

Abstract
Androgen deprivation therapy for prostate cancer leads to a significant increase of HDL, which is generally viewed as beneficial, particularly for cardiovascular disease, but the effect of HDL on prostate cancer is unknown. In this study, we investigated the effect of HDL on prostate cancer cell proliferation, migration, intracellular cholesterol levels, and the role of cholesterol transporters, namely ABCA1, ABCG1 and SR-BI in these processes. HDL induced cell proliferation and migration of the androgen-independent PC-3 and DU145 cells by a mechanism involving ERK1/2 and Akt, but had no effect on the androgen-dependent LNCaP cell, which did not express ABCA1 unlike the other cell lines. Treatment with HDL did not significantly alter the cholesterol content of the cell lines. Knockdown of ABCA1 but not ABCG1 or SR-BI by siRNA, inhibited HDL-induced cell proliferation, migration and ERK1/2 and Akt signal transduction in PC-3 cells. Moreover, after treatment of LNCaP cells with charcoal stripped FBS, ABCA1 was induced approximately 10-fold enabling HDL to induce ERK1/2 activation, whereas siRNA knockdown of ABCA1 inhibited HDL-induced ERK1/2 activation. Simvastatin, which inhibited ABCA1 expression in PC-3 and DU145 cells, attenuated HDL-induced PC-3 and DU145 cell proliferation, migration and ERK1/2 and Akt phosphorylation. In human prostate biopsy samples, ABCA1 mRNA expression was approximately 2-fold higher in the androgen deprivation therapy group than in subjects with benign prostatic hyperplasia or pretreatment prostate cancer groups. In summary, these results suggest that HDL by an ABCA1-dependent mechanism can mediate signal transduction, leading to increased proliferation and migration of prostate cancer cells.
Keywords: HDL, prostate cancer, ABCA1, statin, signal transduction
Introduction
Similar to cardiovascular disease, the morbidity and mortality of prostate cancer is significantly higher in Western countries than in Asian countries (1). The incidence of prostate cancer in Japanese immigrants, who live in United States, is 4 times higher than in Japan (2), which suggests that there are environmental and or nutritional factors that contribute to the development of prostate cancer. A high intake of dietary fat has been implicated as a risk factor for several cancers, including prostate cancer (3, 4). Low density lipoprotein (LDL) and remnant lipoproteins, which increase on high fat diets and promote the delivery of cholesterol to cells, are reported to induce the proliferation of prostate cancer cell lines (5, 6). The synthesis or acquisition of exogenous cholesterol, a key component of cell membranes, is known to be important in cancer cell growth, including prostate cancer. Previous reports suggested a relationship between malignancy and cholesterol levels in prostatic tissue and secretions (7, 8) and in lipid rafts, which are key platforms for cell signaling events (9).
Androgen deprivation therapy in men with prostate cancer often leads to a significant increase of high density lipoprotein (HDL) (10). Androgens are known to suppress the expression of ABCA1 (11), which is necessary for HDL biogenesis (12). ABCA1 is more abundantly expressed in androgen-independent prostate cancer cells than in androgen-dependent cells (13). Besides playing a role in HDL biogenesis and cholesterol efflux, ABCA1 is also known to be involved in HDL signal transduction (14).
In breast cancer and adrenocortical tumor cells, HDL has been shown to be a growth factor, by delivering cholesterol to cells via the SR-BI scavenger receptor (15, 16). For most cell types, HDL promotes the net removal or efflux of cholesterol from cells by ABCA1 and ABCG1 transporters (17). In addition to its role in modulating the level of cellular cholesterol, HDL has many other pleiotrophic effects on cells, which potentially could affect cancer cell growth. HDL, for example, triggers the activation of ERK1/2, and PI3K/Akt (18, 19), that promote cell growth when activated by phosphorylation (20, 21).
In this study, we examined the effect of HDL on the growth of three commonly used human prostate cancer cell lines (22), namely PC-3, DU145, and LNCaP cells. LNCaP cells express the androgen receptor and hence show androgen-dependent growth like most early prostate cancers. In contrast, PC-3 and DU145 cells are androgen-independent and are commonly used as model cell lines for advanced prostate cancer, which often develop castration-resistant after prolong androgen deprivation therapy (22). In this study, we found that HDL in an ABCA1-dependent manner promoted cell proliferation and migration of the androgen independent cell lines by activating ERK1/2 and Akt. These results suggest a previously unrecognized role of HDL and ABCA1 in the pathogenesis of prostate cancer.
Results
HDL induced proliferation and migration of PC-3 and DU145 but not LNCaP cells
First, we examined the effect of HDL on prostate cancer cell proliferation by the MTS assay (Fig. 1A) and by cell counting (Fig. 1B). The viable cell number of PC-3 and DU145 cells grown in serum free media was significantly increased after incubation with HDL in a dose (Fig. 1A) and time-dependent manner (Fig 1B). In contrast, HDL did not induce the proliferation of LNCaP cells (Fig. 1A). We also determined whether HDL could induce the migration of prostate cancer cells in a wound healing assay (Fig 1C and 1D) in the presence of Mitomycin C, to inhibit cell proliferation (Supplementary Fig. S1). Besides increasing cell proliferation, HDL was also found to induce the migration of PC-3 and DU145 cells by approximately 3-fold and 2-fold respectively, but did not alter the cell migration of LNCaP cells.
FIGURE 1. Proliferation and migration of prostate cancer cells by HDL.

A. Cells were incubated in the medium, containing various concentrations of HDL or 10 % FBS. After 48 h, the number of viable cells was measured by the MTS assay. Values are expressed as the mean +SD (n=4). *P<0.01 vs HDL 0 µg/mL. B. Cells were incubated with or without HDL as in Panel A and analyzed for cell growth after the indicated times. The numbers of cells were expressed as fold change over the control (HDL 0 µg/mL, 48 h). Values are expressed as the mean +SD (n=3). *P<0.01 vs HDL 0 µg/mL in each time. C and D. Cells were wounded and then cultured in the medium for 0 or 48 h with or without HDL (PC-3; 100 µg/mL, DU145 and LNCaP; 250 µg/mL). Cell migration into the wound was examined by phase-contrast microscopy. Values are expressed as the mean +SD (n=3). *P<0.01 vs control
HDL did not alter intracellular cholesterol levels in prostate cancer cells
In a dose-dependent manner, HDL was found to both promote cholesterol efflux (Fig. 2A), as well as cholesterol uptake by cells (Fig. 2B). Unlike LDL, which increased total cholesterol levels in PC-3 and DU145 cells, and Methyl-β-cyclodextrin (MBCD), which lowered total cholesterol levels, HDL did not cause a net change in total cholesterol levels in any of the cell lines (Fig. 2C). LNCaP cells appeared to be relatively resistant to any change in total cellular cholesterol levels by the three different treatments (Fig. 2C). After treatment with MBCD, the proliferation of PC-3 cells was significantly reduced, but this response was fully reversed, if MBCD was pre-complexed with cholesterol, or if the cells were co-incubated with LDL (100 µg/mL) or 10 % FBS, which contains lipoproteins (Fig. 2D). In contrast, HDL, at the same dose used for LDL, only partially attenuated the effect of MBCD on PC-3 cell growth.
