

Abstract
Although the oligomers formed by Aβ peptides appear to be the primary cytotoxic species in Alzheimer's disease, detailed information about their structures appears to be lacking. In this article, we use exhaustive replica exchange molecular dynamics and an implicit solvent united-atom model to study the structural properties of Aβ monomers, dimers, and tetramers. Our analysis suggests that the conformational ensembles of Aβ dimers and tetramers are very similar, but sharply distinct from those sampled by the monomers. The key conformational difference between monomers and oligomers is the formation of β-structure in the oligomers occurring together with the loss of intrapeptide interactions and helix structure. Our simulations indicate that, independent of oligomer order, the Aβ aggregation interface is largely confined to the sequence region 10–23, which forms the bulk of interpeptide interactions. We show that the fractions of β structure computed in our simulations and measured experimentally are in good agreement.
Introduction
Aberrant aggregation of polypeptide chains and subsequent amyloid fibril formation are linked to more than 20 various medical disorders, including Alzheimer's, Parkinson's, and Creutzfeldt-Jakob diseases (1). Biomedical experiments and genetic studies suggested that the onset of Alzheimer's disease is related to extracellular aggregation of Aβ peptides (2), which are produced by natural cleavage of the transmembrane amyloid precursor protein. Although these peptides have varying lengths, the 40-residue species, Aβ1–40, are most abundant. Their assembly into amyloid fibrils is a multistage conformational transition, in which mobile oligomeric species appear as intermediates on the pathway leading to mature fibrils (3–7). It has been long known that Aβ fibrils are cytotoxic (8), but recent findings pointed to Aβ oligomers as primary cytotoxic species in Alzheimer's disease (9,10). Furthermore, synaptic structure and function can be impaired even by the smallest Aβ oligomers, dimers (11). According to hydrogen/deuterium exchange experiments, soluble oligomeric species exist in dynamic equilibrium with amyloid fibrils (12). These observations implicate a crucial role played by Aβ oligomers in amyloidogenesis.
A number of experimental studies have been focused on elucidating the structures of Aβ monomers. In aqueous solution, they are largely random, lacking stable secondary or tertiary structures (13–15). Solid-state NMR experiments have probed the endproducts of amyloid assembly, Aβ fibrils, revealing in-registry parallel β-sheet structure (16–18). In contrast to Aβ monomers or fibrils, there is a lack of structural information on Aβ oligomers. At micromolar concentration and normal physiological conditions, Aβ oligomers coexist with monomers (19) and demonstrate a distinctive size distribution existing predominantly as dimers, trimers, or tetramers (20). Higher order (n > 4) oligomers have significantly smaller frequency of occurrence.
Molecular dynamics (MD) simulations, which probe Aβ amyloid formation at all-atom resolution, can provide important structural information supplemental to the experiments (21). For example, conformational ensembles sampled by Aβ1–40 monomer (22,23) and its fragments (24,25) have been investigated. A full-length Aβ1–40 was shown to have several structured regions, including short β-strands, helices, and β-turns, in generally random coil-like ensemble of conformations (22,23). MD simulations of Aβ oligomers, which are more computationally intensive, have been recently reported (26–31). In general, these studies have shown that aggregation leads to conformational changes in Aβ peptides with respect to monomeric species. For example, we have previously shown that interpeptide interactions in Aβ dimers shift the distribution of secondary structure, from helical states toward β-strand conformations (28).
Nevertheless, many questions concerning the oligomer structure remain unexplored. For example (1), what is the distribution of conformations sampled by individual Aβ peptides in the oligomers? It is challenging to obtain this information in the experiments, which report the bulk averages of structural quantities (2). Are there differences between the conformational ensembles of oligomers of different orders and between those of oligomers and monomers? In other words, how does the structure of Aβ peptides change with the progression of aggregation from monomers to, say, tetramers?
To answer these questions, we use exhaustive replica exchange MD (REMD) and an implicit solvent united-atom model to study the structural properties of Aβ monomers, dimers and tetramers. Because dimers and tetramers represent the most abundant Aβ oligomers, some tentative conclusions about Aβ oligomers and their comparison with the monomers can be made. In this work we use the N-terminal truncated fragment of the full-length peptide, Aβ10–40 (Fig. 1 a). According to the experiments (15,17,32,33) and simulations (28,34) truncation of the first nine N-terminal amino acids results in minor changes in the conformational ensembles of Aβ monomers, oligomers, and fibrils (see the Supporting Material for details). Consequently, we use Aβ10–40 as a model of the full-length Aβ1–40 peptide.
Figure 1.
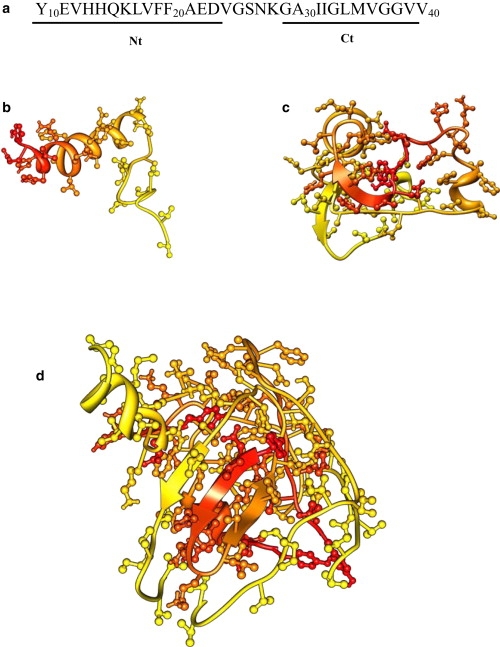
(a) The sequence of Aβ10–40 peptide and the allocation of the N-terminal Nt and C-terminal Ct regions. (b–d) Three Aβ10–40 species studied in our simulations: (b) monomer, (c) dimer, and (d) tetramer. The N- and C-terminals are colored in shades of red and yellow, respectively. The Nt region tends to form most of the interpeptide interactions. The structures are visualized using Chimera (50).
First, we show that the distributions of Aβ peptide conformations are similar in dimers and tetramers, but are sharply different from those sampled in monomeric species.
Second, the oligomer conformational ensemble is characterized by few distinct structural basins, in which the majority of Aβ peptides reside. These basins differ with respect to the distribution of secondary structure and the amount of inter- and intrapeptide interactions.
