

Abstract
Background
Hearing loss with enlarged vestibular aqueduct (EVA) can be inherited as an autosomal recessive trait caused by bi-allelic mutations of SLC26A4. However, many EVA patients have non-diagnostic SLC26A4 genotypes with only one or no detectable mutant alleles.
Methods and results
In this study, the authors were unable to detect occult SLC26A4 mutations in EVA patients with non-diagnostic genotypes by custom comparative genomic hybridisation (CGH) microarray analysis or by sequence analysis of conserved non-coding regions. The authors sought to compare the segregation of EVA among 71 families with two (M2), one (M1) or no (M0) detectable mutant alleles of SLC26A4. The segregation ratios of EVA in the M1 and M2 groups were similar, but the segregation ratio for M1 was significantly higher than in the M0 group. Haplotype analyses of SLC26A4-linked STR markers in M0 and M1 families revealed discordant segregation of EVA with these markers in eight of 24 M0 families.
Conclusion
The results support the hypothesis of a second, undetected SLC26A4 mutation that accounts for EVA in the M1 patients, in contrast to non-genetic factors, complex inheritance, or aetiologic heterogeneity in the M0 group of patients. These results will be helpful for counselling EVA families with non-diagnostic SLC26A4 genotypes.
Enlargement of the vestibular aqueduct (EVA; MIM 600709) is a common radiological inner ear malformation associated with sensorineural hearing loss (SNHL).1 It is the most penetrant finding in autosomal recessive SNHL with goitre (Pendred syndrome (PDS); MIM 274600) but can also be observed without goitre in non-syndromic EVA (NSEVA) (DFNB4; MIM 600791).2 Mutations of SLC26A4, which encodes the pendrin protein, can cause either PDS or NSEVA.3–5 Pendrin is a member of the SLC26A family of transmembrane anion transporters.6
Two mutant alleles of SLC26A4 can be identified in approximately 1/4 of Caucasian patients with EVA. The other 3/4 of those patients carry only one or zero mutant alleles of SLC26A4.7–10 In these latter patients with non-diagnostic genotypes, the aetiology of EVA remains unclear. The pathogenic contribution of mono-allelic SLC26A4 mutations is supported by their detection in up to 44% of NSEVA patients.7 9–13 Azaiez et al11 used the maximum likelihood method14 to estimate that 98% of these apparent heterozygotes have an undetected SLC26A4 mutation in trans configuration.
SLC26A4 mutations are detected more frequently in multiplex EVA families than in simplex families.9 This may reflect a contribution of non-genetic factors to the aetiology of EVA in patients with no SLC26A4 mutations. However, specific non-genetic causes have not been identified to date. Congenital cytomegalovirus infection, for example, is a very common non-genetic cause of childhood hearing loss but accounts for few, if any, cases of EVA.15 It has also been proposed that EVA may be inherited as a digenic or a complex trait caused by a single mutant allele of SLC26A4 in combination with environmental factors or mutations in other genes15 such as FOXI1, a putative upstream regulator of SLC26A4 expression.16 However, other studies indicate that FOXI1 mutations contribute to few, if any, cases of EVA.13 15 17
The aetiology and recurrence risk for EVA in patients with non-diagnostic SLC26A4 genotypes thus remain unknown. In this study, we sought to identify pathogenic copy number variants or mutations of conserved non-coding sequences in these patients. We also analysed the segregation of EVA and SLC26A4 in families with non-diagnostic SLC26A4 genotypes. Since we have never observed abnormal perchlorate discharge results from EVA patients with non-diagnostic SLC26A4 genotypes,7 18 19 we did not include thyroid phenotype in our analyses. Our results provide insight into the aetiology of EVA and estimates of recurrence risk that will be useful when counselling families.
