

Abstract
MicroRNAs (miRNAs) are important regulators of cell fate determination and homeostasis. Expression of these small RNA genes is tightly regulated during development and in normal tissues, but they are often misregulated in cancer. MiRNA expression is also affected by DNA damaging agents, such as radiation. In particular, mammalian miR-34 is upregulated by p53 in response to radiation, but little is known about the role of this miRNA in vivo. Here we show that Caenorhabditis elegans with loss-of-function mutations in the mir-34 gene have an abnormal cellular survival response to radiation; these animals are highly radiosensitive in the soma and radioresistant in the germline. These findings show a role for mir-34 in both apoptotic and non-apoptotic cell death in vivo, much like that of cep-1, the C. elegans p53 homolog. These results have been additionally validated in vitro in breast cancer cells, wherein exogenous addition of miR-34 alters cell survival post-radiation. These observations confirm that mir-34 is required for a normal cellular response to DNA damage in vivo resulting in altered cellular survival post-irradiation, and point to a potential therapeutic use for anti-miR-34 as a radiosensitizing agent in p53-mutant breast cancer.
Keywords: miRNA, DNA damage response, radiation, cancer, C. elegans
The maintenance of genome integrity is important for cellular and organismic health, and for successful reproduction. DNA damage response (DDR) pathways play important roles in genome maintenance through cell cycle arrest followed by DNA repair and/or apoptosis (Ljungman and Lane, 2004). In some cases, cancer risk is enhanced by inherited mutations in genes important in the DDR, such as p53, BRCA, etc. (Miki et al., 1994; Vogelstein et al., 2000). In the majority of tumors, the DDR pathway fails to function properly, although the initial cause of the dysfunction is generally unknown.
MicroRNAs (miRNAs) are a class of non-coding small RNAs that mediate post-transcriptional regulation of specific target mRNAs, and are important in specification and maintenance of cell fate. The relevance of miRNAs to cancer has recently been discovered (reviewed in Esquela-Kerscher and Slack, 2006). Our knowledge of the correlation between miRNAs and cancer-related processes is based on the observations of alterations of normal miRNA expression, and frequent deletion or amplification of miRNA-containing genomic loci in tumor tissues (Calin et al., 2004; Iorio et al., 2005). Moreover, miRNAs are found to regulate pathways important in cancer, such as cell cycle pathways (Johnson et al., 2005, 2007).
The role of miRNAs in the DDR pathway is just emerging, with the evidence that certain miRNAs can modulate the response to cytotoxic therapy, through regulation of genes in the DDR. For example, let-7 miRNA molecules are dramatically downregulated in response to radiation of human lung cells, and exogenous let-7 can radiosensitize lung cancer cells in vitro (Weidhaas et al., 2007). In addition, disruption of normal expression of C. elegans let-7 family miRNAs results in altered responses to radiation in vivo. This alteration is partly through the control of its target, let-60, a homolog of the proto-oncogene, RAS, in humans, as well as C. elegans homologs of RAD51, RAD21, FANCD2 and CDC25, supporting the role of the let-7 family miRNAs in a cancer-related DDR network (Weidhaas et al., 2007). In addition, miRNA expression profiles have been correlated with sensitivity or resistance to certain chemotherapeutic agents (Salter et al., 2008), further showing that miRNA alterations in tumors likely directly impact the DDR and thus the response of tumors to cancer therapy.
Interestingly, a number of miRNAs are upregulated after radiation of human lung cells, including the well-conserved mir-34 family (Weidhaas et al., 2007), which has been shown to be transcriptionally regulated by the p53 gene (He et al., 2007). Several studies found that in mammalian cell lines, DNA damage-induced mir-34 expression was dependent on p53, and that this was followed by induction of cell cycle arrest and/or promotion of apoptosis. However, little is known about the role of miR-34 in the radiation response in vivo. Here, we used C. elegans to determine the role of mir-34 in radiation-induced cell death in vivo.