Third, our simulations suggest that independent of oligomer order n, the Aβ aggregation interface is largely confined to the sequence region 10–23, which forms the bulk of interpeptide interactions. We conclude the article by comparing the computational and experimental distributions of secondary structure.
Methods
Simulation model
To probe the conformational ensembles of Aβ peptides, we employed the CHARMM MD program (35), the united-atom force field CHARMM19, and the SASA implicit solvent model (36). The description of the CHARMM19+SASA model and its testing were reported in our previous studies (28,34,37,38). In recent years, the CHARMM19+SASA model has been used in protein folding simulations (39,40) and to study aggregation of amyloidogenic peptides (41,42). Arguments supporting the use of CHARMM19+SASA implicit solvent model are presented in the Supporting Material.
Three Aβ10–40 systems—monomer, dimer, and tetramer—were considered (Fig. 1). Their description can be found in our previous studies (28,34,43). Briefly, the monomer, dimer, and tetramer systems involve one, two, or four identical unconstrained Aβ10–40 peptides, respectively. The simulation systems utilized spherical boundary condition with the radius Rs = 90 Å and the force constant ks = 10 kcal/(mol Å2). The concentration of Aβ peptides was therefore on the order of mM. Because in vitro Aβ peptides exist predominantly in the form of monomers through tetramers (20), the three in silico systems represent the most abundant Aβ species (excluding the trimers).
Replica exchange simulations
Conformational sampling was performed using replica exchange molecular dynamics (REMD) (44). The description of REMD implementation can be found in our previous studies (28,34). Briefly, 24 replicas were distributed linearly in the temperature range from 300 to 530 K with the increment of 10 K. The temperature range spans the spectrum of Aβ conformational states from aggregated (or collapsed) to completely dissociated (or open). Due to small temperature increments, the energy distributions from neighboring replicas share significant overlap. The exchanges were attempted every 80 ps between all neighboring replicas with the average acceptance rate of 67% (monomer), 54% (dimer), and 38% (tetramer). In all, four (monomer), seven (dimer), or eight (tetramer) REMD trajectories were produced resulting in the cumulative simulation times of 76 (monomer), 134 (dimer), and 154 μs (tetramer). The structures were saved every 40 ps. Between replica exchanges, the system evolved using NVT underdamped Langevin dynamics with the damping coefficient γ = 0.15 ps–1 and the integration step of 2fs. Because the initial parts of REMD trajectories are not equilibrated and must be excluded from thermodynamic analysis, the cumulative equilibrium simulation time was reduced to τsim ≈ 72, ≈113, and ≈126 μs. The REMD trajectories were started with random distributions of peptides in the sphere equilibrated at 600 K. In the initial structures, all peptides were dissociated.
Computation of structural probes
The interactions formed in Aβ peptides and oligomers were analyzed by considering side-chain contacts and hydrogen bonds (HBs). A side-chain contact is formed, if the distance between the centers of mass of side chains is <6.5Å. Two peptides are considered aggregated, if there is at least one side-chain contact between them. Backbone HBs between NH and CO groups were assigned according to Kabsch and Sander (45). We defined two classes of interpeptide backbone HBs. The first includes any HBs between the peptides. The second class is restricted to parallel β-sheet HBs. A parallel HB (pHB) is formed between the residues i and j, if at least one other HB is also present between i + 2 and j or j + 2 (or between i – 2 and j or j – 2). The definition of pHB follows from the structural analysis of parallel β-sheets. Secondary structure in Aβ peptides was computed using the distribution of (ϕ, ψ) backbone dihedral angles. Specific definitions of β-strand and helix states can be found in our previous studies (28).
Throughout this article, angle brackets 〈…〉 imply thermodynamic averages. Because dimer and tetramer include several indistinguishable peptides, we report averages over two and four peptides, respectively. The distributions of states produced by REMD were analyzed using multiple histogram method (46). The convergence of REMD simulations and error analysis were reported in our previous studies (28,43). In particular, the thermodynamic quantities probing inter- and intrapeptide interactions have the errors of 1% (tetramer) and ≲ 4% (dimer or monomer). The errors in computing the conformational states of individual residues did not exceed 7% for the tetramer and monomer and 8% for the dimer.
Cluster analysis of Aβ conformations
The cluster analysis of Aβ peptide conformations is described in the Supporting Material.
Results
The conformational ensembles of Aβ10–40 monomers, dimers, and tetramers were investigated using REMD simulations (Fig. 1). Before presenting the results, it is useful to define the sequence regions in Aβ peptide. Following the experimental Aβ fibril structure (17), we distinguish the N-terminal (Nt, residues 10–23), which corresponds to the first fibril β-strand, and the C-terminal (Ct, residues 29–39), which corresponds to the second fibril β-strand (Fig. 1 a). All thermodynamic quantities for Aβ oligomers and monomers are reported at the temperature 360 K, at which Aβ peptide locks into fibril-like state during fibril growth (37,42). The selection of this temperature facilitates the comparison of conformational changes occurring upon aggregation, up to the deposition of Aβ peptides into the fibril.
Conformational propensities of Aβ oligomers and monomers
To probe the formation of Aβ oligomers we computed the numbers of interpeptide side-chain contacts 〈C(T)〉 and hydrogen bonds (HBs) 〈Nhb(T)〉 formed by a peptide with other chains in the oligomer. Their temperature dependences shown in Fig. 2 indicate that the number of interpeptide interactions increases with the decrease in temperature T. At 360 K, the number of interpeptide contacts 〈C〉 for the tetramer is 55.1, whereas interpeptide HBs (〈Nhb〉 ≈ 6.3) are relatively few (Table 1). Furthermore, tetramer almost completely lacks pHBs, which probe the formation of parallel β-sheets (〈Nphb〉 ≈ 0.8). Aβ peptide in the tetramer also forms a large number of intrapeptide interactions. For example, the numbers of intrapeptide side-chain contacts and HBs are 〈Ci〉 ≈ 25.7 and 〈Nihb〉 ≈ 8.0, respectively (Table 1). The probability of forming a tetramer at 360 K is ≈ 1.0 (see Discussion).
Figure 2.
