

Abstract
We have reported a highly cooperative interaction between leptin and thyrotropin releasing hormone (TRH) in the hindbrain to generate thermogenic responses. Identifying the locus in the hindbrain where leptin and TRH act synergistically to increase thermogenesis will be necessary before we can determine the mechanism(s) by which this interaction occurs. Here, we performed heat-induced epitope recovery techniques and in situ hybridization to determine if neurons or afferent fibers in the hindbrain possess both TRH type 1 receptor and long-form leptin receptor [TRHR1; LepRb, respectively]. LepRb receptors were highly expressed in the solitary nucleus [NST], dorsal motor nucleus of the vagus [DMN] and catecholaminergic neurons of the ventrolateral medulla [VLM]. All neurons that contained LepRb also contained TRHR1. Fibers in the NST and the raphe pallidus [RP] and obscurrus [RO] that possess LepRb receptors were phenotypically identified as glutamatergic type 2 fibers (vglut2). Fibers in the NST and RP that possess TRHR1 receptors were phenotypically identified as serotonergic [i.e., immunopositive for the serotonin transporter; SERT]. Co-localization of LepRb and TRHR1 was not observed on individual fibers in the hindbrain but these two fiber types co-mingle in these nuclei. These anatomical arrangements may provide a basis for the synergy between leptin and TRH to increase thermogenesis.
Keywords: thermoregulation, leptin, TRH, solitary nucleus
1. Introduction
Current models of thermocontrol suggest that hypothalamic-preoptic structures determine a temperature set point. This circuitry, in turn, commands changes in heat gain or loss through connections with the hindbrain, particularly the raphe nuclei and the ventrolateral medulla.
These hindbrain circuits control temperature via connections with sympathetic efferents that regulate brown adipose tissue [BAT] thermogenesis and peripheral blood flow. Activation of caudal raphe neurons by exposure to cold-stress can also increase gastric motility through direct connections with vago-vagal reflex circuits in the dorsal vagal complex. Neurons in the raphe nuclei that contain thyrotropin releasing hormone [TRH] have been implicated in hindbrain control of thermogenesis. TRH release by raphe projection neurons occurs in proportion to the degree of cold stress and TRH drives neurogenic heat production and retention.
CNS thermocontrol is subject to modulation by leptin. Leptin is released by adipocytes into the circulation where it provides the brain with a signal proportional to energy stored as fat. Leptin causes a sympathetically-mediated increase in energy consumption, especially as heat generated by BAT. However, during starvation [with its associated lack of leptin] thermogenesis and other energy-demanding activities are suppressed.
Recent studies show that the hindbrain itself can produce appropriate thermocontrol changes including those triggered by leptin. Our studies in overnight food-deprived animals suggested that leptin, in combination with TRH, significantly increased BAT thermogenesis. Specifically, while low doses of leptin, alone, in the fourth ventricle [4V] had no effect to activate BAT and the presence of TRH, alone, only evoked a small increase in BAT temperature, the combination of leptin plus TRH produced a dramatic increase in thermogenesis. This synergistic effect was preserved in the acutely decerebrated animal; thus, the relevant circuitry for this effect is contained within the hindbrain.
The location(s) of the leptin-TRH interaction in the hindbrain is not yet known. However, the nucleus of the solitary tract [NST] is a good candidate. TRH-ergic input to the NST is provided by medullary raphe neurons that are activated by cold stress sensitive pathways. The NST is also the recipient of vagal afferents that are sensitive to both temperature and endogenous pyrogens.
The most parsimonious explanation for this cooperative effect on thermogenesis would be the demonstration that these two receptors are co-localized on the same cells or fibers in the hindbrain. Therefore, we used both immunohistochemistry (IHC) and in situ hybridization techniques to determine the co-localization of LepRb and TRHR1 receptors on neurons and fibers in the hindbrain. IHC staining for the leptin transduction product phosphorylated STAT3 [signal transducer and activator of transcription 3; PSTAT3] was used to verify leptin activation of neurons in hindbrain structures that contain the LepRb; specifically those that would be activated via 4V route as was used in our physiological experiments on BAT thermogenesis.
2. Results
Localization of TRHR1-immunoreactivity [TRHR1-ir] in the hindbrain of the rat
IHC studies that employed heat-induced epitope retrieval [HIER] techniques [see Methods for details] revealed that the TRHR1 receptor is expressed widely on neuronal cell bodies throughout the NST, VLM, DMN and hypoglossal nuclei (Figures 1, 3, 4, and 6). This is most evident as seen in the extensive dual immunostaining for TRHR1 and neuronal nuclei [NeuN] in Figure 1. Due to the widespread localization of TRHR1 receptors on neuronal cell bodies within these nuclear areas, no attempt to discern the phenotypic identity of these neurons was made.
FIGURE 1. TRHR1 receptors in the dorsal medulla.
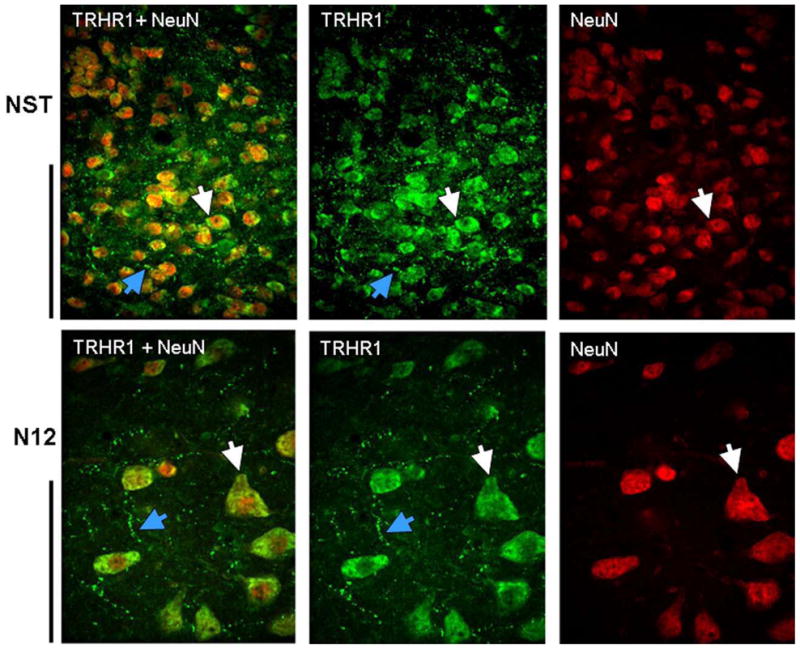
Example of double IHC staining for TRHR1 (green) and NeuN (red) in the NST (top row) and the hypoglossal nucleus (N12; bottom row) indicates that TRHR1-ir is superimposed on neurons (white arrows) and is also present on fibers and varicosities (blue arrows) in these areas. In both rows, NeuN IHC staining alone is seen in the far right; middle panel represents TRHR1 staining alone; and far left panels are merged images of the double stained sections. A similar distribution of TRHR1 receptors was seen in the DMN, VLM, and medullary raphe neurons. Scale bars: NST = 50microns; N12 = 100 microns.
Figure 3. Dual localization of LepRb and TRHR1 receptors on DMN and NST neurons.
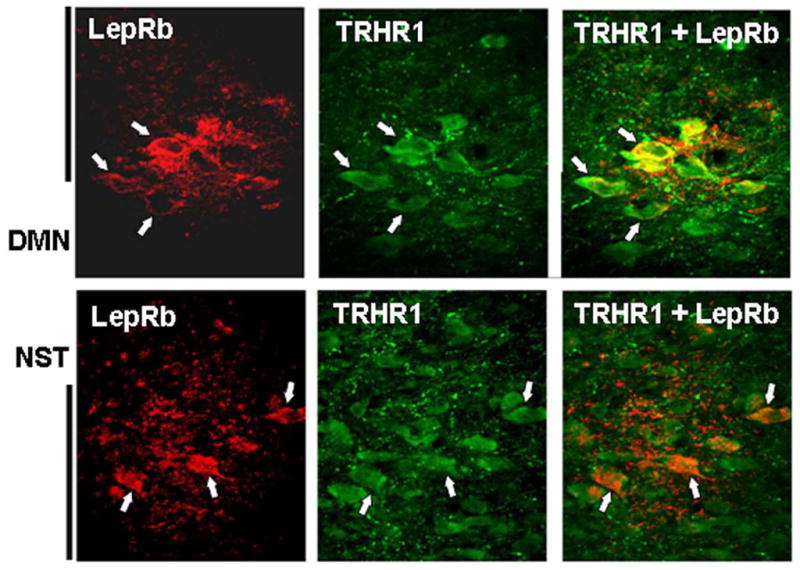
Top row: cluster of neurons (examples indicated at white arrows) in the lateral portion of the dorsal motor nucleus of the vagus [DMN] demonstrate immunoreactivity for both LepRb (red; left panel) and TRHR1 (green; middle panel); merged image seen in right panel. Note the background of LepRb-ir and TRHR1-ir terminals. Fibers and varicosities possessing either of these receptor types are closely apposed but not co-localized.
Bottom row: cluster of neurons in the medial NST (examples indicated at white arrows) show co-localization of LepRb and TRHR1 receptors. As seen in the DMN, LepRb+ and TRHR1+ fibers and varicosities in the NST are adjacent but not co-localized.
Scale bar: DMN = 100 microns; NST = 50 microns.
Figure 4. Distribution of LepRb+ fibers in hindbrain.