First identified in C. elegans, mir-34 is the founding member of an evolutionarily conserved miRNA family found in diverse species, including humans (Figure 1a). This conservation implies an important fundamental role for the mir-34 gene in these organisms. To understand the function of mir-34 in C. elegans, we first examined the expression of mir-34 in vivo by northern blot analysis (Figure 1b). The miR-34 was temporally regulated during development; being extremely low or undetectable in embryos, beginning to be expressed in the first larval stage (L1) animals and showing consistently high expression levels up to the adult stage (Supplementary Information 1a). To determine the tissues expressing miR-34, we established transgenic lines expressing mir-34promoter∷gfp transgene. The gfp signals were widely detected in somatic tissues, such as the pharynx, intestine, head neurons, ventral nerve cord, excretory canal and vulva (Figure 1b), suggesting a broad role for miR-34 throughout the soma of the animal. Expression in the non-vulval tissues seems to be invariable throughout development of C. elegans from the L1 to the early adult stage, but, strikingly, mir-34∷gfp was strongly expressed in all mature vulva cells beginning in the adult stage (Figures 1b and c). This suggests the possibility that mir-34 functions in vulval development, maintenance or function. However, mir-34(gk437) loss-of-function mutants (Figure 3a) did not show obvious morphological defects in the adult vulval cells (Supplementary information 2), indicating that mir-34 is not necessary for vulval cell differentiation under normal laboratory conditions.
Figure 1.

Sequence alignment of mir-34 family MicroRNAs (miRNAs) and the expression of mir-34 in C. elegans (a) Highly conserved regions are highlighted by black (100%) and gray (>50%). The seed region, GGCAGU, is lined. Abbreviations for each organism are shown in Supplementary information 5. (b) Developmental northern blot of mir-34 and gfp signals of mir-34∷gfp. Magnification and exposure time for detecting gfp are shown in each image. (c) mir-34∷gfp expression in vulval cells in the adult stage.
Figure 3.

mir-34 loss-of-function mutants have altered cell survival post-irradiation. (a) Loss of miR-34 expression in mir-34(gk437) was confirmed by northern blot. (b) mir-34 loss-of-function mutants were sensitive to non-apoptotic cell death. (c) mir-34 mutants are resistant to apoptotic cell death. (d) mir-34(gk437); cep-1(gk138) double mutants were significantly more radiosensitive to non-apoptotic cell death than either of the single mutant. Animals were irradiated with 200 Gy. Similar results were obtained from radiation treatment with 400 Gy.
As our miRNA-microarray studies from lung cells (Weidhaas et al., 2007) and other recent studies have shown the alteration of human miR-34 expression after radiation and in tumor cells in mammals (He et al., 2007), we hypothesized that C. elegans mir-34 might play a role in the radiation response as well. To test this, we first examined the effects of radiation on mature miR-34 levels detected by reverse transcriptase PCR in wild-type animals. We found that miR-34 levels rose significantly by 3 hours post-irradiation (Figure 2, Top and Supplementary information 1b), consistent with observations from mammalian cells. Green fluorescent protein signals from the mir-34promoter∷gfp animals also appeared to increase post-irradiation (Supplementary information 1c). These results revealed that mir-34 expression is induced by radiation across multiple phyla.
Figure 2.
