

Abstract
The relationship between ethanol induced oxidative stress and acetylation of histone H3 at lysine 9 (H3AcK9) remains unknown and was therefore investigated in primary cultures of rat hepatocytes. Cells were treated with ethanol and a select group of pharmacological agents and the status of H3AcK9 and reactive oxygen species (ROS) were monitored. When hepatocytes were exposed to ethanol (50 mM, 24 hr) in the presence of N-acetyl cystein (ROS reducer) or dietary antioxidants (quercetin, resveratrol), or NADPH oxidase inhibitor apocynin, ethanol induced increases in ROS and H3AcK9, both were significantly reduced. On the other hand, l-buthionine-sulfoximine (ROS inducer) and inhibitor of mitochondrial complex I (rotenone) and III (antimycin) increased ethanol induced H3AcK9 (p<0.01). Oxidative stress also affected ethanol induced alcohol dehydrogenase 1 (ADH1) mRNA expression. These results demonstrate for the first time that oxidative stress is involved in the ethanol induced histone H3 acetylation in hepatocytes.
Keywords: Histone acetylation, Reactive oxygen species, Hepatocytes
Introduction
Histone acetylation is one of the important epigenetic events with a pivotal role in the control of eukaryotic gene transcription (Jenuwein and Allis, 2001). Transcriptionally active genes are correlated with hyperacetylated histone, while their paucity (hypoacetylated histone) is associated with transcriptionally repressed genes (Smith, 2008). Especially, acetylation of histone H3 at lys9 is considered to be a specific marker of various active genes (Morinobu et al., 2004). Irregular acetylation and deacetylation of histones have been linked to various developmental diseases, eg. leukemia, fragile X-syndrome and Rubinstein-Taybi syndrome (Kundu and Dasgupta, 2007).
Alcohol consumption has long been associated with liver damage (Reuben, 2008). But, the mechanisms underlying deleterious effects of alcohol are not completely understood. They could result from the production of the toxic derivatives from alcohol metabolism or from a direct action of alcohol on cellular components. Recent evidence from this laboratory indicated that ethanol as well as other higher chain surrogate alcohols increased the acetylation of histone H3 at lysine 9 in primary culture of rat hepatocytes (Park et al., 2003; Choudhury and Shukla, 2008) and that this acetylation correlated with ADH1 gene expression (Park et al., 2005). Such histone modifications are proposed to underlie the mechanisms involved in ethanol induced cellular injury (Shukla et al., 2008).
On the other hand, acute and chronic alcohol have been shown to increase the production of reactive oxygen species (ROS), lower cellular antioxidant levels, and enhance oxidative stress in many tissues, especially in the liver. The formation of reactive oxygen species (ROS) such as the superoxide anion (O2 -) and hydrogen peroxide (H2O2) represent an important cause of oxidative injury. Several enzymatic systems, including the CYP2E1-dependent microsomal monoxygenase system, NADPH oxidase, the mitochondrial respiratory chain have been implicated as the sources of ROS during ethanol intoxication (Albano, 2006; Dey and Cederbaum, 2006). Alcohol is metabolized predominantly by ADH1 enzyme in the liver which leads to the production of ROS. Although ethanol induced oxidative stress in the liver is known, the molecular mechanism by which this contributes to the pathogenesis of alcoholic liver disease (ALD) is still incompletely understood.
Oxidative stress and redox status of the cells can regulate nuclear chromatin remodeling (histone acetylation/deacetylation) leading to gene expression (Ito et al., 2004). Oxidative stress also altered histone acetylation/deacetylation which increased the activation of NF-κB and AP-1, leading to the release of the pro-inflammatory cytokine IL-8 in human alveolar epithelial cells (Rahman et al., 2002). ROS generation has been reported to regulate histone acetylation differentially in different cell types (Kang et al., 2003; Ito et al., 2004). At this point in time the relationship between ethanol induced oxidative stress and histone acetylation in the liver remains unknown and was therefore examined here. We designed a series of experiments to pharmacologically manipulate oxidative stress using glutathione modulators, dietary antioxidants and exogenous H2O2 and investigated their effects on ethanol induced H3AcK9 in rat hepatocyte. We have also investigated if mitochondria and/or NADPH oxidase system play any role in the histone acetylation.
Materials and Method
Antibodies for histone H3 protein and polyclonal anti-acetylated histone H3-lysine 9 were purchased from Upstate Biotechnology (Lake Placid, NY). Antibody for β-actin was supplied by Sigma Company (St. Louis, MO). The goat anti-rabbit and anti-mouse immunoglobulin G (IgG) conjugated with horseradish peroxidase (HRP) and Bio-Rad DC protein assay kit were purchased from Bio-Rad Laboratories (Hercules, CA). RNeasy Mini Kit and DCF-DA were bought from Qiagen (Valencia, CA) and Invitrogen (Eugene, Oregon), respectively. RETROscript was purchased from Ambion (Austin, TX). Ethanol (≥98% pure) was acquired from Fisher Scientific (Fair Lawn, NJ). All other chemicals were obtained from Sigma Company (St. Louis, MO).
Isolation and Culture of Rat Hepatocytes
Rat hepatocytes were isolated from male Sprague-Dawley rats (200–250 g) using an in situ liver perfusion with collagenase (Choudhury and Shukla, 2008). Trypan blue exclusion showed 90%-95% hepatocyte viability. Hepatocytes were plated on collagen-coated dishes (7 × 106 cells/ 100 mm dish) in DMEM containing 10% FBS, 2 mM l-glutamine and 100 units /ml penicillin and 100 μg /ml streptomycin. The animal protocols were approved by the University of Mssouri Institutional Animal Care and Use Committee (IACUC).
Treatment of Cells
Hepatocytes were allowed to attach to culture dishes for 2 hr and then treated with different concentrations of modulators, inhibitors or other agents in DMEM containing 0.1% FBS, 2 mM l-glutamine and 100 units/ml penicillin and 100 μg/ml streptomycin for 1 hour. Ethanol was next added as needed for different experiments (see result section). Duplicate sets of culture dishes were used for each treatment. The petridishes were sealed with parafilm and clingwrap to avoid the evaporation of ethanol.
Preparation of Nuclear Extract
Primary culture of hepatocytes were washed twice with cold 1× PBS, scraped, and resuspended in hypotonic lysis buffer containing 20 mM HEPES, 1 mM EDTA, 10 mM NaCl, 1 mM DTT, 1 mM sodium orthovanadate, 2 mM MgCl2, 20 mM glycerophosphate and 1 mM PMSF with 10 μg/ml of leupeptin, aprotinin, and pepstatin A and 0.25% NP-40. Lysates from the duplicate dishes were combined and then subjected to 8 passages through 26-gauge syringe needle. Nucleus were pelleted by centrifugation at 14,000·g for 20 seconds. It was resuspended in the buffer containing 20 mM Hepes, 1 mM EDTA, 420 mM NaCl, 1 mM dithiothreitol (DTT), 20 mM glycerophosphate, 1 mM sodium orthovanadate, 2 mM MgCl2, 1 mM PMSF, 10 μg/ml leupeptin, aprotinin, pepstatin A; 25% glycerol, and 0.25% NP-40, sonicated and re-centrifuged at 14,000g for 20 seconds (4°C). Protein concentration was measured by the Bio-Rad DC protein assay kits.