FIGURE 2. Effect of HDL on cholesterol flux in prostate cancer cells.
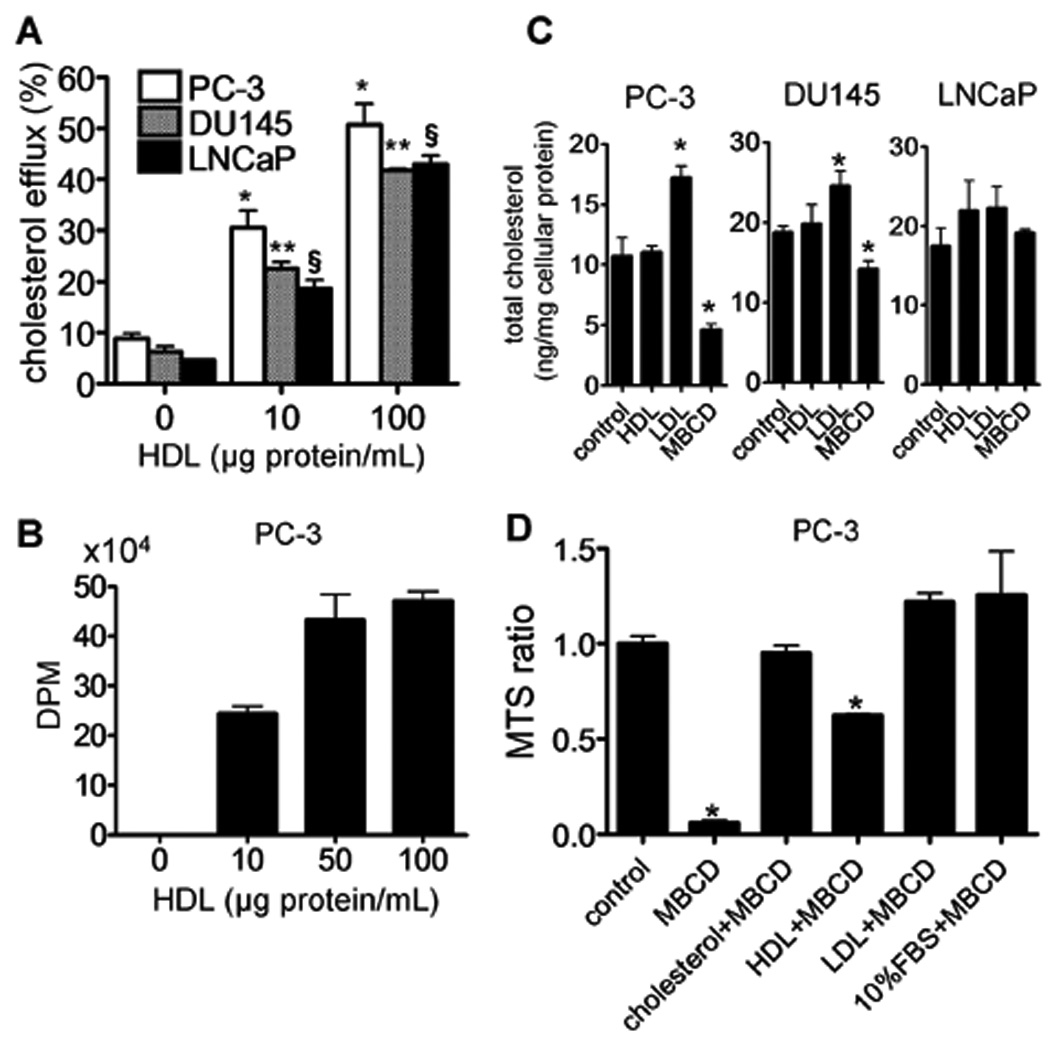
A. Cholesterol efflux to the indicated concentrations of HDL was determined after 18 h. Values are expressed as the mean +SD (n=3). *P<0.01 vs HDL 0 µg/mL (PC-3), **P<0.01 vs HDL 0 µg/mL (DU145), and §P<0.01 vs HDL 0 µg/mL (LNCaP). B. Cholesterol influx after 24 h to PC-3 cells with increasing concentration of HDL was determined after 24 h. Values are expressed as the mean +SD (n=3). C. Total cholesterol mass after 24 h treatment with or without HDL (100 µg/mL), LDL (100 µg/mL) or MBCD (1.25 mM) in the medium. Values are expressed as the mean +SD (n=3). *P<0.05 vs control. D. Effect of MBCD on PC-3 cell proliferation grown in the medium with or without MBCD (1.25 mM), cholesterol (5 µg/mL), HDL (100 µg/mL), LDL (100 µg/mL), or 10% FBS. Values are expressed as the mean +SD. (n=4). *P<0.01 vs control.
HDL activated ERK1/2 and Akt in androgen independent prostate cancer cells
For PC-3 and DU145 cells but not LNCaP cells, HDL at doses greater than 100 ug/mL was found to induce a prompt increase in both ERK1/2 and Akt phosphorylation (Fig 3A and B). PD98059, a MEK inhibitor, LY294002, a PI3K inhibitor, and PTX, a Gi protein inhibitor, were tested for their ability to affect the activation of ERK1/2 and Akt. In PC-3 cells, both PD98059 and PTX reduced HDL-induced phosphorylation of ERK1/2 (Fig. 3C), whereas both LY294002 and PTX reduced HDL-induced phosphorylation of Akt (Fig. 3D). In addition, these inhibitors partially blocked HDL-induced PC-3 proliferation (Fig. 3E) and showed even a greater effect on HDL-induced PC-3 migration (Fig. 3F). Similar results were obtained with DU145 cells (data not shown). The fold increase in Akt phosphorylation by HDL was greater in the presence of LY294002, but this was largely due to the decreased basal level of pAkt (Fig. 3D, lane 6). The overall level of pAkt was significantly decreased after treatment with HDL in the presence of LY294002 (Fig. 3D, lane 6 vs. lane 2).
FIGURE 3. HDL-induced proliferation and migration by MAPK and Akt activation.
