

Abstract
It is now well established that important regulatory interactions occur between the cells in the hematopoietic, immune and skeletal systems (osteoimmunology). B lymphocytes (B cells) are responsible for the generation and production of antibodies or immunoglobulins in the body. Together with T cells these lymphocytes comprise the adaptive immune system, which allows an individual to develop specific responses to an infection and retain memory of that infection, allowing for a faster and more robust response if that same infection occurs again. In addition to this immune function, B cells have a close and multifaceted relationship with bone cells. B cells differentiate from hematopoietic stem cells (HSCs) in supportive niches found on endosteal bone surfaces. Cells in the osteoblast lineage support HSC and B cell differentiation in these niches. B cell differentiation is regulated, at least in part, by a series of transcription factors that function in a temporal manner. While these transcription factors are required for B cell differentiation, their loss causes profound changes in the bone phenotype. This is due, in part, to the close relationship between macrophage/osteoclast and B cell differentiation. Cross talk between B cells and bone cells is reciprocal with defects in the RANKL-RANK, OPG signaling axis resulting in altered bone phenotypes. While the role of B cells during normal bone remodeling appears minimal, activated B cells play an important role in many inflammatory diseases with associated bony changes. This review examines the relationship between B cells and bone cells and how that relationship affects the skeleton and hematopoiesis during health and disease.
Keywords: B cell differentiation, Macrophage/osteoclast differentiation, B cell transcription factors and bone, IL-7, Regulation of hematopoiesis by osteoblast lineage cells, Regulation of B cell development by RANKL-RANK signaling, B cell activation
Introduction
All of the blood borne elements arise from pluripotent hematopoietic stem cells (HSCs) that reside in growth and differentiation supportive structures, known as niches, which are found predominantly on endosteal bone surfaces in contact with the bone marrow. HSC are also in contact with cells that are adherent to the endosteal bone surface. These support cells secrete or express on their cell surface cytokines and growth factors that facilitate HSC growth and differentiation [1–3]. Often these cells are referred to as osteoblasts (OBs) or stromal cells, and although they are in the osteoblast lineage their exact stage of differentiation is unknown. However, it is unlikely that they are mature osteoid secreting osteoblasts.
Long-term reconstituting HSCs (LT-HSC) are multipotential in that they have the capacity for self-renewal as well as producing daughter cells that at each stage of differentiation become more restricted in cell lineage. LT-HSCs give rise to short-term reconstituting HSCs that differentiate to multipotent progenitors (MPPs). It is at this stage that an important separation of the lineages occurs (Fig 1). MPP differentiate into the common myeloid progenitor (CMP), which gives rise to the granulocyte-macrophage progenitor (GMP) and then, in turn, to osteoclasts (OC), macrophages, granulocytes and dendritic cells [4]. The CMP also produces the megakaryocyte-erythrocyte progenitor in a separate lineage. Alternatively, MPPs can also differentiate into the common lymphoid progenitor (CLP) which gives rise to B, T and natural killer cells. Thus, it is at the MPP stage that OC (myeloid) and B cell (lymphoid) differentiation share a common progenitor and critical cell fate decisions are made that dictate whether subsequent differentiation will result in OCs or B cells. These cell fate decisions are made by the expression or repression of proteins under the control of a hierarchy of transcription factors (TFs), which in include PU.1, Ikaros, E2A, Ebf1 and Pax5. These TFs function in a network that regulates B cell differentiation [5]. Whether these TFs function in a similar network to regulate macrophage-OC differentiation remains to be elucidated.
Fig. 1.

Hematopoietic cell differentiation. All hematopoietic cells arise from hematopoietic stem cells (HSC) that give rise to multipotential progenitor cells (MPP, blue). B cells differentiate from the common lymphoid progenitor (CLP, green) and osteoclasts arise from the common myeloid progenitor (CMP, pink). There appears to exist a distinct lineage derived from the MPP that expresses both macrophage and B cell characteristics (B/Mφ bipotent), which can differentiate to B cells, macrophages and possibly osteoclasts (purple). (Adapted from Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA, Weissman IL Ann. Rev. Immunol. 2003; 21:759–806).
B Cell Differentiation
B lymphopoiesis is a highly ordered process proceeding from progenitor cells in the fetal liver, to the bone marrow (BM), to mature B cells in the secondary lymphoid organs. The mature B cell, following activation and T cell derived help, terminally differentiates into immunoglobulin (lg)-secreting plasma cells [6]. B cell development is organized around the assembly of a functional B cell receptor through a process of gene rearrangement called V(D)J recombination [7]. The BM B cell developmental pathway can be divided into several distinct stages based on the recombination status of the lg genes and expression of surface antigens [8]. The earliest characterized committed B cell progenitor (pre-pro-B) expresses the cell surface markers CD45R/B220 and AA4.1 and has its lg heavy (lgH) chain locus in the germ-line configuration (not rearranged) [9]. Subsequent differentiation generates pro-B cells that harbor rearranged lgH D and J genes [10]. As the cells mature, rearrangements occur in V gene segments of the lg heavy chain gene and then finally in the lg light chain genes culminating in a functional surface antigen receptor [7, 11].
The molecular dissection of the B cell differentiation pathway has been greatly facilitated by the identification of TFs required for developmental transitions. Loss of these specific factors precludes the cells from continued maturation, and results in a developmental block of cells at the latest stage of differentiation prior to the arrest. The selective loss of two different basic helix-loop-helix proteins encoded by the E2A gene or the Ebf1 gene has been shown to play a critical role in the initiation of B cell differentiation. Absence of either of these proteins halts B cell development at the earliest stage, before D-J rearrangement of the lgH gene [8, 12, 13]. Commitment and maintenance of progenitors to the B cell lineage is accomplished by the expression of Pax5, [14, 15]. Ebf1and E2A are key factors for the specification and progression of lymphoid progenitors into the B-lymphoid pathway but these function before commitment of the cell, which requires Pax5 expression.
Osteoblast lineage cells support B lymphopoiesis
Osteoblast lineage cells carry out at least three major functions: 1) they make bone; 2) they regulate osteoclast differentiation; and 3) they support hematopoietic cell differentiation and growth. In addition to supporting HSC growth and survival, osteoblast lineage cells also provide support for more differentiated cells in specific lineage niches including B cells [16]. Osteoblasts support commitment and differentiation of all stages of B cell development. Production of B cell precursors from progenitors in vitro required contact with osteoblasts and expression of CXCL12 (SDF-1) and IL-7, which was induced by parathyroid hormone (PTH) [1, 3]. Interestingly, addition of stem cell factor, IL-6 and IL-3 redirected differentiation away from B lymphopoiesis and toward myelopoiesis. Selective elimination of OBs by treatment of Col2.3d-TK transgenic mice with gancyclovir also severely depleted pre-pro B cells from the BM confirming the supportive role of OBs in B cell development [2].