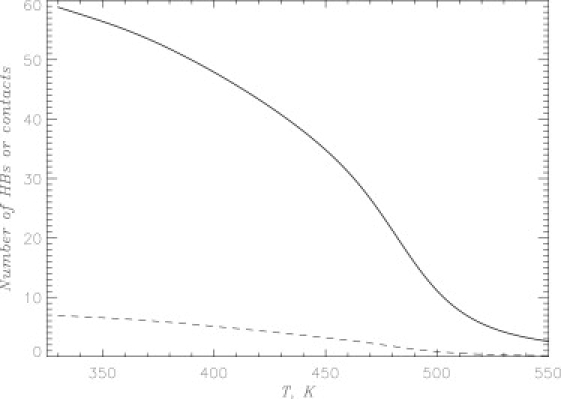
Assembly of Aβ10–40 tetramer is probed by the thermal averages of the numbers of interpeptide side-chain contacts 〈C(T)〉 (solid line) and HBs 〈Nhb(T)〉 (dashed line) as a function of temperature. The plot demonstrates the formation of Aβ10–40 tetramer with the decrease in temperature.
Table 1.
Aβ10–40 structural characteristics
| Aβ species | 〈C〉 | 〈Nhb〉 | 〈S〉∗ | 〈H〉† | 〈Ci〉 | 〈Nihb〉 | Sexp‡ |
|---|---|---|---|---|---|---|---|
| Monomer (1)§ | — | — | 0.24 (0.17,0.29) | 0.32 (0.51,0.15) | 32.2 | 14.1 | 0.24 |
| Dimer (2)§ | 30.0¶ | 3.7 | 0.37 (0.36,0.36) | 0.21 (0.32,0.11) | 24.2 | 9.4 | 0.39 |
| Tetramer (4)§ | 55.1 | 6.3 | 0.39 (0.37,0.38) | 0.20 (0.31,0.10) | 25.7 | 8.0 | 0.45 |
| Fibril (∞)§ | 0.52‖ | 0.57 |
Numbers in parentheses are the fractions of β-structure in the Nt and Ct regions, respectively.
Numbers in parentheses are the fractions of helix in the Nt and Ct regions, respectively.
Experimental fraction of β-structure from Ono et al. (20).
Oligomer order n. Monomer has n = 1.
Data from Takeda and Klimov (34,47).
Data from Takeda and Klimov (42).
With respect to the tetramer, Aβ peptide in the dimer is engaged in fewer interpeptide interactions (Table 1). The numbers of side-chain contacts, 〈C〉, and HBs, 〈Nhb〉, are reduced approximately in half (to 30.0 and 3.7, respectively). However, little difference is observed in the numbers of intrapeptide interactions (side-chain contacts and HBs) in the tetramer and dimer (Table 1). As in the tetramer, pHBs are largely absent in the dimer (〈Nphb〉 ≈ 0.4). The distributions of interpeptide interactions in Aβ oligomers, including the formation of parallel and antiparallel side chain contacts, are analyzed in the Supporting Material. It is also instructive to compare the intrapeptide interactions in the oligomers with those formed in the monomeric Aβ. At 360 K, the monomer contains 〈Ci〉 ≈ 32.2 side-chain contacts and 〈Nihb〉 ≈ 14.1 HBs (Table 1).
The average fractions of β-strand and helix structure, 〈St〉 and 〈Ht〉, in Aβ tetramer are 0.39 and 0.20 (Table 1). The secondary structure distributions for the dimer and monomer were reported in our previous studies (34,47). Using those findings, the changes in the β and helix structure in the tetramer with respect to the dimer are ΔSt–d = 〈St〉 – 〈Sd〉 = 0.02 and ΔHt–d = 〈Ht〉 – 〈Hd〉 = –0.01 (where 〈Sd〉 and 〈Hd〉 are the β-strand and helix fractions in the dimer from Table 1). Similarly, using the β-strand and helix fractions in the monomer 〈Sm〉 and 〈Hm〉 (Table 1), the changes in the secondary structure in the tetramer with respect to the monomer are ΔSt–m = 〈St〉 – 〈Sm〉 = 0.15 and ΔHt–m = 〈Ht〉 – 〈Hm〉 = –0.12. Fig. 3 a presents the distribution of secondary structure, 〈St(i)〉 and 〈Ht(i)〉, in Aβ tetramer and compares it with the dimer and monomer distributions (34,47). Not only the fractions 〈S〉 and 〈H〉 but also the distributions 〈S(i)〉 and 〈H(i)〉 for the tetramer and dimer are almost identical. For example, compared to the dimer, the change in the tetramer β-fraction in the Nt region is ΔSt–d(Nt) = 〈St(Nt)〉 – 〈Sd(Nt)〉 = 0.01, whereas the change in the Ct is ΔSt–d(Ct) = 0.02. According to Table 1 in the Nt, the helix fraction changes by ΔHt–d(Nt) = 〈Ht(Nt)〉 – 〈Hd(Nt)〉 = –0.01 and the same change is observed in the Ct (ΔHt–d(Ct) = –0.01). In contrast, Fig. 3 a implicates profound differences in the secondary structure distributions between the tetramer and monomer. Specifically, the changes in the β-content in the Nt and Ct regions are ΔSt–m(Nt) = 0.20 and ΔSt–m(Ct) = 0.09 (Table 1). For the helix fractions we get ΔHt–m(Nt) = –0.20 and ΔHt–m(Ct) = –0.05 (Table 1).
Figure 3.