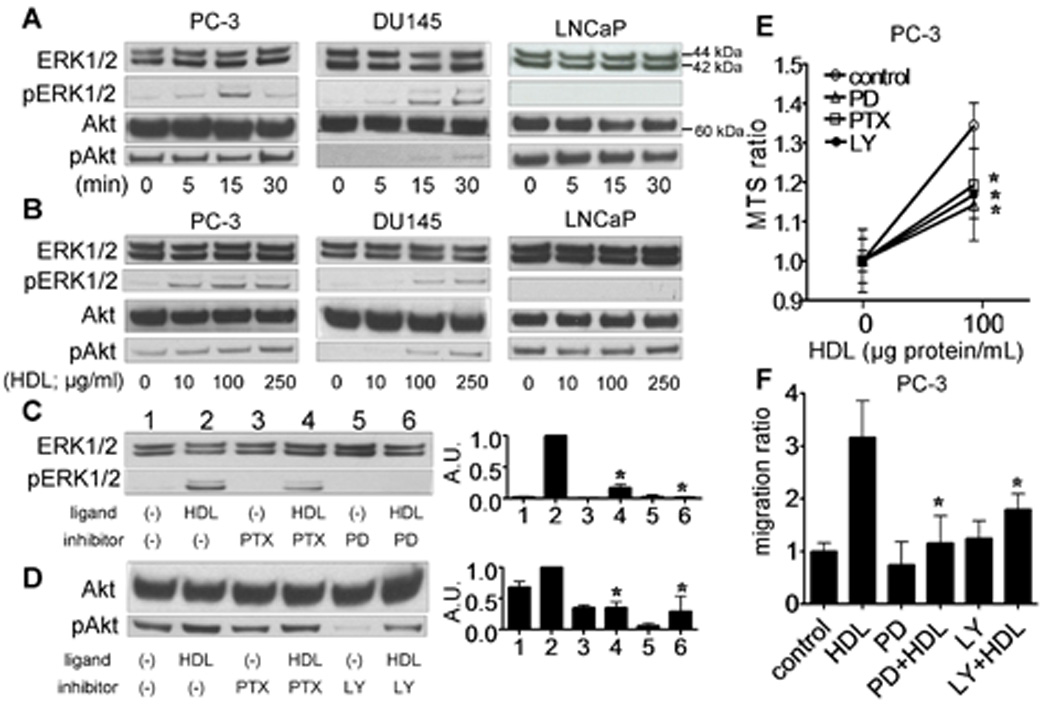
A and B. Cells cultured in the medium for 24 h were analyzed by western blotting after treatment for the indicated times with HDL (Panel A; HDL 100 µg/mL) or after 30 min (Panel B) for the indicated concentration of HDL. C and D. PC-3 cells were preincubated in the absence (−) or presence (+) of PTX (100 ng/mL) for 24 h, PD98059 (2.5 µM) for 1 h or LY294002 (2.5 µM) for 1 h and then incubated with or without HDL (100 µg/ml) for 15 min (C) or 30 min (D). Whole cell lysate was analyzed by western blotting. The graph shows the ratio of pERK1/2 to ERK1/2 in each sample relative to 1.0 for lane 2 (n=3, mean +SD). *P<0.05 vs lane 2. AU; arbitrary unit. E and F. PC-3 cells were preincubated in the absence or presence of PTX (100 ng/mL) for 24 h, PD98059 (2.5 µM) for 1 h or LY294002 (2.5 µM) for 1 h and then incubated with or without HDL (100µg/mL) for 48 h. The number of viable cells was measured by MTS assay. Values are expressed as the ratio of the response between HDL treated and non-treated (n=4, mean ±SD). *Pπ.05 vs HDL (E). Cells were wounded and then cultured for 0 or 48 h with or without HDL (PC-3; 100 µg/mL). Cell migration into the wound was examined by phase-contrast microscopy. Values are expressed as the mean +SD *P<0.05 vs HDL (F).
ABCA1 expression is lower in LNCaP than in PC-3 and DU145 cells
Next, we compared the mRNA expression of three different cell surface receptors that can exchange cholesterol with HDL, namely ABCA1, ABCG1 and SR-BI. ABCA1 mRNA and protein were observed in PC-3 and DU145 cells but was barely detectable in LNCaP cells (Fig. 4A and 4B). ABCG1 mRNA was expressed nearly equally in PC-3 and LNCaP cells but was relatively low in DU145 cells. The expression of SR-BI mRNA was similar in all three cell lines. Androgens are known to suppress the expression of ABCA1 in LNCaP cells (11), which we confirmed by incubating LNCaP cells in charcoal stripped FBS and showing the induction of ABCA1 mRNA and protein (Fig. 4C and 4D). This response could be blocked by when LNCaP cells in charcoal-stripped FBS were treated with R1881 (Methyltrienolone), a synthetic androgen (Fig. 4E). Incubation of the androgenin-dependent cell lines, PC-3 and DU145, in charcoal-stripped FBS had no effect on ABCA1 expression (data not shown). The level of ABCA1 mRNA in human prostate tissue was also examined. No difference was observed in the level of ABCA1 mRNA expression in benign prostatic hyperplasia (BPH) versus prostate cancer tissue, but prostate cancer tissue from patients treated with androgen deprivation therapy was approximately 2-fold higher (Fig. 4F). Like ABCA1, ABCG1 was also induced in prostate cancer tissue from patients treated with androgen deprivation therapy (Supplementary Fig. S2). The only significant difference observed for SR-BI was that patients with advanced prostate cancer and a Gleason score of 8–10 expressed about 40% less SR-BI mRNA (Supplementary Fig. S2).
FIGURE 4. ABCA1 expression in prostate cancer cell lines.
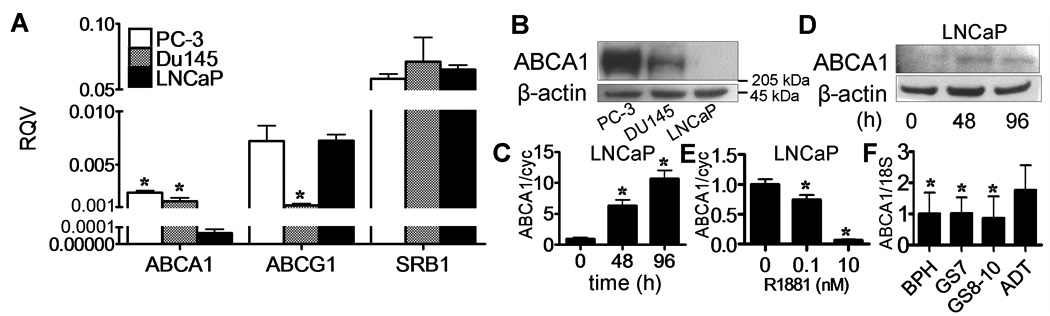
A. mRNA expression of ABCA1, ABCG1 and SR-BI was evaluated by real time PCR, and RQV (relative quantitative volume) is calculated by comparing the expression of cyclopholin A. Values are expressed as the mean +SD (n=4). *P<0.01 vs LNCaP. B. Western blotting of ABCA1 protein levels in prostate cancer cell lines. C and D. Effect of charcoal stripped FBS on the expression of ABCA1 in LNCaP cells. Total RNA (C) and protein (D) were collected after the indicated time. The expression of ABCA1 was evaluated by real time PCR (C) and western blotting (D). The expression of the genes was expressed as the fold change compared to 0 h (C). Values are expressed as the mean +SD (n=3). *P<0.01 vs 0 h (C). E. LNCaP cells were incubated in culture medium with 10 % charcoal stripped FBS for 96 h. After 48h, R1881 were added to the culture medium. Total RNA were collected. The expression of ABCA1 was evaluated by real time PCR. The expression of the genes was expressed as the fold change compared to 0 nM. Values are expressed as the mean +SD (n=3). *P<0.01 vs 0 nM. F. mRNA expression of ABCA1 in benign prostate hypertrophy (BPH; n=20), Gleason score 7 (GS7; n=19), Gleason score 8–10 (GS8-10; n=19) and androgen deprivation therapy group (ADT; n=18) were evaluated by real time PCR. The expression was expressed as the fold change compared to BPH. Values are expressed as the mean +SD *P<0.01 vs ADT.