It is now known that signaling though the PTH/PTH-related peptide receptor (PPR) in osteoblastic cells increases trabecular bone and importantly increase HSCs [1]. PTH is known to increase production of CXCL12 and IL-7 by osteoblastic cells in vitro suggesting that downstream signaling through the PPR could regulate B cell development [1, 3]. Mice made deficient in PTH signaling specifically in osteoblasts by ablation of the G protein α subunit Gsα had a striking decrease in trabecular bone and an almost 50% reduction in BM B cells while other hematopoietic lineages were unaffected [16]. In addition, IL-7 expression was reduced in Gsα deficient osteoblasts, confirming the importance of osteoblast lineage cells in B cell growth and differentiation.
IL-7
IL-7 is a cytokine that has diverse effects on the hematopoietic and immunologic systems and is best known for its non-redundant role in supporting B- and T-lymphopoiesis [17]. IL-7 is the major growth factor for B cells and is apparently expressed by BM stromal cells and osteoblasts [3, 16, 18]. The IL-7 receptor (IL-7R) is expressed on progenitor B cells and is composed of the common γ chain and the IL-7Rα chain [8, 18]. Signals from the IL-7R are required during the pro-B-cell stage for further differentiation, and deficiencies in either IL-7 or the IL-7R result in severe defects in B-cell development. However, both IL-7 and IL-7Rα-deficient mice possess readily detectable numbers of peripheral B cells, indicating that the block in B lymphopoiesis is not absolute in these animals. As stated above, production of IL-7 by osteoblast lineage cells appears critical for normal B-lymphopoiesis [16].
Studies have demonstrated that IL-7 also plays an important role in the regulation of bone homeostasis [19, 20]. However, the precise nature of how IL-7 affects osteoclasts and osteoblasts is controversial, because it has a variety of actions in different target cells. Systemic administration of IL-7 increased osteoclast formation from human peripheral blood cells by increasing osteoclastogenic cytokine production in T cells [21]. Furthermore, mice with global over expression of IL-7 had a phenotype of decreased bone mass with increased osteoclasts and no change in osteoblasts [22]. However, the interpretation of results from in vivo IL-7 treatment studies is complicated by secondary effects of IL-7, which result from the production of bone-resorbing cytokines by T cells in response to activation by this cytokine [21, 23, 24]. Consistent with this conclusion, IL-7 administration did not induce bone resorption or bone loss in T-cell-deficient nude mice [23].
In contrast with previously reported studies, we found differential effects of IL-7 on osteoclastogenesis [19, 21, 23, 25]. IL-7 inhibited osteoclast formation in murine bone marrow cells that were cultured for 5 days with M-CSF and RANKL [25]. We also found that IL-7-deficient mice had markedly increased osteoclast number and decreased trabecular bone mass compared to wild-type controls [26].
The role of IL-7 in the effects of estrogen on bone is also controversial. Treatment of mice with a neutralizing anti-IL-7 antibody inhibited ovariectomy-induced bone loss and the proliferation of early T cell precursors in the thymus [27]. However, we found that trabecular bone loss after ovariectomy was similar in wild type and IL-7-deficient mice [26]. Curiously, IL-7 mRNA levels in bone increase with ovariectomy and this effect may be linked to alterations in osteoblast function, which occur with estrogen withdrawal [20, 28].
Treatment of newborn murine calvaria cultures with IL-7 inhibited bone formation, as did injection of IL-7 above the calvaria of mice in vivo [20]. However, when IL-7 was transgenically over expressed in osteoblasts, trabecular bone mass was increased compared with wild-type mice [29]. Furthermore, targeted expression of IL-7 in the osteoblasts of IL-7-deficient mice rescued the osteopenic bone phenotype of the IL-7-deficient mice [30].
PU.1, Ikaros, E2A, Ebf1 and Pax5 a Network of Transcription Factors
(The TFs are discussed in hierarchical order from the earliest to latest acting in B cell differentiation)
PU.1 is a member of the Ets family of transcription factors and is unique among hematopoietic TFs because it is required for both myeloid and lymphoid lineage development [31]. PU.1−/− mice die during fetal development by day E18.5. They lack B and T cells and fail to develop both macrophages and OCs [32,33] (Fig 2). Due to a lack of OCs their bones are severely osteopetrotic. These data were central in showing that OC differentiation required PU.1 expression early in lineage development and OCs and macrophages were members of the myeloid lineage. In one study PU.1−/− MPP cells isolated from fetal liver (E14.5) were lineage depleted using flow cytometry, then transduced with PU.1 or control virus using retroviral vectors and grown on M-CSF-deficient OP9 stromal cells. PU.1−/− MPPs before tranduction had reduced expression of c-fms, GM-CSFRα and G-CSFR, suggesting a reduced ability to differentiate into all myeloid lineages [34]. PU.1−/− MPP transfected with PU.1 produced 56% macrophages and only 19% pro-B cells [35] while PU.1+/− MPPs transfected with control virus generated 7% macrophages and 86% pro-B cells. PU.1−/− MPPs transfected with control virus failed to grow because they could not respond to added IL-7, due to the absence of IL-7Rs, which are regulated by PU.1 [36]. Failure to respond to IL-7 precludes further B cell differentiation. These data suggest that the increased number of macrophages in the PU.1 rescued cells was not simply due to increased M-CSF. These authors went on to show that higher levels of PU.1 expression promotes macrophage differentiation and blocks B cell development [35]. Conversely, low concentrations of PU.1 protein promote B cell differentiation. These data are consistent with reports showing that wild-type (WT) macrophages express higher levels of PU.1 than wild-type pro-B cells and that PU.1 mRNA is expressed at higher levels in myeloid than in B lymphoid cell lines [35, 37, 38]. In agreement with these data, the amount of PU.1 expressed by OCs is 3 fold more than in BM macrophages (32). PU.1−/− MPPs have impaired expression of Ebf1 and Pax5 [35]. Tranduction of MPPs with an Ebf1 but not Pax5-containing retroviral vector rescued IL-7R expression and pro-B cell development in the PU.1−/− MPPs. Whether these rescued cells can develop into OCs is unknown. Thus, PU.1 is required for the generation of lymphoid progenitors but not for their further differentiation. That is the responsibility of E2A, Ebf1 and Pax5. In the myeloid lineage, PU.1 is required for the generation of myeloid progenitors and the differentiation of macrophages and OCs [32, 34].
Fig. 2.