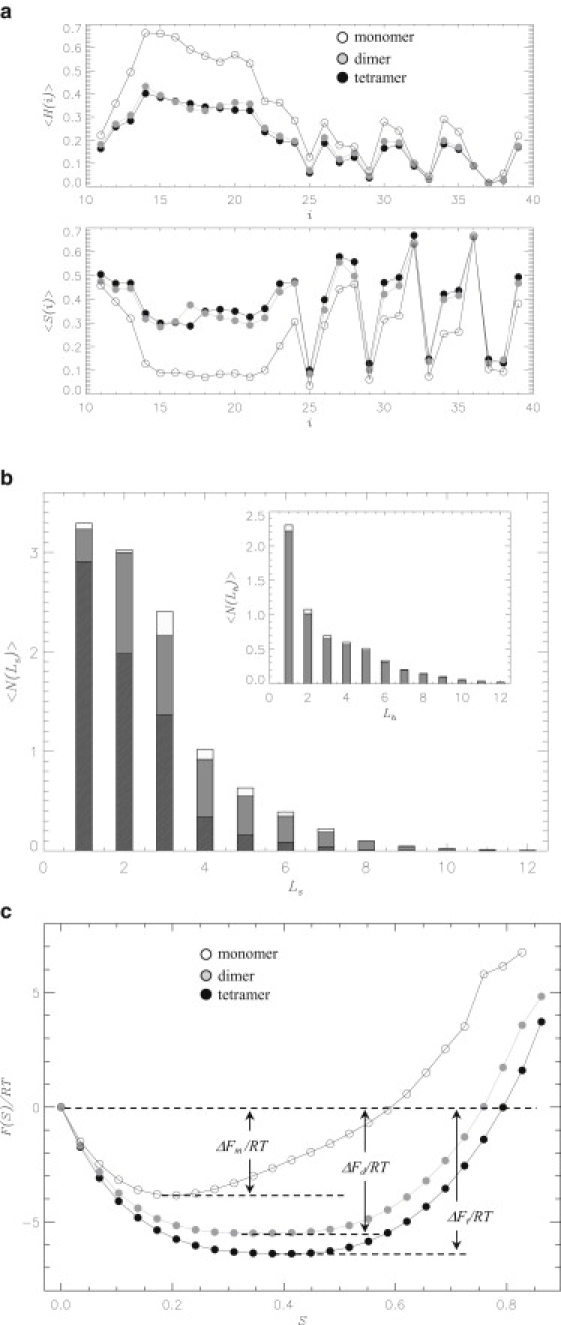
(a) The fractions of helix 〈H(i)〉 and β-strand 〈S(i)〉 structure sampled by residues i in Aβ10–40 tetramer (solid circles), dimer (shaded circles), and monomer (open circles). (b) Distribution of the numbers of residues 〈N(Ls)〉 involved in β-strand fragments of the length Ls. The data for tetramer, dimer, and monomer are shown by open, shaded, and solid bars. (Inset) Distribution of the numbers of residues 〈N(Lh)〉 involved in helix fragments of the length Lh. The data for tetramer and dimer are shown by shaded and open bars. (c) Free energy F(S) of Aβ10–40 peptide as a function of the fraction of residues in the β-strand conformation S: tetramer (solid circles), dimer (shaded circles), and monomer (open circles). The free energy of β structure is ΔF = Fβ – F(S = 0), where Fβ is obtained by integrating over the S states, for which F(S) ≤ Fmin + 1.0RT and Fmin is the minimum in F(S). The free energy F(S) is computed using multiple histogram method (46). This figure suggests that the secondary structure distributions in the tetramers and dimers are similar, but differ sharply from that in the monomer. The plots are computed at 360 K.
Additional information concerning the secondary structure propensities follows from the distributions of β-strand and helix lengths. Fig. 3 b displays the distributions of the number of residues 〈N(Ls)〉 in the β-strand fragments of the length Ls. Consistent with the enhanced β-content in Aβ tetramers and dimers, long β-strands occur more frequently in the oligomers than in the monomer. For example, the average number of residues participating in long β-strands (Ls ≥ 3) is 4.9 for the tetramer, 4.4 for the dimer, and 2.9 for the monomer. (Because 〈N(Ls)〉 is a thermally weighted quantity, it may be smaller or larger than Ls depending on the probability of occurrence of the strands of the length Ls. Same argument applies to 〈N(Lh)〉.) Fig. 3 b reveals that the tetramer and dimer 〈N(Ls)〉 distributions are similar, but both differ from the monomer distribution. As an illustration, consider 〈N(Ls)〉 for Ls = 4. The number of residues involved in the four-residue strands in the tetramer is just 10% larger than in the dimer, but it exceeds threefold the 〈N(Ls = 4)〉 value for the monomer. The inset to Fig. 3 b shows the numbers of residues 〈N(Lh)〉 in helix fragments of the length Lh. Similar to 〈N(Ls)〉, the distributions of helices in the tetramer and dimer are in close agreement. For example, the average number of residues found in “long” helices (Lh ≥ 4) is 1.9 for the tetramer and 2.0 for the dimer. Therefore, Fig. 3, a and b, implicate nearly identical distributions of the secondary structure in the tetramer and dimer, both of which are different from the monomeric one.
Finally, we probe the secondary structure propensities by computing the free energy landscapes for Aβ oligomers and monomers. Fig. 3 c shows the free energies F(S) for the three Aβ species as a function of the fraction of residues in the β-strand conformation S. The β-structure is most stable in the tetramer (ΔFt = –8.6RT) and dimer (ΔFd = –7.8RT). Both oligomer profiles F(S) reveal a broad, almost flat minimum spanning the range of S values from ∼0.2 to 0.6. In contrast, the monomer profile computed earlier (34) is shifted to smaller S and more shallow. As a result, the free energy of monomeric β structure is ΔFm = –5.6RT. Therefore, if the free energy of tetramer β-structure is merely ΔΔFt–d = –0.8RT lower than in the dimer, the free energy difference increases to ΔΔFt–m = –3.0RT when tetramer is compared to the monomer.
Conformational clusters of Aβ oligomers and monomers
To map the conformational distributions we apply the clustering procedure to the equilibrium structures of Aβ monomers, dimers, and tetramers (see Methods, Tables 2–4, and Fig. 4). In this section, we first describe the conformational clusters of Aβ peptides in the tetramer. The dimer and monomer clusters are then compared with the tetramer ones. There are three major conformational clusters in the tetramer, T1–T3, which together comprise 88% of all structures (Fig. 4). The distinctive feature of the most populated cluster T1 is the large fraction of residues in β-strand conformation, which is evenly present in the Nt and Ct regions, and the low helix content (S = 0.46 vs. H = 0.14, Table 2). This cluster is characterized by the largest numbers of interpeptide interactions (C = 60.9 and Nhb = 8.2) and the smallest number of intrapeptide HBs (Nihb = 5.2). Due to elevated β content, T1 is referred to as β-structure cluster. In contrast to T1, the cluster T2 has almost equal fractions of residues sampling β-strand and helix states (S = 0.29 and H = 0.30, Table 2). The helix structure localized in the Nt (H(Nt) = 0.52) exceeds the β-fraction (S(Nt) = 0.16) threefold (Table 2). In T2 there are fewer interpeptide interactions compared to T1 as the number of interpeptide contacts C is reduced almost 20% (to 51.0) and the number of interpeptide HBs is halved (to 4.8). However, compared to the T1 the number of intrapeptide HBs in the T2 is doubled (to 10.8). Consequently, we refer to T2 as helical cluster. The third cluster T3 resembles T2, but has lower helix content, especially in the Nt, where the helix fraction is reduced by a factor of 1.6 to H(Nt) = 0.32. The cluster T3 has also low β-content in the Ct, which is reduced almost twofold compared to T1 or T2. Because T3 forms as much intrapeptide interactions as T2, it represents collapsed Aβ conformations. In all the clusters T1–T3, the Nt is the main aggregation interface. The ratio of the numbers of interpeptide contacts formed by the Nt and Ct, C(Nt)/C(Ct), is the largest in T3 (2.6) followed by T1 (2.0) and is the smallest in T2 (1.6).
