

Abstract
Rationale
Macrophages change their phenotype and biological functions depending on the microenvironment. In atherosclerosis, oxidative tissue damage accompanies chronic inflammation, however, macrophage phenotypic changes in response to oxidatively modified molecules are not known.
Objective
To examine macrophage phenotypic changes in response to oxidized phospholipids that are present in atherosclerotic lesions.
Methods and Results
We show that oxidized phospholipid-treated macrophages develop into a novel phenotype (Mox), strikingly different from the conventional M1 and M2 macrophage phenotypes. Compared to M1 and M2, Mox show a different gene expression pattern, as well as decreased phagocytotic and chemotactic capacity. Treatment with oxidized phospholipids induces both M1 and M2 macrophages to switch to the Mox phenotype. Whole genome expression array analysis and subsequent gene ontology clustering revealed that the Mox phenotype was characterized by abundant over-representation of Nrf2-mediated expression of redox-regulatory genes. In macrophages isolated from Nrf2−/− mice, oxidized phospholipid-induced gene expression and regulation of redox status were compromised. Moreover, we found that Mox macrophages comprise 30% of all macrophages cells in advanced atherosclerotic lesions of LDL receptor knockout mice.
Conclusions
Together, we identify Nrf2 as a key regulator in the formation of a novel macrophage phenotype (Mox) that develops in response to oxidative tissue damage. The unique biological properties of Mox suggest this phenotype may play an important role in atherosclerotic lesion development as well as in other settings of chronic inflammation.
Keywords: Macrophages, Oxidized Phospholipids, Atherosclerosis, Nrf2
Introduction
Functional heterogeneity is a hallmark of cells of the mononuclear phagocyte system. Macrophages have both, pro- and anti-inflammatory properties, as they orchestrate the initiation, but also the resolution phases of inflammation 1, 2. This plasticity in the functional properties of macrophages is determined by the immunological microenvironment. Macrophages, classically activated by IFNγ and/or microbial products display an M1 phenotype that is characterized by high expression of IL-12, iNOS and TNFα and CD86, which triggers type I inflammation and a Th1 response. In contrast, M2 macrophages include various forms of alternatively activated macrophages that were exposed to IL-4, IL-10 or IL-13, immune complexes or glucocorticoid 3. M2 macrophages are defined in mice as cells high in arginase 1 (Arg-1) among other markers, but low in iNOS and IL-12 production 4, 5, and promote a type II inflammation and a Th2 response. In humans different M2 markers have been described 6. Macrophages isolated during the resolution phase of inflammation (rM) were shown to be hybrids of the classically (M1) and alternatively (M2) activated phenotype 2. Tumour-associated macrophages (TAMs) have many features of M2 macrophages; however, the hypoxic environment in tumors may profoundly change their phenotype 7 and thus these cells may represent a separate phenotype 3.
Atherosclerosis is regarded as a chronic inflammatory disease, where accumulation of monocytes in the subendothelial space and differentiation into macrophages are key events in the early stages as well as during propagation of the disease. However, little is known about macrophage phenotypes in atherosclerotic lesions. Macrophage heterogeneity has been reported 8 and both M1 and M2 macrophages have been shown to be present in atherosclerotic lesions 9. The importance of macrophage phenotypic modulation in atherosclerosis has been recently highlighted 10.
During atherogenesis, the oxidation of accumulated low-density lipoprotein (LDL) and apoptotic cell membranes in the subendothelial space11 leads to the enrichment of the tissue with oxidized lipids12. Oxidation of phospholipids such as 1-palmitoyl-2-arachidonoyl-sn-3-glycero-phosphorylcholine (PAPC) yields a series of structurally defined oxidation products (OxPAPC) 13–15, which cause pro- as well as anti-inflammatory effects 16. We have recently shown that oxidized phospholipids are capable of specifically recruiting monocytes/macrophages in vivo, a process that requires the chemokine receptor CCR2 17. However, the effect of an oxidized phospholipid-rich microenvironment on macrophage phenotypic polarization remained unclear. We demonstrate here that phospholipid oxidation products that accumulate in atherosclerotic lesions affect macrophage gene expression and function resulting in development of a unique macrophage phenotype that involves the redox regulated transcription factor Nrf218.
Materials and Methods
For detailed procedures see supplemental Materials provided online at circres.ahajournals.com.
Expression Array Analysis
Macrophages were treated for 3 hours and gene expression was analyzed using Mouse Genome 430 2.0 Arrays (Affymetrix, Santa Clara, CA).
Oxidized phospholipid preparation and analysis
OxPAPC was produced by air oxidation and analyzed by positive ion electrospray mass spectrometry (ESI-MS) as described previously 14, 19.
Generation of bone-marrow-derived macrophages (BMDM) and phenotypic polarization
BMDM were cultured and phenotypically polarized with either 10U/ml IFNγ plus 1μg/ml LPS to generate M1, 10ng/ml IL-4 to generate M2, or 50μg/ml of OxPAPC to generate Mox macrophages.
Statistical analysis
Results are expressed as mean ± SD. Statistical analysis was performed using one-way ANOVA. A p-value of less than 0.05 was considered statistically significant.
Results
Oxidized phospholipids induce a macrophage phenotype (Mox) that is distinct from conventional M1 and M2 phenotypes
To test the effects of oxidized phospholipids on macrophages in vitro, we used primary mouse bone marrow-derived macrophages (BMDM) that had been cultivated in the presence of M-CSF for 7 days. The purity of our cultures was greater than 95%, as assessed by FACS-analysis of CD68 and F4/80 (not shown). Furthermore, the cells expressed only low levels of MHC II, resembling resting, non-activated macrophages (not shown). Incubation of macrophages with concentrations up to 75ug/ml of oxidized phospholipids neither induced apoptotic nor necrotic cell death (Fig. S1). To examine different macrophage phenotypes, cells were either left untreated (M0) or phenotypically polarized in the presence of the M1 stimulus IFNγ plus LPS, the M2 stimulus IL-4, or OxPAPC (Mox).
