

Abstract
Schizophrenia is a severe mental disorder associated with a characteristic constellation of symptoms and neurocognitive deficits. At present, etiological mechanisms remain relatively unknown, although multiple points of convergence have been identified over recent years. One of the primary convergence points is dysfunction of N-methyl-D-aspartate (NMDAR)-type glutamate receptors. Antagonists of NMDAR produce a clinical syndrome that closely resembles, and uniquely incorporates negative and cognitive symptoms of schizophrenia, along with the specific pattern of neurocognitive dysfunction seen in schizophrenia. Genetic polymorphisms involving NMDAR subunits, particularly the GRIN2B subunit have been described. In addition, polymorphisms have been described in modulatory systems involving the NMDAR, including the enzymes serine racemase and D-amino acid oxidase/G72 that regulate brain D-serine synthesis. Reductions in plasma and brain glycine, D-serine and glutathione levels have been described as well, providing potential mechanisms underlying NMDAR dysfunction. Unique characteristics of the NMDAR are described that may explain the characteristic pattern of symptoms and neurocognitive deficits observed in schizophrenia. Finally, the NMDAR complex represents a convergence point for potential new treatment approaches in schizophrenia aimed at correcting underlying abnormalities in synthesis and regulation of allosteric modulators, as well as more general potentiation of pre- and post-synaptic glutamatergic and NMDAR function.
Keywords: Schizophrenia, glutamate, NMDA receptor, glycine, D-serine, visual, auditory, review
The glutamate hypofunction model of schizophrenia was first proposed over two decades ago, based upon the observation that phencyclidine (PCP), ketamine and similarly acting psychotomimetic compounds induced their unique behavioral effects by blocking neurotransmission at N-methyl-D-aspartate-type glutamate receptors (NMDAR) [60]. The ability of these compounds to transiently reproduce key symptoms of schizophrenia by blocking NMDAR led to the concept that symptoms in schizophrenia may reflect underlying dysfunction or dysregulation of NMDAR-mediated neurotransmission.
Since the original promulgation of the glutamate/NMDA hypothesis of schizophrenia in the late 1980’s, significant progress has been made in delineating the potential role of NMDAR dysfunction in the etiology of schizophrenia. In contrast to dopaminergic (DA) models, the wide array of symptoms of schizophrenia, including both positive and negative symptoms, as well as cognitive symptoms and neuropsychological dysfunction are more easily explained by a glutamate/NMDA perspective. Further, many of the findings from etiological investigations, such as genetic association studies, synergize to a much greater degree with glutamatergic than dopaminergic models of the disorders (see Talbot and Arnold, this volume). Overall, therefore, these findings suggest that NMDAR may represent a critical site of convergence for factors that predispose to schizophrenia, as well as a final common pathway for mechanisms leading to the complex constellation of symptoms and neurocognitive abnormalities that most exemplifies chronic, treatment resistant schizophrenia.
The ability of NMDAR antagonists to induce many, if not most, of the symptoms and deficits associated with schizophrenia, suggests that NMDAR dysfunction or dysregulation, of itself, is sufficient to account for these features of the illness. NMDAR, therefore, may be seen as the point at which multiple environmental and genetic factors converge to induce a discrete clinical syndrome. As exemplified by this special issue, a critical goal of current schizophrenia research is to go beyond simple gene/receptor identifications to understand how pathogenic processes lead to the complex behavioral features of schizophrenia. This paper will review the unique properties of NMDAR that make them potential candidates for convergence points in pathogenic mechanisms in schizophrenia, as well as the potential pathways from NMDAR dysfunction to both symptoms and neurocognitive deficits in schizophrenia.
NMDA receptor dysfunction as a convergence point for schizophrenia
NMDARs are a type of receptor for the neurotransmitter glutamate, which is the primary excitatory neurotransmitter in the brain. Glutamate accounts for 100% of pyramidal neurons, virtually all cortico-cortical neurotransmission and approximately 60% of total brain neurons. Glutamate mediates its neurophysiological effects through both ionotropic and metabotropic receptor types. Ionotropic receptors, which are linked to intrinsic ionic channels, include NMDA-, AMPA- and kainite-type receptors in accordance with differential sensitivity to these synthetic glutamate analogs. Metabotropic receptors are divided into groups in accordance with second messenger systems, with Group I receptors (mGluR1 and 5) receptors positively linked to phospholipase C, and Group II (mGluR2 and3) and Group III (mGluR4, 6, 7 and 8) negatively linked to adenylcyclase. In general, Group I receptors potentiate glutamate function, particularly at NMDAR, whereas Group II/III receptors inhibit presynaptic glutamate release and glutamate function [62].
Although NMDAR dysfunction may be due, in part, to impaired overall glutamatergic function, not all glutamatergic perturbations give rise to clinical symptoms resembling schizophrenia. For example, reductions of overall glutamate levels in brain, such as occurs in hepatic insufficiency, leads to an encephalopathic state that does not closely resemble any standard psychiatric disorder [77]. Similarly, Fragile-X syndrome is associated with a peptide that modulates mGluR5 receptors [38]. This pathophysiological process produces a clinical state characterized by mental retardation and autistic-like features, but not a psychosis. A critical question with regard to glutamatergic models of schizophrenia, therefore, is how and why the specificity in clinical syndromes, given the relative lack of specificity of many of the genetic and environmental factors that are presently considered risk factors for the disorder.
Specificity of the clinical syndrome
An appealing feature of the DA model of schizophrenia was that it offered a potential anatomic specificity to account for the specificity of clinical features and neuropsychological deficits in schizophrenia. Dopaminergic systems project preferentially to striatal, frontal and limbic areas, leading to the general concept that specificity might be related to localized dysfunction within those regions, and preserved function elsewhere. Whereas some subjects with schizophrenia might show a localized frontostriatal dysfunction, the large majority show a generalized pattern of deficit on batteries such as the MATRICS Consensus Cognitive Battery (MCCB), arguing against localized dysfunction. In general, therefore, the DA model has not been able to account well for the pattern of cognitive deficit seen in schizophrenia.
The NMDA model offers an alternative form of specificity based upon neural processing. Although NMDAR are diffusely distributed throughout brain (Figure 1), within each brain region, NMDAR receptors participate in only a subset of neural processes. The specific engagement of NMDARs in a subset of processing is conveyed by unique features of the receptor, of which four are most salient: voltage dependence, calcium permeability, slow kinetics, and complex modulatory processes. These properties of NMDAR thus suggest that, at the molecular level, processes that depend upon these aspects of NMDAR function should be impaired, whereas processes that do not depend upon these types of processing should be intact.
Figure 1.

Relative distributions of dopamine (A) and NMDA (B) receptors in rodent brain. From www.brain-atlas.org
On an etiological level, linkages with schizophrenia have been found for NMDAR genes (GRIN1 and GRIN2B), with limited support for polymorphisms affecting other subunits [rev. in7]. Even in the absence of intrinsic NMDAR abnormalities, however, there are multiple schizophrenia associated regulatory pathways that converge on NMDAR, such as the allosteric modulators glycine and D-serine, as well as a polyamine [114] and redox state [112] regulatory site. As reviewed below, this suggests that dysregulation, rather than dysfunction, of NMDAR-mediated neurotransmission might be also etiologic in schizophrenia. NMDAR are also critically dependent on integrity of presynaptic glutamate pathways and postsynaptic scaffolding and regulatory structures (see Hahn et al, this volume).
Intrinsic functional characteristics of NMDAR
Although all neurotransmitter receptors are unique, some are more unique than others. In the case of NMDAR, several features distinguish them from other ligand-gated channel receptors, and make NMDA receptors particularly unique. Structurally, NMDAR are heteroligomers with multiple subunits surrounding a central pore than serves as an intrinsic ion channel. Subunits are divided into NR1, NR2 and NR3 subtypes, with functional receptors containing an NR1 subunit along with multiple additional NR2 and/or NR3 subunits. Four NR2 isoforms (NR2A-D) have been identified, with NR2A and NR2B isoforms dominating in adult brain. Two NR3 isoforms (NR3A-B) have also been identified.
Voltage sensitivity
Perhaps the most unique quality of NMDARs is that the ion channel is blocked in a voltage-dependent fashion by Mg2+ at resting potential. The blockade is only relieved when neurons are partially depolarized. As a result, glutamate-induced NMDAR channel opening does not necessarily lead to current flow through the NMDAR channel. Instead, current flow is condition on concomitant postsynaptic depolarization.
This property of NMDAR makes them ideally suited to generate “conditional” response, such as the surface recorded mismatch negativity (MMN) response, that is elicited only when a given auditory stimulus deviates from a previously established pattern [61]. Other cognitive event-related potential (ERP) components, such as P300, also occur in response to a confluence of events, and so may represent current flow through open, unblocked NMDAR channels.
In addition, at the systems level, the dual ligand- and voltage-sensitivity of NMDAR permits them to play a distinct role in other neurophysiological processes that also show reliable deficits in schizophrenia.
Learning and Memory
The dual ligand- and voltage- sensitivity of NMDARs permits them to function in a “Hebbian” fashion to integrate information from multiple information pathways. In the case of NMDARs, the integration would occur based upon one neural pathway controlling levels of presynaptic glutamate release, and a second controlling pre-existent or coincident postsynaptic membrane potential. NMDAR activation (i.e. depolarizing current flow) occurs only when both pre- and post-synaptic signals coincide. As proposed by Hebb, processes of this type are critical for memory formation.
NMDARs are not the only potential Hebbian elements in brain. Thus, NMDA theories would not of necessity predict that all new learning is impaired. Nevertheless, much cortical and hippocampal plasticity does appear to have strong NMDA contribution, so that broad impairments of learning and plasticity would be expected, as are seen in schizophrenia.
Ensemble selection
The voltage-dependent blockade of NMDARs also creates a situation such that they show highly non-linear activation patterns. Thus, whereas AMPA receptor activation increases linearly in proportion to the degree of presynaptic glutamate release, NMDARs show a non-linear pattern in which no post-synaptic response is seen until a threshold degree of presynaptic activation is obtained. Once the threshold is reached, the voltage-dependent blockade is progressively overcome, at which point no further increase is possible. The non-linearity of NMDAR-based responses can also be manipulated by systems which regulate basal membrane voltage to dynamically select or deselect opposing neural ensembles, permitting “winner take all” type responses in brain. A failure of non-linear gain mechanisms within sensory regions could underlie the pattern of deficit in perceptual organization frequently seen in schizophrenia [27], whereas failure of voltage-dependent ensemble selection mechanisms could underlie both clinic symptoms such as ambivalence and cognitive executive deficits such as impairments in task switching [159] typically seen in schizophrenia.
Filtering
Finally, the dual sensitivity of NMDARs may also render them more sensitive to the temporal patterns of afferent impulse flow that other receptor types. Single presynaptic events induce only small, short duration postsynaptic depolarizations that are insufficient to unblock NMDAR channels. However, following a presynaptic burst, depolarization induced by glutamate released during the initial presynaptic spikes may be sufficient to depolarize subsynaptic membranes to threshold, permitting later spikes in the burst to trigger NMDAR activation. As such, NMDARs may contribute more than other receptor types to basic sensory processes such as luminance contrast enhancement [27], such that they block weak, subthreshold signals (i.e. single bursts), but amplify stronger ones (i.e. presynaptic bursts).