Western Blot Analysis
Equal amounts (40 μg) of nuclear extracts were run on 15% SDS–PAGE and transferred onto nitrocellulose membrane and then blocked with 5% nonfat dried milk for 1.5 hours. After washing the membrane with 1× TBST, membrane was incubated with primary antibody with the dilution of 1:2,000 for anti-H3AcK9. When incubating with specific acetylated lysine H3 antibody, the same membrane was also incubated at the same time with 0.25 μg /ml concentration of antibody for β-actin, overnight at 4°C Membranes were incubated with horseradish-conjugated secondary antibody for 1 hour at room temperature. The horseradish peroxidase was detected by enhanced chemiluminescence (ECL, Pierce). The band density was measured using Bio-Rad quantity-1 software. This band quantification was always done in the linear range of X-ray film sensitivity by selecting appropriate exposure time. This was tested and confirmed using standard amount of protein and various exposure times. The figures in this mansucript show data from 3-4 separate experiments together with western blot image of representative experiments.
Measurement of reactive oxygen species
For the assessment of ROS produced by ethanol, cells were grown in 6 well plates for 23 hr in phenol free DMEM media and the DCF-DA assay was used. The method was slightly modified (instead of HKRB buffer, phenol free media was used) from previously described technique (Sheehan et al, 1997). DCF-DA (10 μM) was added to the wells and incubated for 1hr. During this time they were shielded from light. After 1 hr the media containing excess DCF was removed from the cells and replaced with fresh DMEM media. Fluorescent intensity of the cells was measured at 485 nm excitation and 520 nm emission using a Packard Fusion plate reader (Fig. 1). In subsequent experiments where effects of various agents on ROS production were examined (Fig. 8), we used a Fuji LAS-3000 imager. In this latter case DCF (30 μM) was added to wells in the dark followed by washing and replacement with the fresh DMEM as above. The imager measured fluorescence in the linear range before saturation. The background values were appropriately subtracted using Fuji Multi-gauge software before the data were calculated and analysed.
Figure1. Ethanol induced ROS generation in hepatocytes.
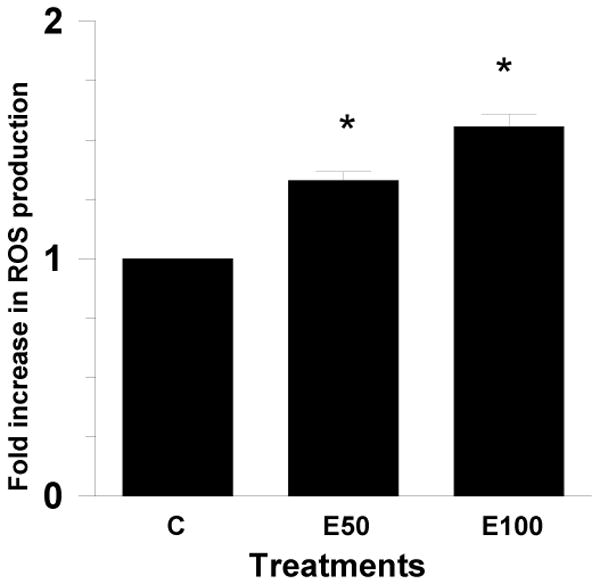
Primary culture of rat hepatocytes were treated with 50 mM or 100 mM ethanol in 6 well plates for 23 hr. DCF-DA (10 μM) was added to the wells. After 1 hr, the media was removed from the wells and replaced with fresh phenol free DMEM media. Fluorescent intensity of the cell was measured at 485 nm excitation and 520 nm emission in a Packard Fusion plate reader. Data represent mean ± SE (bar), n=3 experiments. Values correspond to fold increase compared to control. *p<0.001 (Ethanol compared to Control) (C=Control; E50=50 mM Ethanol; E100=100 mM Ethanol).
Figure 8. Levels of ROS in hepatocytes treated with anti- and pro-oxidants in the presence and absence of ethanol.

Primary culture of hepatocytes were treated with various antioxidants (top panel) or pro-oxidants (bottom panel) for 1 h. Ethanol (50 mM) was then added and cells grown for 23 hr. Levels of ROS were determined using DCF dye as detailed in the Methods section. Results are mean ± SEM (n=3 experiments). The concentration of various antioxidants used in Fig. 8 top panel: Apocyanin, 1 mM; NAC, 10 mM; Quercetin, 5 mM; Reseveratrol, 0.5 mM. Concentrations used for pro-oxidants in Fig. 8 bottom panel were as follows: Antimycin, 25 μM; BSO, 1 mM; H2O2, 50 μM; Rotenone, 1 μM.
RNA isolation and RT-PCR
Hepatocytes were treated with various modulators in the presence or absence of ethanol for 24 h and RNA was isolated using Qiagen RNAeasy kit. Two micrograms of total RNA were reverse transcribed with 100 units of Moloney murine leukemia virus reverse transcriptase at 55°C for 60 min, and 92°C for 10 min. Each cDNA preparation was amplified by polymerase chain reaction (PCR). The PCR conditions for ADH I were 45 s at 94°C, 2 min at 55°C, and 2 min at 70°C for 35 cycles. The sequences for the primers were 5′-ACCATCGAGGACATAGAA-3′ (Forward) and 5′-GTGGAGCCTGGGGTCAC-3′ (Reverse). For consistency, GAPDH mRNA was used as the internal control. The PCR products were visualized by 1.5% agarose-ethidium bromide gel electrophoresis.
Statistical Analysis
Each experiment was performed 3-5 times. Each figure was created from 3 separate experiments. Statistical analyses were performed using a standard one-way ANOVA. Values with p < 0.05 were considered significant.
Results
In order to investigate the role of oxidative stress in ethanol induced histone acetylation, we have employed various experimental approaches. This includes pharmacological agents to increase (eg. 1-buthionine sulfoximine, BSO; H2O2) or decrease (eg. resveratrol; quercetin; N-acetyl cystein, NAC) oxidative stress and study their effects on H3AcK9. We also studied the source of the oxidative stress and its relationship to ADH1 gene expression. The in vivo peripheral blood concentration of ethanol in chronic alcoholics can reach 50 mM (Deitrich and Harris, 1996) and was therefore selected concentration in this study. The concentration ranges of the modulators were chosen from the literature. A range of concentration of the reagents was used in preliminary experiments to select dose(s) for additional studies that was nontoxic to hepatocytes cultures as assessed by trypan blue dye exclusion. Under the treatment conditions described in here, we also did not see any detachment of the cells from the culture dishes, further suggesting that the cell viability, based on these two parameters, was not compromised. Ethanol induced H3 acetylation at lys9 was analyzed by western blotting using anti-acetyl histone H3 lysine 9 antibody. We have reported earlier that treatment of hepatocytes with ethanol for 1 to 24 hr did not alter the protein levels of β-actin (an endogenous housekeeping protein), or histone H3 (Park et al, 2005; Choudhury et al, 2008). As a loading control for western blots, samples were probed with antibodies for β-actin or histone H3 in the present investigation and bands are shown. This further confirmed that the ethanol or the oxidative stress did not change protein levels of β-actin or histone H3.