Initial comparison of macrophage phenotypes demonstrated distinct morphological features as well as different biological functions. Actin (not shown) and tubulin staining revealed striking differences in cytoskeletal organization of the different macrophage phenotypes (Fig. 1A). To test chemoattracting properties, we used conditioned media from different macrophage phenotypes and examined their effect on THP-1 cell migration in modified Boyden chambers. Both M1 and M2 macrophages induced THP-1 cell migration, while Mox were significantly less effective (Fig. 1B). We next analyzed phagocytotic capacity of the different macrophage phenotypes. Engulfment assays were conducted using either 2μm fluorescent carboxylate beads or apoptotic thymocytes. Our results show that phagocytic capacity of Mox was significantly impaired compared to M1 or M2 macrophages (Fig. 1C, D).
Figure 1. Mox macrophages represent a functionally distinct phenotype.

Bone marrow-derived macrophages were polarized with either 10U/ml IFNγ plus 1μg/ml LPS (M1) or 10ng/ml IL-4 (M2), 50μg/ml OxPAPC (Mox) or medium only (M0) for 18 hours. Cells were stained with an anti-tubulin antibody and analyzed by epifluorescence microscopy (A), bar indicates 20μm. (B) Migration of THP-1 cells to supernatants from macrophages treated as indicated, n=2. (C) Phagocytosis of carboxylate beads (left) and apoptotic murine thymocytes (right) by bone marrow-derived macrophages treated for 8 hrs as indicated, n=2.
Whole-genome expression array analysis revealed that treatment of macrophages with oxidized phospholipids induced a specific gene expression pattern, which was strikingly different from that of conventional M1 and M2 phenotypes (Fig. 2A). 1255 genes were exclusively upregulated in M1 (Table S1), 265 in M2 (Table S2) and 119 in Mox (Table S3) macrophages. Genes that were exclusively upregulated in Mox included a cluster of redox-regulated genes such as Srx1, Gclc, Gclm, Txnrd1 and HO-1, but also the nuclear receptor Nr4A2 (Nurr1), VEGF and Trb3. Only 38 genes were upregulated in both M1 and Mox and only 13 genes were overlapping between M2 and Mox (Fig. 2B, Tables S4-S6).
Figure 2. Oxidized phospholipids induce a unique gene expression pattern.

Bone marrow-derived macrophages were stimulated with 10U/ml IFNγ plus 1μg/ml LPS (M1), 10ng/ml IL-4 (M2), 50μg/ml OxPAPC (Mox), or medium only (M0), for 3 hours. (A) Affimetrix gene array analysis and subsequent heat map generation identify unique clustering patterns for each macrophage phenotype. (B) Differentially upregulated genes were identified by selecting for the top 5% of total genes exhibiting greater than 1.5-fold induction. A Venn-diagram of overlapping genes is shown, which represents intersections of probe set IDs. Numbers represent total probe sets including duplicate gene titles as present on the array (refer to supplemental tables).
Array data were confirmed by real-time quantitative PCR, ELISA or FACS for selected genes. OxPAPC neither increased expression of the specific M2 marker arginase 1 nor the specific M1 markers iNOS (Fig. 3A), TNFα, JE/MCP-1, IL-12 or TLR2 (Fig. S2). Among genes that were exclusively upregulated by OxPAPC we confirmed HO-1 (mRNA and protein), VEGF (Fig. 3B-D), and Trb3 (not shown). Furthermore, Srxn1 and Txnrd1 (Fig. 5B, C), Dusp-1, Nurr1, Dusp-5, Thsp, (Fig. S5), GCLM and GSR1 (Fig. S6) were confirmed to be only induced in Mox. It is notable that in addition to the 38 M1/Mox overlapping genes found in the array analysis (Table S4), the pro-inflammatory M1 genes IL-1β (Figs. S2, S4) and COX-2 (Fig. S5), were also upregulated in Mox. However, their expression did not reach statistical significance in the array experiment.
Figure 3. Confirmation of genes upregulated by OxPAPC.

Bone marrow-derived macrophages were stimulated with 10U/ml IFNγ and 1μg/ml LPS (M1), 10ng/ml IL-4 (M2), indicated concentrations of OxPAPC (Mox) or medium only (co). RNA was isolated after 3 hours of stimulation, and reverse transcribed. RT-PCR was performed and expression of iNOS and arginase 1 (A), HO-1 (B) or VEGF (D) was performed. n=4, results are shown as mean average ±SD. p<0.05. For flow cytometric analysis (C), cells were stimulated with 10U/ml IFNγ and 1μg/ml LPS (M1), 10ng/ml IL-4 (M2), OxPAPC (Mox), medium only (co) or with IL-10 as positive control for 18 hours. Macrophages were fixed and stained for HO-1 using an Alexa-488 labeled rabbit anti HO-1 antibody.
Figure 5. Oxidized phospholipids induce redox-regulating genes via Nrf2.
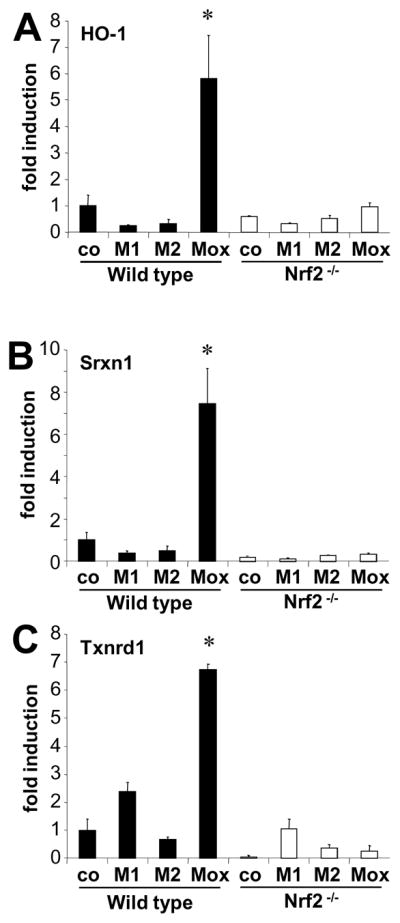
Bone marrow derived macrophages from either C57BL6/J mice (wild type) or Nrf2 deficient mice were stimulated with either 10U/ml IFNγ and 1μg/ml LPS (M1) or 10ng/ml IL-4 (M2), or control medium only (co). RNA was isolated after 3 hours, expression of heme-oxygenase 1 (HO-1, A), Sulfiredoxin 1 (Srxn1, B), or Thioredoxin reductase 1 (Txnrd1, C) was analysed by RT-PCR.