Table 2.
Structural clusters in Aβ10–40 tetramer
| Cluster | p∗ | S† | H‡ | C§ | (C(Nt), C(Ct))¶ | Nhb‖ | Nihb∗∗ |
|---|---|---|---|---|---|---|---|
| T1 | 0.46 | 0.46 (0.48,0.47) | 0.14 (0.21,0.09) | 60.9 | (33.0,16.4) | 8.2 | 5.2 |
| T2 | 0.23 | 0.29 (0.16,0.46) | 0.30 (0.52,0.10) | 51.0 | (26.0,16.4) | 4.8 | 10.8 |
| T3 | 0.19 | 0.33 (0.37,0.27) | 0.23 (0.32,0.19) | 52.6 | (31.1,11.9) | 5.3 | 10.7 |
Fraction of structures included in the cluster, i.e., occurrence probability.
Fractions of β-structure in Aβ peptide and in the Nt and Ct terminals (in parentheses).
Fractions of helix in Aβ peptide and in the Nt and Ct terminals (in parentheses).
Number of interpeptide contacts formed by a peptide in oligomer.
Numbers of interpeptide contacts formed by the Nt and Ct sequence regions.
Number of interpeptide HBs formed by a peptide in oligomer.
Number of intrapeptide HBs in a peptide.
Table 3.
Structural clusters in Aβ10–40 dimer
| Cluster | p∗ | S† | H‡ | C§ | (C(Nt), C(Ct))¶ | Nhb‖ | Nihb∗∗ |
|---|---|---|---|---|---|---|---|
| D1 | 0.47 | 0.44 (0.46,0.45) | 0.15 (0.22,0.09) | 33.1 | (17.5,9.5) | 4.7 | 6.9 |
| D2 | 0.25 | 0.27 (0.15,0.42) | 0.31 (0.52,0.12) | 28.8 | (15.3,8.8) | 2.9 | 11.7 |
| D3 | 0.19 | 0.29 (0.32,0.23) | 0.26 (0.36,0.20) | 27.3 | (15.5,7.1) | 2.7 | 13.1 |
Fraction of structures included in the cluster, i.e., occurrence probability.
Fractions of β-structure in Aβ peptide and in the Nt and Ct terminals (in parentheses).
Fractions of helix in Aβ peptide and in the Nt and Ct terminals (in parentheses).
Number of interpeptide contacts formed by a peptide in oligomer.
Numbers of interpeptide contacts formed by the Nt and Ct sequence regions.
Number of interpeptide HBs formed by a peptide in oligomer.
Number of intrapeptide HBs in a peptide.
Table 4.
Structural clusters in Aβ10–40 monomer
| Cluster | p∗ | S† | H‡ | Nihb§ |
|---|---|---|---|---|
| M1 | 0.55 | 0.26 (0.16,0.35) | 0.31 (0.52,0.15) | 12.9 |
| M2 | 0.33 | 0.17 (0.13,0.24) | 0.37 (0.54,0.21) | 17.4 |
Fraction of structures included in the cluster, i.e., occurrence probability.
Fractions of β-structure in Aβ peptide and in the Nt and Ct terminals (in parentheses).
Fractions of helix in Aβ peptide and in the Nt and Ct terminals (in parentheses).
Number of intrapeptide HBs in a peptide.
Figure 4.
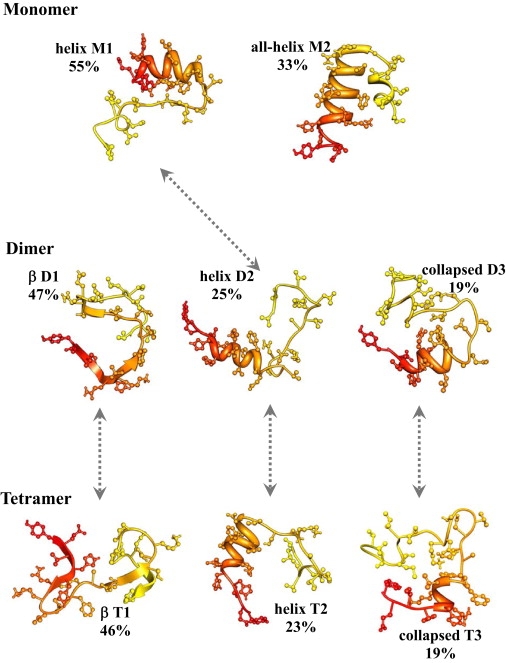
Conformational ensembles of Aβ10–40 peptides in monomers, dimers, and tetramers at 360 K. The figure shows typical peptide structures from populated conformational clusters. Shaded arrows indicate similarities between the clusters. The cluster characteristics are given in Tables 2–4. The N- and C-terminals are colored in shades of red and yellow, respectively. The picture demonstrates that the conformational ensembles of monomers and oligomers are distinct, whereas Aβ10–40 peptides in the dimers and tetramers sample similar structural distributions. The structures are visualized using Chimera (50).
Similar to the tetramer, there are three main clusters in the dimer, D1–D3, which together represent 91% of structures (Table 3 and Fig. 4). The β-structure cluster D1 is almost identical to T1. The cluster D2, which features elevated helix structure in the Nt, resembles the tetramer T2. It follows from Tables 2 and 3 that similarity between the tetramer T1-T2 and dimer D1-D2 clusters is manifested in the distributions of secondary structure and intrapeptide interactions. As expected for the dimer, D1 and D2 form fewer interpeptide interactions compared to the tetramer. Finally, there are close parallels between the collapsed clusters D3 in the dimer and T3 in the tetramer. As T3, the dimer cluster displays extensive intrapeptide interactions and the low Nt helix and Ct strand contents compared to those seen in D2. Table 3 shows that matching dimer and tetramer clusters have almost identical occurrence probabilities. As for the tetramer, a polarized aggregation interface is observed in all dimer clusters. The largest ratio C(Nt)/C(Ct) = 2.2 is found in the collapsed cluster D3, whereas the helix D2 has the lowest ratio (1.7).