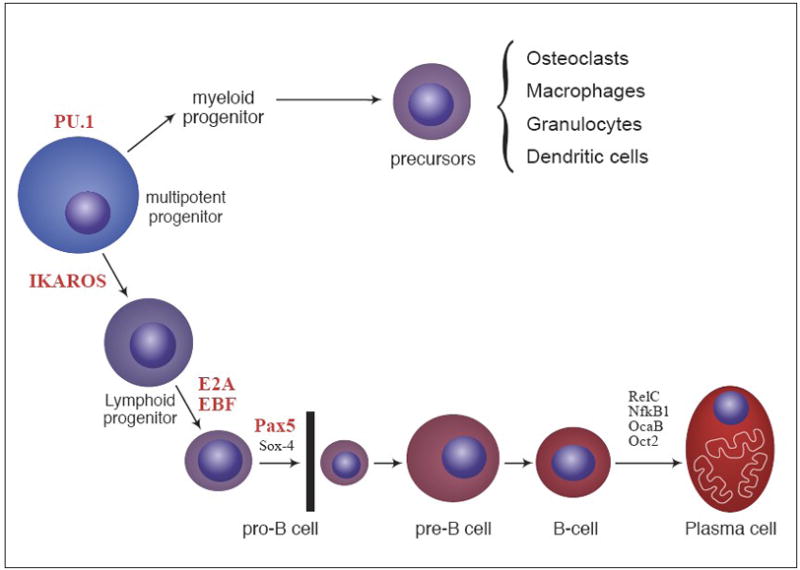
Transcriptional regulation of B cell differentiation. B cell differentiation is regulated, in part, by the expression of a series of transcription factors that function in a temporal manner. These transcription factors include; PU.1, Ikaros, E2A, Ebf1 and Pax5. Loss of these specific factors precludes the cells from continued maturation, and results in a developmental block of cells at the latest stage of differentiation prior to the arrest. In addition to the absence of B cells, mice deficient in PU.1, Ebf1 and Pax5 have profound changes to their skeletons. No data is available on the bone phenotype in mice deficient in Ikaros or E2A.
Ikaros is a member of a Kruppel-like zinc finger family of transcription factors that also includes Helios and Aiolos [39, 40]. The Ikaros gene (Ikzf1) is widely expressed in hematopoietic lineage cells including self-renewing HSCs and MPPs [41]. Mice deficient in Ikzf1 have substantially less HSC activity and their MPPs are inhibited from differentiating into the CLP. This results in a complete failure of the B cell lineage to develop [42, 43]. Ikzf1−/− mice have altered T cell development in the thymus and functional changes in peripheral T cells [44]. Ikaros also regulates cytokine gene expression [44]. Ikaros deficient mice have increased myelopoiesis with increased numbers of Mac-1+ cells in the BM and spleen [43]. To examine hematopoietic progenitor differentiation, BM derived MPPs (lin−Sca-1hic-Kithi) from Ikzf1−/− or WT bone marrow were transfected with a retrovirus encoding EBF-GFP or control virus. Cells were grown on a stromal cell monolayer in the presence of B cell promoting cytokines Flt3 ligand and IL-7. The GFP+ cells were sorted and grown on a fresh stromal cell monolayer in the presence of the cytokines and monitored for differentiation. WT MPPs transduced with control virus developed into CD19+ B cell precursors as expected [45]. Expression of EBF in these cells increased the development of mature B cell progeny. Importantly, Ikzf1−/− MPPs transduced with control virus differentiated not into B cells, but rather gave rise to CD11b+/Mac-1 myeloid progenitors [45]. Transduction of these cells with EBF blocked myelopoiesis and after 2–3 weeks of culture CD19+ B cell precursors developed. These results suggest that Ikzf1 deficiency leads to increased myelopoiesis in vitro and is consistent with the in vivo data. Whether the increase in myelopoiesis reflects an increase in osteoclastogenesis is unknown. At present no data is available on the status of the skeleton of Ikzf1−/− mice.
E2A is a member of the E-protein family of basic helix-loop-helix proteins, which bind E-box elements to regulate transcription. The E2A gene encodes two E proteins, E12 and E47, which are produced by differential splicing [46]. E2A proteins are expressed in HSCs and in different subsets of hematopoietic progenitors. As an example, BM from E2A−/− mice had reduced numbers of long-term HSCs, granulocyte-macrophage progenitors and severe depletion of erythroid progenitors [47]. Although these data suggest E2A−/− mice may have a bone phenotype they remain unexamined. Loss of E2A results in arrest of B cell development at the earliest appearance of B cell lineage genes and before rearrangement of the IgH locus [48]. It appears that E2A and Ebf1 work together to specify progenitors to enter B cell differentiation. To investigate the role E2A plays in myeloid and B cell differentiation MPPs (Lin−c-KithiSca-1hiCD27+) were isolated from BM and transduced with GFP-expressing retroviral constructs encoding E12, E47 or control and cultured in B cell supporting conditions (grown on stromal cells in the presence of SCF, Flt3L, IL-7) [49]. MPPs transduced with control virus produced mainly CD11b/Mac-1+ myeloid cells. MPPs transduced with either E12 or E47 were unable to induce B cell differentiation suggesting that E2A alone is not sufficient to specificy B cell differentiation. In contrast, transduction of MPPs with Ebf1 induced B cell differentiation (see below). These data suggest that E2A alone is not able to shift cell fate allocation from myeloid to B lymphoid.
Ebf1 is the founding member of small multigene family of helix-loop-helix proteins that are evolutionarily conserved with defined roles in cellular differentiation and function. Ebf1 is expressed early in the B cell lineage (fetal liver), in adipocytes, and we have shown it is expressed in osteoblast lineage cells including mesenchymal stem cells [13, 50, 51]. We have reported that all bone formation parameters (osteoblast number, osteoid thickness, bone volume and bone formation rate) are significantly increased in Ebf1−/− mice as compared to their age-matched littermate controls [50]. OC numbers increase in vivo as the mice age. Thus, Ebf1−/− mice have a bone phenotype strikingly different than PU.1−/− mice. B cell development is blocked at its earliest stage in Ebf1−/− mice [50]. To investigate the role Ebf1 plays in myeloid and B cell differentiation MPPs from Ebf1−/− fetal livers were isolated as described for PU.1−/− MPPs. Cells grown in B cell supporting conditions (SCF, Flt3L, IL-7) express a lymphoid phenotype (IL-7R+CD45R/B220+CD19−) [49]. Cells grown in myeloid supporting conditions (SCF, Flt3L, GM-CSF and M-CSF) developed macrophage-like cell morphology and were CD11b/Mac-1+ CD115/ c-fms+. No data is available on whether Ebf1−/− MPPs are altered in their ability to differentiate into OC precursors or functional OCs. To test whether Ebf1 could regulate myeloid lineage development, Ebf1−/− MPPs were transfected with a control GFP retroviral vector or an Ebf1-GFP retroviral vector and the cells cultured in myeloid supporting conditions. Cells transduced with GFP developed CD11b/Mac-1+ cells. In contrast, cells transduced with Ebf1-GFP caused an 85% reduction in CD11b/Mac-1+ cells. These data indicate that Ebf1 negatively regulates myeloid development and suggests that Ebf1 deficient mice should have markedly increased OCs. However, the long bones of young (4 week-old) Ebf1−/− mice actually had a reduced number of OCs although this reduction was lost as the mice age. In addition, there was no difference in in vitro osteoclast differentiation from BM macrophages from Ebf1−/− mice compared to controls. This discrepancy suggests additional regulation of OC differentiation in the Ebf1−/− mice in vivo.