Calcium permeability
A second key feature of NMDARs is their calcium permeability. Because of this, NMDAR activation mediates not only neuronal depolarization, but also a cascade of intracellular events that leads to initiation of long term potentiation (LTP) and depression (LTD) [12]. These processes are the primary physiological events underlying learning and new memory formation in cortex and hippocampus, consistent with the well described memory deficits associated with schizophrenia [67].
Most ligand-gated channels, including most forms of AMPA/kainite or GABAA receptors, permit entrance of ions such as Na+, K+, or Cl− that affect membrane potential but have no metabolic roles in the cell. NMDARs, in contrast, are permeable not only to Na+, but also to Ca2+, which induces metabolic, as well as ionic effects in cells. In particular, Ca2+ entry into neurons serves as a primary trigger for LTP and LTD. Consistent with this role, in hippocampus and cortex, NMDAR activation is required for the initiation, but not maintenance of LTP [125]. This is consistent with the observation that patients with schizophrenia show deficits in memory formation but not retention, and argues against structural damage to the hippocampus itself as the primary cause [67].
The Ca2+ permeability of NMDARs may also provide a link between functional and structural models of schizophrenia. For example, one of the best replicated histological findings in schizophrenia is a downregulation of the GABAergic related protein’s parvalbumin (PV) and GAD67 expression [108]. PV is a calcium binding protein intended to buffer Ca2+ concentration. Reduced PV expression may therefore reflect reduced Ca2+ entry into PV-containing interneurons. Similarly, GAD67 is the primary synthetic enzyme for GABA. NMDAR blockade has a complex interaction with GABA interneurons [55]. Reduced GAD67 expression may therefore reflect reduced excitatory drive from NMDAR. Exposure of cultured GABA interneurons to NMDAR antagonists leads to a reduction of PV and GAD67 expression similar to that seen in schizophrenia both in vitro [88] and in vivo [15]. Similar effects have also recently been reported following prenatal NMDAR antagonist exposure [85, 157], associated with increased postpubertal sensitivity to NMDAR antagonists [1].
Slow kinetics
NMDARs also differs from other types of ligand-gated channel in terms of receptor kinetics. Most ionotropic glutamate receptors show fast onset and offset, with depolarizations lasting on the order of 10s of milliseconds. Conversely, NMDAR show slow onset of depolarization, but also prolonged depolarizing responses potentially lasting 10s to 100s of milliseconds. As with voltage sensitivity, the slow onset kinetics of NMDARs makes them more sensitive to bursts than isolated impulses, which is consistent with gating or information integration [117]. In addition, the complexity of unblocking kinetics makes NMDAR highly sensitive to timing, leading to spike-timing-dependent synaptic plasticity [79].
In visual cortex, the slow kinetics of NMDAR may play a critical role in detection of motion stimuli. Lateral geniculate cells in cat show either lagged or non-lagged responses, based upon degree of NMDAR involvement [53]. Motion is decoded by comparison of lagged vs. non-lagged response to a stimulus, so that NMDAR blockade specifically abolishes both the lagged response profile and the visual cortical response to motion [140]. Deficits in motion detection are among the best replicated physiological impairments in schizophrenia [32, 109, 143] and are associated with deficits in lower-level visual processing [87], consistent with underlying dysfunction of NMDAR-dependent low-level motion detection circuits.
In prefrontal cortex, working memory information is retained, in part, by neurons that remain persistently active during the delay period. NMDAR in prefrontal cortex have recently been shown to have longer kinetics than in sensory regions, potentially related to increased NR2B vs. NR2A composition, consistent with the persistent 10–20 Hz physiological firing rates observed during delay in this region [158].
Non-neuronal localization
Another interesting aspect of NMDAR is that, while they are localized mostly on neurons, non-neuronal locations have also been described. Deficits in myelin structure [11] and gene expression [48] have been demonstrated consistently in schizophrenia, although underlying pathophysiological mechanisms have yet to be determined.
In oligodendrocytes, which give rise to myelin, NMDARs are located preferentially on growth cones but not cell bodies, and stimulate glial growth during development and maintenance of glial integrity throughout the lifespan [29]. This localization has been studied most extensively with respect to excitotoxicity [156], but would also predict that a reduction in NMDAR stimulation would lead to reduced oligodendrocyte growth and proliferation. In utero exposure to NMDAR antagonists, such as PCP, has been shown to arrest progenitor differentiation, leading to reduced numbers of mature oligodendrocytes during adulthood [110], consistent with the observations in schizophrenia.
Regulatory features of NMDAR
In addition to the intrinsic characteristics of NMDAR that make them a potential convergence point for theories of schizophrenia, NMDAR are also subject to complex modulatory processes within brain. Thus, even in the absence of intrinsic abnormalities of NMDAR themselves, dysregulation of NMDAR is possible via a number of convergent mechanisms. An emerging story from the genetics of schizophrenia is the potential contribution of numerous genes of small effect size, many of which converge on regulatory pathways related to NMDAR.
Allosteric modulators
In addition to glutamate, which serves as the primary phasic influence for channel opening, NMDAR are modulated in a more tonic fashion by two separate endogenous brain amino acids, glycine and D-serine, which bind to an allosteric regulatory site on the NMDAR. In addition, separate, physiologically relevant modulatory sites have been identified for polyamines [114] and redox state [112]. Disturbances in any of these modulatory systems could thus potentially lead to specific impairments in NMDAR function in schizophrenia.
Glycine levels in brain are regulated primarily by glycine type 1 (GlyT1) transporters which are co-localized with NMDAR and serve to maintain low, subsaturating glycine levels within the glutamatergic synapse. Although no disturbances of GlyT1 transporters have been described to date in schizophrenia, these transporters may nevertheless represent a potential site for intervention in schizophrenia [66]
D-Serine levels are regulated by serine racemase (SR), which serves as the primary synthetic enzyme, and D-amino acid oxidase (DAAO) which serves as the primary degratory enzyme. Specific transport systems for D-serine have also been described [69]. Replicated susceptibility haplotypes are reported for both DAAO - the enzyme primarily responsible for cerebral D-serine degradation - and G72/G30, also known as D-amino acid oxidase activator (DAOA) - an enzyme that modulates DAAO [e.g. 33, 118]. Other studies, while not finding an overall association, nevertheless found strong association between G72 and neurocognitive function and hippocampal activation [45, 113].
In animals, knockout of SR produces a schizophrenia-like phenotype [14] that can be rescued with either D-serine or clozapine [98]. Reduced SR levels [16] and increased DAAO activity [115] have been demonstrated in postmortem brain tissue in schizophrenia, suggesting potential physiological relevance of the genetic polymorphisms.
Genetic evidence has also been supported over recent years by evidence of altered peripheral and central glycine/D-serine levels. In the case of glycine, reduced plasma levels in schizophrenia correlate with severity of negative symptoms [129, 149] and response to clozapine [150]. Several studies have reported reduced D-serine levels in CSF [16, 49] and plasma [51] in schizophrenia patients, although contrary results are also reported [e.g.59].
Along with the glycine/D-serine, NMDAR are sensitive to modulation by the redox agent glutathione, as well as by compounds such as Zn2+ and polyamines. Of these, redox regulation seems most relevant, as reductions in glutathione have been reported in both plasma and brain in schizophrenia, with first reports occurring in the pre-neuroleptic era [8, 160]. Furthermore, deficits in hippocampal LTP similar to those observed in schizophrenia are induced by experimental glutathione depletion in rodents [145], suggesting functional relevance of the reduced glutathione state.
The existence of modulatory sites also provides potential mechanisms for intervention at NMDARs. To date, both the glycine and more recently the redox sites of the NMDAR have been targeted therapeutically using compounds such as glycine, D-serine, glycine transport inhibitors [66] or N-acetylcysteine [101]. A more recent approach has been to block GLYT1 transporters, leading to reduced glycine clearance from the extracellular space [70]. As with direct NMDAR modulators, these compounds reverse behavioral effects of NMDAR antagonists in animal models [66], suggesting that they may represent a promising new approach for treatment of schizophrenia.
Variable subunit composition
A final feature of NMDARs that perhaps gives opportunity for convergence is the heteromeric nature and variable subunit composition of the receptor. Different subunit compositions can produce receptors with different functional properties. NMDAR subunit composition varies over space and time in the CNS. During development, NR2B subunits predominate in forebrain, and in sensory systems, the switch from NR2B to NR2A is related to sensory experience and coincides with the timing of the critical period for sensory plasticity [161].
Ca2+ conductance is far larger in receptors containing NR2B than NR2A subunits, so the switch decreases probability of subsequent plasticity [135]. Voltage sensitivity is more pronounced in receptors containing NR2A or NR2B subunits than in those containing NR2C or NR2D [132]. Although the glycine/D-serine site is present on the NR1 subunit and is thus is a characteristic feature of most functional NMDARs, affinity of binding is affected by subunit composition with similar, excitatory effects on NMDARs containing NR2 subunits, but opposite effects on receptors containing NR3 subunits, with glycine serving to activate NR3-containing receptors, and D-serine to inhibit them in the presence of glycine [116]. Subtle shifts in subunit compositions can thus have major effects on native receptor function.
NR2 subunit composition does not appear to alter susceptibility to PCP or ketamine, the two channel blockers than have been most widely used to validate the NMDA model in humans, other compounds, such as MK-801, might discriminate between composition types [40, 132]. At present, the clinical syndrome induced by exposure to MK-801 has not been sufficiently characterized to know whether the subunit selectivity enhances or degrades the similarity to schizophrenia.
NR2D levels are typically low in adults, although upregulation has been reported in prefrontal cortex in schizophrenia [6]. In contrast, a more recent study found selective downregulation of NR1, NR2A and NR2C subunits in frontal cortex, and no change in NR2B or NR2D [17], which theoretically might reflect tonic NMDAR underactivation. Nevertheless, given the discrepant results, more studies are needed. Patterns of change may also be different in discrete neuronal components, which would not be detectable using current histological approaches.
In the thalamus, there may be an increase in NR2B transcript expression [35] and decreases in the NR1 exon 22 containing splice variants [124], with no changes in expression of the other subunits [35]. More recently [34], increased expression of NR2B protein and PSD95 (one of the many associated proteins that make up the NMDAR complex) was reported in the dorsomedial thalamus, but not in the ventral thalamus. This finding suggests that there may be abnormal trafficking of a subset of NMDARs in the thalamus in schizophrenia.
Summary
In summary, any explanatory model of schizophrenia needs to account not only for why some brain processes are impaired, but also why others are intact. In the case of NMDAR models, specificity comes from involvement of NMDAR in only a subset of neurophysiological processes within each brain region, based upon its distinctive characteristics. First, because NMDAR are voltage- as well as ligand- (glutamate-) sensitive, they may play a particular role in processes where flexibility of ensemble selection is critical. NMDAR involvement might be particularly important when a less common response pathway is required, for example when a prepotent response must be inhibited and a less common response selected.
Because NMDAR activation serves as a trigger for LTP and LTD, processes that require LTP-based learning such as hippocampal memory formation should be impaired, whereas processes that rely on other forms of plasticity may be intact. Finally, because of the long duration of NMDAR depolarizations, processes requiring integration over time periods tens to a few hundred milliseconds, such as gating or information integration may be particularly impaired. Complex mechanisms of NMDAR regulation have been observed in brain, suggesting that a confluence of processes may impair NMDAR function in the absence of intrinsic abnormalities of the receptors themselves.
