

Abstract
Although the pathophysiological processes involved in dopamine (DA) neuron degeneration in Parkinson's disease (PD) are not completely known, apoptotic cell death has been suggested to be involved and can be modeled in DAergic cell lines using the mitochondrial toxin 1-methyl-4-phenylpyridinium (MPP+). Recently, it has been suggested that histone deacetylase inhibitors (HDACIs) may reduce apoptotic cell death in various model systems. However, their utility in interfering with DA cell death remains unclear. The HDACIs sodium butyrate (NaB), valproate (VPA) and suberoylanilide hydroxamic acid (SAHA) were tested for their ability to prevent MPP+-mediated cytotoxicity in human derived SK-N-SH and rat derived MES 23.5 cells. All three HDACIs at least partially prevented MPP+ -mediated apoptotic cell death. The protective effects of these HDACIs coincided with significant increases in histone acetylation. These results suggest that HDACIs may be potentially neuroprotective against DA cell death and should be explored further in animal models of PD.
Keywords: HDACI (Histone deacetylase Inhibitor), MPP+, Sodium Butyrate, Valproate, SAHA, Dopamine
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterized by progressive loss of dopaminergic substantia nigra pars compacta neurons and depletion of striatal dopamine (DA). Although the precise pathophysiological processes involved in the initiation and perpetuation of the degenerative process that occurs in PD is not completely known, apoptotic cell death has been suggested to be involved in the pathophysiology of PD. Apoptotic cell death has been described in the post-mortem PD brain (Anglade et al., 1997; Mochizuki et al., 1996) as well as in various animal and in vitro models of DA cell death utilizing various neurotoxins, including the methylpyridinium ion, MPP+ (Mochizuki et al., 1994; Speciale, 2002; Tatton and Kish, 1997).
Apoptotic cell death mediated by mitochondrial stress, as occurs following exposure to the mitochondrial complex I inhibitor MPP+, is associated with caspase activation, which may result in histone hypoacetylation (Rouaux et al., 2003). Hypoacetylation does not favor active transcription and may exacerbate the effects of toxin administration in DAergic cells, contributing to cell death (for review see (Kazantsev and Thompson, 2008)). Conversely, hyperacetylation as a result of the administration of histone deacetylase inhibitors (HDACIs) can promote transcription of potential cell survival factors (e.g., MnSOD, catalase (Jenner, 2003; Klivenyi et al., 1998; Petri et al., 2006), anti-apoptotic Bcl-2 family members (Faraco et al., 2006; Kim et al., 2007; Petri et al., 2006), and inhibitors of apoptosis (IAP's) (Ryu et al., 2003)) suggesting the treatment with HDACIs may be beneficial to neuronal survival.
Anti-epileptic drugs (i.e., sodium butyrate (NaB), valproic acid (VPA)) as well as hydroxamate compounds (e.g., suberoylanilide hydroxamic acid (SAHA)) can inhibit histone deacetylases (HDACs), the enzymes responsible for the removal acetyl groups from acetylated proteins, thus promoting the hyperacetylation of target proteins (Phiel et al., 2001; Remiszewski, 2002; Riggs et al., 1977). Histone deacetylases modulate the acetylation state of a variety of proteins including histones and transcription factors (for review see (Haberland et al., 2009; Kazantsev and Thompson, 2008)) and HDACIs may protect cells from a variety of toxic insults including excitotoxicity, inflammation, and oxidative stress (Chen et al., 2006; Chen et al., 2007; Dompierre et al., 2007; Langley et al., 2008; Leng et al., 2008), all of which have been reported to contribute to MPP+-mediated toxicity in DAergic cells (Hirsch et al., 2003; Jenner, 2003; Przedborski and Jackson-Lewis, 1998).
The ability of HDACIs to modulate histones, transcription factors, trophic responses and inflammatory mediators, all of which may play important roles in mediating DAergic cell death in PD, makes these compounds interesting candidates to protect against cell death in PD. The present study was designed to assess the efficacy of different HDACIs (i.e., NaB, VPA, and SAHA) in protecting two distinct DAergic cell lines (human neuroblastoma-derived SK-N-SH and rat mesencephalic-derived MES 23.5), from apoptotic cell death induced by MPP+. The data suggest that HDACIs may mitigate the effects of MPP+ on cell viability and DA neurochemistry while inhibiting expression of apoptotic markers.