METHODS
Subjects
This study was approved by the Combined Neuroscience Institutional Review Board, National Institutes of Health (NIH), Bethesda, Maryland. Written informed consent was obtained from all subjects or their legal guardians. Our cohort comprised 71 probands, 85 siblings (19 affected and 66 unaffected), and 137 parents from 71 families. Two hundred and thirty-one of 293 subjects were evaluated at the NIH Clinical Center, including 132 family members that were previously reported.7 19 The evaluations included medical–otolaryngological history interviews and physical examinations, pure tone and speech audiometry and, in most subjects, computed tomography (CT) and/or magnetic resonance imaging (MRI) of the temporal bones. The phenotypes of 62 subjects were inferred from existing medical records or medical and/or family history interviews. We performed SLC26A4 sequence analysis as described.7
We grouped families according to the number of mutant alleles of SLC26A4 detected in the proband: zero (M0), one (M1), or two (M2). Four hypofunctional variants (p.F335L, p.C565Y, p.L597S, and p.M775T) and two variants (c.−60A>G and c.−3−2A>G) of indeterminate pathogenicity were considered non-pathogenic unless they were in trans configuration with SLC26A4 mutations previously reported to be pathogenic.19 20 Our cohort comprised 43 M0 families, eight M1 families, and 20 M2 families. Three of 20 M2 and 13 of 43 M0 families segregated variants of indeterminate pathogenicity, including one M2 and seven M0 families with p.L597S.7 19
Comparative genomic hybridisation (CGH) microarray analysis
We analysed the DNA of probands from seven M1 families (142, 156, 217, 242, 264, 280 and 293) and two M0 families (182 and 255). We used a custom microarray with 15 bp probe spacing (2006-07-31_HG18_SLC26A4_FT; Nimblegen Sytems Inc, Madison, Wisconsin, USA) representing a 3.8 Mb region encompassing SLC26A4 on chromosome 7 (nt 105,238,316–109,038,316; UCSC version hg18, March 2006). DNA samples from parents were used for hybridisation controls. For M1 families, we used the heterozygous carrier parent as the control.
Sequence analysis of non-coding regions
We designed primers (supplemental table S1) to polymerase chain reaction (PCR) amplify and sequence non-coding regions of SLC26A4 from probands from seven M1 families (142, 156, 217, 242, 264, 280 and 293) and one M0 family (118).7 We sequenced intronic and upstream conserved regions predicted by the Vertebrate Multiz Alignment and PhastCons Conservation (28 species) tool of the University of California Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu/). We also sequenced candidate inner ear specific promoter regions17 and Genscan predicted exons (NT_007933.614) (fig 1) (supplemental table S1). Variants were considered non-pathogenic when they were detected among >1% of normal Caucasian control chromosomes from Coriell Cell Repositories (HD200CAU; Camden, New Jersey, USA).
Figure 1.

Schematic illustration of chromosome 7q31 encompassing the SLC26A4 gene with STR markers shown above. Non-coding regions (labelled “a” to “v”) that are most highly conserved among 28 species were sequenced in seven M1 families and one M0 family. These regions include an inner ear specific positive regulatory element (k) predicted by Adler et al17 and potential additional exons predicted by Genescan (NT_007933.614) (“a”, “m” and “o”).
Zygosity analysis
The zygosity of twin pairs in families 219, 213, and 245 was previously established.7 19 We analyzed 14 unlinked STR markers in twins 1750 and 1751 (family 261) to determine zygosity.
Calculation of segregation ratio
We assumed single incomplete ascertainment to calculate segregation ratios among M0, M1, and M2 groups. We used Weinberg's proband method to correct for ascertainment bias against carrier parents with normal progeny.21 In this method, each proband is considered to be providing information that his or her parents are capable of producing affected progeny. An unbiased estimate of the segregation ratio (p) is calculated from remaining members of the sibship,
where r and s indicate the number of affected offspring and the total number of offspring in each family, respectively. Monozygotic twin pairs were each treated as a single observation in the analysis, reducing the total number of siblings for our analysis from 85 to 83. We first calculated the ratio among 48 siblings from 36 families (22 M0, 5 M1, and 9 M2) that were evaluated at the NIH Clinical Center. We repeated the calculations for the same cohort with 35 additional siblings (from 4 M0, 1 M1 and 8 M2 families) that were not evaluated at the NIH Clinical Center. These siblings were considered to be affected if they had radiologically confirmed EVA or a history of pre- or perilingual onset deafness in the absence of radiologic imaging data. We also calculated segregation ratios among heterozygous mutation carriers in M1 and M2 families.