ROS production in ethanol treated hepatocytes
Ethanol, 50 mM and 100 mM, treatment of hepatocytes for 24 hr increased ROS production by 1.3 and 1.5 fold, respectively, over control (p<0.001) (Fig. 1). These results are in agreement with other reports demonstrating increased ROS by ethanol in hepatocytes (Lee and Shukla, 2005; Cabrales-Romero et al., 2006). It may be mentioned that after 24 hr about 78 % (78.8 mM) of the added ethanol (100 mM) was still present in the medium (Weng et al, 2000).
Role of glutathione modulators in ethanol induced H3AcK9 and ADH1 gene expression
Cellular redox potential is largely determined by glutathione (GSH). GSH is important in protecting the organelle from ROS produced during coupled mitochondrial electron transport and oxidative phosphorylation. We, therefore, investigated the role of oxidative stress in ethanol induced histone acetylation using GSH precursor NAC or GSH depletor BSO (an inhibitor of glutamate-cystein ligase). It is extensively documented that NAC increases GSH and BSO decreases it, in hepatocytes (Zafarullah et al, 2003; Sinbandhit-Tricot et al, 2003; Chen et al, 2005; McVicker et al, 2009). We have shown earlier that antioxidant NAC diminished the ethanol or hydrogen peroxide induced ROS in hepatocytes (Lee and Shukla, 2005) under conditions used here. NAC (10 mM) itself did not affect the histone acetylation, but when hepatocytes were pretreated with NAC for 1 hr followed by 23 hr with 50 mM ethanol, it ameliorated ethanol induced acetylation by 46% (p<0.01) (Fig. 2a). On the other hand, 1 mM BSO significantly increased H3AcK9 on its own by 1.9 fold (p<0.05) and it amplified ethanol induced acetylation by 1.8 fold (ethanol=2.9 fold and ethanol +BSO=4.7 fold) (p<0.001) (Fig. 2b).
Figure 2. Effect of glutathione modulators on ethanol induced histone H3K9 acetylation in hepatocytes.
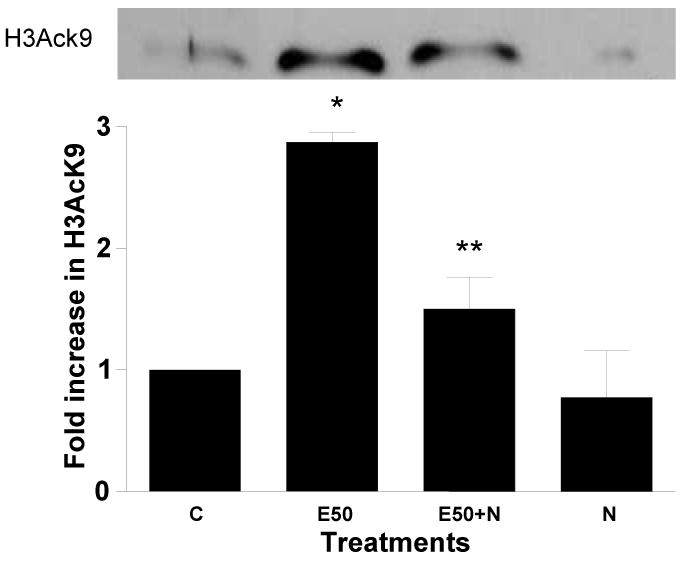
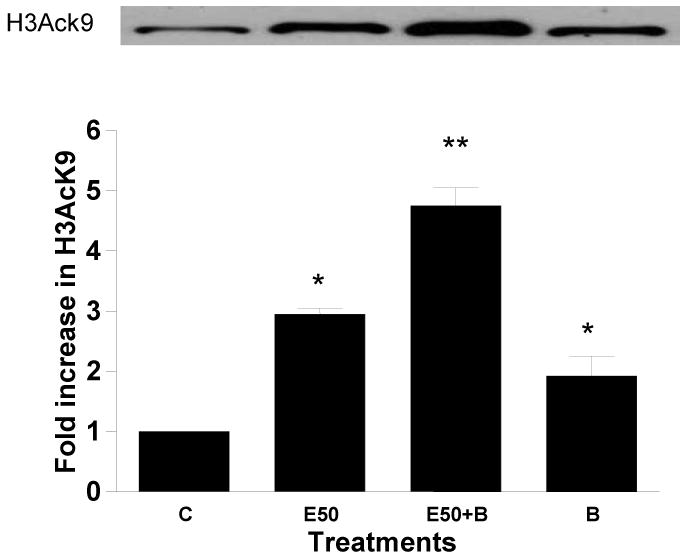
Hepatocytes were pretreated with a) 10 mM NAC or b) 1 mM BSO for 1 hr followed by with or without 50 mM ethanol for 23 hr. Nuclear extracts were prepared for immunoblot analysis to detect acetylated histone H3 at lys9. Identical amounts (40 μg) of extract were subjected to 15% SDS–PAGE and transferred onto nitrocellulose membrane. H3AcK9 levels were monitored using anti-H3 AcK9 antibody and ECL detection. A western blot from a representative experiment is shown. Quantitative analysis of acetylated histone H3 was performed by densitometric analysis and is presented in histogram as mean ± SE (bar), n = 3 experiments. Values represent fold increase compared to control (C=Control; E50=50 mM Ethanol; N=10mM N-acetyl cystein; E50+N=50 mM Ethanol+10 mM N-acetyl cystein; B=1 mM Buthionine -S-sulfoxamine; E50+B=50 mM Ethanol+1 mM Buthionine -S-sulfoxamine). Fig. 2a) *p<0.01(E compared to C) and **p<0.01 (E50 + N compared to E50). Fig. 2b) *p<0.001 (E compared to C), **p<0.001 (E compared to E + B), *p<0.05 (C compared to B).
In a similar approach as above, we also examined the relationship between ethanol induced oxidative stress and ADH1 gene expression. We have previously shown using chromatin immunoprecipitation (CHIP) assay that ethanol induced ADH1 gene expression correlates with histone H3 acetylation at lys9 (Park et al., 2005). We pretreated hepatocyte with 10 mM NAC or 1 mM BSO for 1 hr followed by 50 mM ethanol for 23 hr. Cells were also treated with only 10 mM NAC or 1 mM BSO. Subsequently, we examined the ADH1 mRNA expression with RT-PCR. Ethanol (50 mM) increased the ADH1 gene expression by 1.3 fold and when combined with BSO, ADH1 gene expression went up to 1.8 fold. NAC decreased ethanol induced ADH1 gene expression by 15% and these changes were statistically significant (Fig. 3). The fact that pharmacological manipulations to increase or decrease the ethanol induced oxidative stress showed a concomitant change in both H3AcK9 level and ADH1 gene expression supports a relationship among oxidative stress, H3AcK9 and ADH1 expression.
Figure 3. Effect of NAC and BSO in ADH1 gene expression.