Inhibition of ABCA1 expression decreased HDL-induced ERK1/2 and Akt activation
To further determine whether ABCA1 affects HDL-induced signal transduction, ABCA1 expression was reduced by transfection with a siRNA against ABCA1. ABCA1 mRNA was markedly reduced by siRNA treatment cells (Fig. 5A) and a corresponding decrease in the amount of ABCA1 protein was observed in PC-3 cells (Fig. 5B). The reduced level of ABCA1 in PC-3 cells following siRNA transfection was associated with reased basal cell proliferation (Fig. 5C). HDL-induced phosphorylation of ERK1/2 and Akt (Fig. 5D) and reduced HDL-induced cell proliferation (Fig. 5E) and cell migration (Fig. 5F). In contrast, siRNA knockdown of ABCG1 or SR-BI had no effect on HDL-induced cell proliferation (Supplementary Fig. S3A and B).
FIGURE 5. Effect of ABCA1 siRNA on HDL-induced signal activation.

A and B. Effect of siRNA on ABCA1 expression in PC-3 was evaluated. Mock transfected cells (M) or cells transfected with ABCA1 siRNA (A) or negative control siRNA (N), cells were incubated for 48 h before harvest for real time PCR (A) and western blotting (B). Values are expressed as the mean +SD (n=3). *P<0.01 vs negative (N). C. After transfection, PC-3 cells were cultured in the medium with 1% FBS. After 48 h, the number of viable cells was evaluated by MTS assay. Values are expressed as the mean +SD (n=4). *P<0.01 vs negative. D. After transfection, PC-3 cells were incubated with the medium containing 10 % FBS for 24 h, and medium was switched to serum-free medium. After 24 h, whole cell lysate was analyzed by western blotting after treatment for 15 min (ERK1/2) or 30 min (Akt). The graph shows the ratio of pERK1/2 to ERK1/2 or pAkt to Akt in each sample relative to 1.0 for lane 2 (n=3, mean +SD). *P<0.05 vs lane 2. E. After transfection, PC-3 cells were incubated with the medium containing 10% FBS for 24 h, and medium was switched to serum-free medium. After 24 h, cells were collected, and seeded again to 96-well plates in the medium with HDL (100 µg/mL). The number of viable cells was evaluated by MTS assay at the indicated times. The OD of the cell lysates was expressed as the fold change compared to 0 h in each group. Values are expressed as the mean +SD *P<0.05 vs negative. F. After transfection, PC-3 cells were incubated with the medium containing 10% FBS for 24 h, and medium was switched to serum-free medium. After 24 h, cells were wounded and then cultured for 0 or 48 h with or without HDL (100 µg/mL). Cell migration into the wound was examined by phase-contrast microscopy. Values are expressed as the mean +SD. (n=3). *P<0.05 vs N+HDL. M; mock, N; negative and A; ABCA1.
Overexpression of ABCA1 induced ERK1/2 activation by HDL in LNCaP cells
To evaluate whether up-regulation of ABCA1 can induce HDL-induced signal transductions in LNCaP cells, they were cultured in media containing charcoal-stripped or normal FBS for 96 h. HDL stimulated ERK1/2 phosphorylation in charcoal-stripped FBS treated cells but not in cells grown in FBS (Fig. 6A), which do not express ACBCA1 under these conditions (Fig. 4A and 4B) Phosphorylation of ERK1/2 induced by HDL could be attenuated after transfection with a siRNA for ABCA1 (Fig. 6B). When LNCaP cells were transfected with ABCA1 (Fig. 6C), they were able like PC-3 and DU145 cells to phosphorylate ERK1/2 when stimulated with HDL (Fig. 6D). We confirmed the effect of siRNA for ABCA1 on LNCaP cells by real time PCR (Supplementary Fig. S4)
FIGURE 6. Activation of ERK1/2 in LNCaP by HDL after Overexpression of ABCA1.

A. LNCaP cells were cultured in phenol red free RPMI1640 with 10 % FBS or charcoal stripped FBS for 72 h. Media was aspirated, and switched to phenol red free RPMI1640 with 1% FBS or charcoal stripped FBS for 24 h. Cells were then stimulated with HDL for 15 min. Cells were harvested and cell lysates were prepared for Western blotting. N; normal, CS; charcoal stripped. B. LNCaP cells were transfected with ABCA1 siRNA (A) or negative siRNA (N). After transfection, the cells were cultured in phenol red free RPMI1640 with 10 % charcoal stripped FBS for 72 h. Medium was aspirated, and switched to phenol red free RPMI1640 with 1% charcoal stripped FBS for 24 h. Cells were then stimulated with HDL (250 µg/mL) for 15 min, and harvested for Western blotting. Cells without transfection (M) and negative siRNA-transfected cells (N) were used as controls. The graph shows the ratio of pERK1/2 to ERK1/2 in each sample relative to 1.0 for lane 2 (n=3, means + s.d.).*P<0.05 vs lane 4. C. Effect of ABCA1 vector on ABCA1 expression in LNCaP cells was evaluated. Cells transfected with ABCA1 or control vector were incubated for 48 h before western blotting. D. After transfection of ABCA1 (A) or control (C) vector, LNCaP cells were incubated with the medium containing 10 % FBS for 24 h, and medium was switched to serum-free medium. After 24 h, whole cell lysate was analyzed by western blotting after treatment of HDL (250 µg/mL) for 15 min.