Results
HDAC inhibitors promote acetylation of multiple histone residues in SK-N-SH and MES 23.5 cells
Both cell lines were responsive to HDACIs, which differentially induced acetylation of various histone residues (Fig. 1 A and B). For example, VPA induced acetylation at H4 Lys8 in MES 23.5 cells but not in SK-N-SH cells. However, all three HDACIs induced significant hyperacetylation of histone 3 at Lys9 (p<.01, F= 23.94, 14.58; df =11, 11; SK-N-SH and MES 23.5, respectively) in both cell lines. Additionally, histone H3 Lys 9 hyperacetylation was maintained in MPP+/HDACI-treated SK-N-SH and MES 23.5 cells (Figure 3C, D). Therefore, this residue was used as a marker to assess HDACI-induced hyperacetylation in subsequent experiments.
Figure 1. Administration of histone deacetylase inhibitors induces histone acetylation at multiple residues.
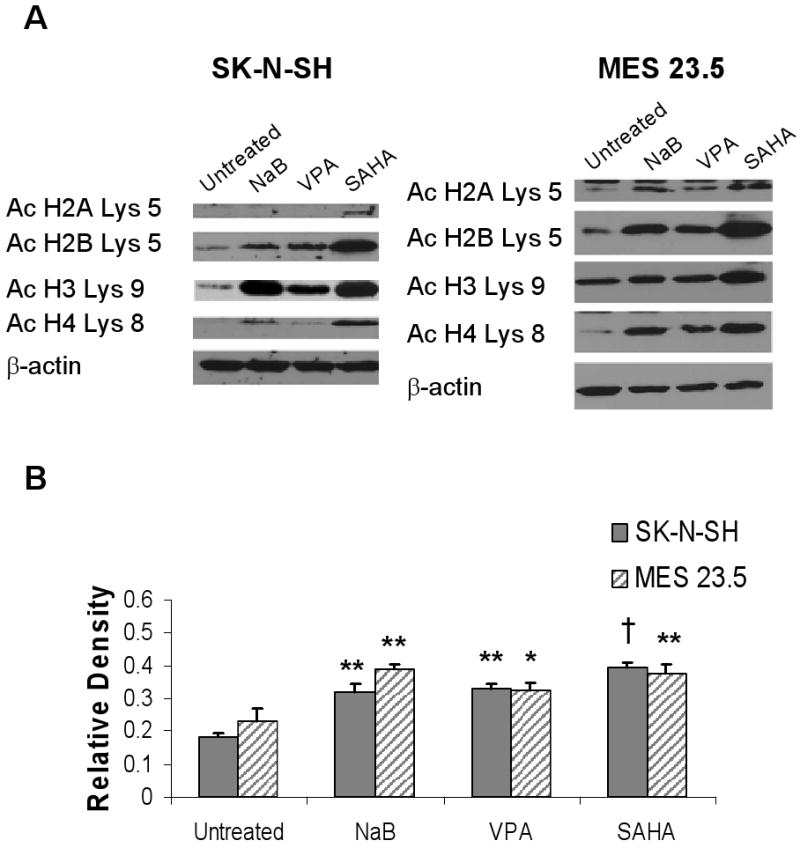
A) Representative western blots of SK-N-SH and MES 23.5 cells treated with HDACIs. Histones 2B, 3, and 4 show increased acetylation compared to untreated cells following incubation with sodium butyrate (NaB), valproate (VPA) and suberoylanilide hydroxamic acid (SAHA), while H2A acetylation was only increased in SAHA treated cells. All membranes were stripped and re-probed for β-actin to verify equal loading and transfer of samples. B) Results of densitometry analysis of acetylated histone H3 (Ac H3 Lys 9) Western blots. Untreated vs. HDACI: *p<05; **p< 0.01; †p< 0.001.
Figure 3. HDACIs improve cell viability in MPP+ treated cells while maintaining histone hyperacetylation.