To demonstrate that the Mox-phenotype can reproducibly be derived from various forms of macrophages, we generated BMDM using three different differentiation methods. Macrophages differentiated in the presence of GM-CSF, M-CSF, or L929 conditioned media, all responded to OxPAPC, as evidenced by upregulation of HO-1, COX-2, IL-1β, and VEGF, but showed no TNFα expression (Fig. S3). To further demonstrate that polarization to the Mox phenotype was not restricted to bone marrow-derived cells, we isolated macrophages from the peritoneal cavity of mice. Phenotypic polarization of peritoneal macrophages in the presence of oxidized phospholipids, M1, or M2 stimuli induced upregulation of HO-1, iNOS and Arg-1, respectively, confirming the results obtained with bone marrow-derived macrophages (Fig. S4).
Together, these results demonstrate that OxPAPC induces macrophage gene expression patterns and changes morphology and biological function towards a novel phenotype (Mox) that is strikingly different from conventional M1 and M2 phenotypes.
Treatment with oxidized phospholipids causes phenotypic switching of the M1 and M2 into the Mox phenotype
To examine the hypothesis that an oxidized lipid-enriched microenvironment can switch gene expression in M1 and/or M2-phenotypes towards the Mox phenotype, we polarized macrophages into M1 or M2, followed by enrichment of the culture medium with different concentrations of OxPAPC or unoxidized PAPC. Profiling gene expression in the resulting cells by RT-PCR revealed that OxPAPC treatment resulted in downregulation of M1 markers IL-1β, iNOS, TNFα and MCP-1 (Fig. 4A), as well as the M2 marker Arg-1 (Fig. 4B), in the respective phenotype. Evidence that OxPAPC changed the existing M1 or M2 phenotypes towards the Mox phenotype is demonstrated by the fact that OxPAPC induced expression of HO-1 in all phenotypes (Fig. 4C). These data show that both M1 and M2 phenotypic polarization in macrophages is reversible in vitro and indicate that the Mox phenotype may also develop in vivo in the presence of oxidized phospholipids.
Figure 4. Oxidized phospholipids skew macrophage phenotypes toward Mox.

Bone marrow-derived macrophages were polarized with either 10U/ml IFNγ and 1μg/ml LPS (M1) or 10ng/ml IL-4 (M2), or control medium only (M0) followed stimulation with either 10 μg/ml PAPC or indicated doses of OxPAPC. RNA was isolated after 3 hours, and gene expression was analyzed using RT-PCR. Typical M1-gene expression (A), as well as arginase-1 expression, a marker for M2 (B) was significantly reduced when stimulated with OxPAPC. HO-1 expression was significantly induced in M0, M1, and M2 macrophages, when stimulated with OxPAPC (C). n=4, results are shown as mean average ±SD, fold over M0; *p<0.05.
Nrf2 regulates gene expression and redox status in Mox
Gene ontology analysis (BABELOMICS) of upregulated genes as obtained from the gene array in Mox, M1, and M2 demonstrated several unique associations with biological functions. While the M1 phenotype was specifically associated with biological functions involved in host defense, Mox was associated with redox regulation and antioxidant activity. Specificity for antioxidant activity associated with Mox was confirmed using chi square test, which demonstrated a highly significant correlation for Mox (p=9.1 ×10−24), while there was no such association with M1 (p=0.3384) or M2 (p=0.6635).
Moreover, ontology testing revealed that gene regulation mediated by the redox-regulated transcription factor Nrf2 was exclusively associated with the Mox phenotype. Indeed, a set of Mox-specific genes contain in their promoter regions one or more antioxidant response elements (ARE), which serve as binding sites for Nrf2 20–22. Known ARE-containing genes that were exclusively upregulated in Mox include Srxn1, Cebpb, Gclc, Gdf15, Gclm, Pde3b, Txnrd1, Pim1, Hmox1, Gadd45a, Dgat1, Gsr, Dusp1, and Tgif1 (Table S3).
To examine whether Nrf2 was involved in transcription of Mox genes, we used macrophages isolated from wild type C57Bl/6 or Nrf2−/− mice. We found that oxidized phospholipid-induced expression of several redox-regulated genes including HO-1, sufiredoxin-1 (Srnx1), and thioredoxin reductase (Txnrd) was dependent on Nrf2 (Fig. 5A-C). In contrast, other Mox-specific genes including VEGF, Dusp-1, Nurr-1, Dusp-5, and Thsp, as well as the M2-specific marker arginase 1, and the M1/Mox overlapping genes IL-1β and COX-2 (Fig. S5), were upregulated independently of Nrf2. Moreover, we show that OxPAPC and the cyclopentenone 15d-PGJ223, but not native PAPC, DMPC or LPS, induce Nrf2-dependent gene expression (GCLM, GSR1 and HO-1) in BMDM (Fig. S6).
The involvement of Nrf2 in the response to OxPAPC strongly indicated a role in redox regulation. Analysis of the redox status in the different macrophage phenotypes revealed that while the total glutathione levels were not significantly different in M0, M1, M2 and Mox macrophages, the GSH/GSSG ratio, an indicator of oxidative stress, was lower in M2 compared to the M1 macrophages (Fig. S7A-C). This is in accordance with previous reports 24 demonstrating that the GSH/GSSG ratio is lower in M2 compared to M1 macrophages. Interestingly, we observed an increased GSH/GSSG ratio in the Mox macrophages, compared to M1 and M2 phenotypes, suggesting that Mox macrophages have the ability to cope better with oxidative stress. Notably, the macrophages had been treated for 6 hours, which provided sufficient time for activation of Nrf2-dependent redox-regulating genes. Consequently, in Nrf2 deficient macrophages, the total glutathione level was significantly lower compared to wild type macrophages, which indicates deficits in redox regulation. There was no significant difference in the GSH/GSSG ratios of M0 and Mox phenotypes in Nrf2 KO macrophages (Fig. S7A-C).