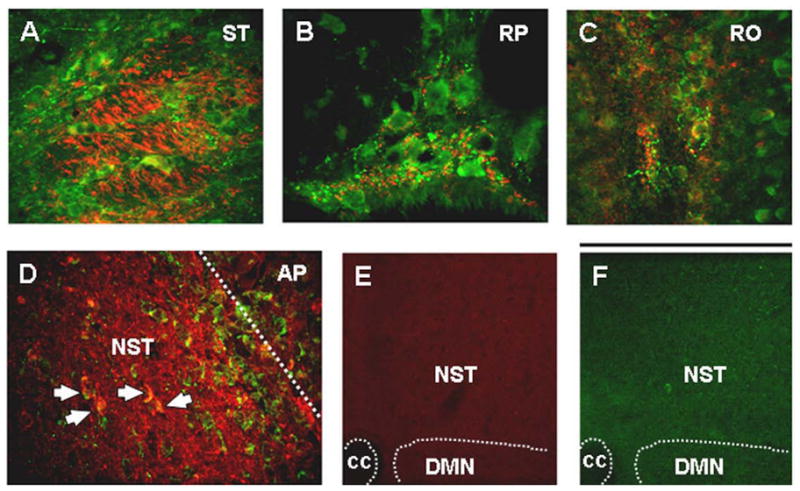
LepRb-ir (red) fibers and varicosities are seen among TRHR1-ir (green) cells and fibers. These red and green fibers are adjacent and co-mingle but do not show co-localization of receptors. This pattern is seen in (A) fascicles of the solitary tract (ST); (B) raphe pallidus (RP), and (C) raphe obscurrus (RO).
(D) Border between the medial solitary nucleus (NST) and the area postrema (AP; white dashed line) showing an abundance of LepRb-ir (red) fibers and neurons (white arrows for selected neurons) in the NST but not the AP.
(E) LepRb-ir staining is suppressed by pretreatment of tissue with LepRb epitope blocking peptide.
(F) TRHR1-ir staining is suppressed by treatment with excess TRHR1.
Scale bar A–D = 100 microns; E, F = 300 microns. cc= central canal
Figure 6. Distribution of LepRb+, TRHR1+ and tyrosine hydroxylase (TH) in the ventrolateral medulla (VLM).
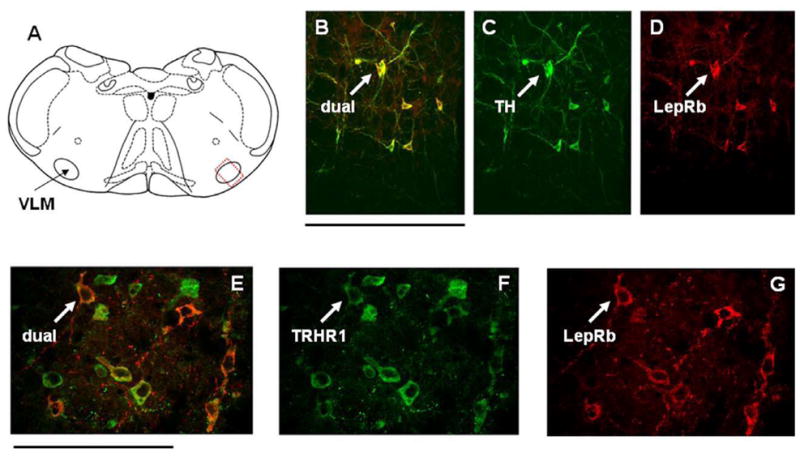
(A) Schematic diagram of the approximate location of VLM neurons with LepRb receptors [dotted rectangle].
(B–D) VLM neurons identified as TH+ appear to possess LepRb receptors.
(E–G) VLM neurons with LepRb receptors also possess TRHR1 receptors. Scale bar B–D = 200 microns; E–G = 100 microns.
TRHR1-ir fibers and varicosities are also prominent in the NST, DMN, raphe pallidus (RP), raphe obscurrus (RO), and in the dorsal portion of the hypoglossal motor nucleus (Figure 1, 2, 3, 4). Phenotypic identification of TRHR1+ fibers in the hindbrain revealed that TRHR1-ir was not localized to fibers and varicosities that were positive for vesicular glutamate transporters 1 and 2 (vglut1, vglut2), or glutamine deaminase (GAD67). However, we observed complete overlap of immunoreactivity for TRHR1 receptors and fibers/varicosities that were also serotonin transporter (SERT) positive in the nuclear areas we examined (see for example, Figure 2). Note that epitope blocking for the TRHR1 antibody with recombinant TRHR1 receptor completely blocked TRHR1 immunoreactivity on both cells and fibers (e.g., Figure 4F).
Figure 2. Phenotype identification of TRHR1-ir fibers.
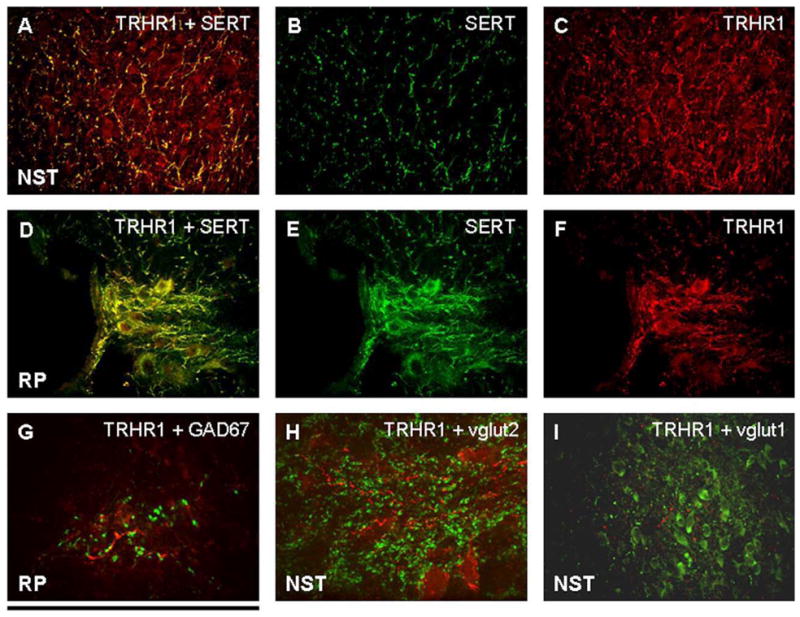
A, B, and C: Merged image (A) demonstrating that practically all SERT-ir fibers (B) in the NST possess TRHR1 receptors (C).
D, E, and F: Merged image (D) of staining in the raphe pallidus demonstrating that nearly all SERT-ir fibers and neurons (E) possess TRHR1 receptors (F).
G: GAD67-ir (GABA phenotype; green label) fibers in the RP are in the vicinity of TRHR1 receptors (red) but are not co-localized.
H: In the NST, vglut 2-ir fibers and varicosities (green) are in the vicinity of TRHR1 receptors (red) but are not co-localized.
I: Similarly, vglut 1-ir fibers and varicosities (green) in the NST are in the vicinity of TRHR1 receptors (red) but are not co-localized.
Scale bar: A–F, I = 150 microns; G, H = 20 microns.
Localization of LepRb-ir to the hindbrain of the rat
The NST, DMN, and VLM regions contained neurons densely expressing LepRb-ir (Figure 3, 4, 6); all neurons in these areas also expressed TRHR1-ir (Figure 1, 3, 4, and 6). The distribution of LepRb-ir neurons in these areas was more discrete than the distribution of TRHR1 receptors on neurons in these same regions. Specifically, LepRb-ir neurons in the NST were concentrated in the medial (i.e., sub-postremal) area as well as the pars centralis (Figure 4, 5). The area postrema appeared to have few LepRb-ir neurons (Figure 4D), while the choroid plexus did not have any cells expressing LepRb-ir (Figure 7). Phenotypic identification of LepRb-ir neurons in the VLM region revealed that these cells were also tyrosine hydroxylase-immunoreactive (TH-ir; Figure 6). This pattern was not seen in the NST. That is, despite the fact that the NST contains a substantial number of TH-ir neurons, only a few TH-ir neurons in the NST were also LepRb-ir (data not shown).
Figure 5.

Representative distribution of LepRb-ir fibers [crosshatch] and neurons [+] at four rostrocaudal levels of the hindbrain. TRHR1 receptors were distributed on neurons throughout the hindbrain but were especially prominent in the NST, DMN, and VLM. Essentially all LepRb+ neurons in these three sites also showed co-localization of TRHR1-ir.
Figure 7. Choroid plexus does not show LepRb-ir.
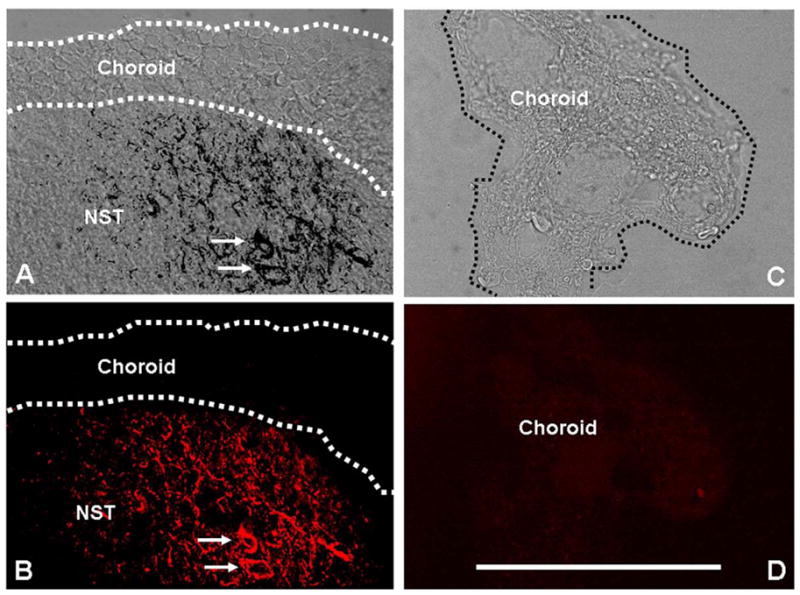
(A) Floor of the fourth ventricle at rostrocaudal level 13.0mm posterior to bregma showing the choroid plexus overlying the NST. Superimposed confocal image of LepRb-ir (black) neurons and fibers in the NST (cells indicated at arrows) onto a differential interference contrast (DIC) image of the NST and the overlying choroid plexus.