The monomeric structures partition into two main clusters, M1 and M2, which combined represent 88% of Aβ conformations (Table 4 and Fig. 4). The most populated cluster M1 resembles the dimer D2 and tetramer T2 clusters. It features similar β-strand and helix fractions (S = 0.26 and H = 0.31). With the helix structure being localized in the Nt region, H(Nt) in M1 is identical to the respective values for D2 and T2 in Tables 2 and 3. Furthermore, the number of intrapeptide HBs in M1 (Nihb = 12.9) is similar to Nihb in D2 and T2 (Tables 2 and 3). The second cluster M2, which is less populated than M1, differs from all dimer or tetramer clusters. It is characterized by elevated helix structure (H = 0.37) and suppressed β-structure (S = 0.17). The distinctive feature of all-helix M2 is the large number of intrapeptide HBs (Nihb = 17.4), which is increased ∼50% compared to M1.
Discussion
Conformations of Aβ peptides in dimers and tetramers are similar
Our REMD simulations suggest that the conformational ensembles of Aβ peptides in the dimers and tetramers are similar. This conclusion is based on the following observations.
Aβ peptides in the dimers and tetramers form similar amount of intrapeptide interactions. For example, the differences in the numbers of intrapeptide HBs 〈Nihb〉 and side-chain contacts 〈Ci〉 are merely 18% and 6% (Table 1).
Although there are more interpeptide interactions formed in the tetramer than in the dimer, their distributions in both oligomers are very similar (see the Supporting Material). Indeed, the number of contacts between the sequence regions s1 and s2 (= Nt, Ct) in the dimer can be obtained by rescaling those formed in the tetramer, i.e., 〈Cd(s1, s2)〉 ≈ 0.6 〈Ct(s1, s2)〉 (see the Supporting Material). More importantly, the Nt terminal forms two-thirds of all interpeptide interactions in the tetramer and dimer (〈C(Nt)〉 ≈ 2 〈C(Ct)〉), implicating the Nt region as the main aggregation interface in Aβ oligomers. It is also noteworthy that there are few parallel HBs in the dimers and tetramers, which are the hallmark of fibril structure (17). No preference for parallel or antiparallel aggregation interface is observed in Aβ oligomers (see the Supporting Material).
There are similarities in the secondary structure. For example, the fractions of β-strand and helix structure in the tetramer and dimer differ by 5% (Table 1). Fig. 3 a demonstrates that the distributions of secondary structure along Aβ sequence are nearly identical in both oligomers. The fractions of β-strand and helix structure in the Nt and Ct do not differ by >5% (the exception is 〈H(Ct)〉 being different by ∼10%). Furthermore, as illustrated in Fig. 3 b the distributions of β-strands and helix fragments in the tetramer and dimer are similar. Finally, Fig. 3 c demonstrates that the free energy of the β-strand structure in the tetramer is only marginally lower than in the dimer (ΔΔFt–d ≲ RT).
Computation of conformational clusters provides the most compelling evidence for the similarity of the dimer and tetramer conformational ensembles (Tables 2 and 3 and Fig. 4). The three conformational clusters T1–T3 in the tetramer match the clusters D1–D3 in the dimer. Importantly, not only are the structural characteristics of these clusters similar, but so are the probabilities of their occurrence. Because the three clusters in the tetramer and dimer comprise ∼90% of Aβ conformations, we suggest that, at least for small oligomers, Aβ conformational ensembles are independent of the oligomer order n.
Our findings indicate that in contrast to monomers the β-structure in Aβ oligomers occurs more frequently than the helix conformations. The enhancement of β-structure in oligomers is driven by the formation of interpeptide interactions, mostly by side-chain contacts with minor contribution from HBs (Table 1). Most β-strands in the oligomers are short (Fig. 3 b) and, because they form few parallel HBs, ordered β-sheets are rare. Therefore, the oligomer structure contains few elements of fibril-like conformations.
Conformations of Aβ peptides in oligomers and monomers are distinct
It follows from our results that the conformational ensembles of Aβ oligomers and monomers are different. Following the same approach used for comparing the dimers and tetramers, we identify the following differences.
Aβ monomers and oligomers differ with respect to the distribution of intrapeptide interactions. For example, compared to the tetramer the number of intrapeptide HBs in the monomer increases 80% coupled with 25% increase in the number of side-chain contacts (Table 1).
The secondary structures in Aβ monomers and oligomers are different. The fractions of β-strand and helix structure in the tetramer are 40% larger and 60% lower, respectively, than in the monomer (Table 1). Fig. 3 a shows that there are striking dissimilarities in the distributions of secondary structure along Aβ sequence. For example, the fraction of β-strand in the Nt regions of the tetramer 〈St(Nt)〉 exceeds that for the monomer 〈Sm(Nt)〉 by more than twofold. The distributions of β-strands in the tetramer and monomer also differ considerably (Fig. 3 b and Results). Consistent with these findings indicating the enhancement of β content in the oligomers, the free energy of the β-strand structure in the monomer is ≈3.0RT higher than in the tetramer.
The analysis of conformational clusters reveals that the oligomer and monomer conformational ensembles are different (Tables 2–4 and Fig. 4). The monomer distribution contains only two clusters, of which only one, M1, bears similarity to the oligomer clusters T2 or D2. Therefore, the monomer samples significant fraction (∼0.3) of conformations (the all-helix cluster M2), which are not seen in the oligomers. Conversely, the oligomers contain the clusters (T1,T3 and D1,D3) with elevated β-strand content (S ≳ 0.3) not observed in the monomers. For example, in T1 and D1 the fraction of β structure approaches 0.5 in both sequence regions Nt and Ct.
In summary, the key difference between the conformational ensembles of monomers and oligomers is the increase in β-structure in the oligomers occurring together with the loss of intrapeptide interactions. These changes are manifested by the disappearance of the monomeric cluster M2, emergence of the new oligomeric clusters T1,T3 (or D1,D3), and decrease in the statistical weight of the cluster M1 (D2,T2) (Tables 2–4 and Fig. 4).