In addition, we calculated the ratios under either of two assumptions: p.F335L, p.C565Y, p.L597S, p.M775T, c.−60A>G and c.−3−2A>G are always or never pathogenic, irrespective of the trans allele genotype.
Haplotype analysis
We performed haplotype analyses of SLC26A4 linked short tandem repeat (STR) markers (D7S496, D7S2459 and D7S2456) on the M0 (n = 17) and M1 (n = 1) families with participating siblings. The results for 11 other families (seven M0, four M1) were previously described.7
Statistical analysis
Fisher's exact test was used to compare segregation ratios among genotype groups. A value of p<0.05 was considered to be significant.
RESULTS
Occult SLC26A4 mutation analyses
CGH microarray analysis revealed only a 338 bp deletion (rs6150268) located 1.56 Mb upstream of SLC26A4 (chr 7: 105,521,227–105,521,564; UCSC version hg18, March 2006) in four of seven M1 probands. We designed primers flanking this copy number variant and detected it by PCR amplification in 46 of 63 Caucasian control subjects. These results provided no evidence for the presence of pathogenic copy number variations in the 3.8 Mb region encompassing SLC26A4.
We did not identify any pathogenic variations in non-coding conserved regions from eight (one M0, seven M1) probands (supplemental table S1 available online).
Segregation analyses
The overall segregation ratio of EVA in 48 siblings evaluated at the NIH Clinical Center was 0.27 (95% confidence interval (CI) 0.17 to 0.41). However, the segregation ratio differed among the genotype groups M0, M1, and M2. The M1 ratio of 0.67 (95% CI 0.29 to 0.90) was significantly higher (p = 0.011) than the M0 ratio of 0.13 (95% CI 0.052 to 0.29), but did not differ significantly (p = 0.62) from 0.45 (95% CI 0.21 to 0.72), the ratio for the M2 group in which EVA is inherited as an autosomal recessive trait. The segregation ratio in the M0 group was significantly lower (p = 0.037) than that in the M2 group (table 1).
Table 1.
Segregation of enlargement of the vestibular aqueduct (EVA) among siblings ascertained at the National Institutes of Health
| Number of SLC26A4 mutations in proband | Number of siblings |
Segregation ratio* (95% CI) | |
|---|---|---|---|
| Affected | Unaffected | ||
| 0 | 4 | 27 | 0.13 (0.052 to 0.29) |
| 1 | 4 | 2 | 0.67 (0.29 to 0.90) |
| 2 | 5 | 6 | 0.45 (0.21 to 0.72) |
| Total | 13 | 35 | 0.27 (0.17 to 0.41) |
CI, confidence interval.
The M1 ratio of 0.67 was significantly higher (p=0.011) than the M0 ratio of 0.13, but did not differ significantly (p=0.62) from the M2 ratio of 0.45. The segregation ratio in the M0 group was significantly lower (p=0.037) than that in the M2 group.
The differences between genotype groups persisted when we calculated ratios for the extended cohort including 35 siblings who were not evaluated at the NIH Clinical Center (table 2). The differences persisted among M0 versus M1 (p = 0.02), M0 versus M2 (p = 0.067), and M1 versus M2 groups (p = 0.230) (table 2), although the difference between the M0 and M2 groups was not statistically significant.
Table 2.
Segregation of enlargement of the vestibular aqueduct (EVA)* among the entire cohort of 83 siblings
| Number of SLC26A4 mutations in proband | Number of siblings |
Segregation ratio† (95% CI) | |
|---|---|---|---|
| Affected | Unaffected | ||
| 0 | 4 | 34 | 0.11 (0.043 to 0.24) |
| 1 | 4 | 4 | 0.50 (0.21 to 0.79) |
| 2 | 10 | 27 | 0.27 (0.15 to 0.43) |
| Total | 18 | 65 | 0.22 (0.14 to 0.32) |
CI, confidence interval.
Includes EVA inferred from outside evaluations and family history interviews.
The M1 ratio of 0.50 was significantly higher (p=0.02) than the M0 ratio of 0.11, but did not differ significantly (p=0.23) from the M2 ratio of 0.27.