In addition to upholding the redox status, Mox-specific genes may have important functions to protect cells from dying in oxidatively damaged tissue. In support of this hypothesis, we show that Nrf2-dependent gene regulation is essential for survival of macrophages. Treatment of Nrf2 deficient macrophages with oxidized phospholipids resulted in increased cell death, while wild type macrophages were protected (Fig. S7D).
Mox represent a distinct subpopulation of macrophages in murine aortic atherosclerotic lesions
Immunohistochemistry of lesion macrophages showed that HO-1 expressing macrophages (Fig. 6A) clearly represent a distinct phenotype, since they neither co-localized with iNOS expressing (M1) nor Arg-1 expressing (M2) macrophages (Fig. 6B, C). Combination staining using HO-1, Srdxn1 and Txdn1 showed that all three Mox markers co-localized within atherosclerotic lesions in mice (Fig. 6D). These data clearly demonstrate that the Mox phenotype is distinguishable from M1 and M2 macrophages.
Figure 6. Immunocytochemistry and FACS analysis of mouse atherosclerotic plaques demonstrate that phenotypic markers for Mox are distinctly different from phenotypic markers for M1 and M2.
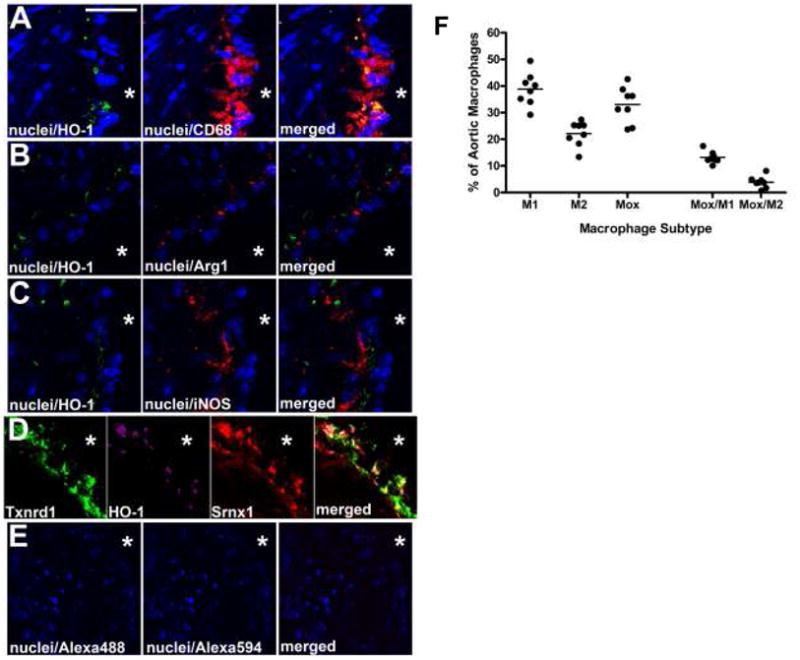
In A, nuclei (blue; DAPI) is co-stained with the Mox marker HO-1 (green; Alexa 488) and the pan-macrophage marker CD68 (red; Alexa 594); in B, nuclei are co-stained with HO-1 (green; Alexa 488) and the M2 phenotypic marker Arg-1 (red); in C, nuclei is co-stained with HO-1 (green; Alexa 488) and the M1 phenotypic marker iNOS (red; Alexa 594). In D, three different Mox phenotypic markers are co-stained (Txnrd1 (green; Alexa 488), HO-1 (magenta; Alexa 405), and Srnx1 (red; Alexa 594)). Control images in E are the secondary antibodies alone. Scale bar in A is 30 μm for A-C, 25 μm for D, and 50 μm for E; * represents the lumen of the vessel in each image. (F) LDLR−/− mice were fed a Western diet for 30 weeks, after which the aortas were harvested for flow cytometry. Tissues were digested with an enzymatic cocktail and the resulting aortic cell suspensions were stained with antibodies against CD11b, CD11c, CD45, CD86, CD206, HO-1 and a viability dye. Data are represented as percentages of total CD11b+/CD11c+ aortic cells. Macrophage subtypes were defined as M1: CD86+, M2: CD206+, Mox: HO-1+, Mox/M1: CD86+ HO-1+ and Mox/M2: CD206+ HO-1+. Each point represents a single animal.
To analyse the relative abundance of the various macrophage phenotypes in established lesions, we used aortas from LDLR−/− mice that had been fed an atherogenic diet for 30 weeks. Lesion macrophages were analyzed by FACS and live CD45+ cells were gated on F4/80+ and CD11b+. For phenotypic characterisation, we used the costimulatory molecule CD86 for M1, the mannose receptor CD206 for M2, and HO-1 for Mox (Online Figure VIII A-G). Of the F4/80+ CD11b+ macrophage population, 34.4% ± 3.53 represented the Mox phenotype, 21.8% ± 2.40 were M2 and 39.2% ± 1.90 were of the M1 phenotype (Fig. 6F). While, as expected, there was some overlap between phenotypes, 65.9% ± 2.49 of the Mox macrophages did not express either of the classical M1 or M2 markers. In all of the aortas, less than 12% of the HO-1+ cells were also positive for another marker (9.6% ± 1.09 were also positive for CD86 and only 2.3% ± 0.35 for CD206). Moreover, HO-1 positive cells showed a significant overlap with thrombospondin-1, another Mox marker (Online Figure VIII H).
Including CD11c in our analysis (Online Figure IX) did not result in a significant change in the proportion of M1, M2 and Mox subpopulations. Consistent with previously published studies, CD11c+ cells represent a small fraction of the CD45+ cells present in the murine aorta25, some of which are CD11b+. Analysis of the general population of CD45+ CD11c+ cells (which includes F4/80+ CD11b+ cells) revealed that less than 2% of these cells were HO-1 positive. Therefore, while we cannot exclude the possibility that a subset of CD11c+ HO-1+ cells exists, in the current data set it appears that HO-1 is predominantly found on F4/80+ CD11b+ CD11c− macrophages. In contrast, approximately 5% of the CD11c+ cells are CD206+ and 55% appear to be CD86 positive (Online Figures IX and X).
These results demonstrate HO-1 clearly distinguishes Mox from M1 or M2 phenotypes and thus can be used as a reliable marker. Our data also indicate that upon encountering an oxidized lipid-rich microenvironment such as present in atherosclerotic lesions, macrophages polarize into the Mox phenotype in vivo.