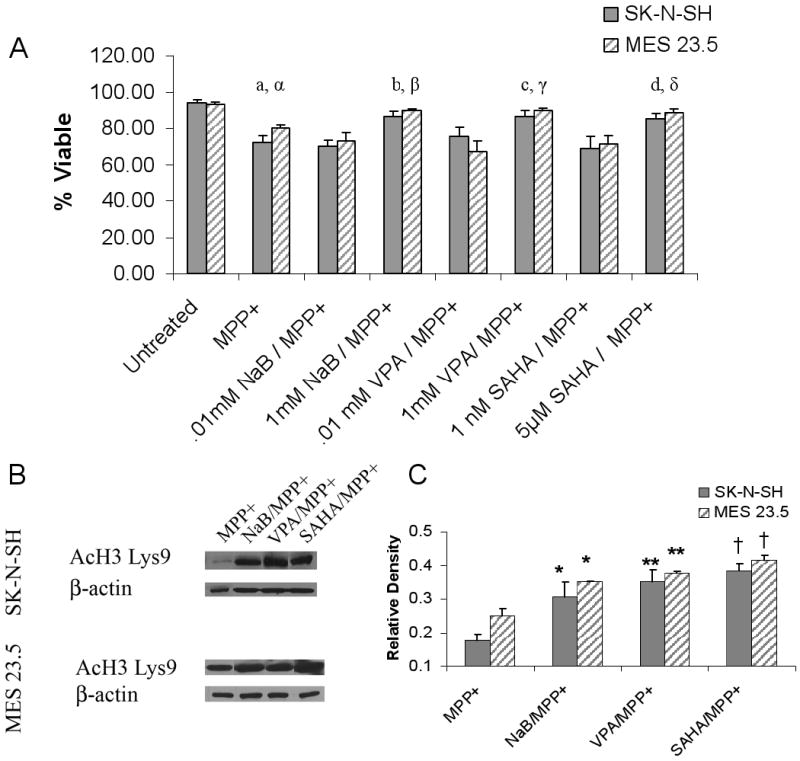
A) Changes in the percent of viable cells estimated by trypan blue exclusion following incubation of cells in MPP+ or MPP+/HDACI. SK-N-SH cells were treated with 100μM MPP+ while MES 23.5 cells were treated with 50μM MPP+. a, α Untreated vs. MPP+ p<0.0001; b, β MPP+ vs. NaB/MPP+ p<0.001; c, γ: MPP+ vs. VPA/MPP+ p<0.001; d, δ: MPP+ vs. SAHA/MPP+ p< 0.01. Incubation of cells with low level HDACIs did not prevent MPP+ mediated decreases in cell viability. B) HDACI-induced hyperacetylation of histone H3 on Lys9 was maintained in the presence of MPP+ (24hr incubation) in both SK-N-SH and MES 23.5 cells. C) Densitometric analysis of acetylated histone H3 Lys9: MPP+ vs. HDACI/MPP+ * p<05; ** p< 0.01; †p< 0.001.
MPP+ Induced Apoptosis/Cell Death is Inhibited by HDACIs
The number of SK-N-SH and MES 23.5 cells with nicked DNA, indicative of cells undergoing apoptosis, was significantly increased (t= 15.2; 6.34, df= 6; 6, respectively; p< 0.001 vs. control in all cases) 24 hrs. after MPP+ exposure (Figure 2A). Caspase-3 activation was not detectable in SK-N-SH cells (similar to what has been described by others (Chetsawang et al., 2007)). Cleaved caspase-3 was absent in normal MES 23.5 cells but it was significantly increased in MES 23.5 cells 24 hrs. after MPP+ exposure (Figure 2B).
Figure 2. MPP+ -mediated induction of apoptotic markers.