Simvastatin inhibits ABCA1 expression in androgen independent prostate cancer cells
Statin type drugs are known to modulate prostate cancer cell growth (23, 24) and are also known in many cell types to modulate the expression of ABCA1 (25, 26). In androgen independent cells, PC-3 and DU145, simvastatin in a time and dose-dependent manner markedly decreased ABCA1 mRNA (Fig. 7A) and protein, which also had a slightly faster migration (Fig. 7B). In contrast, simvastatin increased ABCA1 mRNA (Fig. 7A) in LNCaP cells, but ABCA1 protein in LNCaP cells was still undetectable after simvastatin treatment (data not shown). HDL-induced ERK1/2 and Akt phosphorylation (Fig. 7C), cell proliferation (Fig. 7D), and cell migration (Fig. 7E) were all reduced in PC-3 and DU145 cells by simvastatin treatment. Simvastatin, however, also partially reduced the basal rate of cell proliferation (Fig. 7D), migration (Fig. 7E) and Akt phosphorylation (Supplementary Fig. S5) of PC-3 cells in the absence of HDL.
FIGURE 7. Inhibition of ABCA1 expression in PC-3 and DU145 cells after simvastatin treatment.
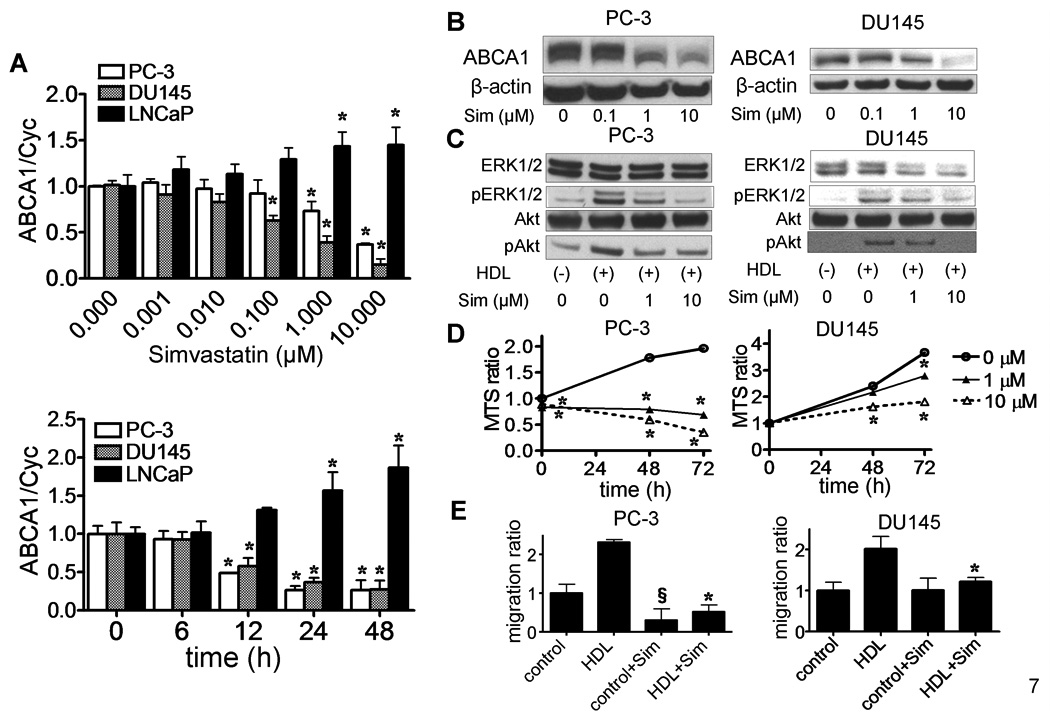
A. Total cell mRNA was extracted after treatment for the indicated concentration of simvastatin (48 h) or the indicated time with simvastatin (10 µM) in the medium containing 10%FBS. ABCA1 mRNA levels were measured by real-time PCR. Values are expressed as the mean +SD *P<0.05 vs 0 µM or 0 h. B. ABCA1 protein levels were measured by Western blotting. Cells were incubated in medium containing 10% FBS and various concentrations of simvastatin. After 48 h, total cell protein was extracted. Sim; simvastatin. C. Cells were incubated with the medium containing 10% FBS and various concentrations of simvastatin for 24 h, and medium was switched to serum-free medium containing various concentrations of simvastatin. After 24 h, the cells were stimulated with HDL (PC-3; 100 µg/mL, DU145; 250 µg/mL) for 15 min (ERK1/2) or 30 min (Akt). Cell lysates were prepared for Western blotting. D. Cells were cultured in culture medium containing various concentrations of simvastatin. After 48 h, HDL was added at 100 µg/mL (PC-3) or 250 µg/mL (DU145). After the indicated time, the number of viable cells was measured by MTS assay. The OD of the cell lysates was expressed as the fold change compared to control (0 h, 0 µM). Values are expressed as the mean +SD (n=4). *P<0.01 vs. simvastatin 0 µM. E. Cells were cultured in the medium with or without simvastatin (10 µM). After 48 h, cells were wounded and then cultured for 0 or 48 h with or without HDL (PC-3; 100 µg/mL, DU145; 250 µg/mL). Cell migration into the wound was examined by phase-contrast microscopy. Values are expressed as the mean +SD (n=3). *P<0.05 vs HDL, §P<0.05 vs control.
Discussion
The main finding of this study was that HDL induced cell proliferation and migration in androgen-independent PC-3 and DU145 cells but not in androgen-dependent LNCaP cells. We initially expected that HDL may inhibit cell growth and migration by removing cholesterol from cells and thus would antagonize the pro-growth effect of LDL on prostate cancer cells (5, 6), but HDL did not cause any net change in the cholesterol content of the three prostate cancer cells examined (Fig. 2D). This is most likely because of the ability of HDL to also donate cholesterol to cells by the SR-BI receptor (27) and the ability of cells to compensate for any loss of cholesterol by de novo synthesis (28). The decreased response of LNCaP cells to HDL-induced cell growth and migration appeared to correlate with their lower level of ABCA1, suggesting a role of ABCA1 in these processes. This was confirmed by siRNA knockdown experiments and in experiments that increased ABCA1 expression (Fig. 6). In cells expressing ABCA1, HDL also promoted the phosphorylation of both ERK1/2 and Akt (Fig. 3A), which are known to stimulate cell growth when activated (20, 21). We previously reported remnant lipoproteins can also promote prostate cancer cell growth by a similar mechanism (6). Another interesting finding was that prostate cancer tissue from subjects treated with androgen deprivation therapy, which raises HDL (10), were found to have increased ABCA1 expression (Fig. 4F).
It has been previously described that HDL is one of the main plasma transporters of sphingosine-1-phosphate (S1P), a potent bioactive signaling lipid (29). Furthermore, HDL has been shown to deliver S1P to cells and stimulate a variety of cell signaling processes, including the phosphorylation of ERK1/2 and Akt (30, 31) The direct treatment of prostate cancer cells with S1P has also been shown to stimulate cell growth (32). Thus, the observed effect of HDL on cell proliferation in this study may be related to its ability to deliver S1P, which is consistent with the PTX inhibitor results shown in Fig. 3. PTX is an inhibitor of G-coupled protein receptors and was found to block the ability of HDL to stimulate the phosphorylation of ERK1/2 and Akt and the proliferation of PC-3 cells. S1P can work with a wide variety of G-coupled protein receptors, such as S1P1-5, that could lead to the phosphorylation of ERK1/2 and Akt (33). Alternatively, ABCA1 after interacting with HDL is also known to mediate other cell signaling events (14), which could also affect cell proliferation and migration.