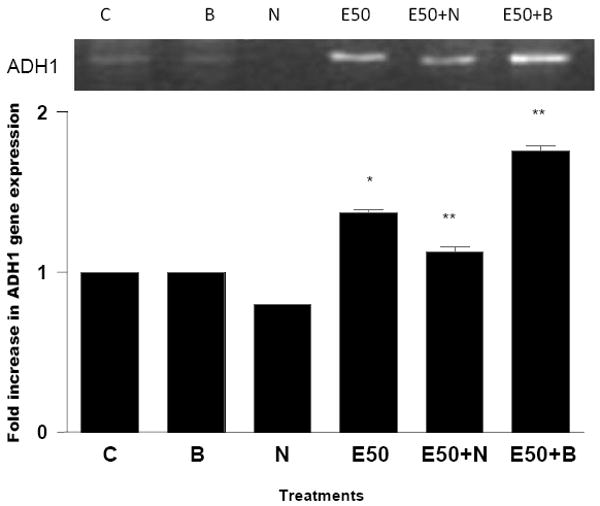
Hepatocytes were pretreated with 10 mM NAC or 1 mM BSO for 1 hr. Then cells were stimulated with 50 mM ethanol for 23 hr. Total RNA was isolated and reverse transcribed to cDNA. cDNA were amplified by PCR with ADH1 primers and PCR products were visualized by 1.5% agarose gel elctrophoresis (C=Control; E50=50 mM Ethanol; N=10 mM N-acetyl cystein; B=1 mM Buthionine -S-sulfoxamine; E50+N=50 mM Ethanol+ 10 mM N-acetyl cystein; E50+B=50 mM Ethanol+1 mM Buthionine-S-sulfoxamine). *p<0.001 (E compared to C), **p<0.001 (E compared to E + B or E + N).
Effects of dietary antioxidants on ethanol induced H3AcK9
Resveratrol or quercetin, used at different concentrations did not show any effect on histone acetylation. When hepatocytes were pretreated (1 hr) with resveratrol (0.5 mM and 5 mM) followed by 50 mM ethanol for 23 hr, histone acetylation was decreased to near basal level (p<0.01) (Fig. 4a). A similar effect was seen when hepatocytes were preincubated (1hr) with 5 mM or 20 mM quercetin followed by treatment with ethanol (50 mM) for 23 hr. Quercetin decreased ethanol induced H3AcK9 by 56% (p<0.001) (Fig. 4b).
Figure 4. Effect of dietary antioxidants on ethanol induced histone H3 acetylation in hepatocytes.
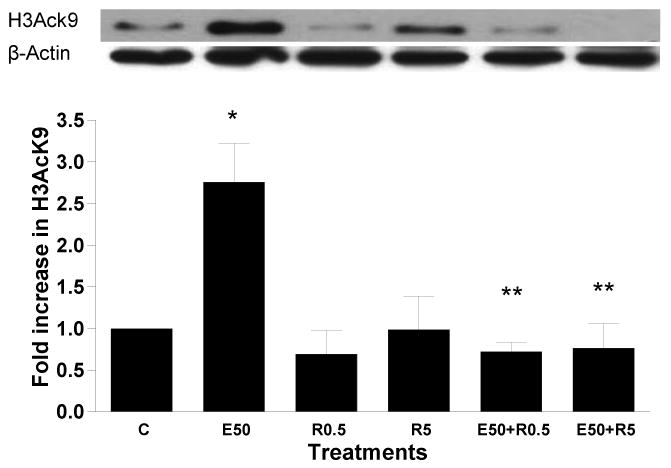
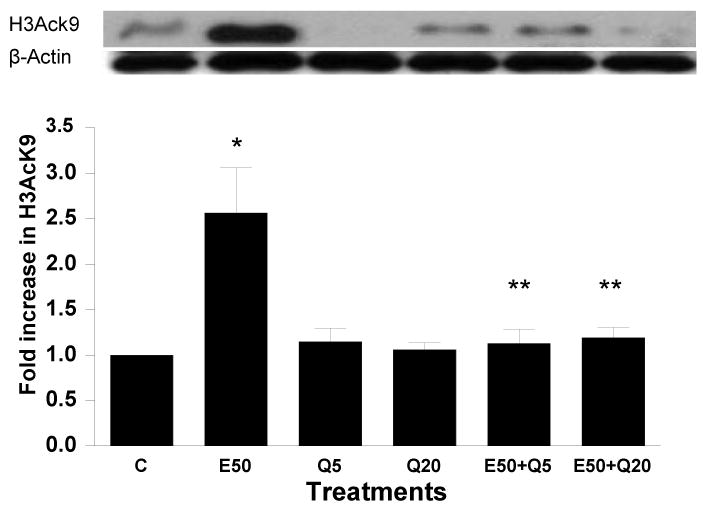
Hepatocytes were pretreated with a) resveratrol (0.5 mM or 5 mM) and b) quercetin (5 or 20 mM) for 1 hr. Then cells were stimulated with 50 mM ethanol for 23 hr. Nuclear extracts were used for Western blot analysis as in Fig. 2. Samples were probed with anti H3Ack9, or antibody for β-actin (loading control). Quantitative analysis of acetylated histone H3 was performed by densitometric analysis and is presented as mean ± SE (bar), n = 3 experiments. A representative blot is also shown. Values represent fold increase compared to control. (C=Control; E50=50 mM Ethanol; R0.5=0.5 mM Resveratrol; R1=1 mM Resveratrol; E50+R0.5=50 mM Ethanol+0.5 mM Resveratrol; E50+R1=50 mM Ethanol+1 mM Resveratrol; Q5=5 mM Quercetin; Q20=20 mM Quercetin; E50+ Q5=50 mM Ethanol+5 mM Quercetin; E50+ Q20=50 mM Ethanol+20 mM Quercetin). Fig. 4a) *p<0.01 (E50 compared to C), **p<0.01 (E50 compared to E50 + R0.5/R5); Fig. 4b) *p<0.01 (E50 compared to C), **p<0.01 (E50 compared to E50 + Q5/Q20).
Effect of exogenous H2O2 on ethanol induced H3AcK9
ROS include oxygen ions, free radicals, and peroxides, both inorganic and organic. Hydrogen peroxide is damaging to the cell because it can easily transform into a hydroxyl radical (via reaction with Fe2+: Fenton reaction), one of the most destructive free radicals. Hepatocytes treated with 50 μM H2O2 for 24 hr showed 2 fold increase in the acetylation of H3K9 (Fig. 5). Hepatocytes preincubated with 50 μM H2O2 (1hr) followed by 50 mM ethanol for 23 hr showed 4.6 fold increase in H3AcK9 (p<0.001, Fig. 5a). In another set of experiment treatment of hepatocytes with H2O2 increased H3AcK9 levels that was attenuated by NAC, but the increases in ADH1 were too small to discern, and hence no affect of NAC was noted on ADH 1 (Fig. 5b). This may be an inherent problem with the use of a highly active agent like H2O2 and the limitations associated with the use of such approaches. It may be noted that H2O2, and the resulting oxidative stress, did not alter levels of histone H3 protein (Fig. 5b).
Figure 5. Effect of exogenous hydrogen peroxide (H2O2) treatment on ethanol induced histone H3 acetylation in hepatocytes.