Discussion
Atherosclerotic vascular complications are major causes of morbidity and mortality worldwide. Several lines of evidence support a central role for macrophage-mediated inflammation in the pathogenesis of atherosclerosis. The local environment in chronically inflamed tissues regulates macrophage phenotypic polarization, and both M1 and M2 macrophages have been shown to be present in fatty streak lesions. However, the endogenous factors that induce macrophage phenotypic polarization in oxidatively damaged tissue are poorly understood. We demonstrate here that oxidized phospholipids that accumulate in atherosclerotic lesions produce a novel macrophage phenotype (Mox) that demonstrates strikingly different gene expression patterns and biological functions compared to conventional M1 or M2 phenotypes. We identify several Mox marker genes, whose expression is largely mediated by the redox-sensitive transcription factor Nrf2. Furthermore, we demonstrate that Mox are formed in vivo and comprise approximately 30% of all CD11b+ cells in established atherosclerotic lesions in mice.
Chemotactic and phagocytic capacity of Mox
The local microenvironment in atherosclerotic lesions determines a monocyte/macrophage-specific inflammatory response, characteristic of chronic inflammation. We found that the monocyte chemotactic activity in Mox supernatants was slightly increased compared to untreated control (M0) macrophages, although it was significantly lower than in M1 or M2 supernatants. It has been previously shown that the M2 macrophage activator IL-4 as well as several M1 mediators, including IL-12, trigger inflammation and macrophage accumulation in atherosclerotic lesions26. Mox also showed a pro-inflammatory phenotype, as evidenced by several upregulated pro-inflammatory genes, including COX-2 and IL-1β, although to a significantly lesser extent than in M1 macrophages. Our data suggest that a microenvironment enriched in oxidized phospholipids leads to the formation of the Mox phenotype and perpetuates the chronic inflammatory response by further attracting cells of the monocyte/macrophage lineage, consistent with “low grade” inflammation that has been reported in chronically inflamed tisues. In support of this hypothesis, we have recently shown that oxidized phospholipids specifically induce macrophage accumulation in a mouse model of inflammation 17.
Decreased capacity of macrophages to clear cell debris and apoptotic cells will result in accumulation of tissue-damaging material, exacerbating inflammation 27. IL-4 is known to increase scavenger receptor expression by macrophages28, facilitating their uptake of apoptotic cells 29. Interestingly, Mox macrophages demonstrated decreased phagocytotic capacity, when compared to M1 or M2 macrophages. We have reported previously that in the context of bacterial infection, decrease of phagocytotic capacity by oxidized phospholipids is detrimental to an adequate host response 30. The inhibited phagocytotic capacity of Mox macrophages may significantly contribute to ongoing tissue damage and pro-inflammatory state which would contribute to progression and/or destabilization of atherosclerotic lesions 31.
Nrf2 mediates Mox phenotypic polarization and controls redox status
While the NFκB signaling pathway plays an essential role in M1 polarization, activation of PPARγ is critically involved in macrophage polarization to the M2 phenotype. The significance of the protective M2 phenotype is underlined by the fact that selective deletion of PPARγ in macrophages exacerbates atherosclerosis 32–34. Here we show that the redox-sensitive transcription factor Nrf2 is a key mediator of macrophage polarization to the Mox phenotype by oxidized phospholipids. Interestingly, negative crosstalk between both Nrf2 and PPARγ and the NFκB pathway has been reported 35.
In response to oxidative stress, Nrf2 escapes ubiniquitinization by dissociating from its negative regulator Keap1 (Kelch-like ECH-associated protein 1), translocates to the nucleus and activates the genes involved in synthesis of antioxidant enzymes36 such as heme oxygenese (HO-1), glutamate-cysteine ligase, modifier subunit (GCLM), glutathione S-transferase (GST) and thioredoxin reductase 1 (Txnrd1) 37, all of which we have identified as specific Mox marker genes. These genes carry a special Nrf2 binding site, called the antioxidant response element (ARE), in their promoter region 38. Nrf2 itself contains an ARE in its promoter, however, its expression was not induced by OxPAPC within 4 hours of stimulation (not shown). Although Nrf2 expression was slightly upregulated after 18 hours, these data indicate that Nrf2 is not transcriptionally regulated by OxPAPC at early timepoints. However, we and others have shown previously that Nrf2 nuclear translocation is induced by oxidized phospholipids 39, 40. Nrf2 is activated by redox sensing either directly or via Keap1 41. In addition, it has been shown that PKC can activate Nrf2 38. Our data clearly demonstrate a requirement for oxidative modification of phospholipids and the presence of specific functional groups for activation of Nrf2-dependent gene expression. Cyclopentenones induce nuclear translocation of Nrf2 by covalently binding to Keap-1 23. The fact that the effects of OxPAPC on Nrf2-dependent gene expression can be mimicked by 15d-PGJ2 indicates a role for cyclopentenones present in OxPAPC (such as PECPC). PGPC, on the other hand, has been shown to induce PKC-dependent signalling 42.
Control of redox status in macrophages by Nrf2 may be important in regulation of various cellular functions that potently influence tissue homeostasis and inflammation 35. Defective redox regulation, due to non-functional or deficient Nrf2, may lead to exacerbated cell death, as seen in chronically inflamed tissue. In atherosclerotic lesions, where cells are constantly exposed to high levels of oxidative stress, macrophages survive for surprisingly long periods of time, despite the toxic environment, implying the upregulation of specific survival mechanisms. The Nrf2 target gene HO-1 was shown to be an important survival factor due to its anti-apoptotic and anti-inflammatory effects. HO-1, which we identified as a specific Mox marker, can be induced by a variety of stimuli, including lipid mediators 41, 43, 44–46, 47, and was previously shown to be expressed in atherosclerotic plaques 48. We found that protection against cell death in Mox was diminished by absence of Nrf2 or inhibition of HO-1 enzymatic activity by tin protoporphyrin (SnPP) (data not shown). The importance of redox regulation in macrophages in atherosclerosis as it relates to survival in an environment of high oxidative stress has been the center of numerous investigations as reviewed previously 49.