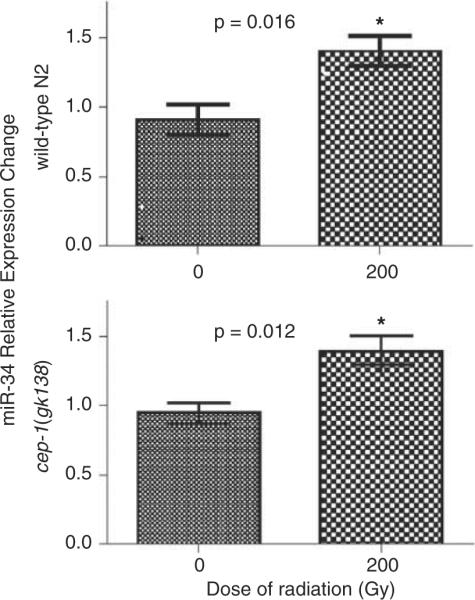
miR-34 expression in C. elegans post-irradiation. Similar increase in miR-34 expression was detected in both wild-type N2 animals and cep-1/p53 mutants at 3 hours post-irradiation. Error bars show s.d. An asterisk indicates statistical significance.
As mir-34 induction post-radiation in mammalian cells has been reported to depend on p53 induction, we next determined whether mir-34 induction post-irradiation is also dependent on cep-1, a p53 homolog in C. elegans. We examined a cep-1 mutant post-irradiation for alterations in mature miR-34 levels by reverse transcriptase PCR, and found similar induction of mature miR-34 in radiated animals in cep-1(gk138) at 3 hours post-irradiation (Figure 2, Bottom and Supplementary information 1b), as observed in wild-type animals. A similar result was obtained from the mir-34promoter∷gfp animals in the cep-1(gk138) (Supplementary information 1c). To further validate this result, these experiments were carried out on two additional cep-1/p53 mutant strains, cep-1(ep347) and cep-1(w40), which also showed mir-34 induction post-radiation (data not shown). These findings seem to indicate that cep-1/p53 is not required for miR-34 induction post-irradiation in C. elegans, unlike that found earlier for mammalian cells.
Nevertheless, as mir-34 was induced post-irradiation, we next tested the role of mir-34 in the DDR using C. elegans, which provides an excellent model to examine the role of miRNAs in the DDR pathway in vivo not only because of the high conservation between miRNAs in C. elegans and mammals, but also because there exists inherently two models of distinct forms of radiation-induced cell death in C. elegans; apoptotic cell death in the germline (Gartner et al., 2000) and non-apoptotic reproductive cell death in the vulva (Weidhaas et al., 2006). Studying both forms of cell death is important, as many genes play roles in both, but alteration of their levels may lead to opposite radiosensitive phenotypes. For example, C. elegans cep-1/p53 has distinct functions in somatic tissues and the germline; whereas cep-1 loss-of-function mutants display protection from radiation-induced apoptotic germ cell death (Derry et al., 2001), they are sensitized to radiation-induced reproductive cell death in the vulva (Weidhaas et al., 2006). In addition, the conserved checkpoint pathway genes, such as rad-5 and mrt-2, are necessary for germ cell apoptosis in C. elegans with their loss resulting in protection from apoptosis in this tissue (Gartner et al., 2000), yet loss-of-function of either of these genes sensitizes animals to radiation-induced reproductive cell death in the vulva (Weidhaas et al., 2006).
We first examined the role of mir-34 in cell survival in vivo in the C. elegans vulval model of radiation-induced reproductive, non-apoptotic cell death (Weidhaas et al., 2006). Radiation sensitivity can be evaluated by scoring the number of animals with abnormal vulva after a sub-lethal dose of radiation (Supplementary information 3e–g). The mir-34 loss-of-function mutant alleles (gk437 and n4276 (data not shown)) were radiosensitive in the vulval model (Figure 3b), indicating that mir-34 is necessary to protect cells from non-apoptotic reproductive cell death. This is the first demonstration that mir-34 plays a role in the response to radiation in vivo and is the first known phenotype associated with mir-34 deletion mutants in C. elegans (Miska et al., 2007) or in any animals.
We next tested the sensitivity of mir-34 loss-of-function animals to apoptosis in the well-established model of radiation-induced apoptosis in the germline (Gartner et al., 2000). We found that, in response to radiation, there were fewer apoptotic bodies in the germline of mir-34(gk437) compared with wild-type N2 animals (Figure 3c, Supplementary information 3a–d)). This observation indicates that mir-34 mutants are resistant to radiation-induced apoptotic cell death and thus, mir-34 is necessary to cause apoptotic cell death in these cells.