To determine the ability of Ebf1 to reconstitute multiple lineages competitive adoptive transfer experiments were performed. Ebf1−/− cells (CD45.2+) that were maintained in B cell promoting conditions were injected, along with WT BM cells (CD45.1+), into lethally irradiated CD45.1+ mice. At 5 weeks the BM and thymus of the recipient mice were examined for lineage reconstitution. Cells derived from Ebf1−/− mice were recognized on the basis of CD45.2 expression by flow cytometry. Ebf1−/− progenitor cells gave rise to Mac-1+ macrophages and CD11c+ dendritic cells in the BM and CD4+CD8+ T cells were detected in the thymus. These data indicate that Ebf1−/− progenitors, even when grown in B cell promoting conditions, are capable of developing myeloid and lymphoid lineage cells [49]. The ability of these cells to differentiate into OCs in vivo remains to be determined.
Pax5 is a member of a multigene family that encodes the paired box (Pax) transcription factors. The Pax5 gene codes for the transcription factor B cell lineage specific activation factor (BSAP) [14]. Pax5 is expressed in the fetal liver where it correlates with the onset of B lymphopoiesis. Within the hematopoietic system, BSAP is expressed exclusively in the B lymphocyte lineage extending from pro-B cells (CD45R/B220+) to mature B cells, but not in terminally differentiated plasma cells [14, 52].
Loss of Pax5 results in an unanticipated massive decrease in trabecular bone in both the tibia and femur of 15-day-old mice [53]. Bone volume (tibia) was reduced by 67% and osteoid volume was reduced by 55%. This was found to be the result of increases in bone resorption and can be accounted for by the >100% increase in the number of OCs in Pax5−/− bone. These data not only indicate a marked increase in the number of OCs but also suggest that they are functional. The number of OBs in the mutant mice was reduced, although not significantly, to that of controls. This implies that the osteopenia was due, in large part, to the increase in OCs. Thus, Pax5−/− mice have a bone phenotype distinct from PU.1 and Ebf1 deficient mice.
Culture of spleen cells from Pax5−/− mice for 10 days results in formation of a lawn of adherent, elongated cells that morphologically appear similar to spleen macrophages [50]. These cells can be recovered from culture, passaged in vitro and developed into cell lines (SCL). The SCLs are 97% CD11b+/Mac-1, 96% CD16/32+/FcγR), and >50% CD115+ (c-fms) and do not express CD19, CD45R/B220, CD117/c-Kit, Ly6A/E/Sca-1, NK1.1 and TCRα/β as determined by flow cytometry. These results indicate that the cells are in the myeloid lineage and are similar to macrophages. Importantly, these cell lines can be passaged in vitro for more than 10 generations in the absence of any added growth factors. WT spleen cells cultured in a similar manner die by the first passage. Culture of SCL with WT BM or spleen cells separated by a cell impermeable membrane (transwells) resulted in both BM and spleen cell proliferation (Horowitz unpublished). These data suggested that a soluble factor secreted by the SCL was responsible for the BM cell proliferation. To address this possibility we cultured WT BM cells with conditioned medium (CM) from the Pax5−/− SCL. SCL CM induced proliferation of the BM cells as measured by 3H-thymidine incorporation. Subsequent treatment of the SCL CM treated cells with RANKL and M-CSF, M-CSF alone or GM-CSF plus IL-4 resulted in the formation of a large number of functional OCs, macrophages and dendritic cells respectively. These data suggest that the Pax5−/− SCL CM induces the expansion of a subset of myeloid progenitors in WT cells. Although more restricted in its developmental plasticity, a parallel can be draw between the SCL and the pro-B cells identified in Pax5−/− BM, which can differentiate into most hematopoietic cell lineages (with the exception of megakaryocytes and red blood cells) [54]. However, the pro-B cells from Pax5−/− BM require growth factor supplementation to proliferate in vitro making the SCL unique in its ability to grow in vitro without added growth factors.
Although OC and B cells arise from different progenitors there are data to support the hypothesis that B cells and macrophages can differentiate from a common progenitor downstream from the MPP. A rare population (0.5%) of B cell progenitors was isolated from adult mouse BM cells and was CD45R/B220−CD19+. When cultured in myeloid stimulating conditions these cells gave rise to macrophages indicating the bi-potential nature of these cells [55]. The loss of Pax5 causes the expression of unwanted genes, including c-fms, the M-CSF receptor that supports macrophage and OC differentiation. Therefore, loss of Pax5 could support the bi-potential progenitor population.
Is the loss of B cells the cause of the bone phenotype?
It is possible that the loss of B cells could be responsible for the bone phenotypes that are seen in Ebf1 and Pax5 deficient mice. However, this seems unlikely because mice lacking B and T cells due to the loss of Rag-1 (C57BL/6-Rag1−/−) one of the two VDJ recombination enzymes have no detectable bone phenotype by histology [53]. Rag genes control B and T cell receptor expression, therefore their loss results in B and T cell deficient mice. Loss of the Rag-1 genes arrests B cell development at approximately the same stage of development as loss of Ebf1 and Pax5. These reports are supported by the data showing that non-obese diabetic-rag−/− (NOD-rag−/−) and NOD-scid IL-2γ−/− mice, which lack B, T and NK cells also do not have a significant bone phenotype compared to age and sex matched NOD control mice as measured by micro-CT (M.Horowitz unpublished). The NOD mouse is a model for type 1 diabetes; however, the bones from the NOD mice were collected well before the onset of diabetes. These data are consistent with the report showing that following ovariectomy T cell deficient mice (nude, Rag2−/−, T cell receptor α−/−) lose trabecular bone mass equivalently to WT controls [56].
It has been reported that μMT heavy chain-deficient mice, which lack mature B cells, are osteopenic due to increased bone resorption, which is caused by a decrease of B cell secreted osteoprotegerin (OPG) [57]. Although no histology is presented, the bone phenotype of these mice appears mild. We have reported that the μMT deficient mice do not have an obvious bone phenotype [53]. In contrast, mice made deficient in OPG have a striking bone phenotype with increased cortical thickness and a “worm-eaten” appearance due to massive osteoclast activity in the cortical bone [58]. The μMT−/− mice reported above have decreased cortical bone. The bone phenotype in the Ebf1 and Pax5 deficient mice is much more severe than that of μMT deficient mice. Loss of Ebf1 results in the loss of B cells as early as B cells can be identified, which could account for as much as 30% of BM cells and these mice have no apparent osteoclast phenotype. Although Pax5 deficient mice do have a demonstrable osteoclast phenotype, it is not caused by the B cell deficiency (Horowitz unpublished). Rag-1 deficiency, which directly affects T and B cell differentiation, results in a failure to develop mature B and T cells and these mice do not have an obvious bone phenotype.