Clinical symptoms as a convergence point for NMDAR models
The ability of PCP or ketamine to induce a schizophrenia-like psychosis has by now become well established [64, 78, 93, 99]. Although PCP psychosis is sometimes suggested as a model for negative-symptom schizophrenia, and amphetamine- or LSD-psychosis, as a model for positive symptoms [46], such a clear distinction does not hold up upon closer inspection. In PCP- or ketamine-induced psychosis, the relative proportions of positive and negative symptoms are highly similar to those observed in both acute and chronic schizophrenia, whereas in amphetamine- or LSD-induced psychosis the level of positive vs. negative symptoms is far in excess of the pattern observed even in acute schizophrenia stages [46, 97]. Thus, most parsimoniously, NMDAR antagonist-induced psychosis produces a psychosis that closely models both the positive and negative symptoms of schizophrenia, whereas amphetamine or LSD produce psychoses possibly resembling the pattern observed in mania, but unlike the pattern observed in typical schizophrenia.
PCP psychosis also uniquely incorporates cognitive symptoms similar to those observed in schizophrenia, including deficits in conceptual disorganization, abstract thinking, mannerisms and poor attention [97]. Although amphetamine has some effect on conceptual disorganization, no effect on cognitive symptoms have been observed. Furthermore, although some additivity between amphetamine and ketamine effects has been observed, the degree of interaction did not reach statistical significance [97], suggesting that dopaminergic hyperactivity is neither necessary or sufficient to produce the types of cognitive symptoms observed in schizophrenia.
A distinction between ketamine-induced symptoms and those of schizophrenia is in the production of hallucinations. During ketamine-induced psychosis, visual perceptual distortions are common but organized auditory hallucinations are rare. This is unlike established schizophrenia, in which auditory hallucinations are far more common. However, the pattern of sensory disturbances seen during ketamine administration does resemble the pattern observed early in the course in schizophrenia [21, 123] where both auditory and visual perceptual disturbances are common, and acute ketamine challenge may be viewed best as a model of prodromal or acute incipient schizophrenia, rather than later, more chronic, phases. Nonetheless, in patients with established schizophrenia, increases in hallucinatory activity are observed during ketamine challenge [100, 119]. Further, in primates, apparent hallucinatory behavior (i.e. threatening non-existent objects) is not observed during acute PCP treatment, but does emerge during chronic administration [111]. There is evidence for an additive effect of amphetamine and ketamine in the production of auditory hallucinations [97], suggesting dual DA and NMDA contributions to positive, as opposed to negative, symptoms.
Positive symptoms
Of all the symptom clusters associated with schizophrenia, positive symptoms are best understood. Positive symptoms are induced not only by NMDAR antagonists, but also reliably by dopaminergic agents, suggesting that the DA system plays a key role. Furthermore, D2 receptor antagonists have a reliable effect on positive symptoms at doses that occupy approximately 60% of striatal D2 receptors, supporting the concept that dopaminergic hyperactivity is sufficient to produce a psychosis-like state [80]. NMDAR antagonists induce increased striatal DA release [144], along with increased positive symptoms [22], consistent with the concept that these agents operate in part through stimulation of frontostriatal dopaminergic systems
Dopaminergic hyperactivity in schizophrenia has also been confirmed by challenge studies showing increased amphetamine-induced DA release in acutely decompensated patients [83] (Figure 2A). Similar effects are induced in normal volunteers by pretreatment with ketamine [84] (Figure 2B), suggesting that underlying disturbances in NMDAR function might underlie the dopaminergic hyperactivity seen in schizophrenia. Similar effects are induced by PCP treatment in rodents and are reversed by concurrent treatment with glycine [68] (Figure 3A), suggesting that NMDAR-based interventions may be effective in reversing positive as well as negative symptoms of schizophrenia (Figure 3B).
Figure 2.
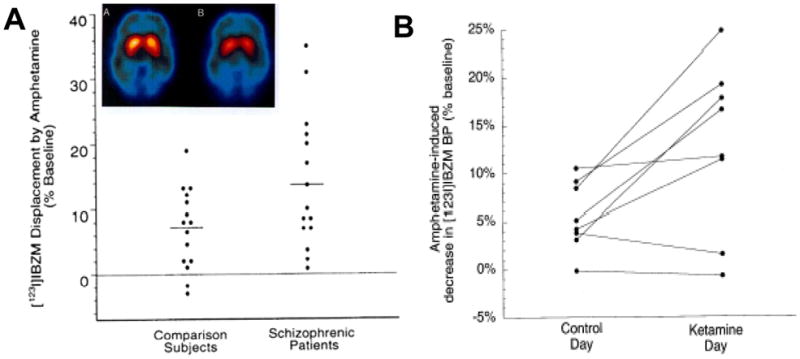
A. Scatter plots showing increased amphetamine-induced dopamine (DA) release in schizophrenia, as measured by displacement of [123I]IBZM. Inset: Image of [123I]IBZM binding in striatum prior to and following amphetamine administration. From [2]. B. Scatter plots showing increased amphetamine-induced DA release in striatum following ketamine administration in normal volunteers. From [83].
Figure 3.

A. Effects of PCP and glycine on amphetamine-induced DA release in frontal cortex and striatum. From [68]. B. Schematic diagram of effects on interactions between NMDA, GABA and DA in striatum.
Despite the strong association of dopaminergic hyperactivity and positive symptoms of schizophrenia, however, there are several nuances to positive symptoms that have yet to be fully incorporated into most operative models of schizophrenia. First, many patients with schizophrenia show persistent positive symptoms even following almost total block of D2 receptors. Second, in detailed in vivo imaging studies, hyperactivity of DA systems has been found only during acute stages of the illness, when correlations with positive symptoms are observed. In contrast, limited dopaminergic hyperactivity is seen during remitted states, despite the persistence of positive symptoms [47, 64].
The persistence of positive symptoms in many patients even after D2 blockade is most consistent with the concept that dopaminergic hyperactivity, of itself, is not the final step leading to psychosis, but rather acts via downstream elements. One critical element appears to be GABAergic outflow neurons from striatum, which, in general, produce behavioral inhibition. These neurons receive both inhibitory innervations via D2 receptors and excitatory innervations via NMDAR receptors. Thus, dopaminergic hyperactivity and NMDAR underactivity would produce similar net effects on output [64, 78].
In terms of treatment, this model would imply that D2 blockade could compensate for underactivity of these neurons even if such underactivity were caused primarily by loss of normal NMDAR-mediated excitatory drive. In such case, however, reduction of activity via D2 blockade would be limited by the degree of basal dopaminergic tone. Thus, as seen in schizophrenia, even total blockade of D2 receptors might be insufficient to compensate for loss of excitatory drive to these neurons. Similarly, in rodent models, antipsychotics show only partial effectiveness in reversal of NMDAR-induced hyperactivity [13].
A second point of convergence of DA and NMDA models is at the level of local regulation of presynaptic DA release. Within striatum and frontal cortex, presynaptic DA release is under control of intrinsic inhibitory GABAergic neurons which, in turn, are activated by NMDAR. Even in studies where no immediate effects of NMDAR antagonists were observed on striatal DA release, significant potentiation of amphetamine-induced DA release has been observed, similar to that observed in schizophrenia [83, 84].
In striatum, regulation of DA release appears to be modulated by GABAB receptors localized to presynaptic DA terminals. GABA release, in turn, is modulated by NMDAR located on GABA interneurons, with stimulation leading to increased GABA release [71]. Furthermore, NMDAR antagonist-induced DA dysregulation can be reversed by NMDAR potentiators, such as glycine or glycine transport inhibitors, suggesting that these may be effective in treatment of positive, as well as negative, symptoms of schizophrenia [65, 68] (Figure 3B).
Negative symptoms
Although NMDAR antagonists reliably produce negative symptoms in normal volunteers, as well as a “dissociative state” in primates, the mechanisms underlying these effects are poorly understood. In dopaminergic models of schizophrenia, attempts were often made to attribute negative symptoms to Parkinsonian-like dopaminergic hypofunction, either longitudinally or cross-sectionally with hypoactivity in some regions coinciding with hyperactivity in others [37]. However, approaches that were effective in treatment of Parkinsons disease have not proven highly efficacious in treatment of negative symptoms, suggesting distinct neurochemical substrates.
Negative symptoms are typically divided into broad domains including blunted affect, anhedonia-asociality, avolition-apathy and alogia/inattention [10, 89]. However, strong intercorrelation is seen between domains, leaving unresolved whether these are separate syndromes or simply different manifestations of the same underlying pathophysiological processes. Furthermore, these concepts continue to evolve clinically, prompting need to develop new neural conceptualizations.
In keeping with DA models, the key conceptualization of negative symptoms in schizophrenia until recently focused on anhedonia, defined primarily as decreased ability to experience pleasure from external events. These deficits were postulated to reflect impairments primarily within mesolimbic dopaminergic systems, and thus were well embedded within standard dopaminergic models. Over recent years, however, detailed studies of anhedonia have failed to confirm decreased hedonic capacity in individuals with schizophrenia. Thus, when asked how much they enjoy a given situation, patients generally report a similar degree of enjoyment as controls [91].
Similarly, patients show similar understanding of internal emotional states as controls [90, 152]. Although subjects often show impaired sensitivity to emotional valence, this deficit has recently been related to impaired ability to process the acoustic [104, 105] or visual [24] properties necessary to extract valence, rather than impaired comprehension of emotion itself.
An alternative explanation for apparent anhedonia and asociality may come from reward theory, in which subjects balance effort, reward and delay. Individuals will, in general, work harder now for greater reward later, but only to a point. Whether or not individuals choose immediate or delayed reward depends in part upon how much extra work is needed (“effort discounting”) and how long the delay is between action and reward (“delay discounting”). In schizophrenia, severity of negative symptoms has been correlated with increased delay discounting, with no difference in processing of reward magnitude [52]. Similarly, using different methodology, patients show normal “in the moment” experience, but reduced correlation between enjoyment and motivation to repeat an action, also suggesting either reduced ability to learn enjoyment/reward associations, or reduced ability to translate pleasure to action [151].
Processes such as delay discounting can be explicitly modeled in rodents. In rats, ketamine specifically increased delay discounting, similar to the pattern observed in schizophrenia, whereas low dose amphetamine had an effect opposite that observed in both schizophrenia and following ketamine administration. By contrast, low doses of amphetamine reduced delay discounting while higher doses increased effort discounting [42], neither of which effects are reported in schizophrenia. Finally, the antipsychotic flupenthixol also increased effort discounting, possibly contributing to avolition observed in schizophrenia. Thus, “weakening of the wellsprings of volition” in schizophrenia may reflect primarily a reduced ability to forego small immediate reward in exchange for greater reward later.
A second key component of negative symptoms is ambivalence, which may underlie the construct of avolition-apathy. In this construct, patients are less able to ignore the negative aspects of enjoyable situations, or positive aspects of aversive situations. Furthermore, increased ambivalence correlated significantly with severity of SANS-rated negative symptoms [152], supporting the importance of the ambivalence concept. As with other aspects of cognitive impairment in schizophrenia, this implies dysfunction of “winner take all” mutually inhibitory circuits, which allows commitment to one of two alternative action patterns.