A large body of evidence demonstrates that failure of expression of Nrf2 leads to various diseases related to oxidative stress, inflammation and xenobiotic metabolism in mice 50–58. Moreover, functional single nucleotide polymorphisms (SNPs) have been detected in Nrf2 that are associated with oxidant-induced acute lung injury and gastric mucosal inflammation in humans 59, 60, 61. Based on these findings, one would expect a protective role of Nrf2 in atherogenesis. Surprisingly, a recent study has shown that Nrf2−/− mice were protected against diet-induced atherosclerosis 62. Whether Nrf2-mediated formation of the Mox macrophage phenotype contributes to the initiation and/or progression of atherosclerotic lesion formation remains to be shown.
Mox develop in vivo in established murine atherosclerotic lesions
It was proposed that the relative abundance of different polarization types of macrophages in the tissue may change dynamically via recruitment of polarized monocytes from the blood and/or through the effects of local factors on macrophages in the tissues 10. In support of the first hypothesis, a recent study suggests that circulating monocytes can be primed for M2 polarization by PPARγ activation 9. Based on our in vitro data demonstrating that the phenotypes can be switched depending on the stimulus present in the culture medium, we hypothesized that in vivo macrophages would have to encounter an oxidized lipid-enriched microenvironment in order to upregulate Mox marker genes. In support of this hypothesis we have previously shown that injection of OxPAPC into the murine air-pouch resulted in accumulation of HO-1-positive macrophages in the air-pouch tissue 17 and i.p. injection of OxPAPC resulted in upregulation of HO-1 gene expression in peritoneal cells (not shown). Moreover, topical application of OxPAPC to murine carotid arteries induced gene expression in vascular cells 63 and Nrf2 activation in endothelial cells 40. Our analysis of established murine aortic lesions shows that about 30% of all CD11b+/CD11c+ cells were of the Mox phenotype, as evidenced by HO-1 expression. M1 (CD86high) and M2 (CD206+) macrophages comprised about 40% and 20% of CD11b+/CD11c+ cells, respectively. Strikingly, there was little overlap between the different phenotypes (~3% with M2 and ~10% with M1), indicating that in established lesions different polarized phenotypes exist, and probably are confined to specific regions within the plaque. Our results using immunohistochemistry show that HO-1 positive macrophages (as determined by CD68 positivity) neither co-localized with M1 (stained with iNOS) nor with M2 (Arg-1) macrophages in the lesions, clearly demonstrating distinct cells. Moreover, three identified Mox markers, HO-1, Srdx1 and Trdx1, co-localized in the lesions, indicating that the Mox phenotype can be characterized by these markers.
Interestingly, the presence of HO-1 expressing macrophages in human atherosclerotic plaques has been reported 64. Oxidative stress can be generated in macrophages by a variety of compounds. Hemoglobin may be the primary inducer of HO-1 at sites of intraplaque hemorrhage, as previously reported 64, 65. Thus, it is very likely that a combination or even the sum of oxidative stress-inducing agents contribute to the formation of the Mox phenotype. In this study we used oxidized phospholipids as model compounds which are abundantly present in atherosclerotic lesions. Together our findings strongly indicate that macrophage phenotypic switching occurs in the tissue after contact with oxidized phospholipids or other oxidative stress-inducing factors.
In summary, our data demonstrate that oxidized phospholipids, which are abundant in atherosclerotic lesions, induce formation of a novel macrophage phenotype (Mox) that is characterised by Nrf2-dependent gene expression and may significantly contribute to pathologic processes in atherosclerotic vessels.
Supplementary Material
Acknowledgments
Sources of funding: This work was supported by NIH grant R01-HL-084422-01 (to N.L.). A.K. was supported by a Max Kade Postdoctoral Fellowship. A.M. is supported by a Postdoctoral Fellowship from the American Heart Association.
Non-standard Abbreviations and Acronyms
- BMDM
bone marrow-derived macrophages
- PAPC
1-palmitoyl-2-arachidonoyl-sn-3- phosphorylcholine
- POVPC
1-palmitoyl-2-oxovaleroyl-sn-3- phosphorylcholine
- PGPC
1-palmitoyl-2-glutaroyl-sn-3- phosphorylcholine
- HO-1
heme oxygenese-1
- Srxn-1
sulforedoxin-1
- Txnrd-1
thioredoxin reductase-1
- GCLM
glutamate-cysteine ligase, modifier subunit
- GST
glutathione S-transferase
- GSR-1
glutathione reductase-1
- OxPAPC
oxidized PAPC
- DMPC
1,2-dimyristoyl-sn-3-phosphorylcholine
- Thsp
thrombospondin
- Dusp
dual specificity phosphatase
- VEGF
vascular endothelial growth factor
- 15dPGJ2
- LPS
lipopolisaccharide
- IL
interleukin
- GSH
glutathione
- GSSG
oxidized glutathione
- lysoPC
lysophosphatidylcholine
- Nrf2
nuclear factor erythroid 2-like 2 (NFE2L2)
Footnotes
Disclosures: None
Reference List
- 1.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005 December;5(12):953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 2.Bystrom J, Evans I, Newson J, Stables M, Toor I, Van RN, Crawford M, Colville-Nash P, Farrow S, Gilroy DW. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood. 2008 November 15;112(10):4117–27. doi: 10.1182/blood-2007-12-129767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez FO, Helming L, Gordon S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu Rev Immunol. 2008 December 23; doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004 December;25(12):677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 5.Mantovani A, Muzio M, Garlanda C, Sozzani S, Allavena P. Macrophage control of inflammation: negative pathways of regulation of inflammatory cytokines. Novartis Found Symp. 2001;234:120–31. doi: 10.1002/0470868678.ch8. [DOI] [PubMed] [Google Scholar]
- 6.Raes G, Van den BR, De BP, Ghassabeh GH, Scotton C, Locati M, Mantovani A, Sozzani S. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol. 2005 June 1;174(11):6561–2. doi: 10.4049/jimmunol.174.11.6561. [DOI] [PubMed] [Google Scholar]