A potential important implication of these findings is that nutritional factors and or drugs that modulate lipids and lipoproteins may affect the formation of prostate cancer and or its growth and metastasis. One report recently described an inverse associations between statin use and the risk of prostate cancer (34), but another study found that statin use was only associated with a reduced risk of advanced disease, especially metastatic or fatal prostate cancer (35). In vitro, statins can inhibit prostate cancer proliferation, possibly by lowering the cholesterol content of rafts and by inhibiting cyclin-dependent-kinase-2 activity (23, 24). In this study, we observed that statins can also decrease the expression of ABCA1 in androgen independent prostate cancer cell lines (Fig. 7). Statins are known to lower the production of LXR ligands, such as 24(S), 25-epoxycholesterol (26), which induce the transcription of ABCA1. The results from this study suggest that the reduced level of ABCA1 expression by simvastatin could contribute to the ability of statins to reduce prostate cancer cell growth. It is important to note, however, that statins have many other pleiotrophic effects, such as altering the prenylation of Ras and RhoA and altering the membrane localization of small GTPases (36, 37), which could also contribute to its ability to decreased cell proliferation. Results from ABCA1 (Fig. 4) and siRNA (Fig. 5) transfection studies and modulation of androgen levels (Fig. 6) are consistent, however, with a role of ABCA1 and ERK1/2 phosphorylation in the ability of HDL to induce cell proliferation in androgen-independent cell lines.
There are no reports on the effect of drugs that raise HDL (38), such as niacin, on prostate cancer, but androgen deprivation therapy used in the treatment of advanced prostate cancer is known to significantly raise HDL. HDL typically increases between 8–20% following androgen deprivation therapy (10). This occurs, at least in part, because androgens can lower HDL levels by increasing the expression of SR-BI (39). As previously reported (11), we also found that androgens inhibit the expression of ABCA1 in LNCaP cells, which are androgen-dependent. In contrast, we found minimal effect of androgens on ABCA1 expression in the androgen-independent PC-3 and DU145 cells. Furthermore, we found that prostate cancer tissue from patients treated with androgen deprivation had higher levels of ABCA1. This suggests the possibility that advanced prostate cancer cells, which become androgen independent following androgen deprivation therapy, may be particularly sensitive to the growth promoting effects of HDL, because of their higher levels of ABCA1 and higher plasma HDL. Statins, which decreased the HDL-induced growth of PC-3 and DU145 by inhibiting ABCA1 and possibly by other mechanisms, may, therefore, be useful in treating hormone refractory prostate cancer after androgen deprivation therapy.
In summary, HDL was found to induce androgen independent prostate cancer cell proliferation and migration by an ABCA1-dependent mechanism. These results suggest that future studies on the effect of diet and lipid-modulating drugs may uncover new approaches for the prevention and treatment of prostate cancer.
Materials and methods
Cells and chemicals
The human prostate cell lines DU145, PC-3 and LNCaP were purchased from American type culture collection (Manassas, VA). DU145 was cultured in DMEM (Sigma, St. Louis, MO); PC-3 and LNCaP in RPMI1640 (Sigma), supplemented with 10 % FBS (Moregate, Bulimba, Australia). Antibodies (rabbit anti-ERK1/2 polyclonal antibody, rabbit anti-phospho-ERK1/2 (Thr202/Tyr204) polyclonal antibody, rabbit anti-Akt polyclonal antibody, rabbit anti-phospho-Akt (Ser473) polyclonal antibody and rabbit anti-human β-actin monoclonal antibody) were purchased from Cell Signaling (Beverly, MA). ABCA1 antibody was purchased from Abcam (Cambridge, MA). The inhibitors PD98059, PTX, and LY294002 and simvastatin were from Calbiochem (San Diego, CA). MBCD, charcoal stripped FBS and R1881 were purchased from Sigma, Invitrogen (Carlsbad, CA) and Perkin Elmer (Waltham, MA), respectively.
Isolation and labeling of lipoproteins
Human plasma was collected from normal healthy volunteers. Lipoproteins (LDL (1.019–1.063 g/mL), HDL (1.063–1.21 g/mL)) were isolated by density gradient centrifugation, as described previously (40). Lipoproteins were dialyzed against 4 L x 3 of of PBS (pH 7.4) and then sterilized, using a 0.22 µm filter unit (Millipore, Billerica, MA). HDL with [1, 2-3H] cholesterol was prepared, as previously described (41, 42).
Cell Proliferation assay of human prostate cancer cells
Cells were seeded into a 96-well microtiter plate in 100 µL of the medium, with 1 % FBS for 48 h. Thereafter, the medium was aspirated and the cells were incubated with the medium containing various concentrations of HDL. After incubation at 37 °C in 5 % CO2 for 48 h, the number of living cells was measured, using an MTS assay (Celltiter 96 Aqueous one solution cell proliferation assay, Promega, Madison, WI). The optical density of the cell lysate was expressed as fold change.
Migration assay
Cells were plated on a 12-well plate and grown to confluence. Thereafter, the medium was aspirated and the cells were incubated with the medium, containing 0.1 % bovine serum albumin (BSA) for 24 h before each experiment. 200 µl tips were used to make a denuded area. Cells were washed twice with PBS and treated with or without HDL for 48 h. Mitomycin C (0.5 µM) was added to the medium for blocking cell proliferation, during the whole 48 h period of the study. Photographs were taken at 0 and 48 h, and the cell migration distance was determined by subtracting the values obtained at 0 h from 48 h. The migration distances were expressed as fold change over the control. A representative experiment of three independent experiments is shown in each figure.
Cholesterol efflux assay
Cells were plated on a 24-well plate and incubated overnight in the medium, containing 10 % FBS. Cells were then washed with PBS and incubated in the medium with 10 % FBS and [1, 2-3H] cholesterol (1 µCi/ml) for 24 h. The cells were then washed with PBS, and incubated with the medium containing 0.1 % BSA and various concentrations of HDL. After 18 h, media was collected, centrifuged and counted for radioactivity. The residual radioactivity in the cell fraction was determined after an extraction with hexane:isopropanol (3:2, v/v). Percentage efflux was calculated by dividing the result by the sum of the radioactive counts in the medium plus the cell fraction.
Cholesterol influx Assay
Cells were plated on a 24-well plate and incubated overnight in the medium, containing 10 % FBS. Cells were then washed with PBS and incubated in the medium containing 0.1 % BSA and various concentrations of HDL with [1, 2-3H] cholesterol for 24 h. The media was collected, centrifuged and counted for radioactivity. The radioactivity in the cell fraction was determined after an extraction with hexane:isopropanol (3:2, v/v). The cellular radioactivity was shown as disintegration per minute (DPM).