Overall brain substrates for negative symptoms remain to be determined. In one recent study, however, NMDAR binding in middle inferior frontal cortex showed a significant correlation with the BPRS negative subscale, suggesting that ketamine may induce negative symptoms through interaction with the NMDAR within this brain region [147].
Cognitive or “disorganized” symptoms
A final classic feature complex of schizophrenia is a disorganization of thought and behavior, evaluated using items such as poor attention, conceptual disorganization, difficulties in abstract thinking and disorientation. Similar deficits are seen following NMDAR antagonist administration, supporting a role of NMDAR in normal attentional and thought organizational processes. The fact that these symptoms cluster together and involve language makes it tempting to localize these types of dysfunctions to brain language regions.
Two ketamine studies have utilized a fine grain approach to analyze thought disorder in patients with schizophrenia relative to normal volunteers. The first used the Thought, Language and Communication scale, which characterizes thought disorder across a range of potential patterns [4]. Both normal volunteers receiving ketamine and schizophrenia patients showed high scores on poverty of speech and content, circumstantiality, loss of goal, perseveration, and tangentiality, all items theoretically linked to ability to maintain short duration memory traces within brain language regions. In contrast, both groups showed low rates on items such as echolalia, semantic or phonetic paraphasias, or neologisms, features that are classically observed following structural damage to posterior language regions.
A second study also noted increased perseveration following both ketamine administration and in schizophrenia [36]. In contrast, patients showed no change in idea density, a deficit that is characteristic of Alzheimer’s disease, again showing relative specificity of the symptoms in schizophrenia relative to other neurocognitive disorders. Thus, as with other features of schizophrenia, cognitive symptoms appear to reflect dysfunction of specific processes within language regions, rather than structural lesions to the regions themselves.
Difficulties in abstract thinking manifests as preferring literal meanings of expressions relative to symbolic meanings. Impaired abstract thinking is one of the best replicated findings following NMDAR antagonist administration [e.g. 96]. As with conceptual disorganization, difficulty in abstract thinking may be seen as difficulty in “switching off” the more common, literal meaning of a word in order to permit associations to be made with the less common, more conceptual meaning. The fact that NMDAR antagonists produce such deficits implicates NMDAR in the selection process. The location of the relevant NMDAR, as well as the local circuitry involved, remains to be determined.
Neuropsychological deficits as a convergence point for NMDAR models
Glutamatergic models provide a framework from which to view the pattern of the core neuropsychological dysfunction [20, 44] associated with schizophrenia. Unlike dopaminergic models of schizophrenia, which predict mainly prefrontal dysfunction, in studies that have utilized comprehensive neuropsychological batteries, similar levels of deficit have been observed across multiple neurocognitive domains, particularly in learning and declarative memory formation [82].
Because glutamatergic systems are widespread, dysfunction of the glutamatergic system predicts widespread dysfunction. To date, NMDAR antagonists such as ketamine have been shown to produce schizophrenia-like cognitive deficits on tests of executive functioning [93–96], attention/vigilance [97, 120, 131, 133], verbal fluency [3, 96, 138], visual and verbal working memory [3, 5, 9, 54, 58, 94, 95, 97, 120, 127, 130], suggesting critical involvement of NMDAR in many of the processes that are impaired in schizophrenia.
In monkeys [146], ketamine treatment reproduces the pattern of task switching impairment seen in schizophrenia [86, 159]. Moreover, in fMRI studies, significant increases are observed in frontal and hippocampal activation [57] even under circumstances where no overt effects are seen on behavior. Thus, regions that have been implicated in cognitive dysfunction in schizophrenia are affected as well in ketamine challenge.
Sensory deficits in schizophrenia
Because NMDAR are located in both sensory and higher cognitive brain regions, a key prediction of NMDAR models is that schizophrenia should be associated with impaired processing within sensory brain regions. Studies over recent years have confirmed such deficits, providing both support for NMDAR models and increased insight into mechanisms underlying cognitive impairment in schizophrenia. To date, studies addressing integrity of sensory systems have been performed primarily in auditory and visual systems, although schizophrenia is known to affect other sensory processes such as weight discrimination [72] and other somatosensory processes [30].
Auditory deficits in schizophrenia
Deficits in auditory processing have been investigated using both behavioral and neurophysiological measures. Behaviorally, patients show deficits in matching of tones following brief delay [148], suggesting dysfunction of the auditory “echoic” sensory memory system, i.e., the brain’s ability to maintain transient representations of simple auditory stimuli.
In patients with schizophrenia, increased tone matching thresholds are observed with no accompanying increase in susceptibility to either visual [76] or auditory distraction [121, 137]. This is a heuristically valuable paradigm, as underlying anatomical substrates have been well characterized in primate and human models. Lesions of auditory sensory cortex, located in the superior temporal lobe, produce elevation deficits in tone matching threshold, e.g., with no accompanying increase in susceptibility to distraction. In contrast, lesions of prefrontal cortex increase distractibility without affecting thresholds [136]. These behavioral findings thus suggest dysfunctional information processing at the level of auditory sensory cortex.
Follow-up studies have investigated the functional consequences of elevated tone matching thresholds to more complex forms of information processing dysfunction. Patients with schizophrenia, for example, show well-established deficits in ability to determine emotion based upon vocal modulation (prosody), which are thought to be rate-limiting in terms of functional outcome [23]. The etiology of such deficits has been poorly understood, as patients show normal emotional responses to happy or sad events, and show intact internal representation of emotion [90], suggesting that failure to detect emotion may be related to underlying failure to utilize sensory cues.
An initial study of prosodic detection in schizophrenia evaluated the relationship between tone matching performance on the one hand, and both auditory and visual emotion detection on the other. Deficits in auditory perceptual performance (tone matching) strongly predicted deficits in auditory, but not visual, emotion detection. Further, although patients showed deficits in both auditory and visual emotion detection, the two sets of deficits were statistically unrelated, suggesting that deficits clustered within, rather than across modalities. These results thus strongly supported the hypothesis that in schizophrenia, deficits in “social cognition” are the consequence of underlying disturbances in underlying tone matching ability [103] rather than specific deficits in the conceptualization of emotion.
A subsequent study demonstrated a similar relationship between tone matching ability and ability to detect attitudinal prosody (sarcasm) [106], as well as non-affective prosody such as ability to differentiate questions from statements (semantic prosody) [104]. Further, severity of deficit across individuals correlated highly with reduced structural integrity within auditory white matter pathways at the level of auditory cortex [104].
Deficits in emotional detection have been found to particularly involve those types of emotional distinctions that depend upon differentiation of pitch [105]. Further, in addition to showing deficits in identifying emotions, patients show deficits in differentiating between emotional intensities, also consistent with inability to process changes in pitch that differentiate emotions [105]. Taken together, these findings suggest that basic deficits in NMDAR-mediated neurotransmission at the level of auditory sensory cortex lead to sensory level disturbances which result in upward disturbances of high level processes such as ability to interpret tone-of-voice.
Auditory function in schizophrenia has also been assessed with event-related potentials (ERP). ERP’s are standardized reactions of the brain to a particular stimulus, and because they index underlying neuronal processes, they allow us to look at the basic functioning of the brain and the root cause of neuropsychological dysfunction. Functioning of the early auditory system can be assessed using ERPs, which trace sequential activation of the auditory system from brainstem through auditory thalamus (medial geniculate nucleus, or MGN) and primary auditory cortex (A1) to auditory association regions. Over recent years, deficits in MMN generation, along with deficits in related phenomena such as auditory N1 generation and 40 Hz responses, have become among the best established biomarkers in schizophrenia [74], consistent with NMDAR models. MMN is elicited in an auditory oddball paradigm in which a sequence of repetitive standard stimuli is infrequently interrupted by stimuli that differ in a physical stimulus dimension such as pitch, duration, intensity or location.
Generators for MMN have been mapped to primary auditory sensory cortex in the region of Heschl’s gyrus [61]. In primary cortex, MMN appears to arise primarily from supragranular cortex, and to represent a selective disinhibition of specific populations of supragranular pyramidal neurons by memory traces established by repetitive standard stimuli. In this model, the disinhibition leads to progressive unblocking of still-closed NMDAR on pyramidal neurons, so that when a deviant stimulus is presented greater NMDAR-mediated current flow occurs then in the absence of disinhibition. Administration of NMDAR antagonists blocks this additional current flow without affecting non-NMDAR-mediated components of the auditory ERP, suggesting that MMN can be considered an index of activity within supragranular auditory layers (Figure 4). Other recently characterized components, in contrast, such as the auditory N1 or 40 Hz response, may arise from infragranular or granular layers, respectively.
Figure 4.

Intracranial recordings from monkey primary auditory cortex showing intracortical correlates of surface mismatch negativity (MMN) generation prior to (A) and following (B) local infusion of the NMDAR antagonist PCP. Boxes show current source density (CSD) correlates of MMN generation. Note reduction in amplitude of supragranular (layer III) responses to deviant stimuli in absence of effect on other intracranial components. From [75].
Deficits in MMN generation were first demonstrated in schizophrenia over 10 years ago and currently represent one of the best replicated neurophysiological findings in schizophrenia [153]. Schizophrenia-like deficits in MMN generation can be induced by local infusion of NMDAR antagonists into primate auditory cortex [61] and by systemic administration of NMDAR antagonists in healthy volunteers [154], suggesting that such deficits may index NMDAR dysfunction at the level of auditory cortex. More recently, MMN-like phenomena have been demonstrated as well in rodents [19] (see also Siegel, this volume), making them useful for drug development studies in schizophrenia [74].
In contrast to effects of NMDAR antagonists, MMN is not modulated via a variety of other psychoactive agents, including the 5-HT2A agonist psilocybin [155] and the D1/D2 agonists bromocriptine and pergolide [107], suggesting both limited dopaminergic involvement and relative specificity of the NMDAR antagonist psychotomimetic effect.
Visual processing deficits
As in the auditory system, NMDARs are located at multiple levels of the early visual system, including retina, lateral geniculate nucleus (LGN) and primary cortex, and studies also have investigated the consequences of NMDAR dysfunction in the early visual system. The early visual system consists of discrete magnocellular and parvocellular pathways that differ in characteristics and function. The magnocellular pathway provides rapid transmission of low-resolution information to cortex, in order to prime attentional systems and “frame” the overall visual scene. The parvocellular pathway, in contrast, provides slower, higher resolution information to fill-in scene details [31]. The magnocellular system, in particular, functions in a non-linear gain mode that is dependent upon NMDAR-mediated neurotransmission. Administration of NMDAR antagonists to cat LGN produces a characteristic reduction in gain that is also observed in schizophrenia [28].
To date, deficits in visual processing have been demonstrated in schizophrenia using both steady-state [26, 28, 92] and transient [25, 43, 142] visual evoked potential approaches. Most recently, deficits in magnocellular processing have been demonstrated as well using fMRI, with patients showing reduced visual activation to low, but not high, spatial frequency stimuli, particularly at low stimulus contrast levels [122] (Figure 5).
Figure 5.

fMRI activation patterns as a function of stimulus spatial frequency and contrast in individuals with schizophrenia (Scz) and controls (Ctl). A. Stimulus used for activation. B. Relative activation as function of spatial frequency (coded by color) shown on unfolded occipital cut. Note reduction in low spatial frequency (red regions) in Scz relative to Ctl, particularly at low contrast, which biases activity toward magnocellular processing. C. Plots of activation densities as a function of spatial frequency for high and low contrast stimuli in Scz vs. Ctl, showing reduced low spatial frequency response. From [122].