- 7.Espey MG. Tumor macrophage redox and effector mechanisms associated with hypoxia. Free Radic Biol Med. 2006 December 1;41(11):1621–8. doi: 10.1016/j.freeradbiomed.2006.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waldo SW, Li Y, Buono C, Zhao B, Billings EM, Chang J, Kruth HS. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol. 2008 April;172(4):1112–26. doi: 10.2353/ajpath.2008.070513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007 August;6(2):137–43. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009 October;29(10):1419–23. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 11.Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, Boren J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002 June 13;417(6890):750–4. doi: 10.1038/nature00804. [DOI] [PubMed] [Google Scholar]
- 12.Lusis AJ. Atherosclerosis. Nature. 2000 September 14;407(6801):233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subbanagounder G, Leitinger N, Schwenke DC, Wong JW, Lee H, Rizza C, Watson AD, Faull KF, Fogelman AM, Berliner JA. Determinants of bioactivity of oxidized phospholipids. Specific oxidized fatty acyl groups at the sn-2 position. Arterioscler Thromb Vasc Biol. 2000 October;20(10):2248–54. doi: 10.1161/01.atv.20.10.2248. [DOI] [PubMed] [Google Scholar]
- 14.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem. 1997 May 23;272(21):13597–607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 15.Watson AD, Subbanagounder G, Welsbie DS, Faull KF, Navab M, Jung ME, Fogelman AM, Berliner JA. Structural identification of a novel pro-inflammatory epoxyisoprostane phospholipid in mildly oxidized low density lipoprotein. J Biol Chem. 1999 August 27;274(35):24787–98. doi: 10.1074/jbc.274.35.24787. [DOI] [PubMed] [Google Scholar]
- 16.Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal. 2009 August 17; doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kadl A, Galkina E, Leitinger N. Induction of CCR2-dependent macrophage accumulation by oxidized phospholipids in the air-pouch model of inflammation. Arthritis and Rheumatism. 2009 doi: 10.1002/art.24448. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009 May 15;284(20):13291–5. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bochkov VN, Kadl A, Huber J, Gruber F, Binder BR, Leitinger N. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002 September 5;419(6902):77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- 20.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999 September 10;274(37):26071–8. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 21.Sekhar KR, Spitz DR, Harris S, Nguyen TT, Meredith MJ, Holt JT, Gius D, Marnett LJ, Summar ML, Freeman ML. Redox-sensitive interaction between KIAA0132 and Nrf2 mediates indomethacin-induced expression of gamma-glutamylcysteine synthetase. Free Radic Biol Med. 2002 April 1;32(7):650–62. doi: 10.1016/s0891-5849(02)00755-4. [DOI] [PubMed] [Google Scholar]
- 22.Lou H, Du S, Ji Q, Stolz A. Induction of AKR1C2 by Phase II Inducers: Identification of a Distal Consensus Antioxidant Response Element (ARE) Regulated by NRF2. Mol Pharmacol. 2006 February 14; doi: 10.1124/mol.105.019794. [DOI] [PubMed] [Google Scholar]
- 23.Niture SK, Kaspar JW, Shen J, Jaiswal AK. Nrf2 signaling and cell survival. Toxicol Appl Pharmacol. 2009 June 16; doi: 10.1016/j.taap.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobashi K, Aihara M, Araki T, Shimizu Y, Utsugi M, Iizuka K, Murata Y, Hamuro J, Nakazawa T, Mori M. Regulation of LPS induced IL-12 production by IFN-gamma and IL-4 through intracellular glutathione status in human alveolar macrophages. Clin Exp Immunol. 2001 May;124(2):290–6. doi: 10.1046/j.1365-2249.2001.01535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006 May 15;203(5):1273–82. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003 September;163(3):1117–25. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gautier EL, Huby T, Witztum JL, Ouzilleau B, Miller ER, Saint-Charles F, Aucouturier P, Chapman MJ, Lesnik P. Macrophage Apoptosis Exerts Divergent Effects on Atherogenesis as a Function of Lesion Stage. Circulation. 2009 March 23; doi: 10.1161/CIRCULATIONAHA.108.806158. [DOI] [PubMed] [Google Scholar]
- 28.Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999 July 22;400(6742):378–82. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez-Boyanapalli RF, Frasch SC, McPhillips K, Vandivier RW, Harry BL, Riches DW, Henson PM, Bratton DL. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood. 2009 February 26;113(9):2047–55. doi: 10.1182/blood-2008-05-160564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knapp S, Matt U, Leitinger N, van der PT. Oxidized phospholipids inhibit phagocytosis and impair outcome in gram-negative sepsis in vivo. J Immunol. 2007 January 15;178(2):993–1001. doi: 10.4049/jimmunol.178.2.993. [DOI] [PubMed] [Google Scholar]
- 31.Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2008 October 25; doi: 10.1194/jlr.R800032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001 January;7(1):48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 33.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Eagle AR, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007 June 28;447(7148):1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005 August;25(8):1647–53. doi: 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- 35.Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, Kong AN. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol. 2008 July 23; doi: 10.1016/j.bcp.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273–86. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- 37.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999 January 1;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38(4):769–89. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 39.Gruber F, Mayer H, Lengauer B, Mlitz V, Sanders JM, Kadl A, Bilban M, de MR, Wagner O, Kensler TW, Yamamoto M, Leitinger N, Tschachler E. NF-E2-related factor 2 regulates the stress response to UVA-1-oxidized phospholipids in skin cells. FASEB J. 2009 August 31; doi: 10.1096/fj.09-133520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jyrkkanen HK, Kansanen E, Inkala M, Kivela AM, Hurttila H, Heinonen SE, Goldsteins G, Jauhiainen S, Tiainen S, Makkonen H, Oskolkova O, Afonyushkin T, Koistinaho J, Yamamoto M, Bochkov VN, Yla-Herttuala S, Levonen AL. Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ Res. 2008 July 3;103(1):e1–e9. doi: 10.1161/CIRCRESAHA.108.176883. [DOI] [PubMed] [Google Scholar]