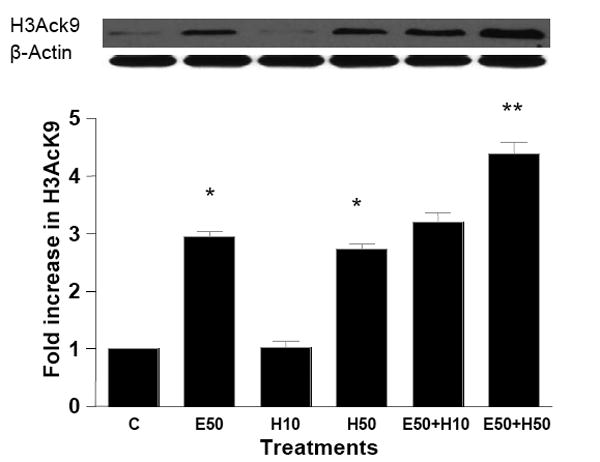
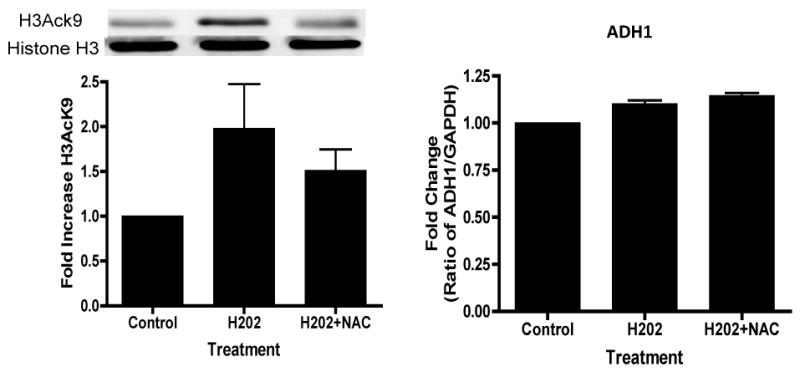
(A) Hepatocytes were pretreated with H2O2 (10 or 50 μM H2O2) for 1 hr. Subsequently, cells were stimulated with 50 mM ethanol for 23 hr. Acetylated histone H3 at lys9 was measured as described in the Materials and Methods and Fig. 2. Values represent the mean of 3 separate experiments, with the ±SE indicated by vertical lines. Values represent as fold increase compared to control (C=Control; E50=50 mM Ethanol; H10=10 μM Hydrogen peroxide; H50=50 μM Hydrogen peroxide; E50+ H10=50 mM Ethanol+10 μM Hydrogen peroxide; E50+ H50=50 mM Ethanol+50 μM Hydrogen peroxide). *p<0.001 (C compared to E50 and H50), **p<0.001 (E compared to E50+ H50). (B) Hepatocytes were treated with 50 μM H2O2 for 24 hrs in the presence or absence of NAC (10 mM) and the levels of H3AcK9 or ADH1 mRNA were monitored.
Role of mitochondrial reactive oxygen species in histone H3 acetylation at lys9
The mitochondrial electron transport chain is the major intracellular source of ROS. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III (Dawson et al., 1993). Generation of this ROS is augmented by selective inhibitors of mitochondrial complex. We used these inhibitors as tools in our study. Hepatocytes were pretreated with the inhibitors of mitochondrial complex I (rotenone) and III (antimycin) for 1 hr and then incubated in the presence or absence of 50 mM ethanol for 23 hr. Hepatocytes incubated with 0.5 μM and 1 μM rotenone increased H3AcK9 by 1.5 fold and 2.9 fold, respectively (Fig. 6a). Though rotenone (0.5 μM) increased ethanol induced acetylation, it is not statistically significant compared to ethanol induced increase in H3AcK9. But, 1 μM rotenone increased ethanol induced acetylation up to 5.2 fold (p<0.01). Similarly, 0.025 mM antimycin also significantly increased ethanol induced H3AcK9 (p<0.01). Antimycin itself showed 2 fold increase in H3AcK9 compared to control (p<0.01) (Fig. 6b).
Figure 6. Effect of mitochondrial respiratory chain inhibitors on ethanol induced histone H3 acetylation in hepatocytes.
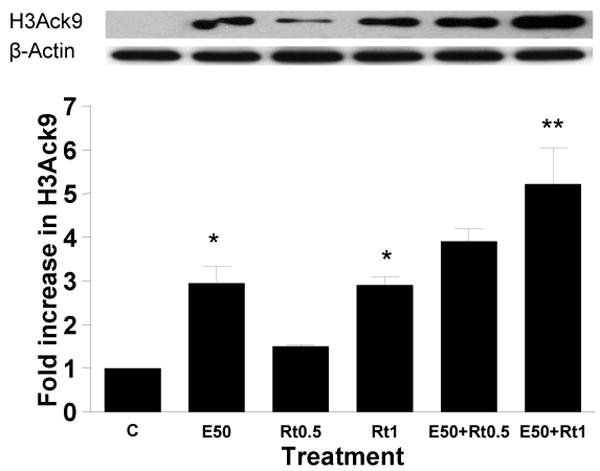
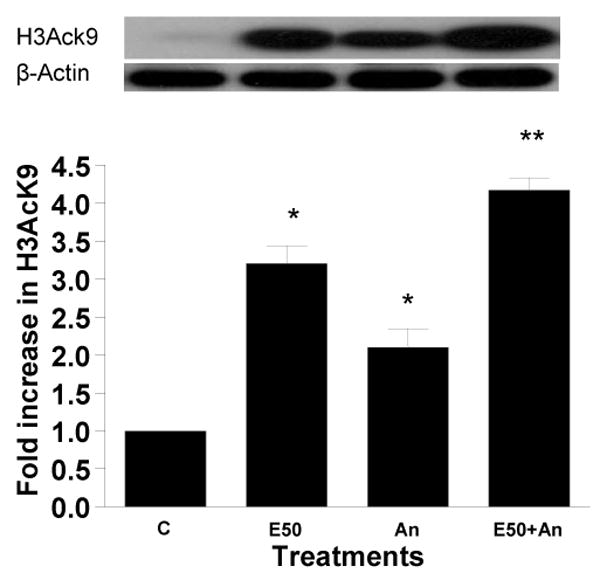
Hepatocytes were pretreated with a) rotenone (0.50 μM or 1 μM) and b) 0.025 mM antimycin for 1 hr. Then cells were treated with 50 mM ethanol for 23 hr. Equal amounts (40 μg) of nuclear extract were used for Western blot analysis to detect acetylated histone H3 at lys9. Quantitative analysis of acetylated histone H3 was performed by densitometric analysis and is presented as mean ± SE (bar), n = 3 experiments. A representative blot is also shown. Values represent as fold increase compared to control (C=Control; E50=50 mM Ethanol; Rt0.5=0.5 μM Rotenone; Rt1=1 μM Rotenone; E50+ Rt0.5=50 mM Ethanol+0.5 μM Rotenone; E50+ Rt1=50 mM Ethanol+1 μM Rotenone; An=0.025 mM Antimycin; E50+An=50 mM Ethanol+0.025 mM Antimycin) a) *p<0.05 (C compared to E and Rt1), **p<0.01 (E compared to E+ Rt1)
Another potential candidate for the ROS generation is NADPH oxidase system in hepatocytes. It is composed of two essential membrane bound components, gp91phox/Nox2 and p22phox, which comprise flavocytochrome b558, and our cytosolic componenets, p47phox, p40phox, p67phox, and the small G protein Rac1/2. Translocation of cytosolic subunits to the membrane occurs and NADPH oxidase generates superoxide anion following stimulation of cells (Babior, 1999; Kono et al., 2000). To explore if NADPH oxidase is involved in the histone acetylation, we used apocynin which has been shown to prevent the translocation of the cytosolic subunits to the membrane bound catalytic subunit, and thereby inhibits the formation of an active NADPH oxidase complex (Reinehr et al., 2005). Hepatocytes incubated with increasing concentration of apocynin decreased ethanol induced H3AcK9. Apocynin at 0.1 mM reduced the ethanol induced acetylation by 31% (p<0.01). Interestingly, at higher concentration, 1 mM apocynin reduced ethanol induced histone acetylation by 72% (p< 0.001; Fig. 7). Apocynin itself did not show any significant effect on the histone acetylation.