(B) Pseudocolored confocal image of LepRb (red) staining in the NST alone.
(C) DIC image of a detached fragment of 4th ventricular choroid.
(D) In situ hybridization for LepRb shows no label in the choroid. While the NST expresses abundant LepRb-ir and in situ hybridization for LepRb, the choroid is devoid of label as one would expect if the immunoreactivity is specific for the long form of the leptin receptor (LepRb). The choroid plexus expresses “short forms” of the leptin receptor, not LepRb. Scale bar = 100 microns.
The NST, DMN, and medullary raphe nuclei were filled with a background of LepRb-ir fibers and varicosities (Figure 3, 4, 5). The staining pattern strongly suggests LepRb-ir labeling of vagal afferents in the solitary tract (ST) that spreads out into the medial NST (Figure 4, 5). Attempts to phenotypically identify LepRb-ir fibers revealed that vglut1, Gad67 or SERT did not co-localize with LepRb-ir. However, vglut2-type fibers in these regions did demonstrate LepRb-ir (Figure 4, 9). While both TRH-ir and LepRb-ir fibers and terminals are prominent in the NST, DMN, RP, and RO, we did not observe any examples of double immunoreactivity for these two receptor types on individual fibers (i.e., no coincident TRHR1-ir and LepRb-ir was observed on fibers or varicosities in the hindbrain; Figure 3, 4, 9).
Figure 9. LepRb+ fibers and varicosities co-localize with vglut2+ but not GAD67 or SERT.
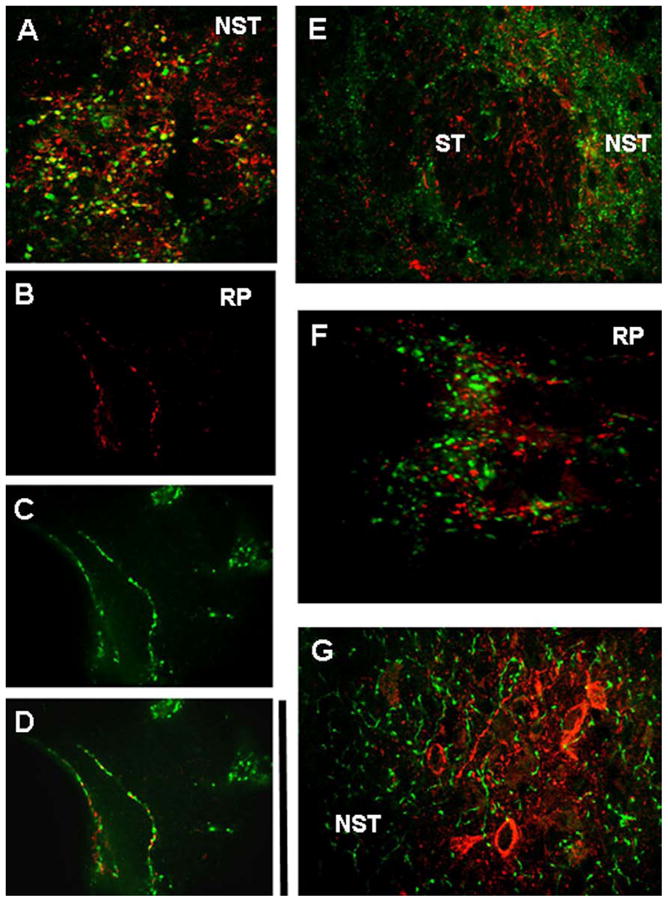
(A) Vglut2+ (green) fibers and varicosities in the medial NST appear to possess LepRb+ (red) receptors (i.e., numerous red/green/yellow overlapping terminals, though the high density of fibers and varicosities here make this difficult to ascertain).
(B) Two isolated fibers in the raphe pallidus (RP) that demonstrate LepRb+ (red).
(C) Same RP fibers as (B); identified as vglut2+ (green)
(D) Same isolated RP fibers as in (B) and (C); co-localization of vglut2+ and LepRp+ on individual fibers.
(E) GAD67+ (green) identified fibers and varicosities in and surrounding the solitary tract (ST) show no evidence of overlap with LepRb receptors (red)
(F) LepRb+ (red) and TRHR1+ (green) fibers and varicosities in the RP are closely adjacent but are not co-localized.
(G) LepRb+ (red) labeled neurons and fibers in the NST do not show co-localization with SERT (green). Scale bar A, B, C, D, F = 20 microns; E = 80 microns; G = 50 microns.
Specificity of the LepRb primary antibody was reinforced by epitope blocking with the specific epitope protein blocker [i.e., recombinant human long form leptin receptor], which eliminated all LepRb-ir staining (Figure 4E). Note, also, that there was no LepRb-ir staining of cells in the choroid plexus (Figure 7) which is in agreement with previous observations that only the short-form of the leptin receptor is expressed in this structure.
Lastly, while the LepRb antibody recognized LepRb receptor in the hypothalamus of the wild type C57BL/6 mouse, as has been previously reported, it did not show any immunoreactivity in the hypothalamus of the db/db mouse [Figure 8]. Recall that db/db mice are mutants that do not possess the long-form leptin receptor LepRb isoform.
Figure 8.
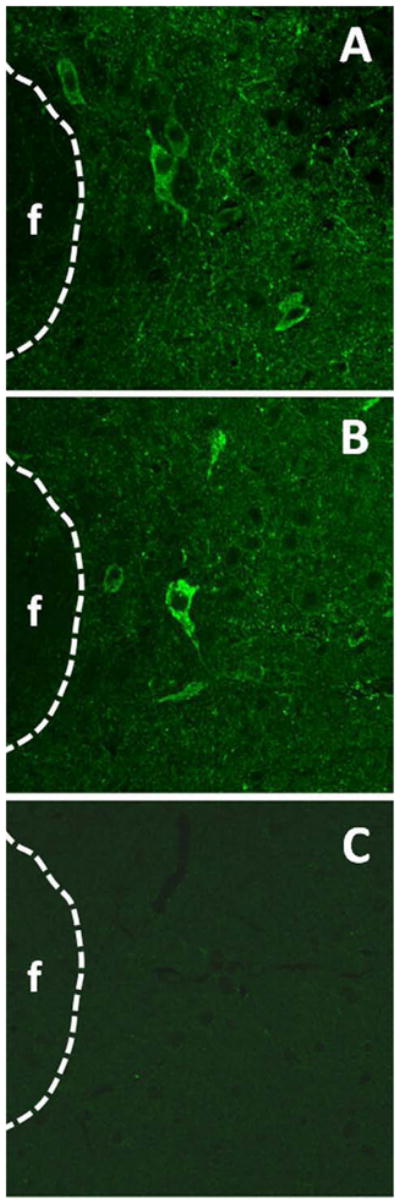
Validation of the specificity of the primary antibody for LepRb. (A) and (B) Positive immunoreactivity staining for LepRb in the perifornical nucleus in the lateral hypothalamus of C57BL/6 wild type mice. (C) Identical IHC processing parameters as used in (A) and (B) above yield no immunoreactivity for LepRb in the db/db mutant mouse. These results, along with the choroid plexus results in Figure 7, demonstrate that this primary antibody specifically recognizes only the long form of the leptin receptor but not the shorter variants. Scale bar = 200 micron.
Double fluorescent in situ hybridization sequences for TRHR1 and LepRb mRNA
Results from our double in situ hybridization corroborate our observations in the IHC studies. Specifically, signals for TRHR1 and LepRb were detected together in cell bodies of the NST, DMN, and VLM (Figure 10).
Figure 10. Double in situ hybridization validation of receptor localization.
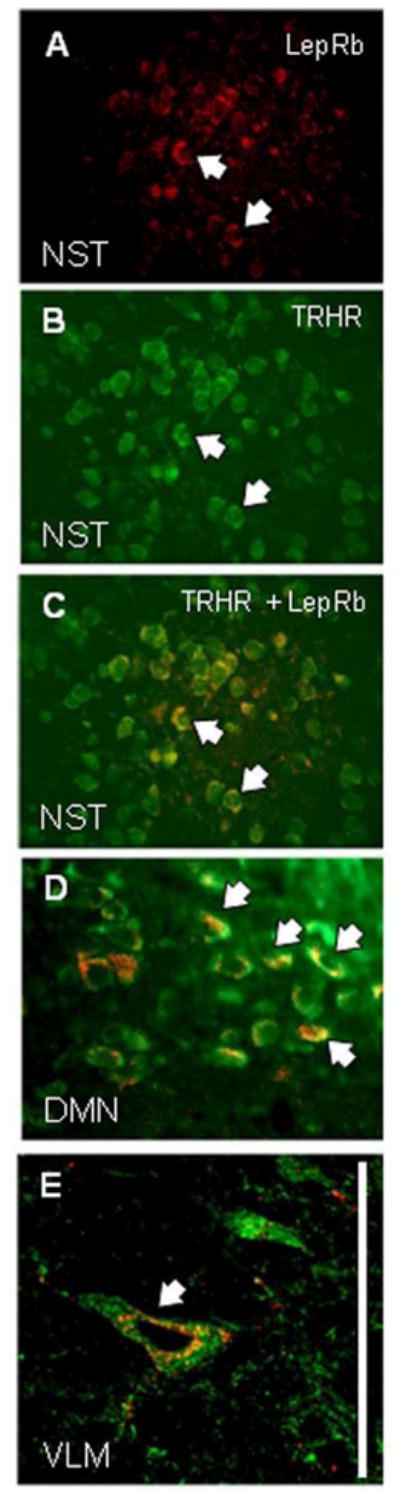
(A) LepRb (red) message is seen in a cluster of neurons in the medial NST. (Arrows indicate the same cells in A–C).