As in the auditory system, deficits in low-level visual processing contribute significantly to higher-level visual impairments. Thus, for example, whereas patients were once considered to have intact reading ability based upon studies with single word reading, more recent studies have demonstrated severe, dyslexia-like impairments in paragraph reading, which correlate with deficits in low level auditory and visual processing [139]. Similarly, deficits in early visual processing produce subsequent impairments on higher order processes such as object identification [39], motion processing [87], emotion recognition [24] and stereopsis [141], suggesting that basic, NMDAR mediated deficits in visual processing significantly affect the way individuals with schizophrenia experience the world, perhaps explaining in part their abnormal reactions to it.
Preserved functions
NMDAR models predict not only that NMDAR related processes should be impaired throughout cortex in schizophrenia, but also that processes that do not require NMDAR processing should be intact. At present, the degree to which NMDAR are involved in specific, high level cortical processes remains relatively unknown. Nevertheless, some processes are well known not to require NMDAR involvement, and have been shown to be normal in schizophrenia.
Key among these include (1) retention of memory following initial learning [128], in contrast to amnestic syndromes resulting from hippocampal damage; (2) retention of information within working [73, 102] and sensory [76] memory systems in schizophrenia; (3) ability to ignore distraction during auditory discrimination – a function that localizes to prefrontal cortex [136]; and (4) preserved switch costs during task switching – a frontoparietal function [81, 86, 159]. Overall theories of schizophrenia, therefore, must account not only for what is impaired, but also for what is preserved. In general, impaired and preserved functions cannot be differentiated anatomically, but may be differentiable neurochemically.
Treatment implications
A final point of convergence for NMDA theories regards treatment. The most obvious drug targets based upon NMDA models are the modulatory sites on the NMDAR itself. These include both the glycine/D-serine [63] and the redox [18, 101] sites. A “second generation” approach is to target the homeostatic mechanisms that regulate brain glycine and D-serine levels, in particular glycine transport inhibitors in the case of glycine [66] or SR or DAAO in the case of D-serine [50].
Other targets are also possible. For example, mGlu2/3 agonists may reverse hyperglutamatergia seen following acute NMDAR inhibition [126, 134], while mGluR5 agonists may enhance NMDAR function, leading to functional improvement [56]. More distantly, interventions that increase presynaptic glutamatergic integrity, or restore impaired postsynaptic NMDAR mechanisms would be expected to be therapeutic, although ideal targets for such interventions remain to be determined.
Finally, if NMDAR dysfunction in schizophrenia is relative, rather than absolute, enhanced practice might be able to overcome reduced plasticity, as has recently been reported for auditory training [41]. Current treatment for schizophrenia focuses on blocking one major receptor. Given the number of convergent mechanisms that may contribute to impaired NMDAR function, ideal treatment in schizophrenia may involve optimizing function within a number of convergent pathways, involving combinations of pharmacological and behavioral interventions, rather than simply targeting the point of convergence.
Conclusion
Over the last 40 years, the DA model has been the leading neurochemical hypothesis of schizophrenia. This model has proven heuristically valuable, with all current medications for schizophrenia functioning primarily to block DA D2 receptors. Yet it remains unlikely that dopaminergic dysfunction, on its own, can fully account for the wide range of symptoms and neurocognitive deficits seen in schizophrenia.
Glutamatergic models provide an alternate approach for conceptualizing the brain abnormalities associated with schizophrenia. As opposed to dopaminergic agonists, NMDAR antagonists produce negative and cognitive symptoms of schizophrenia, along with positive symptoms, and induce neuropsychological and sensory processing deficits that are extremely similar to those observed in schizophrenia. Convergent mechanisms target NMDAR, which in turn contribute directly to negative symptoms and neurocognitive dysfunction directly, as well as to positive symptoms via dysregulation of brain DA systems (Figure 6).
Figure 6.

Summary plot of the proposed role of NMDAR in pathophysiology of schizophrenia. In this model, NMDAR dysfunction or dysregulation reflects final common pathway leading to the complex pattern of symptoms and cognitive deficits seen in schizophrenia, with multiple potential causes of NDMAR dysfunction, including both genetic and environmental contributions.
At present, there are no approved medications for treatment of either negative symptoms or neurocognitive dysfunction. New pharmacological and behavioral approaches aimed at potentiating glutamatergic neurotransmission, particularly at NMDAR-type glutamate receptors, however, offer new hope for future clinical development.
Acknowledgments
Support: Supported in part by USPHS grants R37 MH49334 and R01 DA03383 to DCJ and by the NYU Conte Center (P50 MH086385)
Footnotes
Conflict of interest: Dr. Javitt holds intellectual property rights for use of glycine, D-serine and glycine transport inhibitors in treatment of schizophrenia and related disorders.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abekawa T, Ito K, Nakagawa S, Koyama T. Prenatal exposure to an NMDA receptor antagonist, MK-801 reduces density of parvalbumin-immunoreactive GABAergic neurons in the medial prefrontal cortex and enhances phencyclidine-induced hyperlocomotion but not behavioral sensitization to methamphetamine in postpubertal rats. Psychopharmacology (Berl) 2007;192:303–316. doi: 10.1007/s00213-007-0729-8. [DOI] [PubMed] [Google Scholar]
- 2.Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, van Dyck CH, Charney DS, Innis RB, Laruelle M. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry. 1998;155:761–767. doi: 10.1176/ajp.155.6.761. [DOI] [PubMed] [Google Scholar]
- 3.Adler CM, Goldberg TE, Malhotra AK, Pickar D, Breier A. Effects of ketamine on thought disorder, working memory, and semantic memory in healthy volunteers. Biol Psychiatry. 1998;43:811–816. doi: 10.1016/s0006-3223(97)00556-8. [DOI] [PubMed] [Google Scholar]
- 4.Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D, Breier A. Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry. 1999;156:1646–1649. doi: 10.1176/ajp.156.10.1646. [DOI] [PubMed] [Google Scholar]
- 5.Ahn KH, Youn T, Cho SS, Ha TH, Ha KS, Kim MS, Kwon JS. N-methyl-D-aspartate receptor in working memory impairments in schizophrenia: event-related potential study of late stage of working memory process. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:993–999. doi: 10.1016/S0278-5846(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 6.Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP, Potkin SG, Sandman CA, Bunney WE, Jr, Jones EG. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, Tanzi RE, Bertram L. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nature genetics. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 8.Altschule MD, Siegel EP, Henneman DH. Blood glutathione level in mental disease before and after treatment. A M A. 1952;67:64–68. doi: 10.1001/archneurpsyc.1952.02320130070006. [DOI] [PubMed] [Google Scholar]
- 9.Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A, Krystal JH. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-D- aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57:270–276. doi: 10.1001/archpsyc.57.3.270. [DOI] [PubMed] [Google Scholar]
- 10.Andreasen NC. The scale for the assessment of negative symptoms (SANS) The University of Iowa; Iowa City: 1984. [Google Scholar]
- 11.Ardekani BA, Nierenberg J, Hoptman MJ, Javitt DC, Lim KO. MRI study of white matter diffusion anisotropy in schizophrenia. Neuroreport. 2003;14:2025–2029. doi: 10.1097/00001756-200311140-00004. [DOI] [PubMed] [Google Scholar]
- 12.Artola A, Brocher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- 13.Arvanov VL, Wang RY. Clozapine, but not haloperidol, prevents the functional hyperactivity of N-methyl-D-aspartate receptors in rat cortical neurons induced by subchronic administration of phencyclidine. J Pharmacol Exp Ther. 1999;289:1000–1006. [PubMed] [Google Scholar]
- 14.Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, Jiang ZI, Benneyworth MA, Froimowitz MP, Lange N, Snyder SH, Bergeron R, Coyle JT. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol Psychiatry. 2009;14:719–727. doi: 10.1038/mp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- 16.Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, Agam G. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr Res. 2007;90:41–51. doi: 10.1016/j.schres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33:2175–2186. doi: 10.1038/sj.npp.1301604. [DOI] [PubMed] [Google Scholar]
- 18.Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Judd F, Katz F, Katz P, Ording-Jespersen S, Little J, Conus P, Cuenod M, Do KQ, Bush AI. N-acetyl cysteine as a glutathione precursor for schizophrenia--a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64:361–368. doi: 10.1016/j.biopsych.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Bickel S, Javitt DC. Neurophysiological and neurochemical animal models of schizophrenia: focus on glutamate. Behav Brain Res. 2009;204:352–362. doi: 10.1016/j.bbr.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bilder RM, Lipschutz-Broch L, Reiter G, Geisler S, Mayerhoff D, Lieberman JA. Neuropsychological deficits in the early course of first episode schizophrenia. Schizophr Res. 1991;5:198–199. doi: 10.1016/0920-9964(91)90071-x. [DOI] [PubMed] [Google Scholar]
- 21.Bowers MB., Jr Central dopamine turnover in schizophrenic syndromes. Arch Gen Psychiatry. 1974;31:50–54. doi: 10.1001/archpsyc.1974.01760130034005. [DOI] [PubMed] [Google Scholar]