Measurement of total cholesterol level in vitro
Cells were cultured on a 6-well plate and incubated overnight in the medium, containing 10 % FBS. Cells were then washed with PBS and incubated in the medium containing 0.1% BSA with or without HDL, LDL or MBCD. After 24 h, media was aspirated, and cells were washed with PBS. Cholesterol was extracted by hexane:isopropanol (3:2, v/v), and the solution was transferred to the glass tubes for drying by nitrogen gas. Once the tube was dried, 200 µl of 50 mM Tris, containing 0.1 % TritonX-100 and 10 mM sodium cholate was applied to the tube, and cholesterol concentrations were measured enymatically (Wako, Osaka, Japan). In addition, a solution of 0.1 % SD plus 0.1 N NaOH was applied to wells and the protein concentration was measured by DC protein assay (Bio-Rad). Total cholesterol level was calculated by dividing the result by the total protein concentration.
Quantification of mRNA levels
mRNA levels were quantified, using an 7300 Real Time PCR System (Applied Biosystems, Austin, TX). Total RNA extraction and cDNA synthesis were performed (43) and PCR amplification was done, using 2 µL of cDNA and ABCA1, A B C G 1 a n d S R-BI primer (No. Hs00194045_m1, Hs00245154_m1 and Hs00194092_m1, respectively, Applied Biosystems). Next, PCR was performed for one cycle of 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 60 s at 60 °C. For the internal control, cyclophilin A (No. 4326316E, Applied Biosystems) transcript levels were used. Quantitation of mRNA fold changes were done using the comparative CT(2−Δ ΔCt) cycle (ΔCt) method (44).
Western blotting assays
Cell lysates were prepared in RIPA buffer (Pierce, Rockford, IL), containing 1 mM sodium orthovanadate (Sigma) and protease inhibitors (Complete TM-without EDTA, Roche Diagnostics, Penzberg, Germany). Equal amounts of proteins (30–40 µg/lane) were electrophoresed on 4–12 % SDS–PAGE and transferred onto nitrocellulose membranes. Each membrane was incubated with the primary antibodies described above. Blots were developed with a 1:1000 dilution of the HRP-conjugated secondary antibody (Cell Signaling). Proteins were visualized, using Western Lightning Plus-ECL (Perkin Elmer). For ABCA1, protein lysates (60–100 µg/lane) were electrophoresed on 3–8% SDS-PAGE and transferred onto PVDF membranes. A representative experiment of three independent experiments is shown in each figure.
Prostate biopsy sample analysis
We quantified ABCA1, ABCG1 and SR-BI expression levels in prostate biopsy samples, using a quantitative real-time PCR method. A total of 76 patients, who had undergone prostate biopsy at Gunma University hospital in 2002–2007 were studied. Of these 76 patients, 20 had benign prostatic hyperplasia, 19 had prostate cancer, whose Gleason score is 7, 19 had prostate cancer with Gleason score is from 8 to 10, none of which received any therapy before prostate biopsy. An additional 18 patients with prostate cancer had undergone androgen deprivation therapy for more than 6 months. This study was approved by the Ethical Committee of Gunma University. The following are the primer sequences for 18s RNA: 18s rRNA: forward, 5’-CGG CTA CCA CAT CCA AGG AA-3’, reverse, 5’-GCT GGA ATT ACC GCG GCT GC-3’.
siRNA
Cells were seeded into a 6-well microtiter plate in 2300 µL (for Western blotting) with 10 % FBS. Thereafter, cells were transfected with ON-TARGETplus Non-targeting Pool (No. D-001810-10-05, Dharmacon, Waltham, MA, USA), ON-TARGETplus ABCA1 siRNA (No. L-004128-00, Dharmacon), ABCG1 siRNA (No. L-008615-00, Dharmacon) or SR-BI siRNA (No. L-010592-00, Dharmacon) using DharmaFect (Dharmacon). After transfection, the cells were incubated for 48 h at 37 °C in a 5% CO2 atmosphere.
Overexpression of ABCA1
The pcDNA3.1 vector containing the ABCA1 stop gene and pIRES-hrGFP -1a vector were digested with Not I (New England BioLabs, Ipswich, MA). The digested vectors were run on 1 % SeaKem ME Agarose (FMC Bio Products, Philadelphia, PA) TAE Gel using a Mini Gel Apparatus (Invitrogen). The ABCA1 insert and pIRES-hrGFP-1a vector were cut from the gel and purified with a GeneClean Kit (Bio101, Vista, CA). The purified ABCA1 insert was ligated into the purified pIRES-hrGFP-1a Vector using the T4 DNA Ligase (Roche). 2 µl of the Ligation Reaction was incubated with One Shot Chemically competent E.coli cells (Invitrogen) for 30 minutes. The cell suspension was spread onto a LB agar plate containing 100 µg/ml ampicillin and incubated overnight at 37 °C. Approximately, 25 colonies were picked and grown up for DNA isolation. The DNA was purified by QIAPREP-SPIN MINIPREP KIT (Qiagen). ABCA1 orientation was checked by digesting the various clones with BamH I (New England BioLabs) and running a 1 % SeaKem ME Agarose TAE Gel. pIRES-hrGFP-1a Vector was used as control. We transiently transfected the vectors to LNCaP cells by using Cell Line Nucleofector Kit R (Lonza, Basel, Switzerland).