Figure 7. Effect of NADPH oxidase inhibitor apocynin on ethanol induced histone H3 acetylation in hepatocytes.
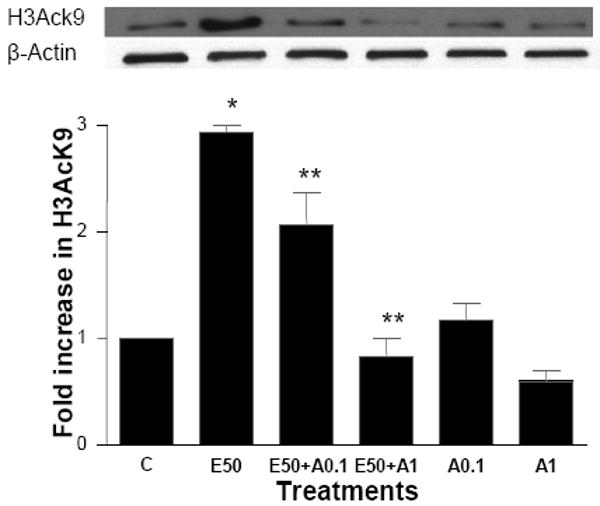
Hepatocytes were pretreated with apocynin (0.1 mM or 1 mM) for 1 hr. Then cells were stimulated with 50 mM ethanol for 23 hr. Nuclear extracts were used for Western blot analysis as in Fig. 2. A representative blot is shown. Quantitative analysis of acetylated histone H3 was performed by densitometric analysis and is presented as mean ± SE (bar), n = 3 experiments. Values represent here as fold increase compared to control (C= control; E50=50 mM Ethanol; E50+A0.1=50 mM Ethanol +0.1mM Apocynin; E50+A1=50 mM Ethanol +1mM Apocynin; 0.1A=0.1mM Apocynin; 1A=1mM Apocynin). *p<0.001 (C compared to E), **p<0.01 (E compared to E + A0.1), **p<0.001 (E compared to E + A1).
Levels of ROS production
In an effort to corelate the changes in H3AcK9 to ROS, we determined the levels of ROS production in primary cultures of hepatocytes exposed to ethanol for 24 h and analysed their levels after treatments with various anti- and pro-oxidants (with and without ethanol). As noted earlier, ethanol induced histone H3 acetylation was monitored at 24 hr, a time that showed optimal response (Park et al 2005). When hepatocytes were treated with ethanol for 24 h the levels of ROS increased by 158.9% over control (control taken as 100%; Fig. 8 top panel) and there was a concomitant rise in H3AcK9 (Fig. 2). In this case, treatment of hepatocytes with antioxidants or reducers of ROS, caused decrease in the basal and ethanol induced elevations in ROS (Fig. 8 top panel), with a concomitant decrease in H3AcK9 (Figs. 2a, 4a, 4b and 7). These data support the relationship between ethanol induced oxidative stress and histone acetylation.
However, when cells were treated with ethanol plus pro-oxidants for 24 h, then an interesting pattern emerged. In this latter case we noted the expected increases in the ROS production with the pro-oxidants alone though it did not show the expected amplification of ethanol induced ROS when combined with pro-oxidants (Fig. 8 bottom panel). On the other hand, treatment of cells for 24 h with pro-oxidants alone and as well as with ethanol, caused augmentation in H3AcK9 (Fig. 2b, 5, and 6). It is possible that treatments with pro-oxidants for 24 h disrupt the ability of the cells to produce ROS and hence a lack of increase in ROS generation in cells treated with pro-oxidants + ethanol. This also cautions the use of pro-oxidants for longer times and that such protocols have limitations. It is our interpretation that once the process of histone acetylation is triggered due to oxidative stress, it continues despite the fact that the cells ability to produce more ROS later may be diminished.
Discussion
Generation of ROS during alcohol exposure is suggested as one of the important mechanisms of alcohol-induced liver injury. ROS are considered hepatotoxic because of their potential to react with macromolecules, inactivate enzymes, cause DNA damage, modify proteins, and induce lipid peroxidation. In the present study, we have established that ethanol generated ROS play role in the histone H3 acetylation and that the ROS generated from mitochondria and NADPH oxidase, both appear to be involved.
Hepatocytes exposed to ethanol, show robust increase in ROS production within 30 min (Cabrales-Romero et al., 2006). As ROS production occurs in cells, a variety of endogenous enzymatic (superoxide dismutase, catalase, glutathione peroxide etc) and nonenzymatic (vitamin C, vitamin E etc) mechanisms have evolved against ROS to maintain the homeostasis. Some of the mechanisms are reported to be impaired after long-term alcohol consumption and may therefore contribute to the liver and other organ damage. In order to investigate the relationship between ROS and histone acetylation we chose 24 hr time point for ethanol treatments, because histone acetylation is highest in hepatocytes at this time (Park et al., 2003). We observed a significant ROS accumulation after 24 hr ethanol treatment together with increased H3AcK9 in hepatocytes. It is possible that ethanol causes an early robust ROS production (Cabrales-Romero et al., 2006) which triggers the process of increasing histone acetylation monitored at 24 hr. Studies using confocal laser scanning microscopic system suggest that active oxidants produced during ethanol metabolism modulate mitochondrial energy synthesis in isolated and cultured hepatocytes (Kurose et al., 1996). In addition, endogenous glutathione-glutathione peroxidase system serve as important antioxidants/ cytoprotective machinery in the hepatocyte exposed to ethanol. Due to its high concentration and its central role in maintaining the cells redox state, glutathione is one of the most important cellular antioxidants. In our study, hepatocytes treated with glutathione precursor NAC decreased ethanol induced histone acetylation and the glutathione depletor BSO showed the opposite result. This clearly implicates oxidative stress in ethanol induced histone H3 acetylation at lysine 9.