The RANK-RANKL Pathway and B cell differentiation
It is well accepted that the interaction between RANKL and its cognate receptor RANK is required for the differentiation of osteoclasts from precursors. However, what is often overlooked is the role the genes that encode these proteins and their downstream effector genes play in regulating B cell development. RANKL deficient mice have severe osteopetrosis due to the inability of OBs to support OC differentiation. These mice are also one of the few mutants that lack all lymph nodes. The number of splenic IgM+sIgD+ or CD45R/B220+sIgM+ B cells was significantly reduced in RANKL−/− mice as compared to controls [59]. To examine B cell differentiation chimeric mice were made by injecting fetal liver cells from RANKL−/− or WT mice into sub-lethally irradiated Rag−/− mice [59]. The BM from Rag−/− mice reconstituted with RANKL−/− cells had normal numbers of CD45R/B220+CD43+ and CD45R/B220+CD25− pro-B cells but significantly reduced numbers of CD45R/B220+CD43−, CD45R/B220+CD25+ and CD45R/B220+sIgM+ B cells, indicating a conspicuous block in the progression from pro-B cells to pre-B cells in the chimeric mice. Therefore, even in the RANKL sufficient environment of the Rag−/− mice, the RANKL−/− cell had a defect in B lymphopoiesis [59].
The question then arises; do other members of the RANK-RANKL pathway affect B cell differentiation or activation? RANK-deficient mice are severely osteopetrotic due to a failure of osteoclast precursors to differentiate to mature osteoclasts. The number of cells in the BM of RANK−/− mice is markedly reduced with few hematopoietic colonies due to the osteopetrosis [60]. As hematopoiesis relocates, the spleens of RANK−/− mice are approximately twice the size of controls but have a greater than 50% reduction inCD45R/ B220+ B cells [60]. Mature, CD45R/B220+IgM+ splenic B cells were also reduced by 50% as compared to controls. Like the RANKL-deficient mice RANK-deficient mice lack all lymph nodes [60].
Following RANKL-RANK interaction intracellular signal transduction is mediated, in part, by TNF receptor-associated factor 6 (TRAF6). TRAF6−/− mice are osteopetrotic [61]. Although TRAF6−/− mice have similar numbers of OCs compared to controls, they fail to resorb bone because they do not develop an attachment zone or ruffled border. Interestingly, splenic B cells from TRAF6−/− mice fail to proliferate in response to anti-CD40 or lipopolysaccharide stimulation suggesting a role for TRAF6 in B cell proliferation to specific signals.
OPG is a naturally circulating decoy protein receptor that binds RANKL, reducing RANKL’s availability to bind RANK, thus regulating OC differentiation. OPG-deficient mice develop severe osteoporosis with associated fractures due to increased numbers of OCs. OPG is a CD40-regulated gene in B cells and dendritic cells. To analyze the effects of OPG deficiency on B cell development, pro-B cells (CD45R/B220+CD43+) were sorted by FACS from BM and cultured with IL-7. Pro-B cells from OPG−/− mice proliferated 2 fold more after IL-7 stimulation than did wild-type cells [62]. FACS analysis revealed that CD45R/B220+CD43+IgM− or CD45R/B220+CD25−IgM− pro-B cells were increased in the BM of OPG−/− mice as compared to WT controls. In addition, the number of CD45R/B220+CD19+ B cells in the spleen and lymph nodes was greater in OPG−/− mice than in controls.
These data show that the loss of OPG increases pro-and mature B cells, while the loss of RANKL or RANK decreases these populations of cells. In the RANKL−/− mice B cell differentiation is arrested between the pro- and pre-B cell stage of differentiation. The stage of differentiation arrest in the RANK−/− mice is unknown.
Estrogen and B cells
Partially purified populations of B-lymphocytes from murine bone marrow are reported to form osteoclasts in vitro when they were treated with M-CSF and RANKL [63–66]. In addition, production of osteoclastogenic activity in these populations was increased after ovariectomy. However, when isolated to very high purity, purified B cells failed to differentiate into osteoclasts in vitro [67]. These results demonstrate that the osteoclastogenic potential of B cell populations in murine bone marrow is dependent on contaminating cells. Most recently, we and others have found that trabecular bone loss after ovariectomy was similar in wild type mice and mice, which were deficient in the majority of there mature B cells [26, 56, 68]. Estrogens are potent regulators of B-lymphopoiesis at a very early stage [69]. In mice the absolute number of B cells (defined by expression of the antigen CD45R/B200) in bone marrow roughly doubles after ovariectomy [70]. Because B cells at multiple stages of differentiation can express RANKL, it is possible that changes in early B cells with estrogen withdrawal may affect osteoclastogenesis and subsequent bone loss [71]. However, this response does not appear involved in the bone loss that occurs after estrogen withdrawal in humans because increases in B cells were not found in postmenopausal women who had their estrogen replacement stopped [72].
Activated B cells
Activation of B cells is involved in the development of inflammatory arthritis as well as periodontal disease [73, 74]. Activated B cells can produce RANKL as well as other cytokines that are involved in bone resorption and bone formation [73, 75] (Fig 3). In addition, a subpopulation of memory B cells also expresses RANKL [76]. A variety of B cell-related malignancies produce factors that regulate bone cells. Multiple myeloma is a malignancy of plasma cells, which derive from B cells. It often causes enhanced bone resorption and decreased bone formation [77]. Derangements of the RANK/RANKL/OPG system are frequent in this condition and may be due to production of factors by the tumor that stimulates RANKL production in the bone microenvironment or because of direct production of RANKL by the myeloma cells [78, 79]. In addition, a variety of additional factors including MIP-1α and DKK1 are produced by myeloma cells and directly affect bone cell function [77]. B cell lymphomas can also produce RANKL and through this mechanism cause bone loss and hypercalcemia [80].
Fig. 3.