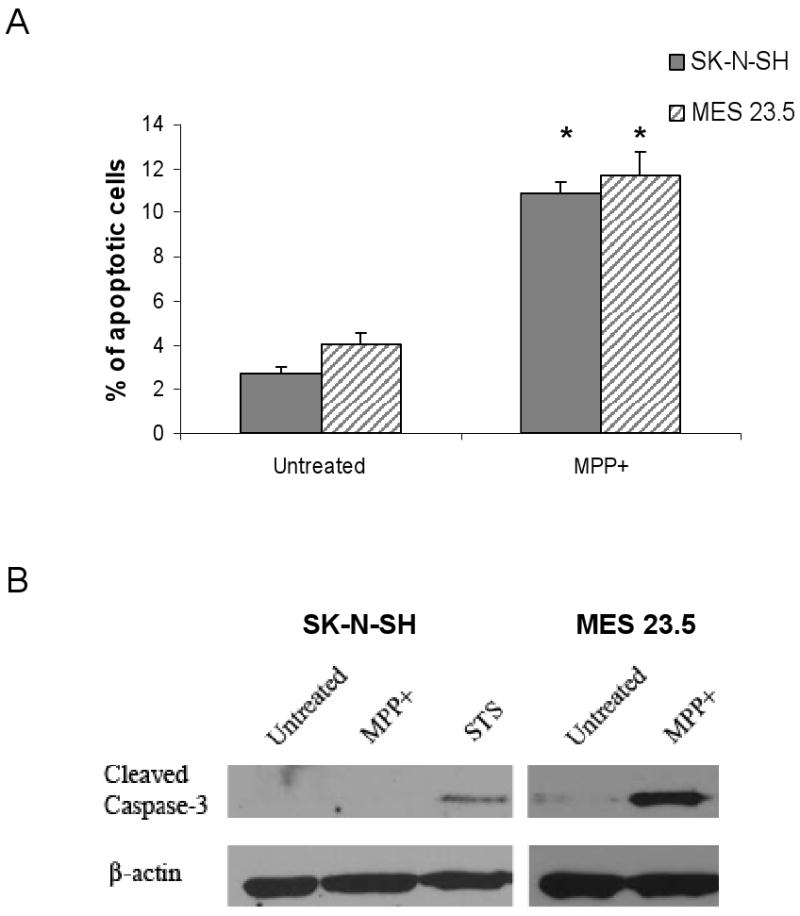
A) Percent of anti-Apo ssDNA positive cells compared to Hoechst labeled nuclei following 24 hr. MPP+ exposure (SK-N-SH: 100 μM; MES 23.5: 50 μM). *Untreated vs. MPP+ p< 0.001. B) Western blots showing the presence of cleaved caspase-3 following 24hrs of MPP+ treatment in MES 23.5 but not SK-N-SH cells. An increase in cleaved caspase-3 is shown with a positive control, 24 hr exposure to 1 μM staurosporine (STS). β-actin blots show equal loading and transfer of samples.
Exposure to MPP+ resulted in a 27.6 ± 3.9% decrease in SK-N-SH cell viability and a 19.8 ± 1.9% decrease MES 23.5 cell viability, as assessed by trypan blue exclusion (Figure 3A). Treatment with HDACIs (beginning 24 hrs prior to and throughout the MPP+ exposure period) resulted in significant improvement in cell viability compared to control cultures exposed to MPP+ alone (p<0.01; F= 32.90, 10.74; df= 29, 23, SK-N-SH and MES 23.5 cells, respectively) (Figure 3A). No protection was observed in cells treated with concentrations of HDACIs that did not induce H3 hyperacetylation (Figure 3A, Supplemental Figure 1). Treatment with HDACIs alone had no effect on cell viability (Supplemental Figure 2).
The enhanced cell viability in HDACI-treated MES 23.5 cells was accompanied by a significant decrease in caspase-3 activation with VPA treatment (31.2 ± 1.1% decrease vs. MPP+ alone, p < 0.05) and complete inhibition of caspase-3 activation with NaB and SAHA treatments (Figure 4).
Figure 4. HDACIs reduce cleavage of caspase-3 in MES 23.5 cells.

Representative western blot demonstrates decreased cleaved caspase-3 expression by pre-incubation with VPA and blocked caspase-3 expression by pre-incubation with NaB and SAHA. β-actin blots show equal loading and transfer of samples.
Exposure to MPP+ resulted in a 32.0 ± 1.5 % and 35.7 ± 1.2 % decrease in the number of SK-N-SH and MES 23.5 TH+ cells, respectively (P<0.001; F= 6.4,12.67; df= 30,18 SK-N-SH and MES 23.5 respectively). In comparison, exposure of SK-N-SH cells to MPP+ together with NaB, VPA or SAHA resulted in TH+ cell losses of only 17.4% ± 2.6, 13.3% ± 4.4, and 1.1% ± 2.1, respectively (P<0.05; F=6.83; df =14, compared to cells treated with MPP+ alone) (Figure 5). Similarly, exposure of MES 23.5 cells to MPP+ and NaB, VPA or SAHA resulted in loss of TH+ cells of only 12.5 ± 7.9 %, 12.8 ± 1.9 %, and 5.8 ± 8.4 %, respectively (P<0.05; F=8.08; df =12, compared to cells treated with MPP+ alone) (Figure 5).
Figure 5. HDACIs inhibit MPP+-induced TH+ cell loss.