- 22.Breier A, Adler CM, Weisenfeld N, Su TP, Elman I, Picken L, Malhotra AK, Pickar D. Effects of NMDA antagonism on striatal dopamine release in healthy subjects: application of a novel PET approach. Synapse. 1998;29:142–147. doi: 10.1002/(SICI)1098-2396(199806)29:2<142::AID-SYN5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 23.Brekke J, Kay DD, Lee KS, Green MF. Biosocial pathways to functional outcome in schizophrenia. Schizophr Res. 2005;80:213–225. doi: 10.1016/j.schres.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Butler PD, Abeles IY, Weiskopf N, Tambini A, Jalbrzikowski M, Legatt ME, Zemon V, Loughead J, Gur RC, Javitt DC. Sensory contributions to impaired emotion processing in schizophrenia. Schiz Bull. 2009;35 doi: 10.1093/schbul/sbp109. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butler PD, Martinez A, Foxe JJ, Kim D, Zemon V, Silipo G, Mahoney J, Shpaner M, Jalbrzikowski M, Javitt DC. Subcortical visual dysfunction in schizophrenia drives secondary cortical impairments. Brain. 2007;130:417–430. doi: 10.1093/brain/awl233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler PD, Schechter I, Zemon V, Schwartz SG, Greenstein VC, Gordon J, Schroeder CE, Javitt DC. Dysfunction of early-stage visual processing in schizophrenia. Am J Psychiatry. 2001;158:1126–1133. doi: 10.1176/appi.ajp.158.7.1126. [DOI] [PubMed] [Google Scholar]
- 27.Butler PD, Silverstein SM, Dakin SC. Visual perception and its impairment in schizophrenia. Biol Psychiatry. 2008;64:40–47. doi: 10.1016/j.biopsych.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butler PD, Zemon V, Schechter I, Saperstein AM, Hoptman MJ, Lim KO, Revheim N, Silipo G, Javitt DC. Early-stage visual processing and cortical amplification deficits in schizophrenia. Arch Gen Psychiatry. 2005;62:495–504. doi: 10.1001/archpsyc.62.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butt AM. Neurotransmitter-mediated calcium signalling in oligodendrocyte physiology and pathology. Glia. 2006;54:666–675. doi: 10.1002/glia.20424. [DOI] [PubMed] [Google Scholar]
- 30.Chang BP, Lenzenweger MF. Somatosensory processing and schizophrenia liability: proprioception, exteroceptive sensitivity, and graphesthesia performance in the biological relatives of schizophrenia patients. J Abnorm Psychol. 2005;114:85–95. doi: 10.1037/0021-843X.114.1.85. [DOI] [PubMed] [Google Scholar]
- 31.Chen CM, Lakatos P, Shah AS, Mehta AD, Givre SJ, Javitt DC, Schroeder CE. Functional anatomy and interaction of fast and slow visual pathways in macaque monkeys. Cereb Cortex. 2007;17:1561–1569. doi: 10.1093/cercor/bhl067. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Palafox GP, Nakayama K, Levy DL, Matthysse S, Holzman PS. Motion perception in schizophrenia. Arch Gen Psychiatry. 1999;56:149–154. doi: 10.1001/archpsyc.56.2.149. [DOI] [PubMed] [Google Scholar]
- 33.Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, Bougueleret L, Barry C, Tanaka H, La Rosa P, et al. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci U S A. 2002;99:13675–13680. doi: 10.1073/pnas.182412499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clinton SM, Haroutunian V, Meador-Woodruff JH. Up-regulation of NMDA receptor subunit and post-synaptic density protein expression in the thalamus of elderly patients with schizophrenia. J Neurochem. 2006;98:1114–1125. doi: 10.1111/j.1471-4159.2006.03954.x. [DOI] [PubMed] [Google Scholar]
- 35.Clinton SM, Meador-Woodruff JH. Abnormalities of the NMDA Receptor and Associated Intracellular Molecules in the Thalamus in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology. 2004;29:1353–1362. doi: 10.1038/sj.npp.1300451. [DOI] [PubMed] [Google Scholar]
- 36.Covington MA, Riedel WJ, Brown C, He C, Morris E, Weinstein S, Semple J, Brown J. Does ketamine mimic aspects of schizophrenic speech? J Psychopharmacol. 2007;21:338–346. doi: 10.1177/0269881107077729. [DOI] [PubMed] [Google Scholar]
- 37.Davis KL, Kahn RS, Ko GN, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–1486. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 38.Dolen G, Bear MF. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol. 2008;586:1503–1508. doi: 10.1113/jphysiol.2008.150722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doniger GM, Foxe JJ, Murray MM, Higgins BA, Javitt DC. Impaired visual object recognition and dorsal/ventral stream interaction in schizophrenia. Arch Gen Psychiatry. 2002;59:1011–1020. doi: 10.1001/archpsyc.59.11.1011. [DOI] [PubMed] [Google Scholar]
- 40.Dravid SM, Erreger K, Yuan H, Nicholson K, Le P, Lyuboslavsky P, Almonte A, Murray E, Mosely C, Barber J, French A, Balster R, Murray TF, Traynelis SF. Subunit-specific mechanisms and proton sensitivity of NMDA receptor channel block. J Physiol. 2007;581:107–128. doi: 10.1113/jphysiol.2006.124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fisher M, Holland C, Merzenich MM, Vinogradov S. Using neuroplasticity-based auditory training to improve verbal memory in schizophrenia. Am J Psychiatry. 2009;166:805–811. doi: 10.1176/appi.ajp.2009.08050757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Floresco SB, Tse MT, Ghods-Sharifi S. Dopaminergic and glutamatergic regulation of effort- and delay-based decision making. Neuropsychopharmacology. 2008;33:1966–1979. doi: 10.1038/sj.npp.1301565. [DOI] [PubMed] [Google Scholar]
- 43.Foxe JJ, Doniger GM, Javitt DC. Early visual processing deficits in schizophrenia: impaired P1 generation revealed by high-density electrical mapping. Neuroreport. 2001;12:3815–3820. doi: 10.1097/00001756-200112040-00043. [DOI] [PubMed] [Google Scholar]
- 44.Gold S, Arndt S, Nopoulos P, O’Leary DS, Andreasen NC. Longitudinal study of cognitive function in first-episode and recent- onset schizophrenia. Am J Psychiatry. 1999;156:1342–1348. doi: 10.1176/ajp.156.9.1342. [DOI] [PubMed] [Google Scholar]
- 45.Goldberg TE, Straub RE, Callicott JH, Hariri A, Mattay VS, Bigelow L, Coppola R, Egan MF, Weinberger DR. The G72/G30 gene complex and cognitive abnormalities in schizophrenia. Neuropsychopharmacology. 2006;31:2022–2032. doi: 10.1038/sj.npp.1301049. [DOI] [PubMed] [Google Scholar]
- 46.Gouzoulis-Mayfrank E, Heekeren K, Neukirch A, Stoll M, Stock C, Obradovic M, Kovar KA. Psychological effects of (S)-ketamine and N,N-dimethyltryptamine (DMT): a double-blind, cross-over study in healthy volunteers. Pharmacopsychiatry. 2005;38:301–311. doi: 10.1055/s-2005-916185. [DOI] [PubMed] [Google Scholar]
- 47.Guillin O, Abi-Dargham A, Laruelle M. Neurobiology of dopamine in schizophrenia. Int Rev Neurobiol. 2007;78:1–39. doi: 10.1016/S0074-7742(06)78001-1. [DOI] [PubMed] [Google Scholar]
- 48.Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA. Genome-wide expression analysis reveals dysregulation of myelination- related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hashimoto K, Engberg G, Shimizu E, Nordin C, Lindstrom LH, Iyo M. Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:767–769. doi: 10.1016/j.pnpbp.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 50.Hashimoto K, Fujita Y, Horio M, Kunitachi S, Iyo M, Ferraris D, Tsukamoto T. Co-administration of a D-amino acid oxidase inhibitor potentiates the efficacy of D-serine in attenuating prepulse inhibition deficits after administration of dizocilpine. Biol Psychiatry. 2009;65:1103–1106. doi: 10.1016/j.biopsych.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, Nakazato M, Kumakiri C, Okada S, Hasegawa H, Imai K, Iyo M. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572–576. doi: 10.1001/archpsyc.60.6.572. [DOI] [PubMed] [Google Scholar]
- 52.Heerey EA, Robinson BM, McMahon RP, Gold JM. Delay discounting in schizophrenia. Cogn Neuropsychiatry. 2007;12:213–221. doi: 10.1080/13546800601005900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heggelund P, Hartveit E. Neurotransmitter receptors mediating excitatory input to cells in the cat lateral geniculate nucleus. I. Lagged cells. J Neurophysiol. 1990;63:1347–1360. doi: 10.1152/jn.1990.63.6.1347. [DOI] [PubMed] [Google Scholar]
- 54.Hetem LA, Danion JM, Diemunsch P, Brandt C. Effect of a subanesthetic dose of ketamine on memory and conscious awareness in healthy volunteers. Psychopharmacology (Berl) 2000;152:283–288. doi: 10.1007/s002130000511. [DOI] [PubMed] [Google Scholar]
- 55.Homayoun H, Moghaddam B. NMDA Receptor Hypofunction Produces Opposite Effects on Prefrontal Cortex Interneurons and Pyramidal Neurons. J Neuroscience. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Homayoun H, Stefani MR, Adams BW, Tamagan GD, Moghaddam B. Functional Interaction Between NMDA and mGlu5 Receptors: Effects on Working Memory, Instrumental Learning, Motor Behaviors, and Dopamine Release. Neuropsychopharmacology. 2004;29:1259–1269. doi: 10.1038/sj.npp.1300417. [DOI] [PubMed] [Google Scholar]
- 57.Honey GD, Honey RA, O’Loughlin C, Sharar SR, Kumaran D, Suckling J, Menon DK, Sleator C, Bullmore ET, Fletcher PC. Ketamine disrupts frontal and hippocampal contribution to encoding and retrieval of episodic memory: an fMRI study. Cereb Cortex. 2005;15:749–759. doi: 10.1093/cercor/bhh176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Honey RA, Turner DC, Honey GD, Sharar SR, Kumaran D, Pomarol-Clotet E, McKenna P, Sahakian BJ, Robbins TW, Fletcher PC. Subdissociative dose ketamine produces a deficit in manipulation but not maintenance of the contents of working memory. Neuropsychopharmacology. 2003;28:2037–2044. doi: 10.1038/sj.npp.1300272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hons J, Zirko R, Ulrychova M, Cermakova E, Libiger J. D-serine serum levels in patients with schizophrenia: relation to psychopathology and comparison to healthy subjects. Neuro endocrinology letters. 2008;29:485–492. [PubMed] [Google Scholar]
- 60.Javitt DC. Negative schizophrenic symptomatology and the phencyclidine (PCP) model of schizophrenia. Hillside J Psychiatry. 1987;9:12–35. [PubMed] [Google Scholar]
- 61.Javitt DC. Intracortical mechanisms of mismatch negativity dysfunction in schizophrenia. Audiol Neurootol. 2000;5:207–215. doi: 10.1159/000013882. [DOI] [PubMed] [Google Scholar]
- 62.Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry. 2004;9:984–997. 979. doi: 10.1038/sj.mp.4001551. [DOI] [PubMed] [Google Scholar]
- 63.Javitt DC. Is the glycine site half saturated or half unsaturated? Effects of glutamatergic drugs in schizophrenia patients. Curr Opin Psychiatry. 2006;19:151–157. doi: 10.1097/01.yco.0000214340.14131.bd. [DOI] [PubMed] [Google Scholar]
- 64.Javitt DC. Glutamate and Schizophrenia: Phencyclidine, N-Methyl-D-Aspartate Receptors, and Dopamine-Glutamate Interactions. Int Rev Neurobiol. 2007;78:69–108. doi: 10.1016/S0074-7742(06)78003-5. [DOI] [PubMed] [Google Scholar]
- 65.Javitt DC. Glycine transport inhibitors and the treatment of schizophrenia. Biol Psychiatry. 2008;63:6–8. doi: 10.1016/j.biopsych.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 66.Javitt DC. Glycine transport inhibitors for the treatment of schizophrenia: symptom and disease modification. Current opinion in drug discovery & development. 2009;12:468–478. [PubMed] [Google Scholar]