(B) TRHR1 (green) message is seen in a wider distribution of neurons in the NST.
(C) Superimposition of images A and B to show both messages in single NST neurons.
(D) Double in situ hybridization labeling of LepRb and TRHR1 message in scattered neurons in the lateral DMN.
(E) Example of double in situ hybridization labeling for LepRb and TRHR1 in the VLM. Scale bar A–D = 50 microns; E = 20 microns.
PSTAT3 expression in the medulla following 4V application of leptin
One goal of this study was to investigate a potential locus within the hindbrain where leptin may have its effect to gate thermogenesis evoked by TRH in our previous studies. This was done by tracking the evoked expression of PSTAT3, which is a marker of leptin activation of downstream transduction mechanisms. Therefore, we replicated the route and dose of administration that was used in those studies (i.e., 4V application of 5ug) and observed that a large increase in the expression of PSTAT3 was localized only in the NST (Figure 11).
Figure 11. PSTAT3 evoked only in the NST after 4V application of leptin.
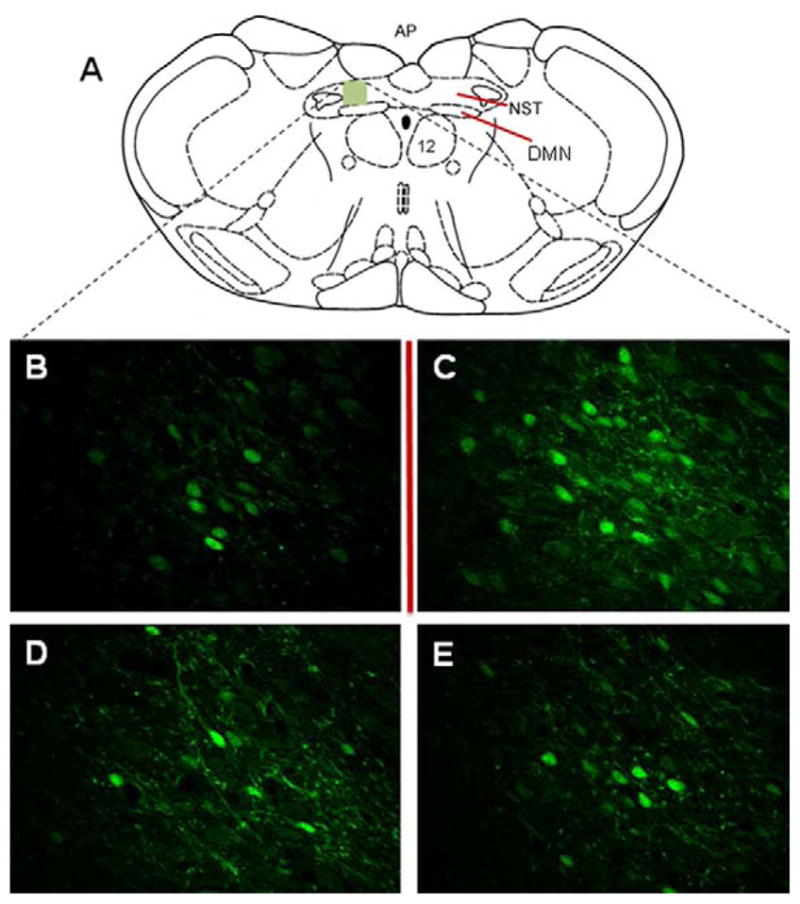
(A) Schematic drawing shows approximate area that contains PSTAT3-activated NST neurons in response to 5ug leptin 4V (same dose as used in previous studies to gate hindbrain thermogenic mechanisms). Control PBS 4V produced no PSTAT3 activation (data not shown).
(B–E) NST neurons PSTAT3-activated (green) in response to 4V leptin application; confocal images taken at ~10micron increments through NST. Scale bar = 100 microns.
3. DISCUSSION
HIER methods, immunostain sensitivity and selectivity
Immunostaining for the localization and identification of receptors such as TRHR1 and LepRb has been challenging and attempts have met with mixed success and controversy. The present studies used stringent criteria for validating the specificity of our LepRb and TRHR1 primary antibodies:
immunoreactivity was eliminated by preincubation with excess epitope;
results from our double ISH for mRNA for LepRb and TRHR1 validated our observations in the IHC studies;
an internal validation, in the rat, was made by the observation that the choroid plexus [which contains short form (LepRa) but not the long form of leptin receptor (LepRb)] does not show labeled cells and, lastly,
LepRb antibody specificity was further validated by its ability to label neurons in the hypothalamus of wild-type C57BL/6 but not db/db mutant mouse.
Additionally, our observation of functional LepRb receptors in the NST (discussed below) was validated by the upregulation of PSTAT3 in response to 4V leptin and corroborates previous reports of LepRb localization to the NST.
Localization of TRHR1 in the hindbrain
Our IHC and ISH results show that the TRHR1 receptor is localized to practically all neuronal cell bodies in the NST, VLM, DMN, and hypoglossal nucleus [Table 1]. Indeed, every NeuN labeled cell in these structures appears to express the TRHR1 receptor. These results are in good agreement with earlier results demonstrating high density TRH receptor binding in these regions of the hindbrain. Given the wide spread distribution of TRHR1 receptors on neurons within these regions, we made no further attempt to phenotypically identify the neurons involved.
Table 1.
Summary of results
| Cells | Fibers/varicosities | |
|---|---|---|
|
IHC: LepRb+ |
NST | NST – vglut2+ |
| DMN | DMN – vglut2+ | |
| medullary raphe – vglut2+ | ||
| VLM | ||
| TRHR1+ | NST | NST – SERT+ |
| DMN | DMN – SERT+ | |
| medullary raphe | medullary raphe – SERT+ | |
| VLM | ||
| hypoglossal | hypoglossal – SERT+ | |
| LepRb+ co-localized with TRHR1+ | NST | |
| DMN | none | |
| VLM | ||
| pSTAT3+ (after ICV leptin) | NST | not applicable |
|
In situ: LepRb+ co-localized with TRHR1+ |
NST | |
| DMN | not applicable | |
| VLM | ||
Previous work has shown that the TRHR1 receptor is highly concentrated in the NST, DMN, and hypoglossal nucleus. While the VLM region of the medulla was not specifically mentioned as a locus of intense TRHR1 expression, the region in and around the nucleus ambiguus [which adjoins the VLM], as well as scattered cells in the medullary reticular formation were found to express high levels of TRHR1. This distribution of the TRH receptor in neurons in the autonomic hindbrain help to explain the array of CNS functions that have been associated with TRH.
Additionally, our results demonstrate TRHR1-ir on fibers and varicosities, especially in the NST, DMN, and the medullary raphe. The TRH receptor is closely associated with the serotonin [5HT] positive fibers as identified by dual IHC for the serotonin transporter [SERT; Table 1]. This result is not surprising, as a very high percentage of TRHergic neurons are also serotonergic. A number of studies have shown that TRH can be a potent releasing stimulus for 5HT. Our results suggest that TRH could produce this increase in 5HT release via direct action at presynaptic terminals.
Localization of LepRb-ir in the hindbrain
LepRb-ir neurons are localized in loose clusters of cells in the DMN, NST, and the VLM [Table 1]. The localization of the LepRb leptin receptor to hindbrain structures has been controversial, with some earlier studies claiming strong immunostaining and message expression in the NST and DMN while other studies had suggested that this receptor is only weakly expressed in the hindbrain. However, more recent work by Elmquist’s laboratory using Cre-EYFP reporter mice as well as in situ hybridization work from Grill’s laboratory indicates that LepRb receptor is, in fact, densely distributed in the hindbrain, particularly in the NST and DMN.
Leptin receptor activation can cause a range of effects on different neuronal populations. Within the DVC, in vitro studies show that both NST and DMN neurons are inhibited by leptin through an ATP-dependent opening of potassium conductances. However, in vivo recordings from physiologically-identified NST neurons that form part of the gastric vago-vagal reflex path show that leptin activates these neurons. Recordings from other locations in the neuraxis show similar heterogeneity of responses to leptin and potential mechanisms of action.
Additionally, evidence suggests that leptin may potently activate primary vagal afferent neurons. The immediate downstream result of this is to increase the sensitivity of vagal afferents to other modes of stimulation and to increase glutamatergic vagal afferent input to second order visceral sensory neurons in the NST. Our finding of substantial LepRb-ir on vagal afferent fibers in the ST and NST provides a structural basis for these physiological observations. LepRb-ir fibers within the NST as well as the raphe pallidus were identified to be Vglut2-ir.
Hindbrain neurons activated by 4V application of leptin as evidenced by PSTAT3 expression
Fourth ventricular application of leptin positively modulates the effectiveness of TRH to elicit increases in BAT thermogenesis. Using the same dose and route of leptin application, PSTAT3 expression was evoked in neurons of the NST but not other sites in the hindbrain [Table 1.] Although PSTAT3 expression in the NST induced by systemic injections of leptin has been reported previously, the fact that the NST was the only locus to respond to our 4V application of leptin, replicates our earlier physiological studies and suggests that the NST is the target for the observed synergistic effects of leptin and TRH on BAT thermogenesis.
Expression of PSTAT3 in the NST is not surprising since leptin and other large signal molecules in the circulation have ready access to this nucleus through a weakened blood brain barrier as well as via specific transport mechanisms. Further, dendrites from neurons in the NST extend into the area postrema, a structure well outside the vascular diffusion barrier and in direct contact with the fourth ventricle.