Cells were treated with MPP+ (SK-N-SH: 100μM MPP+ for 48 hrs; MES 23.5: 50μM MPP+ for 24 hrs) and the number of tyrosine hydroxylase immuno-positive (TH+) cells was determined using a stereological procedure. The number of TH+ cells per well were compared to control wells not receiving MPP+ to determine the % of control. a, α Untreated/Control vs. Untreated/MPP+ p<0.001; b, β Untreated/MPP+ vs. NaB/MPP+ p<0.05; c, γ: Untreated/MPP+ vs. VPA/MPP+ p<0.05; d, δ: Untreated/MPP+ vs. SAHA/MPP+ p< 0.01.
Although cell death from MPP+ requires uptake of the toxin through the DA transporter [23], it seems unlikely that inhibition of transporter activity and uptake of MPP+ significantly contributed to the protective effects observed with the different HDACIs. Specific cellular uptake of DA was not significantly altered by treatment with either NaB or VPA but was decreased by 66.0 + 5.5% in SK-N-SH cells following SAHA treatment (P<0.001 vs. control). Thus, partial inhibition of DA transporter function could have contributed to the protective effects seen with SAHA but this mechanism does not appear to underlie the protective effects observed with NaB or VPA.
Discussion
The present results suggest that the HDACIs NaB, VPA and SAHA at least partially protect SK-N-SH and MES 23.5 DAergic cell lines from MPP+-induced apoptotic cell death. Although exposure of these cell lines to MPP+ apparently stimulated different forms of apoptotic-like cell death (i.e.: non-caspase-3 (SK-N-SH) and caspase-3-mediated (MES 23.5)), nonetheless, treatment with HDACIs improved cell survival in both cell lines. HDACIs enhanced cell viability following MPP+ exposure, resulting in a greater number of TH+ cells than in MPP+ - only control cultures and, in MES 23.5 cells, additionally reduced cleaved caspase-3 expression. These protective effects of HDACIs coincided with histone H3 hyperacetylation and in the cases of NaB and VPA, were not related to any alteration in DA transporter function.
HDACIs may protect against a wide range of toxic insults by decreasing expression of inflammatory mediators (Chen et al., 2006; Chen et al., 2007; Peng et al., 2005) as well as by reducing excitotoxicity (Leng et al., 2008; Oliveira et al., 2006) and oxidative stress (Langley et al., 2008; Ryu et al., 2003) and enhancing neurotrophic factor expression [29]. Many of the actions of HDACIs in the nervous system have been hypothesized to be mediated at least in part via microglia or astrocytes (Chen et al., 2006; Chen et al., 2007; Peng et al., 2005; Wu et al., 2008). However, as neither microglia nor astrocytes were present in the present culture systems, HDACI-related protection against MPP+ toxicity was mediated through glial-independent mechanisms. Sodium butyrate and VPA may directly reduce oxidative stress and modulate the transcription of free radical scavengers (MnSOD, SOD1 and catalase) (Ryu et al., 2003). Additionally, HDAC inhibition may activate pro-survival pathways (such as the p21waf/cip pathway) (Langley et al., 2008). Maintenance of histone hyperacetylation in the presence of MPP+ suggests that transcriptional activation, possibly involving the targets described above, may contribute to the protective effects observed in the present study.
In contrast to the present findings, Wang et al. (Wang et al., 2009) reported that the administration of low levels of the hydroxamate HDACI trichostatin A (TSA) exacerbated the effects MPP+ and rotenone on cell viability in several DAergic cell lines. These data are difficult to interpret since the concentration of TSA used induced apoptosis independent of MPP+ administration. Additionally, the concentrations of MPP+ used (200 μM with N27 cells and 400 μM with SH-SY5Y cells) were higher than those used in the present study and the mechanism of cell death was not assessed. Langley et al (Langley et al., 2008) showed that pulsed administration rather than constant exposure to TSA was able to protect primary cortical cells from excitotoxicity with little if any intrinsic cytotoxic effects. Together, these data suggest that care must be taken in choosing an appropriate HDACI and an appropriate exposure paradigm for evaluating potential protective effects of these drugs.
In summary, the HDACIs NaB, VPA, and SAHA were able to reduce MPP+-mediated toxicity in both the human SK-N-SH and rat MES 23.5 DAergic cell lines and the protective actions of these drugs coincided with the hyperacetylation of histones. However, the effects of HDACIs may be drug, dose and cell type dependent. Additional studies of these compounds utilizing in vivo models of DAergic neuron degeneration are needed to further evaluate the therapeutic potential HDACI-mediated protection of DAergic neurons.