- 41.Oh JY, Giles N, Landar A, rley-Usmar V. Accumulation of 15-deoxy-delta(12,14)-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochem J. 2008 April 15;411(2):297–306. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Birukov KG, Leitinger N, Bochkov VN, Garcia JG. Signal transduction pathways activated in human pulmonary endothelial cells by OxPAPC, a bioactive component of oxidized lipoproteins. Microvasc Res. 2004 January;67(1):18–28. doi: 10.1016/j.mvr.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 43.Lim HJ, Lee KS, Lee S, Park JH, Choi HE, Go SH, Kwak HJ, Park HY. 15d-PGJ2 stimulates HO-1 expression through p38 MAP kinase and Nrf-2 pathway in rat vascular smooth muscle cells. Toxicol Appl Pharmacol. 2007 August 15;223(1):20–7. doi: 10.1016/j.taap.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 44.Weigert A, Tzieply N, von KA, Johann AM, Schmidt H, Geisslinger G, Brune B. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol Biol Cell. 2007 October;18(10):3810–9. doi: 10.1091/mbc.E06-12-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weigert A, Johann AM, von KA, Schmidt H, Geisslinger G, Brune B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood. 2006 September 1;108(5):1635–42. doi: 10.1182/blood-2006-04-014852. [DOI] [PubMed] [Google Scholar]
- 46.Weis N, Weigert A, von KA, Brune B. Heme Oxygenase-1 Contributes to an Alternative Macrophage Activation Profile Induced by Apoptotic Cell Supernatants. Mol Biol Cell. 2009 January 7; doi: 10.1091/mbc.E08-10-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res. 2008 April 25;102(8):950–8. doi: 10.1161/CIRCRESAHA.107.170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ijas P, Nuotio K, Saksi J, Soinne L, Saimanen E, Karjalainen-Lindsberg ML, Salonen O, Sarna S, Tuimala J, Kovanen PT, Kaste M, Lindsberg PJ. Microarray analysis reveals overexpression of CD163 and HO-1 in symptomatic carotid plaques. Arterioscler Thromb Vasc Biol. 2007 January;27(1):154–60. doi: 10.1161/01.ATV.0000251991.64617.e7. [DOI] [PubMed] [Google Scholar]
- 49.Gieseg SP, Leake DS, Flavall EM, Amit Z, Reid L, Yang YT. Macrophage antioxidant protection within atherosclerotic plaques. Front Biosci. 2009;14:1230–46. doi: 10.2741/3305. [DOI] [PubMed] [Google Scholar]
- 50.Aleksunes LM, Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol Pathol. 2007;35(4):459–73. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- 51.Iizuka T, Ishii Y, Itoh K, Kiwamoto T, Kimura T, Matsuno Y, Morishima Y, Hegab AE, Homma S, Nomura A, Sakamoto T, Shimura M, Yoshida A, Yamamoto M, Sekizawa K. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells. 2005 December;10(12):1113–25. doi: 10.1111/j.1365-2443.2005.00905.x. [DOI] [PubMed] [Google Scholar]
- 52.Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008 July 1;181(1):680–9. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- 53.Li J, Stein TD, Johnson JA. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol Genomics. 2004 August 11;18(3):261–72. doi: 10.1152/physiolgenomics.00209.2003. [DOI] [PubMed] [Google Scholar]
- 54.Li YJ, Takizawa H, Azuma A, Kohyama T, Yamauchi Y, Takahashi S, Yamamoto M, Kawada T, Kudoh S, Sugawara I. Disruption of Nrf2 enhances susceptibility to airway inflammatory responses induced by low-dose diesel exhaust particles in mice. Clin Immunol. 2008 September;128(3):366–73. doi: 10.1016/j.clim.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Mochizuki M, Ishii Y, Itoh K, Iizuka T, Morishima Y, Kimura T, Kiwamoto T, Matsuno Y, Hegab AE, Nomura A, Sakamoto T, Uchida K, Yamamoto M, Sekizawa K. Role of 15-deoxy delta(12,14) prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am J Respir Crit Care Med. 2005 June 1;171(11):1260–6. doi: 10.1164/rccm.200406-755OC. [DOI] [PubMed] [Google Scholar]
- 56.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001 March 13;98(6):3410–5. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001 October;60(4):1343–53. doi: 10.1046/j.1523-1755.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- 58.Yu X, Kensler T. Nrf2 as a target for cancer chemoprevention. Mutat Res. 2005 December 11;591(1–2):93–102. doi: 10.1016/j.mrfmmm.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 59.Arisawa T, Tahara T, Shibata T, Nagasaka M, Nakamura M, Kamiya Y, Fujita H, Hasegawa S, Takagi T, Wang FY, Hirata I, Nakano H. The relationship between Helicobacter pylori infection and promoter polymorphism of the Nrf2 gene in chronic gastritis. Int J Mol Med. 2007 January;19(1):143–8. [PubMed] [Google Scholar]
- 60.Marzec JM, Christie JD, Reddy SP, Jedlicka AE, Vuong H, Lanken PN, Aplenc R, Yamamoto T, Yamamoto M, Cho HY, Kleeberger SR. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007 July;21(9):2237–46. doi: 10.1096/fj.06-7759com. [DOI] [PubMed] [Google Scholar]
- 61.Arisawa T, Tahara T, Shibata T, Nagasaka M, Nakamura M, Kamiya Y, Fujita H, Hasegawa S, Takagi T, Wang FY, Hirata I, Nakano H. The relationship between Helicobacter pylori infection and promoter polymorphism of the Nrf2 gene in chronic gastritis. Int J Mol Med. 2007 January;19(1):143–8. [PubMed] [Google Scholar]
- 62.Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, Polotsky VY, Biswal S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE. 2008;3(11):e3791. doi: 10.1371/journal.pone.0003791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Furnkranz A, Schober A, Bochkov VN, Bashtrykov P, Kronke G, Kadl A, Binder BR, Weber C, Leitinger N. Oxidized phospholipids trigger atherogenic inflammation in murine arteries. Arterioscler Thromb Vasc Biol. 2005 March;25(3):633–8. doi: 10.1161/01.ATV.0000153106.03644.a0. [DOI] [PubMed] [Google Scholar]
- 64.Boyle JJ, Harrington HA, Piper E, Elderfield K, Stark J, Landis RC, Haskard DO. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol. 2009 March;174(3):1097–108. doi: 10.2353/ajpath.2009.080431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Philippidis P, Mason JC, Evans BJ, Nadra I, Taylor KM, Haskard DO, Landis RC. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ Res. 2004 January 9;94(1):119–26. doi: 10.1161/01.RES.0000109414.78907.F9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.