Leptin and TRH interactions and homeostasis
Thermogenic responses to cold stress are “permitted” depending on the nutritional state of the animal. For example, even brief periods of starvation depress BAT thermogenesis in response to cold. Restoration of leptin can dis-inhibit the effects on thermogenesis that are caused by starvation. Similarly, lowered leptin levels depress thermogenesis, resulting in thermal torpor. Disruption of leptin signaling, as seen in db/db mice [i.e., no LepRb receptors] or ob/ob mice [i.e., no leptin], is associated with an inability to mount an acute, non-shivering thermogenic response to cold stress. Indeed, it has been suggested that one of leptin’s most important, and phylogenetically oldest functions, is to “gate” or at least positively modulate thermogenesis in proportion to energy availability. One clear advantage of such a “gating” arrangement would be the ability to suppress physiological attempts at neurogenic thermogenesis at times when the metabolic fuel supply may not be able to sustain it.
Our previous physiological experiments demonstrated that while 4V application of TRH modestly increased BAT temperature, the co-presence of leptin greatly augmented the thermogenic response to TRH. Further, it is clear that the isolated hindbrain is fully competent to produce this synergistic effect on temperature. One of the transduction events that is initiated by activation of the leptin receptor involves activation of phosphatidyl inositol-3 kinase (PI3K) which results in the production of phosphadiylinositol (3,4,5) trisphosphate (PIP3). This intermediate can produce downstream changes in neuronal excitability but can also directly and positively modulate the Gq protein-phospholipase C (PLC) transduction mechanism that underlies the cellular effects of TRH. Here, tyrosine kinase activated by PIP3 causes the phosphorylation of PLC at the SH2 regulatory site. Thus, using such a mechanism, leptin could amplify the effects of TRH by modulating its signal transduction (i.e., activation of PLC). Our recent physiologic study supports this view in that wortmannin (PI3kinase antagonist) as well as PP2 (SH2-tyrosine kinase inhibitor) were able to uncouple the synergistic effects to activate BAT thermogenesis that would normally be evoked by leptin plus TRH. Clearly, for this hypothesis of cross talk between signal transduction pathways of leptin and TRH to be the basis for the synergistic outcomes that we have previously observed in thermogenesis, the receptors for these two ligands must be present on the same cell. In the current studies, such co-localization of TRHR1 and LepRb receptors was observed in the NST, DMN, and VLM [Table 1].
Additionally, these studies revealed that NST neurons receive input from glutamatergic [vglut2] fibers that also possess LepRb receptors, in parallel with, incoming serotonergic fibers that possess TRHR1 receptors. It is likely that many, if not most, of the vglut2/LepRb fibers in the NST are vagal afferents as it is clear that both markers are present on fibers in the ST [Figures 4A and 9] and these receptors are functional on the vagal cell bodies of origin in the nodose ganglion. The potential interactions between TRH affecting 5HT-releasing terminals and leptin modulating glutamate-releasing terminals are undoubtedly complex. As mentioned above, the nature of the interactions is likely to be different in different loci. However, one potential outcome of such an arrangement could be a mutual increase in the pre-synaptic release of serotonin and glutamate caused by TRH and leptin, respectively. The post-synaptic consequences of this interaction can yield profound changes in autonomic functions such as cardiovascular control via actions in the NST.
Further, not unlike the NST, medullary raphe (RP and RO) also receives 5HT and glutamatergic afferent fibers that possessTRHR1 and LepRb receptors, respectively. As discussed above, the combined action of TRH and leptin could have significant effects on these raphe neurons as a result of increases in pre-synaptic release of 5HT and glutamate, respectively. Presynaptic regulation of 5HT and glutamate inputs to components of brainstem thermoregulatory circuits should have a significant impact on BAT temperature.
We find that TH-ir neurons in the ventrolateral medulla (VLM) express receptors for both LepRb and TRHR1. These catecholamine neurons probably belong to the A1 noradrenergic cell group which innervates a wide variety of structures dealing with the integrated control of autonomic, endocrine and behavioral functions tied to homeostasis. Areas receiving A1 input include the hypothalamus (especially the paraventricular region), amygdala, ventral tegmentum as well as solitary nucleus, rostral VLM, and the raphe pallidus. The integration of information by the VLM about the need to generate heat [i.e., carried by TRHergic projections] and information about the availability of fuel [i.e., carried by circulating leptin levels] could affect a vast array of homeostatic processes including but certainly not limited to thermogenesis. However, our results also suggest that these putative A1 VLM neurons may not integrate leptin and TRH signals after all. That is, while TRHergic projections and, therefore, TRH release are pervasive in the brainstem, access of leptin to the raphe and the VLM would have to depend on transport from the vasculature or diffusion from the CSF. Note that we did not observe an upregulation of PSTAT3 expression in the raphe or the VLM in response to 4V application of leptin. Thus, our results suggest that neither the raphe nor the VLM may be a primary detector of leptin
The NST, on the other hand, has a much weaker vascular diffusion barrier than the VLM or raphe. The “convergence” of leptin signals from the circulation with synaptic TRH signals from brainstem nuclei signaling heat production may not be possible in the VLM or the raphe but could be triggered through action in the NST. This view is supported by our finding that 4V leptin applied in amounts sufficient to augment thermogenesis only cause PSTAT3, a marker for leptin action, to be expressed in medial NST neurons. A recent report supports the view that the NST and the medullary VLM are important players in the brainstem activation of BAT thermogenesis.
While the TH+ phenotype of neurons in the VLM express abundant LepRb, only a few TH+ neurons in the medial NST contain the receptor. This sort of phenotype “mismatch” is not uncommon in that the LepRb receptor may be found on neurons of one phenotype in one region, but not another. For example, only about half of medial basal hypothalamic POMC neurons co-express LepRb: ~80% of retrochiasmatic-POMC neurons show co-localization with LepRb receptors, whereas only ~30% of the arcuate-POMC neurons show such co-localization. Further, while TH-neurons in the ventral tegmental area co-localize LepRb receptors, TH neurons in the locus coeruleus do not possess these receptors. Thus, it appears that LepRb receptors are expressed on neurons involved in specific functions and pathways rather than on neurons of a specific phenotype.
Model for combined leptin-TRH control of BAT thermogenesis in the hindbrain [Figure 12]
Figure 12. Summary schematic of histological observations and hypothetical model of hindbrain regulation of thermogenesis.

Our previous physiological studies showed a synergistic relationship between leptin and TRH on hindbrain thermogenic circuits. Our current IHC observations on the localization of receptors for TRHR1 and LepRb offer several means by which leptin and TRH might interact to augment thermogenesis. The co-localization of these two receptors on individual neurons in the NST affords the possibility that signal transduction pathways of these two ligands might interact directly, as suggested in our previous studies. Further, leptin action on glutamatergic fibers and TRH action on serotonin fibers could, together, powerfully modulate synaptic inputs to circuit elements important to regulating thermogenesis.
The medullary raphe nuclei responsible for descending control of sympathetically mediated thermogenesis are spontaneously active and may be strongly modulated by descending hypothalamic afferent pathways activated by cold exposure. These raphe medullary projections to the spinal cord are, in turn, responsible for activating sympathetic thermogenesis may be TRH-ergic. The medullary raphe nuclei also send TRH-ergic projections to the solitary nucleus. TRH can either inhibit or excite NST neurons, depending on the phenotype of the neurons in question [e.g., vs. ]. However, it is well known that raphe TRH-ergic input to the NST is activated by cold stress.
Even though raphe neurons are spontaneously active and can be powerfully excited by descending forebrain “cold-stress pathways”, they are also under significant GABA-ergic inhibition. Indeed, one of the most potent manipulations to provoke an increase in BAT thermogenesis is the application of GABA-A receptor antagonists directly to the medullary raphe. Clearly then, suppression of GABAergic input to the medullary raphe would be a physiologically efficient way to activate thermogenic raphe projections to the spinal cord utilizing hindbrain neural pathways alone. Indeed, both the NST and medullary VLM have recently been shown to control BAT thermogenesis through a GABA- disinhibitory mechanism.
Leptin and TRH could converge within the NST. Leptin can gain access to the NST through the action of specific transporters as well as by direct diffusion because this structure possesses fenestrated capillaries that admit proteins and peptide hormones. TRH-ergic projections from the raphe to the NST are dense. Perhaps leptin and TRH, together, cause the activation of neurons in the NST that, in turn, disinhibit thermogenic raphe neurons. The functional consequence of such a mechanism would be that, unless there were a demand for heat production (as signaled by TRH release onto the NST) and there were sufficient adipose storage (as signaled by leptin), energy would not be spent in heat production. A similar concept has been put forward by the works of DiRocco and Grill (1979) and Sue Ritter’s lab, i.e., they have shown that the hindbrain contains the neural circuitry to both detect needs (e.g., metabolic or heat) and ameliorate that need through the release of stored fuels when metabolic fuels are in abundance. Follow-up anatomical and neurophysiological studies will be necessary to show the validity of our model of hindbrain modulation of thermogenesis.
4. Experimental Procedures
Animals
Long-Evans rats [250–450g; body weight; either sex; N = 35] obtained from the breeding colony located at Pennington Biomedical Research Center, were used in these studies. All animals were maintained in a room with a 12:12 hour light-dark cycle with constant temperature and humidity, and given food and water ad libitum. All experimental protocols were performed according to the guidelines set forth by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committees at the Pennington Biomedical Research Center.
Naïve rats were deeply anesthetized with thiobutabarbital [Inactin,Sigma, St Louis, MO; 150mg/kg, ip.] and transcardially perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde. The brainstem was post fixed at room temperature in 4% paraformaldehyde overnight prior to being cryoprotected by soaking in 30% sucrose solution. Sections were cut 40μm thick on a Microm HM430 freezing microtome.
As an additional measure to verify the specificity of the primary antibody for LepRb, hypothalamic regions from C57BL/6 and db/db mice that had been similarly processed (as described above) were graciously provided by Dr H. Muenzberg. All subsequent IHC processing for LepRb-ir proceeded as described below for the IHC processing of rat hindbrain histological sections.