Experimental Procedures
Cell Culture Conditions
SK-N-SH cells were obtained from ATCC and were maintained in DMEM media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (PenStrep). MES 23.5 cells (provided by Dr. Weidong Le, Baylor College of Medicine) were maintained in DMEM F12 media containing HEPES, Sato's salts, 2% NBS, 5% FBS and 1% PenStrep. Serum was obtained from Atlanta Biologicals Inc., Lawrenceville, GA; all other reagents were obtained from Invitrogen Inc., Carlsbad, California. Both cell lines were maintained at 37°C with 5% CO2.
Measurement of Histone Acetylation
Cells were grown to 70% confluence in T25 flasks. Histone deacetylase inhibitors (HDACIs) were applied to cells at the following concentrations for 24hrs unless otherwise specified: 1mM sodium butyrate (NaB; Sigma Inc., St. Louis, MO), 1mM valproic acid (VPA; Sigma) and 5 μM suberoylanilide hydroxamic acid (SAHA; Cayman Chemicals Inc., Ann Arbor, MI). These concentrations were selected for their ability to induce significant histone hyperacetylation without inducing cytotoxicity, as determined in preliminary studies. Cells were exposed to for HDACIs for 24hrs as shorter time points (1, 3, 6, and 12 hrs) did not induce significant histone hyperacetylation. Cells were collected by trypsinization 24hrs after addition of HDACIs and pelleted by centrifugation. Cell pellets were lysed in a hypotonic lysis buffer (10mM HEPES, 1.5mM MgCl2, 10mM KCl containing HALT protease inhibitor (Pierce, Thermo Fisher Scientific, Inc., Waltham, MA)). Hydrochloric acid was added to a final concentration of 0.2M and cells were incubated at 4°C for 30 min. Cellular debris and non-acid soluble protein were collected by centrifugation and the supernatant was used for standard western blot detection of acetylated histone residues. Briefly, 10 μg of protein was loaded onto a 4-12% gradient Bis-Tris polyacrilamide gel (Invitrogen, Inc.) and protein was transferred to a 0.22 μm nitrocellulose membrane (Biorad, Inc.). Membranes were blocked in 5% non-fat milk in TBS containing Tween-20 for 1 hr and primary antibody (1:1,000 Ac H2A Lys5, AcH2B Lys5, AcH4 Lys8; Cell Signaling Technology and AcH3 Lys9 (1:10,000; Millipore, Inc.) was added for 1hr at room temperature. Secondary antibody (1:20,000, horseradish peroxidase conjugated goat anti-rabbit, Pierce, Inc.) was added for 1 hr at room temperature. After multiple washes, membranes were developed using the Pierce Pico chemiluminescent detection kit. Membranes were then stripped in TBS buffer containing SDS, blocked in milk and re-probed with β-actin (Imgenex, Inc.) to ensure equal loading and transfer of all samples.
Apo ssDNA Apoptosis Assay
Cells were plated at 5,000 cells/well (SK-N-SH) or 3,000 cells/well (MES 23.5) in 8 well chamber slides (Nunc™) and allowed to grow for 48hrs. Cells were then treated with 100 μM MPP+ (SK-N-SH) or 50 μM MPP+ (MES 23.5) 24 hrs. These toxin concentrations were previously shown to result in apoptosis in the cell lines currently used [25, 26]. Cells were fixed in a methanol-based fixative (Cell Technology, Inc.) at -20 °C for 24 hrs and processed for detection of nicked DNA (Apo ssDNA kit, Cell Technology, Inc.). Cells were then incubated with Hoechst 33342 (0.5% v/v, Immunochemistry Technologies, LLC) in sterile PBS for 10 min at room temperature. Following several PBS washes, slides were cover-slipped with AquaPerm mounting media (Immunon) and examined using a BX-60 microscope. Cells undergoing apoptosis were visualized as having discrete, condensed masses co-localized with Hoechst nuclear stain.