We observed the same pattern of relative differences in the segregation ratios among genotype groups under the assumption that the four hypofunctional variants and two variants of indeterminate pathogenicity were pathogenic, irrespective of trans allele genotype: M0 versus M1 (0.04 vs 0.38; p = 0.003), M0 versus M2 (0.04 vs 0.27; p = 0.013), and M1 versus M2 (0.38 vs 0.27; p = 0.53). The observed pattern of differences persisted when the same six variants are assumed to be non-pathogenic: M0 versus M1 (0.11 vs 0.45, p = 0.018), M0 versus M2 (0.11 vs 0.26, p = 0.079), and M1 versus M2 (0.45 vs 0.26, p = 0.28).
The segregation ratio among the four heterozygous carrier siblings from M1 families was 1.00. As expected, the ratio among the five heterozygous carrier siblings from M2 families was 0.00, since they do not carry the second mutant allele segregating in their respective families.
We found no bias in the gender of the heterozygous carrier parents (two males, two females) transmitting the mutant allele of SLC26A4 to their affected M1 offspring. We detected no audiologic or radiologic evidence of vertical transmission of EVA in any of our M0 or M1 families.7
Haplotype analyses of SLC26A4 linked STR markers in 24 M0 and five M1 families with participating siblings revealed concordant segregation with EVA in 21 families and discordant segregation in eight families (fig 2). In families 144, 213, 255, 259, 261, and 283, we observed unaffected siblings and the proband carrying the same SLC26A4 linked haplotypes. This result could reflect either discordant inheritance or non-penetrance. Non-penetrance (bilateral) of EVA is not unexpected from the frequent observation of unilateral non-penetrance (that is, unilateral EVA) in M0 patients.7 In contrast, families 133 and 219 each have affected sibling pairs with different SLC26A4 linked haplotypes. Families 133 and 219 were considered to be M1 in our previously published haplotype analysis,7 but we now recognise their variants (p.R776C and c.-3-2A>G) as likely to be benign polymorphisms.19 20 22 We did not observe discordant segregation of SLC26A4 linked STR markers with EVA in any of five M1 families (fig 3).
Figure 2.

Discordant segregation of SLC26A4 linked short tandem repeat (STR) marker haplotypes with enlargement of the vestibular aqueduct (EVA). The haplotypes within each family are arbitrarily represented as A, B, C, or D—for example, an A haplotype in one family does not necessarily imply the same combination of genotypes in the A haplotype for another family. Families 133, 144, 213, and 219 were previously described by Pryor et al.7 Haplotypes are underlined when linked to a SLC26A4 sequence variant. p.V609G is a common polymorphism (dbSNP rs17154335). If p.R776C, c. -3-2A>G, p.L597S and c.-60A>G are considered non-pathogenic in trans with a wild type allele,19 20 22 all of these eight families are M0 families (tables 1 and 2).
Figure 3.
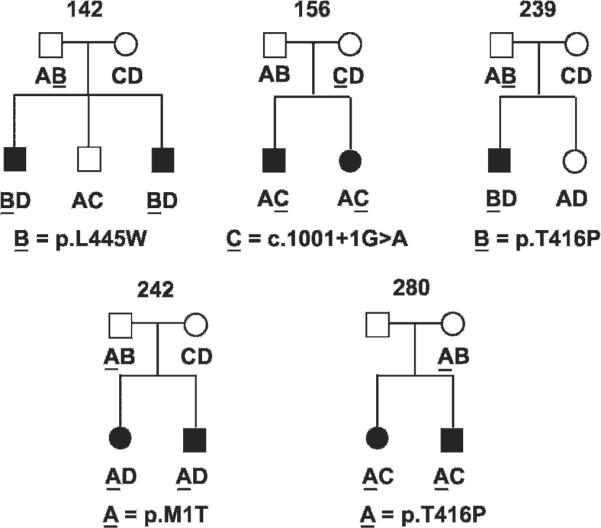
Segregation of SLC26A4 linked short tandem repeat (STR) marker haplotypes and enlargement of the vestibular aqueduct (EVA) in M1 families. The haplotypes within each family are arbitrarily represented as A, B, C, or D. Haplotypes are underlined when linked to an SLC26A4 mutation. We did not include families 133 and 219 which we formerly considered to be M17 but are now considered M0.