Double immunofluorescent staining for LepRb and TRHR1 and phenotypic identification of fibers and cells
Sections taken from any given hindbrain [N = 16] were processed at the same time to ensure uniformity of immunostaining. To perform heat-induced epitope recovery [HIER] prior to immunostaining, free-floating tissue sections were immersed in a Biocare (Concord, CA) Rodent Decloaking buffer and placed in the Biocare Decloaker for 30min at 80°C. After cooling, sections were rinsed with Tris-Buffered Saline (TBS) containing 0.05% Triton X-100 (Sigma Aldrich, St Louis, MO); then incubated in 1% sodium borohydride (Sigma Aldrich; to neutralize any remaining fixative) for 20min followed by several TBS rinses. Sections were placed in a non-specific antigen blocking solution of Rodent Block R (Biocare) for 30min, followed by a rinse with TBS. All incubation and rinse steps were under constant agitation. After another TBS rinse, the tissue sections were placed in a cocktail of the primary antibodies for LepRb plus TRHR1 or tyrosine hydroxylase (TH), glutamine deaminase (GAD67), serotonin transporter (SERT), vesicular glutamate transporter 1 (vglut1) and vesicular glutamate transporter 2 (vglut2); see Table 1. Tissue sections were exposed to primary antibodies plus 0.3% Triton X100 over night at room temperature. The following day, sections were rinsed several times with TBS buffer prior to being immersed in appropriate secondary antibodies for each phenotype (refer to Table 1) tagged with either Alexa 488 or rhodamine red X for 2hrs at room temperature. The tissue sections were rinsed with TBS buffer; mounted on Plus slides and coverslipped with ProLong Gold Anti-fade Reagent (Invitrogen, Carlsbad, CA) for immunofluorescence.
As controls for the specificity of the LepRb and TRHR1 antibodies used in these IHC studies, epitope blocking was performed on subsets of perfused hindbrain slices. Specifically, for LepRb primary antibody, the specific epitope blocker [i.e, recombinant human long form LepRb; Neuromics] was combined with the primary antibody at a ratio of 5:1 during the incubation with the tissue sections. Similarly, the TRHR1 primary antibody was incubated with a 5:1 excess of rat TRHR1. The rest of the IHC procedure proceeded as described above.
PSTAT3 immunofluorescence
In a separate subset of studies, tissue was IHC stained for identification of activated cells that were positive for PSTAT3 (i.e., leptin transduction molecule) following fourth ventricular application of leptin. As described in our previous studies, animals (N=8 total) were deeply anesthetized with thiobutabarbital and secured in the stereotaxic frame; the floor of the 4V was exposed. Either leptin (5ug in 2uL PBS; N = 6) or PBS (2uL; N = 6) was applied 4V. This dose of leptin was shown in our previous studies to act as a potent gating condition for thermogenic responses. Because we wanted to see which cells were activated by leptin as evidenced by up regulation of PSTAT3, we allowed 30min from the time of agonist stimulation to the time of transcardial perfusion and brain harvest. IHC protocol for the presence of PSTAT3 was followed as described above with the exceptions that HIER was not done and 100% methanol was substituted for the borohydride step. The remaining steps of the IHC process proceeded as above (refer to Table 2 for antibody details).
Table 2.
Immunohistochemical reagents used
| Primary antibodies (dilutions) | Source; cat. no. | Secondary antibody tags | Source; type |
|---|---|---|---|
| NeuN 1:500 | Chemicon; cat#MAB377; mouse | Rhodamine-RedX (red) | Jackson ImmunoResearch Labs donkey anti-mouse |
| TRHR1 5ug/ml | Imgenex; cat#71813; rabbit | Alexa 488 (green) OR Rhodamine-RedX (red) | Invitrogen donkey anti-rabbit ************************************* Jackson ImmunoResearch Labs donkey anti-rabbit |
| LepRb 1:500 | Neuromics; cat#CH14014; chicken | Rhodamine-RedX (red) | Jackson ImmunoResearch Labs goat anti-chicken |
| TH 1:500 | Immunostar; cat#22941; mouse | Alexa 488 (green) | Invitrogen donkey anti-mouse |
| GAD67 2ug/ml | Neuromics; cat#GT5142; goat | Alexa 488 (green) | Invitrogen donkey anti-goat |
| SERT 1:500 | Immunostar: cat #24330; rabbit | Alexa 488 (green) | Invitrogen donkey anti-rabbit |
| vglut1 1:1000 | Millipore; cat#AB5905; guinea pig | Alexa 488 (green) | Invitrogen donkey anti-guinea pig |
| vglut2 1:1000 | Millipore; cat#MAB5504; mouse | Alexa 488 (green) | Invitrogen donkey anti-mouse |
| PSTAT3 1:100 | Cell Signaling; cat#9131; rabbit | Alexa 488 (green) | Invitrogen donkey anti-rabbit |
| β-actin 1:1000 | Cell signaling cat#4967; rabbit | HRP conjugated | Cell Signaling Goat anti-rabbit |
In situ hybridization for TRHR1 and LepRb
Following standard transcardial perfusions with saline and 4% paraformaldehyde, tissue processing and sectioning as described above, in situ hybridization for the identification and localization of TRH and leptin receptors was performed on the hindbrain tissue of four rats. Specifically tagged probes for in situ hybridization identification of the TRHR1 receptors (5DigN/CTTTTCCTCCTACCCTTACTCA) and the long form leptin receptor, LepRb, (5Biosg/GCTTTCTCTCCCACCCACAACT) were obtained from Equixon (Woburn, MA). Hindbrain histological sections were treated with 0.25% and 0.5% acetic anhydride for 5min prior to being treated with 1ug/ml of Proteinase K (Ambion, Austin TX) for 30min at 37°C. Sections were rinsed with glycine (2mg/ml in 0.1M PBS) and incubated with a pre-hybridization buffer obtained from Biognostik (Gottingen, Germany) for 1 hour at 47°C. After pre-hybridization, the sections were hybridized (i.e., pre-hybridization buffer plus tagged specific probes) overnight at 47°C. On the following day, sections were washed for 15min with the 5X SSC (RNase-free, sodium chloride, sodium citrate; Ambion, Foster City, CA) at 47°C, followed by two washes with 0.2X SSC for 30min each at 47°C; and a 5min wash with 0.1M PBS at room temperature. The hybridized probes and targets were then visualized via fluorescent label (i.e., fluorescein Avidin DCS diluted in ISH buffer [Vector Laboratory [Burlingame, CA] for the biotinylated probe of the LepRb receptor = green fluorescence) or IHC via monoclonal antibody generated against digoxigenin (i.e., rhodamine tagged anti-Dig diluted in 5% sheep serum for the TRHR1 receptor = red fluorescence; Jackson ImmunoResearch, West Grove, PA). Processed sections were cover slipped with ProLong Gold Antifade Reagent (Invitrogen, Carlsbad, CA) for immunofluorescence.
In situ Controls
As a means of verifying the quality of our tissue, poly(dT) probes were used to detect total mRNA poly A tails. The strong poly(dT) signal was indicative that the tissue RNA had not degraded and that the chance of detecting the specific mRNAs of interest in this tissue were optimized. As negative in situ controls, tissue sections were probed with the sense-strand probes of the same size and specific activity [synthesized by Equixon]. Positive labeling was never encountered in these experiments.
Microscope
Processed tissue sections were examined with a Nikon E800 microscope equipped with a Perkin Elmer CSU21 spinning disk laser confocal system. Images were collected with a Hamamatsu ORCA CCD camera using Ultraview system integration software. All graphic images presented here were not electronically manipulated beyond uniform adjustments of brightness, contrast, or color balance, i.e., no information in the original images were obscured or eliminated.