Detection of Cleaved Caspase-3
Cells were grown to 70% confluence in T25 flasks and treated with media containing HDACIs or control media for 24hrs. Cells were then exposed to 100 μM MPP+ (SK-N-SH), 50 μM MPP+ (MES 23.5), 1 μM Staurosporine (STS, positive control) or control media for 24 hrs. Cells were collected using trypsin and nuclear and cyotplasmic proteins were extracted (Ne-Per kit, Pierce, Inc.). Protein was quantified using BCA reagent and 20 μg of protein was loaded onto a 4-12% gradient Bis-Tris polyacrylamide gel and transferred to a 0.22 μm nitrocellulose membrane. Membranes were blocked in 5% non-fat milk in TBS containing Tween-20, (T-TBS), for 1 hr and incubated in primary antibody (Rabbit anti-cleaved caspase-3 fragment, 1:1,000; Cell Signaling Technologies) overnight at 4°C. Following several rinses in T-TBS, secondary antibody (horseradish peroxidase conjugated goat anti-rabbit, 1:20,000) was added for 1 hr at room temperature. After multiple washes, membranes were developed using the Pierce Dura chemiluminescent detection kit. Membranes were stripped and re-probed for β-actin (Imgenex) to ensure equal loading and transfer of all samples.
Measurement of Cell Viability
Cell viability was assessed by trypan blue exclusion. Cells were grown to 70% confluence in 6 well plates. HDACIs were added as previously described or at lower levels that did not promote H3 hyperacetylation (0.01 mM NaB and VPA and 1 nM SAHA) for 24 hrs followed by exposure to 100 μM MPP+ (SK-N-SH) or 50 μM MPP+ (MES 23.5) for 24 hrs. Cells were collected using trypsin and cell pellets were collected and resuspended in 1 mL Hank's Balanced Salt solution (HBSS). 100 μL of cell suspension was combined with 250 μL of 0.4% trypan blue and 150 μL HBSS and incubated for 10 min. at room temperature. Cells were counted using a hemocytometer. The number of blue (dead cells) was subtracted from the total cell number which was then divided by the total number of cells counted to estimate % viability.
Tyrosine Hydroxylase (TH) immunochemistry
Cells were plated at 5,000 cells/well and allowed to grow for 24 hrs. Plating densities were determined empirically for each cell line and were chosen to produce 60% confluence at the time of HDACI treatment. HDACIs and MPP+ were added as described above. After overnight fixation in 4% paraformeldahyde, endogenous peroxidase activity was blocked using a 1% peroxide solution in PBS and non-specific staining was blocked using 2.5% bovine serum albumin in PBS containing 0.3% triton X-100 (PBS-T) for 1 hr at room temperature. Primary antibody (rabbit anti-TH, 1:500 in PBS-T; Pel Freez) was then added and after overnight incubation and several washes, secondary antibody (biotinylated goat anti-rabbit, 1:1,000; Jackson Immunoresearch) was added for 1 hr followed by incubation in avidin-biotin complex (ABC Kit, Vector). 3,3′-Diaminobenzidine (DAB) was used to visualize TH positive (TH+) cells. An optical fractionater-based stereological estimate of the number of TH+ cells was obtained (StereoInvestigator, MBF biosciences). The region of interest (ROI) was set as the entire slide chamber (0.8 cm2) at low magnification (4×). A square grid (1.3 × 1.3 mm) was randomly placed over the ROI by the computer and a counting frame measuring 140 × 140 μm was used to count the number of TH+ cells at high magnification (40×). A cell was counted if it had an identifiable nucleus and was within the counting frame and/or touching the top or right border of the counting frame. All sampling parameters were determined empirically to generate a reproducible estimate of the number of cells in each ROI.
DA Uptake Assay
Cells were plated in 48 well plates at 5,000 cells per well and allowed to grow for 72 hrs. HDACIs or control media was added for 24 hrs in order to induce hyperacetylation prior to monitoring DA uptake. Media was then removed and cells were exposed to 10nM 3H DA (54.2 Ci/mmol, Perkin Elmer, Inc.) in Krebs Ringers buffer (16mM sodium phosphate, 119mM NaCl, 4.7mM KCl, 1.8mM CaCl2, 1.2mM MgSO4, 1.3mM EDTA, 5.6 mM glucose, pH = 7.4) for 20 min at 37°C. Cells were then placed on ice and washed 3 times in ice-cold buffer before being digested with 1M NaOH for 1 hr at room temperature. Tritium was counted using a liquid scintillation counter (Packard TriCarb 2200). Specific uptake was measured by subtracting background, non-specific uptake in the presence of 10 μM mazindol (Sigma, Inc.).
