raines D.E. Kinetics of anesthetic-induced conformational transitions in a four-α-helix bundle protein. Biochemistry. 2006;45:1435–1441. doi: 10.1021/bi052206o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris G.M., Goodsell D.S., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 29.Dundas J., Ouyang Z., Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z., Xu Y., Tang P. Parametrization of 2-bromo-2-chloro-1,1,1-trifluoroethane (halothane) and hexafluoroethane for nonbonded interactions. J. Phys. Chem. A. 2004;108:781–786. [Google Scholar]
- 31.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 33.Nose S. A unified formulation of the constant-temperature molecular-dynamics methods. J. Chem. Phys. 1984;81:511–519. [Google Scholar]
- 34.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 35.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 36.Smart O.S., Neduvelil J.G., Sansom M.S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996;14:354–360. doi: 10.1016/s0263-7855(97)00009-x. 376. [DOI] [PubMed] [Google Scholar]
- 37.Roth S.H., Miller K.W., Fink B.R. Plenum Medical; New York: 1986. Molecular and Cellular Mechanisms of Anesthetics. [Google Scholar]
- 38.Tassonyi E., Charpantier E., Bertrand D. The role of nicotinic acetylcholine receptors in the mechanisms of anesthesia. Brain Res. Bull. 2002;57:133–150. doi: 10.1016/s0361-9230(01)00740-7. [DOI] [PubMed] [Google Scholar]
- 39.Liu L.T., Willenbring D., Tang P. General anesthetic binding to neuronal α4β2 nicotinic acetylcholine receptor and its effects on global dynamics. J. Phys. Chem. B. 2009;113:12581–12589. doi: 10.1021/jp9039513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L.T., Haddadian E.J., Tang P. Higher susceptibility to halothane modulation in open- than in closed-channel α4β2 nAChR revealed by molecular dynamics simulations. J. Phys. Chem. B. 2010;114:626–632. doi: 10.1021/jp908944e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y., Barkley M.D. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 42.Alcala J.R., Gratton E., Prendergast F.G. Fluorescence lifetime distributions in proteins. Biophys. J. 1987;51:597–604. doi: 10.1016/S0006-3495(87)83384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kash T.L., Jenkins A., Harrison N.L. Coupling of agonist binding to channel gating in the GABAA receptor. Nature. 2003;421:272–275. doi: 10.1038/nature01280. [DOI] [PubMed] [Google Scholar]
- 44.Xiu X., Hanek A.P., Dougherty D.A. A unified view of the role of electrostatic interactions in modulating the gating of cys loop receptors. J. Biol. Chem. 2005;280:41655–41666. doi: 10.1074/jbc.M508635200. [DOI] [PubMed] [Google Scholar]
- 45.Jha A., Cadugan D.J., Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the cys-loop and M2-M3 linker. J. Gen. Physiol. 2007;130:547–558. doi: 10.1085/jgp.200709856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee W.Y., Sine S.M. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- 47.Sala F., Mulet J., Criado M. Charged amino acids of the N-terminal domain are involved in coupling binding and gating in α7 nicotinic receptors. J. Biol. Chem. 2005;280:6642–6647. doi: 10.1074/jbc.M411048200. [DOI] [PubMed] [Google Scholar]
- 48.Pratt M.B., Husain S.S., Cohen J.B. Identification of sites of incorporation in the nicotinic acetylcholine receptor of a photoactivatable general anesthetic. J. Biol. Chem. 2000;275:29441–29451. doi: 10.1074/jbc.M004710200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.