Comparing experimental and computational Aβ conformational ensembles
Recent experiments employing photoinduced chemical crosslinking technique probed the structures of Aβ aggregated species (20). The CD analysis of secondary structure revealed that the most profound conformational change occurs upon the conversion of monomers into dimers. Table 1 shows that this conversion results in the increase in the β-content from Smexp = 0.24 (monomer) to Sdexp = 0.39 (dimer). The difference in the fractions of β-structure in the dimer and tetramer is relatively small (Sdexp = 0.39 vs. Stexp = 0.45 in Table 1). Therefore, monomer-to-dimer conversion results in ≈60% increase in β-fraction, whereas there is only 15% additional increase in the β-structure upon forming the tetramer.
To test the accuracy of our simulations, Fig. 5 compares the experimental and in silico fractions of β-structure. This comparison is facilitated by including the data for Aβ fibril peptides determined experimentally or computed in our recent studies using the same simulation model (Table 1) (20,42). Fig. 5 shows that the experimental and computational β contents Sexp(n) and 〈S(n)〉 are in good agreement for all available values of oligomer order n. This lends support to the in silico conformational ensembles obtained in our study.
Figure 5.
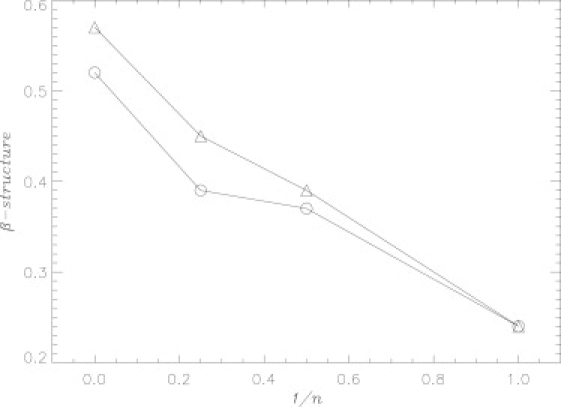
The experimental β content Sexp(n) (20) (triangles) and the in silico β content 〈S(n)〉 (open circles) are plotted as a function of the inversed oligomer order n. The plot suggests that simulations reproduce fairly well experimental estimates of secondary structure.
It should be mentioned that CHARMM19+SASA model predicts higher helix content in Aβ species (monomers through tetramers) than the experiments (20). There are two possible reasons for this discrepancy.
First, we use truncated Aβ10–40 peptides, in which first nine N-terminal residues are deleted. Our previous study (34) has shown that the truncation results in 19% increase in the fraction of helix residues in the monomers (from 0.27 in Aβ1–40 to 0.32 in Aβ10–40).
Second, we cannot rule out that the SASA implicit solvent model overestimates the fraction of helix structure in Aβ species due to approximate treatment of hydration effects. Hence, further studies will be needed to determine the exact cause of this discrepancy.
It is also important to comment on the experimental and in silico oligomer-size distributions. A continuous distribution of Aβ species, from monomers to tetramers, is observed in the experiments (20). In our simulations, Aβ dimers or tetramers are formed with the probability ∼1.0. To resolve this difference, one needs to recall that the in vitro experiments probing Aβ aggregation are performed at micromolar concentrations, whereas a millimolar Aβ concentration is used in our study to accelerate sampling of aggregated states. Consequently, in simulations the thermodynamic equilibrium is expected to shift to higher order oligomers.
Qualitatively similar changes in the secondary structure were observed in the oligomers formed by α-synuclein (48). The Raman spectroscopy showed that the small α-synuclein oligomers possess significant amount of helix structure (47%), which decreases upon aggregation progression. Simultaneously, the fraction of β-sheet conformations grows from 29% in small oligomers to 54% in protofilaments. Therefore, the conversion of helix structure into β in the oligomers is likely to be a generic feature of aggregation.
A computational study relevant to our simulations has investigated the oligomers formed by human islet amyloid polypeptide (hIAPP) (49). It has been shown that hIAPP trimers do not contain parallel or antiparallel in-registry β-structure characteristic of seeded hIAPP fibrils. Moreover, in agreement with our data, implicit solvent MD study of Aβ1–39 dimer also found no evidence of fibril-like conformations (30). Those findings are qualitatively consistent with our results on Aβ oligomers.
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging or the National Institutes of Health.
This work was supported by grant No. R01 AG028191 from the National Institute on Aging, National Institutes of Health, Bethesda, MD.
Supporting Material
References
- 1.Selkoe D.J. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 4.Kirkitadze M.D., Bitan G., Teplow D.B. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J. Neurosci. Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 5.Murphy R.M. Peptide aggregation in neurodegenerative disease. Annu. Rev. Biomed. Eng. 2002;4:155–174. doi: 10.1146/annurev.bioeng.4.092801.094202. [DOI] [PubMed] [Google Scholar]
- 6.Nelson R., Sawaya M.R., Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makin O.S., Atkins E., Serpell L.C. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA. 2005;102:315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshiike Y., Akagi T., Takashima A. Surface structure of amyloid-β fibrils contributes to cytotoxicity. Biochemistry. 2007;46:9805–9812. doi: 10.1021/bi700455c. [DOI] [PubMed] [Google Scholar]
- 9.Kayed R., Head E., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 10.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 11.Shankar G.M., Li S., Selkoe D.J. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carulla N., Caddy G.L., Dobson C.M. Molecular recycling within amyloid fibrils. Nature. 2005;436:554–558. doi: 10.1038/nature03986. [DOI] [PubMed] [Google Scholar]
- 13.Esler W.P., Felix A.M., Maggio J.E. Activation barriers to structural transition determine deposition rates of Alzheimer's disease a beta amyloid. J. Struct. Biol. 2000;130:174–183. doi: 10.1006/jsbi.2000.4276. [DOI] [PubMed] [Google Scholar]
- 14.Riek R., Güntert P., Wüthrich K. NMR studies in aqueous solution fail to identify significant conformational differences between the monomeric forms of two Alzheimer peptides with widely different plaque-competence, A β(1-40)(ox) and A β(1-42)(ox) Eur. J. Biochem. 2001;268:5930–5936. doi: 10.1046/j.0014-2956.2001.02537.x. [DOI] [PubMed] [Google Scholar]
- 15.Hou L., Shao H., Zagorski M.G. Solution NMR studies of the A β(1-40) and A β(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 16.Burkoth T.S., Benzinger T., Lynn D.G. Structure of the β-Amyloid(10–35) fibril. J. Am. Chem. Soc. 2000;122:7883–7889. [Google Scholar]