Data Analyses
Mean and standard error were calculated for all samples, run at least in triplicate. A one way ANOVA with Bonferroni posthoc t-test was used to determine statistically significant differences between treatments (i.e., p < 0.05).
Supplementary Material
Acknowledgments
This research was supported by the F.M. Kirby Foundation and NIEHS training grant T32 ES07282.
List of Abbreviations
- DA
Dopamine
- PD
Parkinson's disease
- MPP+
1-methyl-4-phenylpyridinium
- HDACs
Histone deacetylases
- HDACIs
Histone deacetylase inhibitors
- NaB
Sodium butyrate
- VPA
Valproic acid
- SAHA
Suberoylanilide hydroxamic acid
- IAP's
Inhibitors of Apoptosis
- TH
Tyrosine Hydroxylase
- DAB
3,3′-Diaminobenzidine
- ROI
Region of interest
- DAT
Dopamine transporter
- TSA
Trichostatin-A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, Wilson B, Lu RB, Gean PW, Chuang DM, Hong JS. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–25. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- Chen PS, Wang CC, Bortner CD, Peng GS, Wu X, Pang H, Lu RB, Gean PW, Chuang DM, Hong JS. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience. 2007;149:203–212. doi: 10.1016/j.neuroscience.2007.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetsawang J, Govitrapong P, Chetsawang B. Melatonin inhibits MPP+-induced caspase-mediated death pathway and DNA fragmentation factor-45 cleavage in SK-N-SH cultured cells. J Pineal Res. 2007;43:115–20. doi: 10.1111/j.1600-079X.2007.00449.x. [DOI] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–83. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70:1876–84. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson's disease. Ann N Y Acad Sci. 2003;991:214–28. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol. 2003;53 3:S26–36. doi: 10.1002/ana.10483. discussion S36-8. [DOI] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–68. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone Deacetylase Inhibitors Exhibit Anti-Inflammatory and Neuroprotective Effects in a Rat Permanent Ischemic Model of Stroke: Multiple Mechanisms of Action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, St Clair D, Wermer M, Yen HC, Oberley T, Yang L, Beal MF. Manganese Superoxide Dismutase Overexpression Attenuates MPTP Toxicity. Neurobiology of Disease. 1998;5:253–258. doi: 10.1006/nbdi.1998.0191. [DOI] [PubMed] [Google Scholar]
- Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–76. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci. 2008;28:2576–88. doi: 10.1523/JNEUROSCI.5467-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki H, Nakamura N, Nishi K, Mizuno Y. Apoptosis is induced by 1-methyl-4-phenylpyridinium ion (MPP+) in ventral mesencephalic-striatal co-culture in rat. Neurosci Lett. 1994;170:191–4. doi: 10.1016/0304-3940(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Goto K, Mori H, Mizuno Y. Histochemical detection of apoptosis in Parkinson's disease. J Neurol Sci. 1996;137:120–3. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- Oliveira JMA, Chen S, Almeida S, Riley R, Goncalves J, Oliveira CR, Hayden MR, Nicholls DG, Ellerby LM, Rego AC. Mitochondrial-Dependent Ca2+ Handling in Huntington's Disease Striatal Cells: Effect of Histone Deacetylase Inhibitors. J Neurosci. 2006;26:11174–11186. doi: 10.1523/JNEUROSCI.3004-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng GS, Li G, Tzeng NS, Chen PS, Chuang DM, Hsu YD, Yang S, Hong JS. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Brain Res Mol Brain Res. 2005;134:162–9. doi: 10.1016/j.molbrainres.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–9. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord. 1998;13 1:35–8. [PubMed] [Google Scholar]
- Remiszewski SW. Recent advances in the discovery of small molecule histone deacetylase inhibitors. Curr Opin Drug Discov Devel. 2002;5:487–99. [PubMed] [Google Scholar]
- Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268:462–4. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–49. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:4281–6. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speciale SG. MPTP: insights into parkinsonian neurodegeneration. Neurotoxicol Teratol. 2002;24:607–20. doi: 10.1016/s0892-0362(02)00222-2. [DOI] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience. 1997;77:1037–48. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang X, Liu L. HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci Lett. 2009;467:212–6. doi: 10.1016/j.neulet.2009.10.037. [DOI] [PubMed] [Google Scholar]
- Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008:1–12. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.