Taken together, mir-34 loss-of-function mutants are more sensitive to reproductive cell death in the vulva, but more resistant to radiation-induced apoptosis in the germline than wild-type animals, similar to cep-1/p53, rad-5 and mrt-2 mutants (Gartner et al., 2000; Weidhaas et al., 2006). These findings support the hypothesis that mir-34 is involved in both apoptotic and non-apoptotic cell death in C. elegans, and also suggest that mir-34 and cep-1 may act together or in similar cell survival pathways, although mir-34 induction post-irradiation did not require cep-1/p53 activity, unlike in mammalian cells.
To better understand the relationship, if any, between mir-34 and cep-1 in the DDR, we tested the effect of double mutations in both of these genes on cell survival in C. elegans. The mir-34(gk437); cep-1(gk138) double mutants were significantly more sensitive to non-apoptotic cell death in the vulva than either of the single mutant (Figure 3d). As at least the mir-34 mutation is a null allele, these observations suggest that mir-34 and cep-1 function in parallel in the DDR pathway to modulate the response to DNA damage in C. elegans.
To see whether mir-34 also plays a role in the DDR and survival post-radiation in mammalian cells, we looked for cell lines with altered miR-34 levels. We found that miR-34a levels were particularly low in breast cancer cell lines representing the triple negative and mesenchymal subgroup compared with other breast cancer cell lines, such as Her-2 (Figure 4a and Supplementary information 4a). Although this has not been reported earlier, this could be explained by common mutations in p53 in these subtypes of breast cancer (Runnebaum et al., 1991; Hui et al., 2006), perhaps preventing p53-mediated transcription of mir-34.
Figure 4.

Post-irradiation breast cancer cell survival is impacted by mir-34 alterations. (a) miR-34a levels were lower in triple negative (TN) and mesenchymal breast cancer cell lines compared with normal epithelial lines and Her-2+ lines. Details are shown in Supplementary information 4. (b) Radiation sensitivity of breast cancer cell lines. The cells treated with radiation (2.5 Gy) were plated in clonogenic assays compared with non-irradiated samples. Error bars represent s.d. (c) Breast cell lines HMEC, MCF10A and MDAMB-231 did not undergo apoptotic cell death post-irradiation in contrast to what was seen in Jurkat cells. The fold-increase in CPP32 activity post-irradiation was compared with the un-irradiated controls. (d) miR-34a levels alter cell survival in MDA-MB-231 breast cancer cell lines in response to increasing radiation doses.
To understand the relationship between miR-34a levels and radiation sensitivity in these cells, we compared sensitivity between a normal breast epithelial line (HMEC), a Her-2 + line (UACC812) and a mesenchymal line, MDA-MB-231. We found that the MDA-MB-231 cell line (with low miR-34a levels) was significantly more radiosensitive than the HMECs or UACC-812 (with high miR-34a levels) (Figure 4b), suggesting that in these lines mir-34 is necessary to protect from radiation-induced cell death. To test the hypothesis that these cells undergo non-apoptotic cell death after radiation, we treated two non-cancerous breast cell lines (MCF10A and HMEC) and MDA-MB-231 with radiation, and carried out assays to determine whether these cells underwent apoptosis, including Caspase-3/CPP32 colorimetric assays (Figure 4c) and PARP cleavage assays (data not shown). Compared with Jurkat cells, which are known to undergo apoptosis, these two normal and one cancerous breast lines did not undergo apoptosis post-irradiation, even at higher doses of radiation, supporting that these breast cells likely inherently undergo non-apoptotic cell death.