Twinning
There were three dizygotic (DZ) and two monozygotic (MZ) twin pairs among 77 live births in M0 families (supplemental table S1 available online). Three DZ twin births but no MZ twin births were reported among 73 live births in the combined group of M1 and M2 families; all three M1/M2 twin births were in one M2 family (family 188).7
DISCUSSION
We were unable to identify any previous published reports of segregation ratios for EVA.
Our segregation ratios for M1 families confirm the pathogenic contribution of detected mono-allelic mutations to EVA and support the hypothesis that EVA is inherited as a mono- or digenic recessive trait in those families. Although our M1 segregation ratio was greater than the expected Mendelian result of 0.25, it was similar to the ratio observed among the M2 families in whom EVA was inherited as a monogenic autosomal recessive trait. The lack of a significant difference between M1 and M2 ratios might be due to the small number of families, especially M1 families. However, the high M1 ratio seems unlikely to reflect dominant inheritance because we have not observed vertical transmission of EVA in M0 or M1 families. The ratios for all of the genotype groups likely reflect an ascertainment bias for multiplex sibships in the families referred to our study.
If the hypofunctional alleles p.R776C and c.-3-2A>G are considered to be non-pathogenic,19 22 there is no longer evidence for digenic or complex inheritance among our M1 families. The lack of discordant segregation of EVA with SLC26A4 linked STR markers in M1 families is thus consistent with the hypothesis of autosomal recessive inheritance of bi-allelic mutations of SLC26A4, although we cannot exclude the possibility of digenic inheritance in M1 families.
Although we were unable to find evidence of pathogenic copy number variations or non-coding sequence mutations, they may reside elsewhere in intronic or regulatory regions that we did not evaluate. Occult mutant alleles of SLC26A4 would be predicted to encode some residual pendrin since the thyroid and auditory phenotypes in M1 patients are generally less severe than in M2 patients. Abnormal perchlorate discharge results are observed almost exclusively among M2 patients,7 18 unilateral EVA is more prevalent among M1 patients whereas EVA is almost always bilateral in M2 patients, and M1 patients tend to have better hearing thresholds than M2 patients.7 11 23
Given the probable influence of ascertainment bias on our segregation ratios, the actual M0 ratio may be closer to zero than our observed ratio of approximately 0.1. This low ratio and discordant segregation of EVA with SLC26A4 in eight of 24 M0 families are consistent with a non-genetic or complex aetiology, or aetiologic heterogeneity that may include Mendelian inheritance in some families. However, we did not detect phenotypic evidence for etiologic heterogeneity among the M0 group since there was no significant difference in hearing thresholds between familial and sporadic patients (not shown).
The twin pairs in our M0 cohort raise the possibility of a causal relationship or a shared aetiology with twinning such as that described for other phenotypes including Beckwith–Wiedemann24 25 and Goldenhar syndromes26 and symmelia.27 However, we identified no patterns among the M0 twin pregnancies to indicate a causal relationship between twinning and EVA (supplemental table S2 available online).
This is the first study to estimate the recurrence risk of EVA, which will be useful for counselling families segregating EVA and one or no detectable mutations of SLC26A4. Families with EVA and one detectable mutation of SLC26A4 seem likely to be segregating EVA as a trait caused by that mutation in combination with a second occult mutant allele of SLC26A4 or another autosomal gene. In contrast, EVA appears to be a non-genetic or complex trait with a significantly lower recurrence rate in families with no detectable SLC26A4 mutations. The discovery of a source of occult mutations in families with non-diagnostic SLC26A4 genotypes could clarify the aetiology and recurrence risk of EVA in those families.
Key points.
-
▶
We calculated segregation ratios and analysed the segregation of SLC26A4 linked markers in enlargement of the vestibular aqueduct (EVA) families with non-diagnostic SLC26A4 genotypes.
-
▶
Our results support the hypothesis of a second, undetected SLC26A4 mutation in M1 patients, whereas EVA appears to be caused by mutations in other genes, non-genetic factors, or both as a complex trait in M0 patients.
-
▶
These results will be helpful for recurrence risk counselling of EVA families with non-diagnostic SLC26A4 genotypes.