- 67.Javitt DC. When doors of perception close: bottom-up models of disrupted cognition in schizophrenia. Annual review of clinical psychology. 2009;5:249–275. doi: 10.1146/annurev.clinpsy.032408.153502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Javitt DC, Balla A, Burch S, Suckow R, Xie S, Sershen H. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29:300–307. doi: 10.1038/sj.npp.1300313. [DOI] [PubMed] [Google Scholar]
- 69.Javitt DC, Balla A, Sershen H. A novel alanine-insensitive D-serine transporter in rat brain synaptosomal membranes. Brain Res. 2002;941:146–149. doi: 10.1016/s0006-8993(02)02557-x. [DOI] [PubMed] [Google Scholar]
- 70.Javitt DC, Frusciante M. Glycyldodecylamide, a phencyclidine behavioral antagonist, blocks cortical glycine uptake: implications for schizophrenia and substance abuse. Psychopharmacology (Berl) 1997;129:96–98. doi: 10.1007/s002130050168. [DOI] [PubMed] [Google Scholar]
- 71.Javitt DC, Hashim A, Sershen H. Modulation of Striatal Dopamine Release by Glycine Transport Inhibitors. Neuropsychopharmacology. 2005;30:649–656. doi: 10.1038/sj.npp.1300589. [DOI] [PubMed] [Google Scholar]
- 72.Javitt DC, Liederman E, Cienfuegos A, Shelley AM. Panmodal processing imprecision as a basis for dysfunction of transient memory storage systems in schizophrenia. Schiz Bull. 1999;25:763–775. doi: 10.1093/oxfordjournals.schbul.a033417. [DOI] [PubMed] [Google Scholar]
- 73.Javitt DC, Shelley AM, Silipo G, Lieberman JA. Deficits in auditory and visual context-dependent processing in schizophrenia: defining the pattern. Arch Gen Psychiatry. 2000;57:1131–1137. doi: 10.1001/archpsyc.57.12.1131. [DOI] [PubMed] [Google Scholar]
- 74.Javitt DC, Spencer KM, Thaker GK, Winterer G, Hajos M. Neurophysiological biomarkers for drug development in schizophrenia. Nature reviews. 2008;7:68–83. doi: 10.1038/nrd2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci U S A. 1996;93:11962–11967. doi: 10.1073/pnas.93.21.11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Javitt DC, Strous RD, Grochowski S, Ritter W, Cowan N. Impaired precision, but normal retention, of auditory sensory (“echoic”) memory information in schizophrenia. J Abnorm Psychol. 1997;106:315–324. doi: 10.1037//0021-843x.106.2.315. [DOI] [PubMed] [Google Scholar]
- 77.Javitt DC, Zukin SR. The role of excitatory amino acids in neuropsychiatric illness. J Neuropsychiatry Clin Neurosci. 1990;2:44–52. doi: 10.1176/jnp.2.1.44. [DOI] [PubMed] [Google Scholar]
- 78.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 79.Kampa BM, Clements J, Jonas P, Stuart GJ. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol. 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kapur S, Remington G. Dopamine D(2) receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biol Psychiatry. 2001;50:873–883. doi: 10.1016/s0006-3223(01)01251-3. [DOI] [PubMed] [Google Scholar]
- 81.Karayanidis F, Nicholson R, Schall U, Meem L, Fulham R, Michie PT. Switching between univalent task-sets in schizophrenia: ERP evidence of an anticipatory task-set reconfiguration deficit. Clin Neurophysiol. 2006;117:2172–2190. doi: 10.1016/j.clinph.2006.06.716. [DOI] [PubMed] [Google Scholar]
- 82.Keefe RS, Bilder RM, Harvey PD, Davis SM, Palmer BW, Gold JM, Meltzer HY, Green MF, Miller del D, Canive JM, Adler LW, Manschreck TC, Swartz M, Rosenheck R, Perkins DO, Walker TM, Stroup TS, McEvoy JP, Lieberman JA. Baseline neurocognitive deficits in the CATIE schizophrenia trial. Neuropsychopharmacology. 2006;31:2033–2046. doi: 10.1038/sj.npp.1301072. [DOI] [PubMed] [Google Scholar]
- 83.Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, Cooper TB, Carlsson A, Laruelle M. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry. 2000;48:627–640. doi: 10.1016/s0006-3223(00)00976-8. [DOI] [PubMed] [Google Scholar]
- 84.Kegeles LS, Martinez D, Kochan LD, Hwang DR, Huang Y, Mawlawi O, Suckow RF, Van Heertum RL, Laruelle M. NMDA antagonist effects on striatal dopamine release: positron emission tomography studies in humans. Synapse. 2002;43:19–29. doi: 10.1002/syn.10010. [DOI] [PubMed] [Google Scholar]
- 85.Keilhoff G, Becker A, Grecksch G, Wolf G, Bernstein HG. Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience. 2004;126:591–598. doi: 10.1016/j.neuroscience.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 86.Kieffaber PD, Kappenman ES, Bodkins M, Shekhar A, O’Donnell BF, Hetrick WP. Switch and maintenance of task set in schizophrenia. Schizophr Res. 2006;84:345–358. doi: 10.1016/j.schres.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 87.Kim D, Wylie G, Pasternak R, Butler PD, Javitt DC. Magnocellular contributions to impaired motion processing in schizophrenia. Schizophr Res. 2006;82:1–8. doi: 10.1016/j.schres.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kirkpatrick B, Fenton WS, Carpenter WT, Jr, Marder SR. The NIMH-MATRICS consensus statement on negative symptoms. Schizophr Bull. 2006;32:214–219. doi: 10.1093/schbul/sbj053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kring AM, Barrett LF, Gard DE. On the broad applicability of the affective circumplex: representations of affective knowledge among schizophrenia patients. Psychol Sci. 2003;14:207–214. doi: 10.1111/1467-9280.02433. [DOI] [PubMed] [Google Scholar]
- 91.Kring AM, Kerr SL, Smith DA, Neale JM. Flat affect in schizophrenia does not reflect diminished subjective experience of emotion. J Abnorm Psychol. 1993;102:507–517. doi: 10.1037//0021-843x.102.4.507. [DOI] [PubMed] [Google Scholar]
- 92.Krishnan GP, Vohs JL, Hetrick WP, Carroll CA, Shekhar A, Bockbrader MA, O’Donnell BF. Steady state visual evoked potential abnormalities in schizophrenia. Clin Neurophysiol. 2005;116:614–624. doi: 10.1016/j.clinph.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 93.Krystal JH, Bennett A, Abi-Saab D, Belger A, Karper LP, D’Souza DC, Lipschitz D, Abi-Dargham A, Charney DS. Dissociation of ketamine effects on rule acquisition and rule implementation: possible relevance to NMDA receptor contributions to executive cognitive functions. Biol Psychiatry. 2000;47:137–143. doi: 10.1016/s0006-3223(99)00097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Krystal JH, D’Souza DC, Karper LP, Bennett A, Abi-Dargham A, Abi-Saab D, Cassello K, Bowers MB, Jr, Vegso S, Heninger GR, Charney DS. Interactive effects of subanesthetic ketamine and haloperidol in healthy humans. Psychopharmacology (Berl) 1999;145:193–204. doi: 10.1007/s002130051049. [DOI] [PubMed] [Google Scholar]
- 95.Krystal JH, Karper LP, Bennett A, D’Souza DC, Abi-Dargham A, Morrissey K, Abi-Saab D, Bremner JD, Bowers MB, Jr, Suckow RF, Stetson P, Heninger GR, Charney DS. Interactive effects of subanesthetic ketamine and subhypnotic lorazepam in humans. Psychopharmacology (Berl) 1998;135:213–229. doi: 10.1007/s002130050503. [DOI] [PubMed] [Google Scholar]
- 96.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 97.Krystal JH, Perry EB, Jr, Gueorguieva R, Belger A, Madonick SH, Abi-Dargham A, Cooper TB, Macdougall L, Abi-Saab W, D’Souza DC. Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine: implications for glutamatergic and dopaminergic model psychoses and cognitive function. Arch Gen Psychiatry. 2005;62:985–994. doi: 10.1001/archpsyc.62.9.985. [DOI] [PubMed] [Google Scholar]
- 98.Labrie V, Fukumura R, Rastogi A, Fick LJ, Wang W, Boutros PC, Kennedy JL, Semeralul MO, Lee FH, Baker GB, Belsham DD, Barger SW, Gondo Y, Wong AH, Roder JC. Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum Mol Genet. 2009 doi: 10.1093/hmg/ddp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- 100.Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–467. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- 101.Lavoie S, Murray MM, Deppen P, Knyazeva MG, Berk M, Boulat O, Bovet P, Bush AI, Conus P, Copolov D, Fornari E, Meuli R, Solida A, Vianin P, Cuenod M, Buclin T, Do KQ. Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology. 2008;33:2187–2199. doi: 10.1038/sj.npp.1301624. [DOI] [PubMed] [Google Scholar]
- 102.Lee J, Park S. Working memory impairments in schizophrenia: a meta-analysis. J Abnorm Psychol. 2005;114:599–611. doi: 10.1037/0021-843X.114.4.599. [DOI] [PubMed] [Google Scholar]
- 103.Leitman DI, Foxe JJ, Butler PD, Saperstein A, Revheim N, Javitt DC. Sensory contributions to impaired prosodic processing in schizophrenia. Biol Psychiatry. 2005;58:56–61. doi: 10.1016/j.biopsych.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 104.Leitman DI, Hoptman MJ, Foxe JJ, Saccente E, Wylie GR, Nierenberg J, Jalbrzikowski M, Lim KO, Javitt DC. The neural substrates of impaired prosodic detection in schizophrenia and its sensorial antecedents. Am J Psychiatry. 2007;164:474–482. doi: 10.1176/ajp.2007.164.3.474. [DOI] [PubMed] [Google Scholar]
- 105.Leitman DI, Laukka P, Juslin PN, Saccente E, Butler P, Javitt DC. Getting the Cue: Sensory Contributions to Auditory Emotion Recognition Impairments in Schizophrenia. Schizophr Bull. 2008 doi: 10.1093/schbul/sbn115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leitman DI, Ziwich R, Pasternak R, Javitt DC. Theory of Mind (ToM) and counterfactuality deficits in schizophrenia: misperception or misinterpretation? Psychol Med. 2006;36:1075–1083. doi: 10.1017/S0033291706007653. [DOI] [PubMed] [Google Scholar]
- 107.Leung S, Croft RJ, Baldeweg T, Nathan PJ. Acute dopamine D(1) and D(2) receptor stimulation does not modulate mismatch negativity (MMN) in healthy human subjects. Psychopharmacology (Berl) 2007;194:443–451. doi: 10.1007/s00213-007-0865-1. [DOI] [PubMed] [Google Scholar]
- 108.Lewis DA, Volk DW, Hashimoto T. Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology (Berl) 2004;174:143–150. doi: 10.1007/s00213-003-1673-x. [DOI] [PubMed] [Google Scholar]
- 109.Li CS. Impaired detection of visual motion in schizophrenia patients. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:929–934. doi: 10.1016/s0278-5846(02)00207-5. [DOI] [PubMed] [Google Scholar]
- 110.Lindahl JS, Kjellsen BR, Tigert J, Miskimins R. In utero PCP exposure alters oligodendrocyte differentiation and myelination in developing rat frontal cortex. Brain Res. 2008;1234:137–147. doi: 10.1016/j.brainres.2008.06.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Linn GS, O’Keeffe RT, Schroeder CE, Lifshitz K, Javitt DC. Behavioral effects of chronic phencyclidine in monkeys. Neuroreport. 1999;10:2789–2793. doi: 10.1097/00001756-199909090-00017. [DOI] [PubMed] [Google Scholar]
- 112.Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, Bankston LA. Cysteine regulation of protein function--as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- 113.Liu YL, Fann CS, Liu CM, Chang CC, Wu JY, Hung SI, Liu SK, Hsieh MH, Hwang TJ, Chan HY, Chen JJ, Faraone SV, Tsuang MT, Chen WJ, Hwu HG. No association of G72 and D-amino acid oxidase genes with schizophrenia. Schizophr Res. 2006;87:15–20. doi: 10.1016/j.schres.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 114.Lynch DR, Guttmann RP. NMDA receptor pharmacology: perspectives from molecular biology. Curr Drug Targets. 2001;2:215–231. doi: 10.2174/1389450013348434. [DOI] [PubMed] [Google Scholar]