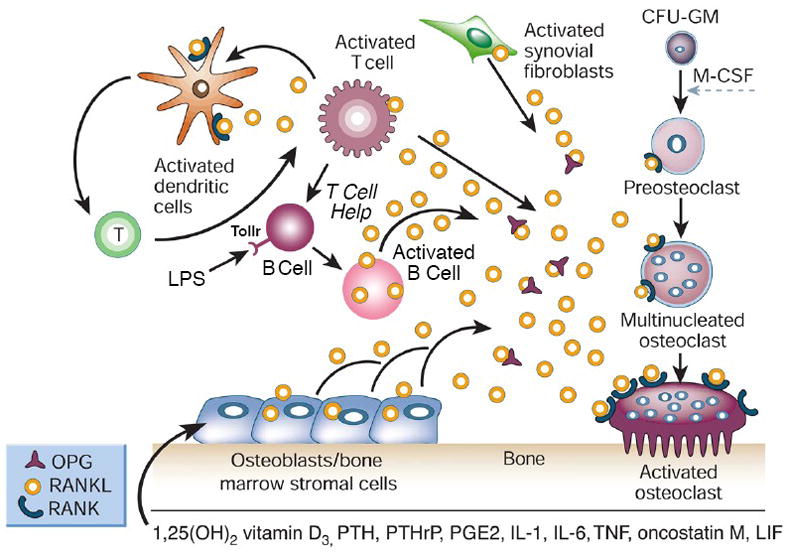
Activated B cells induce osteoclast differentiation. B cells activated by the adaptive immune system (antigen specific) or through the innate immune system (LPS-Toll receptor) results in B cells that secrete or express on their cell surface molecules like RANKL that induce osteoclastogenesis. (Adapted from Boyle WJ, Simonet WS, Lacey DL. Nature 2003; 423:337–342).
Conclusions
Cells in the osteoblast lineage support HSC and B cell differentiation and growth in endosteal bone niches. Support of B cell differentiation requires IL-7, secreted at least in part by osteoblast lineage cells.
The role of IL-7 in regulating bone homeostasis remains controversial.
A network of TFs is required for early B cell differentiation and function in a hierarchical order. Deletion of these transcription factors results in mice with striking bone phenotypes. Importantly, the bone phenotypes are markedly different with PU.1 and Pax5 regulating OC development while Ebf1 regulates OB and adipocyte differentiation. One of the major functions of Ebf1 and Pax5 is the repression of unwanted genes, which helps maintain B lineage fidelity and may be the mechanism underlying the bone phenotypes. This suggests that analysis of Ikaros and E2A deficient animals will also provide important insights into the regulation of bone by the TFs.
In BM a series of interactions occur, some of which are mediated by B cell TFs that regulate the balance between B cell and macrophage differentiation from progenitors. Loss of these key regulatory TFs (e.g. Ikaros, Ebf1 and Pax5) results in lineage allocation being shifted away from B cell differentiation and towards myeloid lineage differentiation.
Loss of members of the RANK, RANKL, OPG pathway not only affects OC differentiation but also B cell development.
B and T cells do not have a major regulatory role in early bone development and normal bone remodeling. However, activated B and T cells can be potent regulators of bone resorption.
Acknowledgments
Writing of this review and experimental work was enabled by support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases/National Institutes of Health grants RO1AR047342, RO1AR049190, RO1AR052690 to MCH; the Yale Core Center for Musculoskeletal Disorders P30AR046032; RO1AR048714 and RO1AR052690 to JAL, and the Department of Orthopaedics and Rehabilitation, Yale University School of Medicine, New Haven, CT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 2.Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aquila HL. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004;103:3258–64. doi: 10.1182/blood-2003-11-4011. [DOI] [PubMed] [Google Scholar]
- 3.Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 2007;109:3706–3712. doi: 10.1182/blood-2006-08-041384. [DOI] [PubMed] [Google Scholar]
- 4.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Sherer DC, Beilhack GF, et al. Biology of hematopoietic stem cells and progenitors: Implications for clinical application. Ann Rev Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 5.Medina KL, Pongubala JMR, Reddy KL, Lancki DW, DeKoter R, Kieslinger M, et al. Assembling a gene regulatory network for specification of the B cell fate. Dev Cell. 2004;7:607–617. doi: 10.1016/j.devcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Ghia P, ten Boekel E, Rolink AG, Melchers F. B-cell development: a comparison between mouse and man. Immunol Today. 1998;19:480–485. doi: 10.1016/s0167-5699(98)01330-9. [DOI] [PubMed] [Google Scholar]
- 7.Hesslein DG, Schatz DG. Factors and forces controlling V(D)J recombination. Adv Immunol. 2001;78:169–232. doi: 10.1016/s0065-2776(01)78004-2. [DOI] [PubMed] [Google Scholar]
- 8.Hardy RR, Hayakawa K. B cell development pathways. Ann Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 9.Li YS, Wasserman R, Hayakawa K, Hardy RR. Identification of the earliest B lineage stage in mouse bone marrow. Immunity. 1996;5:527–535. doi: 10.1016/s1074-7613(00)80268-x. [DOI] [PubMed] [Google Scholar]
- 10.Rolink A, Haasner D, Melchers F, Anderson J. The surrogate light chain in mouse B-cell development. Int Rev Immuno. 1996;13:341–356. doi: 10.3109/08830189609061757. [DOI] [PubMed] [Google Scholar]
- 11.Nemazee D. Receptor editing in B cells. Adv Immunol. 2000;74:89–126. doi: 10.1016/s0065-2776(08)60909-8. [DOI] [PubMed] [Google Scholar]
- 12.Bain G, Maandag EC, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BD, et al. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 13.Lin H, Grosschedl R. Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature. 1995;376:263–267. doi: 10.1038/376263a0. [DOI] [PubMed] [Google Scholar]
- 14.Adams B, Dorfler P, Aguzzi A, Kozmik Z, Urbanek P, Maurer-Fogy I, et al. Pax5 encodes the transcription factor BSAP and is expressed in B-lymphocytes, the developing CNS, and adult testis. Genes Dev. 1992;6:1589–1607. doi: 10.1101/gad.6.9.1589. [DOI] [PubMed] [Google Scholar]
- 15.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-Lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 16.Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gsα-dependent signaling pathways. Proc Natl Acad Sci U S A. 2008;105:16976–16981. doi: 10.1073/pnas.0802898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Namen AE, Lupton S, Hjerrild K, Wignall J, Mochizuki DY, Schmierer A, et al. Stimulation of B-cell progenitors by cloned murine interleukin-7. Nature. 1988;333:571–573. doi: 10.1038/333571a0. [DOI] [PubMed] [Google Scholar]
- 18.Mazzucchelli RM, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. 2007;7:144–154. doi: 10.1038/nri2023. [DOI] [PubMed] [Google Scholar]
- 19.Miyaura C, Onoe Y, Inada M, Maki K, Ikuta K, Ito M, et al. Increased B-lymphopoiesis by interleukin 7 induces bone loss in mice with intact ovarian function: similarity to estrogen deficiency. Proc Natl Acad Sci U S A. 1997;94:9360–9365. doi: 10.1073/pnas.94.17.9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110:1643–1650. doi: 10.1172/JCI15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R. Interleukin-7 stimulates osteoclast formation by up-regulating the T- cell production of soluble osteoclastogenic cytokines. Blood. 2000;96:1873–1878. [PubMed] [Google Scholar]