We next tested the potential of miR-34a alteration to impact cell survival in MDA-MB-231 post-irradiation through transfection of pre-miR-34a to increase miR-34a levels and of anti-miR-34a to further lower miR-34a levels before radiation. Using clonogenic assays, we found that increasing levels of miR-34a protected the MDA-MB-231cells from radiation-induced cell death, and lowering levels of miR-34a sensitized cells to radiation-induced cell death (Figure 4d). We saw the same effect of miR-34a alterations in the Hs578T cell line (data not shown), which also had low miR-34a (Supplementary information 4a). These findings show that mir-34 is necessary to protect cells from non-apoptotic cell death in MDA-MB-231 mammalian cells, identical to how mir-34 impacts non-apoptotic cell survival in C. elegans. Although, these observations indicate that mir-34 manipulation in mammalian cells can alter cell survival post-irradiation, our results also suggest that the impact of mir-34 may depend largely on a cell's innate form of death post-irradiation; cells that inherently undergo non-apoptotic cell death will be sensitized by lowering miR-34a levels and protected by miR-34a overexpression.
Although it remains largely unknown how miRNA expression modulates oncogenic activity and response to DNA damage, there is little doubt that miRNAs are critical regulators in this process. Here, we have shown that mir-34 is a critical component of the radiation response in vivo in C. elegans and in vitro in breast cancer cell lines. We show that mir-34 is involved in both forms of cell death in vivo, apoptotic and non-apoptotic, much like cep-1/p53, and that alteration of miR-34 levels in human breast cell lines can also impact cell survival post-irradiation. Further, we show for the first time that the impact of mir-34 on cell survival largely depends on the cell's innate mode of cell death post-irradiation, as mir-34 impacts both apoptotic and non-apoptotic cell death, yet in opposite directions. Our results also suggest that anti-miR-34a molecules might prove useful in radiosensitizing breast tumors for better treatment.
Supplementary Material
Acknowledgements
We thank the CGC for strains. MK was supported by a postdoctoral fellowship from the Uehara Life Science Foundation. JW was supported by the Breast Cancer Alliance and a K08 grant (CA124484) from the NIH and by an American Cancer Society Institutional Research Grant to the Yale Cancer Center and by the ASTRO Junior Faculty Career Research Training Award. FS was supported by a grant from the Ellison Medical Foundation. FS and JW were supported by a grant from the NIH (CA131301-01A1).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry WB, Putzke AP, Rothman JH. Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science. 2001;294:591–595. doi: 10.1126/science.1065486. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO. A conserved checkpoint pathway mediates DNA damage-induced apoptosis and cell cycle arrest in C. elegans. Mol Cell. 2000;5:435–443. doi: 10.1016/s1097-2765(00)80438-4. [DOI] [PubMed] [Google Scholar]
- He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network–another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene. 2006;25:7305–7310. doi: 10.1038/sj.onc.1209735. [DOI] [PubMed] [Google Scholar]
- Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67:7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Ljungman M, Lane DP. Transcription—guarding the genome by sensing DNA damage. Nat Rev Cancer. 2004;4:727–737. doi: 10.1038/nrc1435. [DOI] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, et al. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runnebaum IB, Nagarajan M, Bowman M, Soto D, Sukumar S. Mutations in p53 as potential molecular markers for human breast cancer. Proc Natl Acad Sci USA. 1991;88:10657–10661. doi: 10.1073/pnas.88.23.10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter KH, Acharya CR, Walters KS, Redman R, Anguiano A, Garman KS, et al. An integrated approach to the prediction of chemotherapeutic response in patients with breast cancer. PLoS ONE. 2008;3:e1908. doi: 10.1371/journal.pone.0001908. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, et al. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111–11116. doi: 10.1158/0008-5472.CAN-07-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidhaas JB, Eisenmann DM, Holub JM, Nallur SV. A Caenorhabditis elegans tissue model of radiation-induced reproductive cell death. Proc Natl Acad Sci USA. 2006;103:9946–9951. doi: 10.1073/pnas.0603791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.