- 115.Madeira C, Freitas ME, Vargas-Lopes C, Wolosker H, Panizzutti R. Increased brain D-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr Res. 2008;101:76–83. doi: 10.1016/j.schres.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 116.Madry C, Betz H, Geiger JR, Laube B. Supralinear potentiation of NR1/NR3A excitatory glycine receptors by Zn2+ and NR1 antagonist. Proc Natl Acad Sci U S A. 2008;105:12563–12568. doi: 10.1073/pnas.0805624105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Magleby KL. Modal gating of NMDA receptors. Trends Neurosci. 2004;27:231–233. doi: 10.1016/j.tins.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 118.Maier W. Common risk genes for affective and schizophrenic psychoses. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 2):37–40. doi: 10.1007/s00406-008-2008-z. [DOI] [PubMed] [Google Scholar]
- 119.Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17:141–150. doi: 10.1016/S0893-133X(97)00036-5. [DOI] [PubMed] [Google Scholar]
- 120.Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, Breier A. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14:301–307. doi: 10.1016/0893-133X(95)00137-3. [DOI] [PubMed] [Google Scholar]
- 121.March L, Cienfuegos A, Goldbloom L, Ritter W, Cowan N, Javitt DC. Normal time course of auditory recognition in schizophrenia, despite impaired precision of the auditory sensory (“echoic”) memory code. J Abnorm Psychol. 1999;108:69–75. doi: 10.1037//0021-843x.108.1.69. [DOI] [PubMed] [Google Scholar]
- 122.Martinez A, Hillyard SA, Dias EC, Hagler DJ, Jr, Butler PD, Guilfoyle DN, Jalbrzikowski M, Silipo G, Javitt DC. Magnocellular pathway impairment in schizophrenia: evidence from functional magnetic resonance imaging. J Neurosci. 2008;28:7492–7500. doi: 10.1523/JNEUROSCI.1852-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Brit J Med Psychol. 1961;34:103–116. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- 124.Meador-Woodruff JH, Clinton SM, Beneyto M, McCullumsmith RE. Molecular abnormalities of the glutamate synapse in the thalamus in schizophrenia. Ann N Y Acad Sci. 2003;1003:75–93. doi: 10.1196/annals.1300.005. [DOI] [PubMed] [Google Scholar]
- 125.Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100:433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- 126.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 127.Morgan CJ, Mofeez A, Brandner B, Bromley L, Curran HV. Acute effects of ketamine on memory systems and psychotic symptoms in healthy volunteers. Neuropsychopharmacology. 2004;29:208–218. doi: 10.1038/sj.npp.1300342. [DOI] [PubMed] [Google Scholar]
- 128.Morris RGM. Synaptic plasticity and learning: selective impairment of learning in rats and blockade of long-term potentiation in vivo by the N-methyl-D-aspartate receptor antagonist AP5. J Neuroscience. 1989;9:3040–3057. doi: 10.1523/JNEUROSCI.09-09-03040.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Neeman G, Blanaru M, Bloch B, Kremer I, Ermilov M, Javitt DC, Heresco-Levy U. Relation of plasma glycine, serine, and homocysteine levels to schizophrenia symptoms and medication type. Am J Psychiatry. 2005;162:1738–1740. doi: 10.1176/appi.ajp.162.9.1738. [DOI] [PubMed] [Google Scholar]
- 130.Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- 131.Oranje B, van Berckel BN, Kemner C, van Ree JM, Kahn RS, Verbaten MN. The effects of a sub-anaesthetic dose of ketamine on human selective attention. Neuropsychopharmacology. 2000;22:293–302. doi: 10.1016/S0893-133X(99)00118-9. [DOI] [PubMed] [Google Scholar]
- 132.Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 133.Passie T, Karst M, Wiese B, Emrich HM, Schneider U. Effects of different subanesthetic doses of (S)-ketamine on neuropsychology, psychopathology, and state of consciousness in man. Neuropsychobiology. 2005;51:226–233. doi: 10.1159/000085724. [DOI] [PubMed] [Google Scholar]
- 134.Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 135.Philpot BD, Cho KK, Bear MF. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron. 2007;53:495–502. doi: 10.1016/j.neuron.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rabinowicz EF, Silipo G, Goldman R, Javitt DC. Auditory sensory dysfunction in schizophrenia: imprecision or distractibility? Arch Gen Psychiatry. 2000;57:1149–1155. doi: 10.1001/archpsyc.57.12.1149. [DOI] [PubMed] [Google Scholar]
- 137.Rabinowitz J, Reichenberg A, Weiser M, Mark M, Kaplan Z, Davidson M. Cognitive and behavioural functioning in men with schizophrenia both before and shortly after first admission to hospital. Cross-sectional analysis. Br J Psychiatry. 2000;177:26–32. doi: 10.1192/bjp.177.1.26. [DOI] [PubMed] [Google Scholar]
- 138.Radant AD, Bowdle TA, Cowley DS, Kharasch ED, Roy-Byrne PP. Does ketamine-mediated N-methyl-D-aspartate receptor antagonism cause schizophrenia-like oculomotor abnormalities? Neuropsychopharmacology. 1998;19:434–444. doi: 10.1016/S0893-133X(98)00030-X. [DOI] [PubMed] [Google Scholar]
- 139.Revheim N, Schechter I, Kim D, Silipo G, Allingham B, Butler P, Javitt DC. Neurocognitive and symptom correlates of daily problem-solving skills in schizophrenia. Schizophr Res. 2006;83:237–245. doi: 10.1016/j.schres.2005.12.849. [DOI] [PubMed] [Google Scholar]
- 140.Rivadulla C, Sharma J, Sur M. Specific roles of NMDA and AMPA receptors in direction-selective and spatial phase-selective responses in visual cortex. J Neurosci. 2001;21:1710–1719. doi: 10.1523/JNEUROSCI.21-05-01710.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schechter I, Butler PD, Jalbrzikowski M, Pasternak R, Saperstein AM, Javitt DC. A New Dimension of Sensory Dysfunction: Stereopsis Deficits in Schizophrenia. Biol Psychiatry. 2006 doi: 10.1016/j.biopsych.2006.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schechter I, Butler PD, Zemon VM, Revheim N, Saperstein AM, Jalbrzikowski M, Pasternak R, Silipo G, Javitt DC. Impairments in generation of early-stage transient visual evoked potentials to magno- and parvocellular-selective stimuli in schizophrenia. Clin Neurophysiol. 2005 doi: 10.1016/j.clinph.2005.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schwartz B, Winstead DK. Visual perception in medicated schizophrenic patients. Am J Psychiatry. 1997;154:585–587. [PubMed] [Google Scholar]
- 144.Smith GS, Schloesser R, Brodie JD, Dewey SL, Logan J, Vitkun SA, Simkowitz P, Hurley A, Cooper T, Volkow ND, Cancro R. Glutamate modulation of dopamine measured in vivo with positron emission tomography (PET) and 11C-raclopride in normal human subjects. Neuropsychopharmacology. 1998;18:18–25. doi: 10.1016/S0893-133X(97)00092-4. [DOI] [PubMed] [Google Scholar]
- 145.Steullet P, Neijt HC, Cuenod M, Do KQ. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience. 2006;137:807–819. doi: 10.1016/j.neuroscience.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 146.Stoet G, Snyder LH. Effects of the NMDA Antagonist Ketamine on Task-Switching Performance: Evidence for Specific Impairments of Executive Control. Neuropsychopharmacology. 2006;31:1675–1681. doi: 10.1038/sj.npp.1300930. [DOI] [PubMed] [Google Scholar]
- 147.Stone JM, Erlandsson K, Arstad E, Squassante L, Teneggi V, Bressan RA, Krystal JH, Ell PJ, Pilowsky LS. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: a [(123)I]CNS-1261 SPET study. Psychopharmacology (Berl) 2008;197:401–408. doi: 10.1007/s00213-007-1047-x. [DOI] [PubMed] [Google Scholar]
- 148.Strous RD, Cowan N, Ritter W, Javitt DC. Auditory sensory (“echoic”) memory dysfunction in schizophrenia. Am J Psychiatry. 1995;152:1517–1519. doi: 10.1176/ajp.152.10.1517. [DOI] [PubMed] [Google Scholar]
- 149.Sumiyoshi T, Anil AE, Jin D, Jayathilake K, Lee M, Meltzer HY. Plasma glycine and serine levels in schizophrenia compared to normal controls and major depression: relation to negative symptoms. Int J Neuropsychopharmacol. 2004;7:1–8. doi: 10.1017/S1461145703003900. [DOI] [PubMed] [Google Scholar]
- 150.Sumiyoshi T, Jin D, Jayathilake K, Lee M, Meltzer HY. Prediction of the ability of clozapine to treat negative symptoms from plasma glycine and serine levels in schizophrenia. Int J Neuropsychopharmacol. 2005;8:451–455. doi: 10.1017/S1461145705005237. [DOI] [PubMed] [Google Scholar]
- 151.Tremeau F, Antonius D, Cacioppo JT, Ziwich R, Butler PD, Malaspina D, Javitt DC. Processing of positive affect in schizophrenia. doi: 10.1016/j.schres.2009.10.019. (submitted) [DOI] [PubMed] [Google Scholar]
- 152.Tremeau F, Antonius D, Cacioppo JT, Ziwich R, Jalbrzikowski M, Saccente E, Silipo G, Butler P, Javitt D. In support of Bleuler: objective evidence for increased affective ambivalence in schizophrenia based upon evocative testing. Schizophr Res. 2009;107:223–231. doi: 10.1016/j.schres.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 153.Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res. 2005;76:1–23. doi: 10.1016/j.schres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 154.Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57:1139–1147. doi: 10.1001/archpsyc.57.12.1139. [DOI] [PubMed] [Google Scholar]
- 155.Umbricht D, Vollenweider FX, Schmid L, Grubel C, Skrabo A, Huber T, Koller R. Effects of the 5-HT2A agonist psilocybin on mismatch negativity generation and AX-continuous performance task: implications for the neuropharmacology of cognitive deficits in schizophrenia. Neuropsychopharmacology. 2003;28:170–181. doi: 10.1038/sj.npp.1300005. [DOI] [PubMed] [Google Scholar]
- 156.Verkhratsky A, Kirchhoff F. NMDA Receptors in glia. Neuroscientist. 2007;13:28–37. doi: 10.1177/1073858406294270. [DOI] [PubMed] [Google Scholar]
- 157.Wang CZ, Yang SF, Xia Y, Johnson KM. Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology. 2008;33:2442–2455. doi: 10.1038/sj.npp.1301647. [DOI] [PubMed] [Google Scholar]
- 158.Wang H, Stradtman GG, 3rd, Wang XJ, Gao WJ. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc Natl Acad Sci U S A. 2008;105:16791–16796. doi: 10.1073/pnas.0804318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wylie GR, Clark EA, Butler PD, Javitt DC. Schizophrenia Patients Show Task Switching Deficits Consistent With N-Methyl-D-Aspartate System Dysfunction But Not Global Executive Deficits: Implications for Pathophysiology of Executive Dysfunction in Schizophrenia. Schizophr Bull. 2008 doi: 10.1093/schbul/sbn119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yao JK, Leonard S, Reddy R. Altered glutathione redox state in schizophrenia. Disease markers. 2006;22:83–93. doi: 10.1155/2006/248387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]