- 17.Petkova A.T., Yau W.-M., Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luhrs T., Ritter C., Riek R. 3D structure of Alzheimers amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narayanan S., Reif B. Characterization of chemical exchange between soluble and aggregated states of β-amyloid by solution-state NMR upon variation of salt conditions. Biochemistry. 2005;44:1444–1452. doi: 10.1021/bi048264b. [DOI] [PubMed] [Google Scholar]
- 20.Ono K., Condron M.M., Teplow D.B. Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. USA. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma B., Nussinov R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Chem. Biol. 2006;10:445–452. doi: 10.1016/j.cbpa.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 22.Sgourakis N.G., Yan Y., Garcia A.E. The Alzheimer's peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J. Mol. Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang M., Teplow D.B. Amyloid β-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 2008;384:450–464. doi: 10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cecchini M., Curcio R., Caflisch A. A molecular dynamics approach to the structural characterization of amyloid aggregation. J. Mol. Biol. 2006;357:1306–1321. doi: 10.1016/j.jmb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Baumketner A., Shea J.-E. Folding landscapes of the Alzheimer amyloid-β(12-28) peptide. J. Mol. Biol. 2006;362:567–579. doi: 10.1016/j.jmb.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y., Derreumaux P., Wei G. Thermodynamics and dynamics of amyloid peptide oligomerization are sequence dependent. Proteins. 2009;75:954–963. doi: 10.1002/prot.22305. [DOI] [PubMed] [Google Scholar]
- 27.Masman M.F., Eisel U.L.M., Luiten P.G. In silico study of full-length amyloid β 1-42 tri- and penta-oligomers in solution. J. Phys. Chem. B. 2009;113:11710–11719. doi: 10.1021/jp901057w. [DOI] [PubMed] [Google Scholar]
- 28.Takeda T., Klimov D.K. Interpeptide interactions induce helix to strand structural transition in Aβ peptides. Proteins. 2009;77:1–13. doi: 10.1002/prot.22406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chebaro Y., Mousseau N., Derreumaux P. Structures and thermodynamics of Alzheimer's amyloid-beta Aβ(16–35) monomer and dimer by replica exchange molecular dynamics simulations: implication for full-length Aβ fibrillation. J. Phys. Chem. B. 2009;113:7668–7675. doi: 10.1021/jp900425e. [DOI] [PubMed] [Google Scholar]
- 30.Anand P., Nandel F.S., Hansmann U.H.E. The Alzheimer beta-amyloid (Aβ (1–39)) dimer in an implicit solvent. J. Chem. Phys. 2008;129:195102. doi: 10.1063/1.3021062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urbanc B., Betnel M., Teplow D.B. Elucidation of amyloid β-protein oligomerization mechanisms: discrete molecular dynamics study. J. Am. Chem. Soc. 2010;132:4266–4280. doi: 10.1021/ja9096303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paravastu A.K., Petkova A.T., Tycko R. Polymorphic fibril formation by residues 10–40 of the Alzheimer's β-amyloid peptide. Biophys. J. 2006;90:4618–4629. doi: 10.1529/biophysj.105.076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bitan G., Vollers S.S., Teplow D.B. Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 2003;278:34882–34889. doi: 10.1074/jbc.M300825200. [DOI] [PubMed] [Google Scholar]
- 34.Takeda T., Klimov D.K. Probing the effect of amino-terminal truncation for Aβ1–40 peptides. J. Phys. Chem. B. 2009;113:6692–6702. doi: 10.1021/jp9016773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1982;4:187–217. [Google Scholar]
- 36.Ferrara P., Apostolakis J., Caflisch A. Evaluation of a fast implicit solvent model for molecular dynamics simulations. Proteins. 2002;46:24–33. doi: 10.1002/prot.10001. [DOI] [PubMed] [Google Scholar]
- 37.Takeda T., Klimov D.K. Probing energetics of Aβ fibril elongation by molecular dynamics simulations. Biophys. J. 2009;96:4428–4437. doi: 10.1016/j.bpj.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeda T., Klimov D.K. Side chain interactions can impede amyloid fibril growth: replica exchange simulations of Aβ peptide mutant. J. Phys. Chem. B. 2009;113:11848–11857. doi: 10.1021/jp904070w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrara P., Caflisch A. Folding simulations of a three-stranded antiparallel β -sheet peptide. Proc. Natl. Acad. Sci. USA. 2000;97:10780–10785. doi: 10.1073/pnas.190324897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiltpold A., Ferrara P., Caflisch A. Free energy surface of the helical peptide Y(MEARA)6. J. Phys. Chem. B. 2000;104:10080–10086. [Google Scholar]
- 41.Cecchini M., Rao F., Caflisch A. Replica exchange molecular dynamics simulations of amyloid peptide aggregation. J. Chem. Phys. 2004;121:10748–10756. doi: 10.1063/1.1809588. [DOI] [PubMed] [Google Scholar]
- 42.Takeda T., Klimov D.K. Replica exchange simulations of the thermodynamics of Aβ fibril growth. Biophys. J. 2009;96:442–452. doi: 10.1016/j.bpj.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim S., Takeda T., Klimov D.K. Globular state in the oligomers formed by Aβ peptides. J. Chem. Phys. 2010;132:225101. doi: 10.1063/1.3447894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;114:141–151. [Google Scholar]
- 45.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 46.Ferrenberg A.M., Swendsen R.H. Optimized Monte Carlo data analysis. Phys. Rev. Lett. 1989;63:1195–1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
- 47.Takeda T., Klimov D.K. Computational backbone mutagenesis of Aβ peptides: probing the role of backbone hydrogen bonds in aggregation. J. Phys. Chem. B. 2010;114:4755–4762. doi: 10.1021/jp911533q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Apetri M.M., Maiti N.C., Anderson V.E. Secondary structure of α-synuclein oligomers: characterization by Raman and atomic force microscopy. J. Mol. Biol. 2006;355:63–71. doi: 10.1016/j.jmb.2005.10.071. [DOI] [PubMed] [Google Scholar]
- 49.Mo Y., Lu Y., Derreumaux P. Structural diversity of the soluble trimers of the human amylin(20–29) peptide revealed by molecular dynamics simulations. J. Chem. Phys. 2009;130:125101. doi: 10.1063/1.3097982. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.