- 22.Salopek D, Grcevic D, Katavic V, Kovacic N, Lukic IK, Marusic A. Increased bone resorption and osteopenia are a part of the lymphoproliferative phenotype of mice with systemic over-expression of interleukin-7 gene driven by MHC class II promoter. Immunol Lett. 2008;121:134–139. doi: 10.1016/j.imlet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Toraldo G, Roggia C, Qian WP, Pacifici R, Weitzmann MN. IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor kappa B ligand and tumor necrosis factor alpha from T cells. Proc Natl Acad Sci U S A. 2003;100:125–130. doi: 10.1073/pnas.0136772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gendron S, Boisvert M, Chetoui N, Aoudjit F. Alpha1beta1 integrin and interleukin-7 receptor up-regulate the expression of RANKL in human T cells and enhance their osteoclastogenic function. Immunology. 2008;125:359–369. doi: 10.1111/j.1365-2567.2008.02858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SK, Kalinowski JF, Jastrzebski SL, Puddington L, Lorenzo JA. Interleukin-7 is a direct inhibitor of in vitro osteoclastogenesis. Endocrinology. 2003;144:3524–3531. doi: 10.1210/en.2002-221057. [DOI] [PubMed] [Google Scholar]
- 26.Lee SK, Kalinowski JF, Jacquin C, Adams DJ, Gronowicz G, Lorenzo JA. Interleukin-7 influences osteoclast function in vivo but is not a critical factor in ovariectomy-induced bone loss. J Bone Miner Res. 2006;21:695–702. doi: 10.1359/jbmr.060117. [DOI] [PubMed] [Google Scholar]
- 27.Ryan MR, Shepherd R, Leavey JK, Gao Y, Grassi F, Schnell FJ, et al. An IL-7-dependent rebound in thymic T cell output contributes to the bone loss induced by estrogen deficiency. Proc Natl Acad Sci U S A. 2005;102:16735–16740. doi: 10.1073/pnas.0505168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato T, Watanabe K, Masuhara M, Hada N, Hakeda Y. Production of IL-7 is increased in ovariectomized mice, but not RANKL mRNA expression by osteoblasts/stromal cells in bone, and IL-7 enhances generation of osteoclast precursors in vitro. J Bone Miner Metab. 2007;25:19–27. doi: 10.1007/s00774-006-0723-y. [DOI] [PubMed] [Google Scholar]
- 29.Lee S, Kalinowski JF, Adams DJ, Aguila HL, Lorenzo JA. Osteoblast specific overexpression of human interleukin-7 increases femoral trabecular bone mass in female mice and inhibits in vitro osteoclastogenesis. J Bone Miner Res. 2004;19:S410. doi: 10.1002/jbmr.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S, Kalinowski JF, Adams DJ, Aguila HL, Lorenzo JA. Osteoblast specific overexpression of human interlukin-7 rescues the bone phenotype of interleukin-7 deficient female mice. J Bone Miner Res. 2005;20:S48. doi: 10.1002/jbmr.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 32.Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, et al. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386:81–84. doi: 10.1038/386081a0. [DOI] [PubMed] [Google Scholar]
- 33.Scott EW, Fisher RC, Olson MC, Kehrli EW, Simon MC, Singh H. PU.1 functions in a cell-autonomous manner to control the differentiation of multipotential lymphoid-myeloid progenitors. Immunity. 1997;6:427–447. doi: 10.1016/s1074-7613(00)80287-3. [DOI] [PubMed] [Google Scholar]
- 34.DeKoter RP, Walsh JC, Singh H. Pu.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J. 1998;17:4456–4468. doi: 10.1093/emboj/17.15.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeKoter RP, Singh H. Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science. 2000;288:1439–1441. doi: 10.1126/science.288.5470.1439. [DOI] [PubMed] [Google Scholar]
- 36.DeKoter RP, Lee H-J, Singh H. Pu.1 Regulates expression of the interleukin-7 receptor in lymphoid progenitors. Immunity. 2002;16:297–309. doi: 10.1016/s1074-7613(02)00269-8. [DOI] [PubMed] [Google Scholar]
- 37.Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA. The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell. 1990;61:113–124. doi: 10.1016/0092-8674(90)90219-5. [DOI] [PubMed] [Google Scholar]
- 38.Ross IL, Dunn Tl, Yue X, Roy S, Barnett CJ, Hume Da. Comparison of the expression and function of the transcription factor PU.1 (Spi-1 proto-oncogene) between murine macrophages and B lymphocytes. Oncogene. 1994;9:121–132. [PubMed] [Google Scholar]
- 39.Georgopoulos K. Haematopoietic cell-fate decisions, chromatin regulation and Ikaros. Nat Rev Immunol. 2002;2:162–174. doi: 10.1038/nri747. [DOI] [PubMed] [Google Scholar]
- 40.Cobb BS, Smale ST. Ikaros-family proteins: in search of molecular functions during lymphocytes development. Curr Top Microbiol Immunol. 2005;290:29–47. doi: 10.1007/3-540-26363-2_3. [DOI] [PubMed] [Google Scholar]
- 41.Kelley CM, Ikeda T, Koipally J, Avitahl N, Wu L, Georgopoulos K, et al. Helios, a novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Curr Biol. 1998;8:508–515. doi: 10.1016/s0960-9822(98)70202-7. [DOI] [PubMed] [Google Scholar]
- 42.Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79:143–156. doi: 10.1016/0092-8674(94)90407-3. [DOI] [PubMed] [Google Scholar]
- 43.Wang JH, Nichogiannopoulou A, Wu L, Sun L, Sharpe AH, Bigby M, et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity. 1996;5:537–549. doi: 10.1016/s1074-7613(00)80269-1. [DOI] [PubMed] [Google Scholar]
- 44.Georgopoulos K, Winady S, Avitahl N. The role of the Ikaros gene in lymphocyte development and homeostasis. Annu Rev Immunol. 1997;15:155–176. doi: 10.1146/annurev.immunol.15.1.155. [DOI] [PubMed] [Google Scholar]
- 45.Reynaud D, Demarco IA, Reddy KL, Schjerven H, Bertolino E, Chen Z, et al. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol. 2008;9:927–936. doi: 10.1038/ni.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 47.Semerad CL, Mercer EM, Inlay MA, Weissman IL, Murre C. E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc Natl Acad Sci U S A. 2009;106:1930–1935. doi: 10.1073/pnas.0808866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhuang Y, Jackson A, Pan L, Shen K, Dai M. Regulation of E2A gene expression in B-lymphocyte development. Mol Immunol. 2004;40:1165–1177. doi: 10.1016/j.molimm.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 49.Pongubala JM, Northrup DL, Lancki DW, Medina KL, Treiber T, Bertolino E, et al. Transcription factor EBF restricts alternative lineage options and promotes B cell fate commitment independently of Pax5. Nat Immunol. 2008;9:203–215. doi: 10.1038/ni1555. [DOI] [PubMed] [Google Scholar]