Acknowledgments
This work was supported by NIH grants NS55866 and DK56373. Hypothalamic regions from C57BL/6 and db/db mice were graciously provided by Dr H. Muenzberg
Comprehensive list of abbreviations
- 4V
fourth ventricle
- 5HT
5-hydroxytryptamine; serotonin
- AP
area postrema
- BAT
brown adipose tissue
- cc
central canal
- DMN
dorsal motor nucleus of the vagus
- GABA
gamma- aminobutyric acid
- GAD67
glutamine deaminase
- HIER
heat-induced epitope retrieval
- HRP
horseradish peroxidase
- IHC
immunohistochemistry
- -ir
immunoreactivity
- LepRb
long-form leptin receptor
- N12
hypoglossal nucleus
- NeuN
neuronal nuclei
- NST
nucleus of the solitary tract
- PBS
phosphate buffered saline
- PI3K
phosphatidyl inositol-3 kinase
- PIP3
phophatidylinositol (3,4,5)-triphophate
- PLC
phospholipase C
- PP2
SH2-tyrosine kinase inhibitor
- PSTAT3
phosphorylated STAT3 [signal transducer and activator of transcription 3]
- RIPA
radioimmunoprecipitation assay
- RO
raphe obscurrus
- RP
raphe pallidus
- SERT
serotonin transporter
- SSC
RNase free, sodium chloride, sodium citrate
- ST
solitary tract
- TBS
tris-buffered saline
- TBST
tris-buffered saline-tween20
- TH
tyrosine hydroxylase
- TRH
thyrotropin releasing hormone
- TRHR1
TRH type 1 receptor
- vglut
vesicular glutamate transporter
- VLM
ventrolateral medulla
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- Arancibia S, Rage F, Astier H, Tapia-Arancibia L. Neuroendocrine and autonomous mechanisms underlying thermoregulation in cold environment. Neuroendocrinology. 1996;64:257–67. doi: 10.1159/000127126. [DOI] [PubMed] [Google Scholar]
- Arvidsson U, Cullheim S, Ulfhake B, Luppi PH, Kitahama K, Jouvet M, Hokfelt T. Quantitative and qualitative aspects on the distribution of 5-HT and its coexistence with substance P and TRH in cat ventral medullary neurons. J Chem Neuroanat. 1994;7:3–12. doi: 10.1016/0891-0618(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Assadian H, Ishikawa Y, Shimatsu A, Tanoh T, Imura H. Serotoninergic denervation suppresses the sympathetic outflow induced by thyrotropin-releasing hormone in conscious rats. J Auton Nerv Syst. 1991;35:193–8. doi: 10.1016/0165-1838(91)90097-m. [DOI] [PubMed] [Google Scholar]
- Bamshad M, Song CK, Bartness TJ. CNS origins of the sympathetic nervous system outflow to brown adipose tissue. Am J Physiol. 1999;276:R1569–78. doi: 10.1152/ajpregu.1999.276.6.R1569. [DOI] [PubMed] [Google Scholar]
- Bates SH, Dundon TA, Seifert M, Carlson M, Maratos-Flier E, Myers MG., Jr LRb-STAT3 signaling is required for the neuroendocrine regulation of energy expenditure by leptin. Diabetes. 2004;53:3067–73. doi: 10.2337/diabetes.53.12.3067. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C, Elmquist JK, Michl P, Ahima RS, van Bueren A, McCall AL, Flier JS. Expression of leptin receptor isoforms in rat brain microvessels. Endocrinology. 1998;139:3485–91. doi: 10.1210/endo.139.8.6154. [DOI] [PubMed] [Google Scholar]
- Blumberg MS, Deaver K, Kirby RF. Leptin disinhibits nonshivering thermogenesis in infants after maternal separation. Am J Physiol. 1999;276:R606–10. doi: 10.1152/ajpregu.1999.276.2.R606. [DOI] [PubMed] [Google Scholar]
- Boulant JA. Role of the preoptic-anterior hypothalamus in thermoregulation and fever. Clin Infect Dis. 2000;31(Suppl 5):S157–61. doi: 10.1086/317521. [DOI] [PubMed] [Google Scholar]
- Broadwell RD, Sofroniew MV. Serum proteins bypass the blood-brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol. 1993;120:245–263. doi: 10.1006/exnr.1993.1059. [DOI] [PubMed] [Google Scholar]
- Cano G, Passerin AM, Schiltz JC, Card JP, Morrison SF, Sved AF. Anatomical substrates for the central control of sympathetic outflow to interscapular adipose tissue during cold exposure. J Comp Neurol. 2003;460:303–326. doi: 10.1002/cne.10643. [DOI] [PubMed] [Google Scholar]
- Cao WH, Madden CJ, Morrison SF. Inhibition of brown adipose tissue thermogenesis by neurons in the ventrolateral medulla and in the nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 2010;299:R277–R290. doi: 10.1152/ajpregu.00039.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SC, Kochan JP, Campfield LA, Burn P, Smeyne RJ. Splice variants of the OB receptor gene are differentially expressed in brain and peripheral tissues of mice. J Recept Signal Transduct Res. 1999;19:245–66. doi: 10.3109/10799899909036649. [DOI] [PubMed] [Google Scholar]
- Crespi F, Keane PE, Morre M. In vivo evaluation by differential pulse voltammetry of the effect of thyrotropin-releasing hormone (TRH) on dopaminergic and serotoninergic synaptic activity in the striatum and nucleus accumbens of the rat. Exp Brain Res. 1986;62:329–34. doi: 10.1007/BF00238852. [DOI] [PubMed] [Google Scholar]
- Dekin MS, Richerson GB, Getting PA. Thyrotropin-releasing hormone induces rhythmic bursting in neurons of the nucleus tractus solitarius. Science. 1985;229:67–9. doi: 10.1126/science.3925552. [DOI] [PubMed] [Google Scholar]
- DiMicco JA, Sarkar S, Zaretskaia MV, Zaretsky DV. Stress-induced cardiac stimulation and fever: common hypothalamic origins and brainstem mechanisms. Auton Neurosci. 2006;126–127:106–19. doi: 10.1016/j.autneu.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Dimicco JA, Zaretsky DV. The dorsomedial hypothalamus: a new player in thermoregulation. Am J Physiol Regul Integr Comp Physiol. 2007;292:R47–63. doi: 10.1152/ajpregu.00498.2006. [DOI] [PubMed] [Google Scholar]
- Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–81. [PubMed] [Google Scholar]
- Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–47. [PubMed] [Google Scholar]
- Elmquist JK. Hypothalamic pathways underlying the endocrine, autonomic, and behavioral effects of leptin. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S78–82. doi: 10.1038/sj.ijo.0801918. [DOI] [PubMed] [Google Scholar]
- Eskay RL, Long RT, Palkovits M. Localization of immunoreactive thyrotropin releasing hormone in the lower brainstem of the rat. Brain Res. 1983;277:159–62. doi: 10.1016/0006-8993(83)90919-8. [DOI] [PubMed] [Google Scholar]
- Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995a;1:1311–4. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- Frederich RC, Lollmann B, Hamann A, Napolitano-Rosen A, Kahn BB, Lowell BB, Flier JS. Expression of ob mRNA and its encoded protein in rodents. Impact of nutrition and obesity. J Clin Invest. 1995b;96:1658–63. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM. Is my antibody-staining specific? How to deal with pitfalls of immunohistochemistry. Eur J Neurosci. 2008;28:2365–70. doi: 10.1111/j.1460-9568.2008.06552.x. [DOI] [PubMed] [Google Scholar]
- Funk D, Post RM, Pert A. Role of central dopaminergic and 5-hydroxytryptaminergic projections in the behavioral responses elicited by thyrotropin-releasing hormone in rats. Psychopharmacology (Berl) 1997;133:356–62. doi: 10.1007/s002130050414. [DOI] [PubMed] [Google Scholar]
- Gary KA, Sevarino KA, Yarbrough GG, Prange AJ, Jr, Winokur A. The thyrotropin-releasing hormone (TRH) hypothesis of homeostatic regulation: implications for TRH-based therapeutics. J Pharmacol Exp Ther. 2003;305:410–6. doi: 10.1124/jpet.102.044040. [DOI] [PubMed] [Google Scholar]
- Geiser F, Kortner G, Schmidt I. Leptin increases energy expenditure of a marsupial by inhibition of daily torpor. Am J Physiol. 1998;275:R1627–32. doi: 10.1152/ajpregu.1998.275.5.R1627. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Kaplan JM. The neuroanatomical axis for control of energy balance. Front Neuroendocrinol. 2002;23:2–40. doi: 10.1006/frne.2001.0224. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 1990;259:R1131–R1138. doi: 10.1152/ajpregu.1990.259.6.R1131. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Wainman DS, Shaver SW. Subregional topography of capillaries in the dorsal vagal complex of rats: II. Physiological properties. J Comp Neurol. 1991;306:83–94. doi: 10.1002/cne.903060107. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. Central noradrenergic neurons: the autonomic connection. Prog Brain Res. 1991;88:365–380. doi: 10.1016/s0079-6123(08)63823-6. [DOI] [PubMed] [Google Scholar]
- Harris RB, Kelso EW, Flatt WP, Bartness TJ, Grill HJ. Energy expenditure and body composition of chronically maintained decerebrate rats in the fed and fasted condition. Endocrinology. 2006;147:1365–76. doi: 10.1210/en.2005-1156. [DOI] [PubMed] [Google Scholar]
- Helke CJ, Sayson SC, Keeler JR, Charlton CG. Thyrotropin-releasing hormone-immunoreactive neurons project from the ventral medulla to the intermediolateral cell column: partial coexistence with serotonin. Brain Res. 1986;381:1–7. doi: 10.1016/0006-8993(86)90682-7. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Nasse JS, Rogers RC. Alpha-1 adrenergic input to solitary nucleus neurones: calcium oscillations, excitation and gastric reflex control. J Physiol. 2005;562:553–68. doi: 10.1113/jphysiol.2004.076919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Barnes MJ, Rogers RC. Leptin and thyrotropin-releasing hormone: cooperative action in the hindbrain to activate brown adipose thermogenesis. Brain Res. 2006;1117:118–24. doi: 10.1016/j.brainres.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC. TNFalpha: a trigger of autonomic dysfunction. Neuroscientist. 2008;14:53–67. doi: 10.1177/1073858407305725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer H, Schafer MK, O’Donnell D, Walker P, Bauer K. Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J Comp Neurol. 2000;428:319–36. [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–10. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Huang J, Pickel VM. Differential distribution of 5HT2A and NMDA receptors in single cells within the rat medial nucleus of the solitary tract. Synapse. 2002;44:64–75. doi: 10.1002/syn.10056. [DOI] [PubMed] [Google Scholar]
- Huo L, Gamber KM, Grill HJ, Bjorbaek C. Divergent leptin signaling in proglucagon neurons of the nucleus of the solitary tract in mice and rats. Endocrinology. 2008;149:492–7. doi: 10.1210/en.2007-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- I’Anson H, Sundling LA, Roland SM, Ritter S. Immunotoxic destruction of distinct catecholaminergic neuron populations disrupts the reproductive response to glucoprivation in female rats. Endocrinology. 2003;144:4325–31. doi: 10.1210/en.2003-0258. [DOI] [PubMed] [Google Scholar]