To pursue further the role of the oxidative stress in histone acetylation, we examined the effect of dietary antioxidants on ethanol induced H3AcK9. Resveratrol and quercetin, polyphenol compounds present in several dietary products, reduced both ethanol induced histone acetylation and ROS production. This further supports a role for ethanol induced oxidative stress in histone acetylation. It was demonstrated that resveratrol protects against oxidative stress in ethanol induced lipid peroxidation in rats (Kasdallah-Grissa et al., 2006). Notably, resveratrol activates sirtuin family of NAD-dependent histone deacetylases involved in regulation of various cellular processes including gene transcription, DNA repair and apoptosis (Howitz et al., 2003) and also alleviates alcoholic fatty liver (Ajmo et al, 2008). Quercetin also showed protective effect in case of iron overload liver injury (Zhang et al, 2006) or dimethylnitrosamine induced liver damage (Lee et al., 2003). Superoxide anion is primarily converted to H2O2 by superoxide dismuatse. Our results indicated that H2O2 also increased ethanol induced acetylation. The effect persisted up to 24 hr treatment with 50 μM exogenous H2O2. It is relevant to note that H2O2 (200 μM) has been shown to induce prolonged histone acetylation in bronchial epithelial cells (Tomita et al., 2003).
At this stage the molecular mechanism by which ethanol or its metabolites eg. acetaldehyde or acetate, elicit histone H3-acetylation remains unknown. The present study highlights a role for the ethanol induced oxidative stress in this response. The mechanism by which ROS elicits increase in histone acetylation is also not known. Several possible mechanisms can be envisaged. For example, ROS can mediate activation of histone acetyl transferase (HAT), or decrease the histone deacetylase (HDAC) activity. This may be via ROS modulation of signaling pathways. In this context it has been shown that MAP kinase is involved in the ethanol induced histone H3 acetylation (Park et al 2005), and ROS is known to affect MAP Kinase and other signaling pathways. It may be noted that, in hepatocytes, ethanol increased HAT activity (Park et al, 2005). The inhibition of ethanol-induced increase in H3AcK9 by the antioxidants resveratrol or quercetin, may be related to their influence on the GSH metabolism or on signaling pathways. It will be useful in future to identify the influence of ROS on individual HAT or HDAC, or on the co-activator/repressor components associated with these histone modifying enzymes. The present study paves the way for such investigations.
It is known that inhibition of ethanol metabolism to acetaldehyde (by 4-methyl pyrazole) or to acetate (by cynamide) reduces the acetylation of histone (Park et al, 2003). This suggested that ethanol metabolism, known to generate ROS, is involved in the histone acetylation. The present report provides evidence for the involvement of ROS. It should be mentioned that acetate, a product of ethanol metabolism, also induces histone acetylation in hepatocytes, but its mechanism may be different from ethanol induced histone acetylation (Park et al, 2005).
The mechanism by which ethanol triggers an increase in reactive oxygen species in the liver is not well understood but mitochondria may play a major role (Bailey and Cunningham, 2002; Dey and Cederbaum, 2006). It is recognized that mitochondrial oxidative stress contributes to the injury observed in several liver diseases including alcoholic liver disease (Cahill et al., 1997; Bailey et al., 1999; Bailey and Cunningham, 2002). Liver pathologies are associated with significant alterations in the functional state of the mitochondrial respiratory chain (Krahenbuhl and Reichen, 1992). Though these electron transporters are combined of four complexes (complex I-IV), data in the literature concerning mitochondrial respiratory chain function in hepatitis and cirrhosis showed that complex I and III are the key sources of ROS in the cell (Barja and Herrero 1998; Barja 1999; Bailey and Cunningham, 2002). It is assumed that complex II is the most resistant complex which is not damaged even in liver cirrhosis (Cederbaum et al., 1974). If the electron flow along the respiratory chain is interrupted (by inhibitors like complex I inhibitor rotenone and complex III inhibitor antimycin) then ROS formation increases as a result of autooxidation of the reduced electron transport components. It has been shown that both rotenone and antimycin increased hepatocyte mitochondrial ROS production, and it was further enhanced by the addition of ethanol (Bailey and Cunningham, 1998; Bailey et al., 1999). Consistent with this profile, rotenone and antimycin also increased histone acetylation and it was enhanced by ethanol (Fig. 6). This argues in favor of mitochondrial ROS as a source of ethanol induced histone acetylation.
Though hepatocytes express components of phagocytic and nonphagocytic forms of NADPH oxidase, the inhibitory action of apocynin on ethanol induced histone acetylation in rat hepatocytes earn some comment. Previous studies have shown that inhibition of NADPH oxidase by apocynin is not mediated by apocynin itself but by a compound derived from apocynin in a peroxidase-dependent way (Stolk et al., 1994). Due to this mechanism, apocynin was shown to inhibit NADPH oxidase in phagocytic cells, whereas in some (peroxidase-deficient) nonphagocytic cells, such as fibroblasts, apocynin stimulates, rather than inhibits, ROS formation (Vejrazka et al., 2003). Hepatocytes contain peroxidases and exhibit endogenous H2O2 formation, which are prerequisites for the inhibitory action of apocynin on NADPH oxidase. This may explain why in ethanol treated hepatocytes (preincubated with apocynin), histone acetylation was inhibited. Since apocynin abolished the ethanol induced histone acetylation, it suggests that NADPH oxidase system plays a role in the oxidative stress induced histone H3 acetylation. Taken together, this study demonstrates for the first time that oxidative stress, apparently generated by both mitochondria and NADPH oxidase systems, is involved in the ethanol induced acetylation of histone H3 at Lys9 in the primary cultures of rat hepatocytes.