Supplementary Material
Acknowledgements
We thank the study families for their participation, Tom Friedman and Barb Biesecker for support, and Tom Friedman and Rob Morell for critical review of the manuscript.
Funding This work was supported by NIH intramural research funds Z01-DC-000039, Z01-DC-000060 and Z01-DC-000064 and the Intramural Research Program of the National Human Genome Research Institute.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
REFERENCES
- 1.Valvassori GE, Clemis JD. The large vestibular aqueduct syndrome. Laryngoscope. 1978;88:723–8. doi: 10.1002/lary.1978.88.5.723. [DOI] [PubMed] [Google Scholar]
- 2.Phelps PD, Coffey RA, Trembath RC, Luxon LM, Grossman AB, Britton KE, Kendall-Taylor P, Graham JM, Cadge BC, Stephens SG, Pembrey ME, Reardon W. Radiological malformations of the ear in Pendred syndrome. Clin Radiol. 1998;53:268–73. doi: 10.1016/s0009-9260(98)80125-6. [DOI] [PubMed] [Google Scholar]
- 3.Usami S, Abe S, Weston MD, Shinkawa H, Van Camp G, Kimberling WJ. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet. 1999;104:188–92. doi: 10.1007/s004390050933. [DOI] [PubMed] [Google Scholar]
- 4.Li XC, Everett LA, Lalwani AK, Desmukh D, Friedman TB, Green ED, Wilcox ER. A mutation in PDS causes non-syndromic recessive deafness. Nat Genet. 1998;18:215–7. doi: 10.1038/ng0398-215. [DOI] [PubMed] [Google Scholar]
- 5.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–22. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 6.Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet. 1999;21:440–3. doi: 10.1038/7783. [DOI] [PubMed] [Google Scholar]
- 7.Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA, Griffith AJ. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005;42:159–65. doi: 10.1136/jmg.2004.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coyle B, Reardon W, Herbrick JA, Tsui LC, Gausden E, Lee J, Coffey R, Grueters A, Grossman A, Phelps PD, Luxon L, Kendall-Taylor P, Scherer SW, Trembath RC. Molecular analysis of the PDS gene in Pendred syndrome. Hum Mol Genet. 1998;7:1105–12. doi: 10.1093/hmg/7.7.1105. [DOI] [PubMed] [Google Scholar]
- 9.Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, Karniski LP, Sheffield VC, Smith RJ. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat. 2001;17:403–11. doi: 10.1002/humu.1116. [DOI] [PubMed] [Google Scholar]
- 10.Albert S, Blons H, Jonard L, Feldmann D, Chauvin P, Loundon N, Sergent-Allaoui A, Houang M, Joannard A, Schmerber S, Delobel B, Leman J, Journel H, Catros H, Dollfus H, Eliot MM, David A, Calais C, Drouin-Garraud V, Obstoy MF, Tran Ba Huy P, Lacombe D, Duriez F, Francannet C, Bitoun P, Petit C, Garabedian EN, Couderc R, Marlin S, Denoyelle F. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet. 2006;14:773–9. doi: 10.1038/sj.ejhg.5201611. [DOI] [PubMed] [Google Scholar]
- 11.Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ, Kimberling WJ, Smith RJ. Genotype-phenotype correlations for SLC26A4-related deafness. Hum Genet. 2007;122:451–7. doi: 10.1007/s00439-007-0415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, Kim HN, Moon SK, Abe S, Tukamoto K, Riazuddin S, Kabra M, Erdenetungalag R, Radnaabazar J, Khan S, Pandya A, Usami SI, Nance WE, Wilcox ER, Griffith AJ. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40:242–8. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pera A, Villamar M, Vinuela A, Gandia M, Meda C, Moreno F, Hernandez-Chico C. A mutational analysis of the SLC26A4 gene in Spanish hearing-impaired families provides new insights into the genetic causes of Pendred syndrome and DFNB4 hearing loss. Eur J Hum Genet. 2008;16:888–96. doi: 10.1038/ejhg.2008.30. [DOI] [PubMed] [Google Scholar]