- Irani BG, Le Foll C, Dunn-Meynell A, Levin BE. Effects of leptin on rat ventromedial hypothalamic neurons. Endocrinology. 2008;149:5146–54. doi: 10.1210/en.2008-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan D. Vagal control of the heart: central serotonergic (5-HT) mechanisms. Exp Physiol. 2005;90:175–81. doi: 10.1113/expphysiol.2004.029058. [DOI] [PubMed] [Google Scholar]
- Korotkova TM, Brown RE, Sergeeva OA, Ponomarenko AA, Haas HL. Effects of arousal- and feeding-related neuropeptides on dopaminergic and GABAergic neurons in the ventral tegmental area of the rat. Eur J Neurosci. 2006;23:2677–85. doi: 10.1111/j.1460-9568.2006.04792.x. [DOI] [PubMed] [Google Scholar]
- Lechan RM, Fekete C. The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res. 2006;153:209–35. doi: 10.1016/S0079-6123(06)53012-2. [DOI] [PubMed] [Google Scholar]
- Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–5. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- Li YW, Bayliss DA. Presynaptic inhibition by 5-HT1B receptors of glutamatergic synaptic inputs onto serotonergic caudal raphe neurones in rat. J Physiol. 1998;510(Pt 1):121–34. doi: 10.1111/j.1469-7793.1998.121bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LH, Talman WT. Vesicular glutamate transporters and neuronal nitric oxide synthase colocalize in aortic depressor afferent neurons. J Chem Neuroanat. 2006;32:54–64. doi: 10.1016/j.jchemneu.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Liu X, Ye K. Src homology domains in phospholipase C-gamma1 mediate its anti-apoptotic action through regulating the enzymatic activity. J Neurochem. 2005;93:892–8. doi: 10.1111/j.1471-4159.2005.03064.x. [DOI] [PubMed] [Google Scholar]
- Lynn RB, Kreider MS, Miselis RR. Thyrotropin-releasing hormone-immunoreactive projections to the dorsal motor nucleus and the nucleus of the solitary tract of the rat. J Comp Neurol. 1991;311:271–88. doi: 10.1002/cne.903110208. [DOI] [PubMed] [Google Scholar]
- Madden CJ, Morrison SF. Endogenous activation of spinal 5-hydroxytryptamine (5-HT) receptors contributes to the thermoregulatory activation of brown adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2010;298:R776–83. doi: 10.1152/ajpregu.00614.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–61. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- Manaker S, Rizio G. Autoradiographic localization of thyrotropin-releasing hormone and substance P receptors in the rat dorsal vagal complex. J Comp Neurol. 1989;290:516–26. doi: 10.1002/cne.902900406. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Hunt SP. Thyrotropin-releasing hormone (TRH) receptors. Localization by light microscopic autoradiography in rat brain using [3H][3-Me-His2]TRH as the radioligand. J Neurosci. 1985;5:551–61. doi: 10.1523/JNEUROSCI.05-02-00551.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maolood N, Meister B. Protein components of the blood-brain barrier (BBB) in the brainstem area postrema-nucleus tractus solitarius region. J Chem Neuroanat. 2009;37:182–95. doi: 10.1016/j.jchemneu.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Martinez V, Wang L, Tache Y. Central TRH receptor 1 antisense blocks cold-induced gastric emptying but not brain c-Fos induction. Peptides. 2001;22:81–90. doi: 10.1016/s0196-9781(00)00359-4. [DOI] [PubMed] [Google Scholar]
- McCann MJ, Hermann GE, Rogers RC. Thyrotropin-releasing hormone: effects on identified neurons of the dorsal vagal complex. J Auton Nerv Syst. 1989;26:107–112. doi: 10.1016/0165-1838(89)90158-6. [DOI] [PubMed] [Google Scholar]
- Mercer JG, Moar KM, Hoggard N. Localization of leptin receptor (Ob-R) messenger ribonucleic acid in the rodent hindbrain. Endocrinology. 1998;139:29–34. doi: 10.1210/endo.139.1.5685. [DOI] [PubMed] [Google Scholar]
- Morrison SF, Sved AF, Passerin AM. GABA-mediated inhibition of raphe pallidus neurons regulates sympathetic outflow to brown adipose tissue. Am J Physiol. 1999;276:R290–7. doi: 10.1152/ajpregu.1999.276.2.R290. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. 2003;144:2121–31. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- Nagashima K, Nakai S, Tanaka M, Kanosue K. Neuronal circuitries involved in thermoregulation. Auton Neurosci. 2000;85:18–25. doi: 10.1016/S1566-0702(00)00216-2. [DOI] [PubMed] [Google Scholar]
- Nelson RJ. Leptin: the “skinny” on torpor. Am J Physiol Regul Integr Comp Physiol. 2004;287:R6–7. doi: 10.1152/ajpregu.00164.2004. [DOI] [PubMed] [Google Scholar]
- Nogueira MI, de Rezende BD, do Vale LE, Bittencourt JC. Afferent connections of the caudal raphe pallidus nucleus in rats: a study using the fluorescent retrograde tracers fluorogold and true-blue. Ann Anat. 2000;182:35–45. doi: 10.1016/s0940-9602(00)80118-1. [DOI] [PubMed] [Google Scholar]
- Palkovits M, Mezey E, Eskay RL, Brownstein MJ. Innervation of the nucleus of the solitary tract and the dorsal vagal nucleus by thyrotropin-releasing hormone-containing raphe neurons. Brain Res. 1986;373:246–251. doi: 10.1016/0006-8993(86)90338-0. [DOI] [PubMed] [Google Scholar]
- Pan W, Kastin AJ. Diurnal variation of leptin entry from blood to brain involving partial saturation of the transport system. Life Sci. 2001;68:2705–14. doi: 10.1016/s0024-3205(01)01085-2. [DOI] [PubMed] [Google Scholar]
- Peters JH, Karpiel AB, Ritter RC, Simasko SM. Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology. 2004;145:3652–7. doi: 10.1210/en.2004-0221. [DOI] [PubMed] [Google Scholar]
- Peters JH, Simasko SM, Ritter RC. Modulation of vagal afferent excitation and reduction of food intake by leptin and cholecystokinin. Physiol Behav. 2006;89:477–85. doi: 10.1016/j.physbeh.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Raab M, Neuhuber WL. Vesicular glutamate transporter 2 immunoreactivity in putative vagal mechanosensor terminals of mouse and rat esophagus: indication of a local effector function? Cell Tissue Res. 2003;312:141–8. doi: 10.1007/s00441-003-0721-5. [DOI] [PubMed] [Google Scholar]
- Rameh LE, Rhee SG, Spokes K, Kazlauskas A, Cantley LC, Cantley LG. Phosphoinositide 3-kinase regulates phospholipase Cgamma-mediated calcium signaling. J Biol Chem. 1998;273:23750–7. doi: 10.1074/jbc.273.37.23750. [DOI] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes KJ, Trimmer JS. Antibodies as valuable neuroscience research tools versus reagents of mass distraction. J Neurosci. 2006;26:8017–20. doi: 10.1523/JNEUROSCI.2728-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L, Card JP, Schwaber JS, Miselis RR. Ultrastructural demonstration of a gastric monosynaptic vagal circuit in the nucleus of the solitary tract in rat. J Neurosci. 1989;9:1985–96. doi: 10.1523/JNEUROSCI.09-06-01985.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L. Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. J Neurosci. 2003;23:10084–10092. doi: 10.1523/JNEUROSCI.23-31-10084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, McCann MJ. Intramedullary connections of the gastric region in the solitary nucleus: a biocytin histochemical tracing study in the rat. J Auton Nerv Syst. 1993;42:119–130. doi: 10.1016/0165-1838(93)90043-t. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE, Travagli RA. Brainstem control of gastric function. In: Johnson LR, editor. Physiology of the gastrointestinal tract. Elsevier Academic Press; 2005. pp. 851–875. [Google Scholar]
- Rogers RC, Barnes MJ, Hermann GE. Leptin “gates” thermogenic action of thyrotropin-releasing hormone in the hindbrain. Brain Res. 2009:135–41. doi: 10.1016/j.brainres.2009.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasek CA, Wessendorf MW, Helke CJ. Evidence for co-existence of thyrotropin-releasing hormone, substance P and serotonin in ventral medullary neurons that project to the intermediolateral cell column in the rat. Neuroscience. 1990;35:105–19. doi: 10.1016/0306-4522(90)90125-n. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Moran TH. Leptin and neuropeptide y have opposing modulatory effects on nucleus of the solitary tract neurophysiological responses to gastric loads: implications for the control of food intake. Endocrinology. 2002;143:3779–84. doi: 10.1210/en.2002-220352. [DOI] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–32. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley LJ, O’Malley D, Irving AJ, Ashford ML, Harvey J. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J Physiol. 2002;545:933–44. doi: 10.1113/jphysiol.2002.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda S, Funahashi H, Nakajo S, Yada T, Maruta O, Nakai Y. Immunohistochemical localization of leptin receptor in the rat brain. Neurosci Lett. 1998;243:41–4. doi: 10.1016/s0304-3940(98)00082-2. [DOI] [PubMed] [Google Scholar]
- Szekely M, Balasko M, Kulchitsky VA, Simons CT, Ivanov AI, Romanovsky AA. Multiple neural mechanisms of fever. Auton Neurosci. 2000;85:78–82. doi: 10.1016/S1566-0702(00)00223-X. [DOI] [PubMed] [Google Scholar]
- Trayhurn P. Thermoregulation in the diabetic-obese (db/db) mouse. The role of non-shivering thermogenesis in energy balance. Pflugers Arch. 1979;380:227–32. doi: 10.1007/BF00582901. [DOI] [PubMed] [Google Scholar]
- Ukropec J, Anunciado RV, Ravussin Y, Kozak LP. Leptin is required for uncoupling protein-1-independent thermogenesis during cold stress. Endocrinology. 2006;147:2468–80. doi: 10.1210/en.2005-1216. [DOI] [PubMed] [Google Scholar]
- Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–7. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- Williams KW, Zsombok A, Smith BN. Rapid inhibition of neurons in the dorsal motor nucleus of the vagus by leptin. Endocrinology. 2007;148:1868–81. doi: 10.1210/en.2006-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky DV, Zaretskaia MV, DiMicco JA. Stimulation and blockade of GABA(A) receptors in the raphe pallidus: effects on body temperature, heart rate, and blood pressure in conscious rats. Am J Physiol Regul Integr Comp Physiol. 2003;285:R110–6. doi: 10.1152/ajpregu.00016.2003. [DOI] [PubMed] [Google Scholar]