Acknowledgments
This work was supported by NIH grant AA016347.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albano E. Alcohol, oxidative stress and free radical damage. Proc Nutr Soc. 2006;65:278–290. doi: 10.1079/pns2006496. [DOI] [PubMed] [Google Scholar]
- Ajmo J, Liang X, Rogers CQ, Pennock B, You M. Reseveratrol alleviates alcoholic fatty liver in mice. Am J Physiol. 2008;295:G833–G842. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- Bailey SM, Cunningham CC. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology. 1998;28:1318–1326. doi: 10.1002/hep.510280521. [DOI] [PubMed] [Google Scholar]
- Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2000;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- Barja G, Herrero A. Localization at complex I and mechanism of the higher free radical production of brain nonsynaptic mitochondria in the short-lived rat than in the longevous pigeon. J Bioenerg Biomembr. 1998;30:235–243. doi: 10.1023/a:1020592719405. [DOI] [PubMed] [Google Scholar]
- Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- Cabrales-Romero Mdel P, Márquez-Rosado L, Fattel-Fazenda S, Trejo-Solís C, Arce-Popoca E, Alemán-Lazarini L, Villa-Treviño S. S-adenosyl-methionine decreases ethanol-induced apoptosis in primary hepatocyte cultures by a c-Jun N-terminal kinase activity-independent mechanism. World J Gastroenterol. 2006;12:1895–1904. doi: 10.3748/wjg.v12.i12.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill A, Wang X, Hoek JB. Increased Oxidative Damage to Mitochondrial DNA Following Chronic Ethanol Consumption. Biochem Biophys Res Commun. 1997;235:286–290. doi: 10.1006/bbrc.1997.6774. [DOI] [PubMed] [Google Scholar]
- Cederbaum AI, Lieber CS, Rubin E. Effects of chronic ethanol treatment of mitochondrial functions damage to coupling site I. Arch Biochem Biophys. 1974;165:560–569. doi: 10.1016/0003-9861(74)90283-5. [DOI] [PubMed] [Google Scholar]
- Chen T, Pearce LI, Peterson J, Stoyanovsky D, Billiar T. Glutathione depletion renders rat hepatocytes sensitive tonitric oxide donor-mediated toxicity. Hepatology. 2005;42:598–607. doi: 10.1002/hep.20813. [DOI] [PubMed] [Google Scholar]
- Choudhury M, Shukla SD. Surrogate alcohols and their metabolites modify histone H3 acetylation: involvement of histone acetyl transferase and histone deacetylase. Alcohol Clin Exp Res. 2008;32:829–839. doi: 10.1111/j.1530-0277.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- Dawson TL, Gores GJ, Nieminen AL, Herman B, Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol. 1993;264:961–967. doi: 10.1152/ajpcell.1993.264.4.C961. [DOI] [PubMed] [Google Scholar]
- Deitrich RA, Harris RA. How much alcohol should I use in my experiments? Alcohol Clin Exp Res. 1996;20:1–2. doi: 10.1111/j.1530-0277.1996.tb01033.x. [DOI] [PubMed] [Google Scholar]
- Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:63–74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression, role of tyrosine nitration. Biochem Biophys Res Commun. 2004;315:240–245. doi: 10.1016/j.bbrc.2004.01.046. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kang J, Zhang Y, Chen J, Chen H, Lin C, Wang Q, Ou Y. Nickel-induced histone hypoacetylation: the role of reactive oxygen species. Toxicol Sci. 2003;74:279–286. doi: 10.1093/toxsci/kfg137. [DOI] [PubMed] [Google Scholar]
- Kasdallah-Grissa A, Mornagui B, Aouani E, Hammami M, Gharbi N, Kamoun A, El-Fazaa S. Protective effect of resveratrol on ethanol-induced lipid peroxidation in rats. Alcohol Alcohol. 2006;41:236–239. doi: 10.1093/alcalc/agh256. [DOI] [PubMed] [Google Scholar]
- Kono H, Rusyn I, Yin M, Gäbele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krähenbühl S, Reichen J. Adaptation of mitochondrial metabolism in liver cirrhosis. Different strategies to maintain a vital function. Scand J Gastroenterol Suppl. 1992;193:90–96. doi: 10.3109/00365529209096012. [DOI] [PubMed] [Google Scholar]
- Kundu, Dasgupta . Subcellular Biochemistry: Chromatin and Disease. Springer Publisher; 2007. [Google Scholar]
- Kurose I, Higuchi H, Kato S, Miura S, Ishii H. Ethanol-induced oxidative stress in the liver. Alcohol Clin Exp Res. 1996;20:77–85. doi: 10.1111/j.1530-0277.1996.tb01736.x. [DOI] [PubMed] [Google Scholar]
- Lee ES, Lee HE, Shin JY, Yoon S, Moon JO. The flavonoid quercetin inhibits dimethylnitrosamine-induced liver damage in rats. J Pharm Pharmacol. 2003;55:1169–1174. doi: 10.1211/0022357021396. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Shukla SD. Pro- and anti-apoptotic roles of c-Jun N-terminal kinase (JNK) in ethanol and acetaldehyde exposed rat hepatocytes. Eur J Pharmacol. 2005;508:31–45. doi: 10.1016/j.ejphar.2004.12.006. [DOI] [PubMed] [Google Scholar]
- McVicker BL, Tuma PL, Kharbanda KK, Lee SML, Tuma DJ. Relationship between oxidative stress and hepatic glutathione levels in ethanol-mediated apoptosis of polarized hepatic cells. World J Gastroenterol. 2009;15:2609–2616. doi: 10.3748/wjg.15.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinobu A, Kanno Y, O'Shea JJ. Discrete roles for histone acetylation in human T helper 1 cell-specific gene expression. J Biol Chem. 2004;279:40640–40646. doi: 10.1074/jbc.M407576200. [DOI] [PubMed] [Google Scholar]
- Park PH, Miller R, Shukla SD. Acetylation of histone H3 at lysine 9 by ethanol in rat hepatocytes. Biochem Biophys Res Commun. 2003;306:501–504. doi: 10.1016/s0006-291x(03)01040-4. [DOI] [PubMed] [Google Scholar]
- Park PH, Lim RW, Shukla SD. Involvement of histone acetyltransferase (HAT) in ethanol-induced acetylation of histone H3 in hepatocytes: potential mechanism for gene expression. Am J Physiol Gastrointest Liver Physiol. 2005;289:1124–1136. doi: 10.1152/ajpgi.00091.2005. [DOI] [PubMed] [Google Scholar]
- Rahman I, Gilmour PS, Jimenez LA, MacNee W. Oxidative stress and TNF-α induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002;234-235:239–248. [PubMed] [Google Scholar]
- Reinehr R, Becker S, Eberle A, Grether-Beck S, Häussinger D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem. 2005;280:27179–2794. doi: 10.1074/jbc.M414361200. [DOI] [PubMed] [Google Scholar]
- Reuben A. Alcohol and the liver. Curr Opin Gastroenterology. 2008;l24:328–338. doi: 10.1097/MOG.0b013e3282fbceca. [DOI] [PubMed] [Google Scholar]
- Sheehan JP, Swerdlow RH, Mller SW, Davis RE, Parks JK, Parker WD, Tuttle JB. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer's disease. J Neurosci. 1997;17:4612–4622. doi: 10.1523/JNEUROSCI.17-12-04612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SD, Velazquez J, French S, Lu S, Ticku M, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32:1525–1534. doi: 10.1111/j.1530-0277.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- Sinbandhit-Tricot S, Cillard J, Chevanne M, Morel I, Cillard P, Sergent O. Glutathione depletion increases nitric oxide-induced oxidative stress in primary rat hepatocyte cultures: involvement of low molecular weight iron. Free Radical Biol Med. 2003;34:1283–1294. doi: 10.1016/s0891-5849(03)00108-4. [DOI] [PubMed] [Google Scholar]
- Smith CL. A shifting paradigm: histone deacetylases and transcriptional activation. Bioessays. 2008;30:15–24. doi: 10.1002/bies.20687. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- Tomita K, Barnes PJ, Adcock IM. The effect of oxidative stress on histone acetylation and IL-8 release. Biochem Biophys Res Commun. 2003;301:572–577. doi: 10.1016/s0006-291x(02)03029-2. [DOI] [PubMed] [Google Scholar]
- Vejrazka M, Micek R, Stipek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim Biophys Acta. 2005;1722:143–147. doi: 10.1016/j.bbagen.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Weng Y, Shukla SD. Ethanol alters Angiotensin II stimulated MAP kinase in hepatocytes: agonist selectivity and ethanol metabolic dependence. Eur J Pharm. 2000;398:323–331. doi: 10.1016/s0014-2999(00)00313-7. [DOI] [PubMed] [Google Scholar]
- Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li H, Zhao Y, Gao Z. Dietary supplementation of baicalin and quercetin attenuates iron overload induced mouse liver injury. Eur J Pharmacol. 2006;535:263–269. doi: 10.1016/j.ejphar.2006.01.067. [DOI] [PubMed] [Google Scholar]