- 14.Kimberling WJ. Estimation of the frequency of occult mutations for an autosomal recessive disease in the presence of genetic heterogeneity: application to genetic hearing loss disorders. Hum Mutat. 2005;26:462–70. doi: 10.1002/humu.20221. [DOI] [PubMed] [Google Scholar]
- 15.Pryor SP, Demmler GJ, Madeo AC, Yang Y, Zalewski CK, Brewer CC, Butman JA, Fowler KB, Griffith AJ. Investigation of the role of congenital cytomegalovirus infection in the etiology of enlarged vestibular aqueducts. Arch Otolaryngol Head Neck Surg. 2005;131:388–92. doi: 10.1001/archotol.131.5.388. [DOI] [PubMed] [Google Scholar]
- 16.Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4) Am J Hum Genet. 2007;80:1055–63. doi: 10.1086/518314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adler L, Efrati E, Zelikovic I. Molecular mechanisms of epithelial cell-specific expression and regulation of the human anion exchanger (pendrin) gene. Am J Physiol Cell Physiol. 2008;294:C1261–76. doi: 10.1152/ajpcell.00486.2007. [DOI] [PubMed] [Google Scholar]
- 18.Madeo AC, Manichaikul A, Reynolds JC, Sarlis NJ, Pryor SP, Shawker T, Griffith AJ. Evaluation of the thyroid in patients with hearing loss and enlarged vestibular aqueducts. Arch Otolaryngol Head Neck Surg. 2009;135:670–6. doi: 10.1001/archoto.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi BY, Stewart AK, Madeo AC, Pryor SP, Lenhard S, Kittles R, Eisenman D, Jeffrey Kim H, Niparko J, Thomsen J, Arnos KS, Nance WE, King KA, Zalewski CK, Brewer CC, Shawker T, Reynolds JC, Butman JA, Karniski LP, Alper SL, Griffith AJ. Hypo-Functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: genotype-phenotype correlation or coincidental polymorphisms? Hum Mutat. 2009;30:599–608. doi: 10.1002/humu.20884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pera A, Dossena S, Rodighiero S, Gandia M, Botta G, Meyer G, Moreno F, Nofziger C, Hernandez-Chico C, Paulmichl M. Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA. Proc Natl Acad Sci USA. 2008;105:18608–13. doi: 10.1073/pnas.0805831105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher R. The effects of methods of ascertainment upon the estimation of frequencies. Ann Eugen. 1934;6:13–25. [Google Scholar]
- 22.Pfarr N, Borck G, Turk A, Napiontek U, Keilmann A, Muller-Forell W, Kopp P, Pohlenz J. Goitrous congenital hypothyroidism and hearing impairment associated with mutations in the TPO and SLC26A4/PDS genes. J Clin Endocrinol Metab. 2006;91:2678–81. doi: 10.1210/jc.2006-0142. [DOI] [PubMed] [Google Scholar]
- 23.Madden C, Halsted MJ, Hopkin RJ, Choo DI, Benton C, Greinwald JH., Jr. Temporal bone abnormalities associated with hearing loss in Waardenburg syndrome. Laryngoscope. 2003;113:2035–41. doi: 10.1097/00005537-200311000-00034. [DOI] [PubMed] [Google Scholar]
- 24.Bose B, Wilkie RA, Madlom M, Forsyth JS, Faed MJ. Wiedemann-Beckwith syndrome in one of monozygotic twins. Arch Dis Child. 1985;60:1191–2. doi: 10.1136/adc.60.12.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leonard NJ, Bernier FP, Rudd N, Machin GA, Bamforth F, Bamforth S, Grundy P, Johnson C. Two pairs of male monozygotic twins discordant for Wiedemann-Beckwith syndrome. Am J Med Genet. 1996;61:253–7. doi: 10.1002/(SICI)1096-8628(19960122)61:3<253::AID-AJMG9>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 26.Lawson K, Waterhouse N, Gault DT, Calvert ML, Botma M, Ng R. Is hemifacial microsomia linked to multiple maternities? Br J Plast Surg. 2002;55:474–8. doi: 10.1054/bjps.2002.3902. [DOI] [PubMed] [Google Scholar]
- 27.Davies J, Chazen E, Nance WE. Symmelia in one of monozygotic twins. Teratology. 1971;4:367–78. doi: 10.1002/tera.1420040312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.