- 50.Hesslein DGT, Fretz JA, Xi Y, Nelson T, Zhou S, Lorenzo JA, et al. Ebf1 dependent control of the osteoblast and adipocyte lineages. Bone. 2009;44:537–546. doi: 10.1016/j.bone.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akerblad P, Lind U, Liberg D, Bamberg K, Sigvardsson M. Early B-cell factor (O/E-1) is a promoter of adipogenesis and involved in control of genes important for terminal adipocyte differentiation. Mol Cell Biol. 2002;22:8015–8025. doi: 10.1128/MCB.22.22.8015-8025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urbanek P, Wang Z, Fetka I, Wagner E, Busslinger M. Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell. 1994;79:901–912. doi: 10.1016/0092-8674(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 53.Horowitz MC, Xi Y, Pflugh DL, Hesslein DGT, Schatz DG, Lorenzo JA, et al. Pax5 deficient mice exhibit early onset osteoporosis with increased osteoclast progenitors. J Immunol. 2004;173:6583–6591. doi: 10.4049/jimmunol.173.11.6583. [DOI] [PubMed] [Google Scholar]
- 54.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-Lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 55.Montecino-Rodriguez E, Leathers H, Dorshkind K. Bipotential B-macrophage progenitors are present in adult bone marrow. Nat Immunol. 2001;2:83–88. doi: 10.1038/83210. [DOI] [PubMed] [Google Scholar]
- 56.Lee S-K, Kadono Y, Okada F, Jacquin C, Koczon-Jaremko B, Gronowicz G, et al. T lymphocyte-deficient mice lose trabecular bone mass with ovariectomy. J Bone Miner Res. 2006;21:1704–1712. doi: 10.1359/jbmr.060726. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian W-P, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007;109:3839–3848. doi: 10.1182/blood-2006-07-037994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kong Y-Y, Yoshida H, Sarosi I, Tan H-L, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 60.Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes & Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lomaga MA, Yeh W-C, Sarosi I, Duncan GS, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes & Dev. 1999;15:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yun TJ, Tallquist MD, Aicher A, Rafferty KL, Marshall AJ, Moon JJ, et al. Osteoprotegerin, a crucial regulator of bone metabolism, also regulates B cell development and function. J Immunol. 2001;166:1482–1491. doi: 10.4049/jimmunol.166.3.1482. [DOI] [PubMed] [Google Scholar]
- 63.Blin-Wakkach C, Wakkach A, Rochet N, Carle GF. Characterization of a novel bipotent hematopoietic progenitor population in normal and osteopetrotic mice. J Bone Miner Res. 2004;19:1137–1143. doi: 10.1359/JBMR.040318. [DOI] [PubMed] [Google Scholar]
- 64.Kanematsu M, Sato T, Takai H, Watanabe K, Ikeda K, Yamada Y. Prostaglandin E 2 induces expression of receptor activator of nuclear factor-kappaB ligand/osteoprotegrin ligand on pre-B cells: Implications for accelerated osteoclastogenesis in estrogen deficiency. J Bone Miner Res. 2000;15:1321–1329. doi: 10.1359/jbmr.2000.15.7.1321. [DOI] [PubMed] [Google Scholar]
- 65.Katavic V, Grcevic D, Lee SK, Kalinowski J, Jastrzebski S, Dougall W, et al. The surface antigen CD45R identifies a population of estrogen-regulated murine marrow cells that contain osteoclast precursors. Bone. 2003;32:581–590. doi: 10.1016/s8756-3282(03)00097-8. [DOI] [PubMed] [Google Scholar]
- 66.Sato T, Shibata T, Ikeda K, Watanabe K. Generation of bone-resorbing osteoclasts from B220+ cells: its role in accelerated osteoclastogenesis due to estrogen deficiency. J Bone Miner Res. 2001;16:2215–2221. doi: 10.1359/jbmr.2001.16.12.2215. [DOI] [PubMed] [Google Scholar]
- 67.Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res. 2006;21:67–77. doi: 10.1359/JBMR.051007. [DOI] [PubMed] [Google Scholar]
- 68.Li Y, Li A, Yang X, Weitzmann MN. Ovariectomy-induced bone loss occurs independently of B cells. J Cell Biochem. 2007;100:1370–1375. doi: 10.1002/jcb.21121. [DOI] [PubMed] [Google Scholar]
- 69.Kincade PW, Medina KL, Payne KJ, Rossi MI, Tudor KS, Yamashita Y, et al. Early B-lymphocyte precursors and their regulation by sex steroids. Immunol Rev. 2000;175:128–137. [PubMed] [Google Scholar]
- 70.Masuzawa T, Miyaura C, Onoe Y, Kusano K, Ohta H, Nozawa S, et al. Estrogen deficiency stimulates B lymphopoiesis in mouse bone marrow. J Clin Invest. 1994;94:1090–1097. doi: 10.1172/JCI117424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anderson DM, Maraskovsky E, Billingsley WL, Cougall WC, Tometsko ME, Roux ER, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 72.Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111:1221–1230. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schett G. Osteoimmunology in rheumatic diseases. Arthritis Res Ther. 2009;11:210. doi: 10.1186/ar2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, et al. B and T lymphocytes are the primary aources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006;169(3):987–998. doi: 10.2353/ajpath.2006.060180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hayer S, Polzer K, Brandl A, Zwerina T, Kireva T, Smolen JS, et al. B-cell infiltrates induce endosteal bone formation in inflammatory arthritis. J Bone Miner Res. 2008;23:1650–1660. doi: 10.1359/jbmr.080508. [DOI] [PubMed] [Google Scholar]
- 76.Ehrhardt GR, Hijikata A, Kitamura H, Ohara O, Wang JY, Cooper MD. Discriminating gene expression profiles of memory B cell subpopulations. J Exp Med. 2008;205:1807–1817. doi: 10.1084/jem.20072682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roodman GD. Pathogenesis of myeloma bone disease. J Cell Biochem. 2010;109:283–291. doi: 10.1002/jcb.22403. [DOI] [PubMed] [Google Scholar]
- 78.Pearse RN, Sordillo EM, Yaccoby S, Wong BR, Liau DF, Colman N, et al. Multiple myeloma disrupts the TRANCE/ osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci U S A. 2001;98:11581–11586. doi: 10.1073/pnas.201394498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sezer O, Heider U, Jakob C, Zavrski I, Eucker J, Possinger K, et al. Immunocytochemistry reveals RAANKL expression of myeloma cells. Blood. 2002;99:4646–4647. doi: 10.1182/blood-2002-01-0148. [DOI] [PubMed] [Google Scholar]
- 80.Shibata H, Abe M, Hiura K, Wilde J, Moriyama K, Sano T, et al. Malignant B-lymphoid cells with bone lesions express receptor activator of nuclear factor-kappaB ligand and vascular endothelial growth factor to enhance osteoclastogenesis. Clin Cancer Res. 2005;11:6109–6115. doi: 10.1158/1078-0432.CCR-05-0181. [DOI] [PubMed] [Google Scholar]