Statistical analysis
All data unless otherwise indicated are expressed as the mean + SD of at least triplicates. Differences between the values were evaluated by one-way analysis of variance (one-way ANOVA) with Tukey’s post-hoc analysis. In all analyses, p values of less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
Research by ART and YS was supported by intramural NHBLI funding. The authors thank ML Sampson (Department of Laboratory Medicine, Clinical Center, National Institutes of Health, Bethesda, MD) and Naomi Takase (Gunma University, Maebashi, Japan) for technical assistance.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Reference
- 1.Hsing AW, Tsao L, Devesa SS. International trends and patterns prostate cancer incidence and mortality. Int J Cancer. 2000;85:60–67. doi: 10.1002/(sici)1097-0215(20000101)85:1<60::aid-ijc11>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 2.Grönberg H. Prostate cancer epidemiology. Lancet. 2003;361:859–864. doi: 10.1016/S0140-6736(03)12713-4. [DOI] [PubMed] [Google Scholar]
- 3.Mills PK, Beeson WL, Phillips RL, Fraser GE. Cohort study of diet, lifestyle, and prostate cancer in Adventist men. Cancer. 1989;64:598–604. doi: 10.1002/1097-0142(19890801)64:3<598::aid-cncr2820640306>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Giovannucci EM, Rimm EB, Colditz GA, et al. A prospective study of dietary fat and risk of prostate cancer. J Natl Cancer Inst. 1993;85:1571–1579. doi: 10.1093/jnci/85.19.1571. [DOI] [PubMed] [Google Scholar]
- 5.Hughes-Fulford M, Chen Y, Tjandrawinata RR. Fatty acid regulates gene expression and growth of human prostate cancer PC-3 cells. Carcinogenesis. 2001;22:701–707. doi: 10.1093/carcin/22.5.701. [DOI] [PubMed] [Google Scholar]
- 6.Sekine Y, Koike H, Nakano T, Nakajima K, Suzuki K. Remnant lipoproteins stimulate proliferation and activate MAPK and Akt signaling pathways via G protein-coupled receptor in PC-3 prostate cancer cells. Clin Chim Acta. 2007;383:78–84. doi: 10.1016/j.cca.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 7.Swyer GIM. The cholesterol content of normal and enlarged prostates. Cancer Res. 1942;2:372–375. [Google Scholar]
- 8.Acevedo HF, Campbell EA, Saier EL, et al. Urinary cholesterol. V. Its excretion in men with testicular and prostatic neoplasms. Cancer. 1973;32:196–205. doi: 10.1002/1097-0142(197307)32:1<196::aid-cncr2820320130>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 9.Oh HY, Lee EJ, Yoon S, Chung BH, Cho KS, Hong SJ. Cholesterol level of lipid raft microdomains regulates apoptotic cell death in prostate cancer cells through EGFR-mediated Akt and ERK signal transduction. Prostate. 2007;67:1061–1069. doi: 10.1002/pros.20593. [DOI] [PubMed] [Google Scholar]
- 10.Saylor PJ, Smith MR. Metabolic complications of androgen deprivation therapy for prostate cancer. J Urol. 2009;181:1998–2008. doi: 10.1016/j.juro.2009.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuchi J, Hiipakka RA, Kokontis JM, et al. Androgenic suppression of ATP-binding cassette transporter A1 expression in LNCaP human prostate cancer cells. Cancer Res. 2004;64:7682–7685. doi: 10.1158/0008-5472.CAN-04-2647. [DOI] [PubMed] [Google Scholar]
- 12.Remaley AT, Amar M, Sviridov D. HDL-replacement therapy: mechanism of action, types of agents and potential clinical indications. Expert Rev Cardiovasc Ther. 2008;6:1203–1205. doi: 10.1586/14779072.6.9.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ni J, Pang ST, Yeh S. Differential retention of alpha-vitamin E is correlated with its transporter gene expression and growth inhibition efficacy in prostate cancer cells. Prostate. 2007;67:463–471. doi: 10.1002/pros.20517. [DOI] [PubMed] [Google Scholar]
- 14.Imaizumi S, Miura S, Nakamura K, et al. Antiarrhythmogenic effect of reconstituted high-density lipoprotein against ischemia/reperfusion in rats. J Am Coll Cardiol. 2008;51:1604–1612. doi: 10.1016/j.jacc.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 15.Cao WM, Murao K, Imachi H, et al. A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer res. 2004;64:1515–1521. doi: 10.1158/0008-5472.can-03-0675. [DOI] [PubMed] [Google Scholar]
- 16.Murao K, Imachi H, Cao W, et al. High-density lipoprotein is a potential growth factor for adrenocortical cells. Biochem Biophys Res Commun. 2006;344:226–232. doi: 10.1016/j.bbrc.2006.03.131. [DOI] [PubMed] [Google Scholar]
- 17.Yancey PG, Bortnick AE, Kellner-Weibel G, de la Llera-Moya M, Phillips MC, Rothblat GH. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–719. doi: 10.1161/01.ATV.0000057572.97137.DD. [DOI] [PubMed] [Google Scholar]
- 18.O’Connell BJ, Genest J., Jr High-density lipoproteins and endothelial function. Circulation. 2001;104:1978–1983. doi: 10.1161/hc3901.096667. [DOI] [PubMed] [Google Scholar]
- 19.Nofer J, Assmann G. Atheroprotective affects of high-density lipoprotein-associated lysosphingolipids. Trends Cardiovasc Med. 2005;15:265–271. doi: 10.1016/j.tcm.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Chuanhai G, Louis ML, David TP. Mitogenic signaling in androgen sensitive and insensitive prostate cancer cell lines. J Urol. 2000;163:1027–1032. [PubMed] [Google Scholar]
- 21.Kane LP, Mollenauer MN, Xu Z, Turck CW, Weiss A. Akt-dependent phosphorylation specifically regulates Cot induction of NF-kappa B-dependent transcription. Mol Cell Biol. 2002;22:5962–5974. doi: 10.1128/MCB.22.16.5962-5974.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sobel RE, Sadar MD. Cell lines used in prostate cancer research: a compendium of old and new lines. J Urol. 2005;173:342–359. doi: 10.1097/01.ju.0000141580.30910.57. [DOI] [PubMed] [Google Scholar]
- 23.Sivaprasad U, Abbas T, Dutta A. Differential efficacy of 3-hydroxy-3-methylglutaryl CoA reductase inhibitors on the cell cycle of prostate cancer cells. Mol Cancer Ther. 2006;5:2310–2316. doi: 10.1158/1535-7163.MCT-06-0175. [DOI] [PubMed] [Google Scholar]
- 24.Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sone H, Shimano H, Shu M, et al. Statins downregulate ATP-binding-cassette transporter A1 gene expression in macrophages. Biochem Biophys Res Commun. 2004;316:790–794. doi: 10.1016/j.bbrc.2004.02.121. [DOI] [PubMed] [Google Scholar]
- 26.Wong J, Quinn CM, Gelissen IC, Jessup W, Brown AJ. The effect of statins on ABCA1 and ABCG1 expression in human macrophages is influenced by cellular cholesterol levels and extent of differentiation. Atherosclerosis. 2008;196:180–189. doi: 10.1016/j.atherosclerosis.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 27.Acton S, Rigotti A, Landschulz KT. Identification of Scavenger Receptor SR-BI as a High Density Lipoprotein Receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 28.Freeman MR, Solomon KR. Cholesterol and prostate cancer. J Cell Biochem. 2004;91:54–69. doi: 10.1002/jcb.10724. [DOI] [PubMed] [Google Scholar]
- 29.Kimura T, Sato K, Kuwabara A, et al. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J Biol Chem. 2001;276:31780–31785. doi: 10.1074/jbc.M104353200. [DOI] [PubMed] [Google Scholar]
- 30.Miura S, Fujino M, Matsuo Y, et al. High density lipoprotein-induced angiogenesis requires the activation of Ras/MAP kinase in human coronary artery endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:802–808. doi: 10.1161/01.ATV.0000066134.79956.58. [DOI] [PubMed] [Google Scholar]
- 31.Mineo C, Yuhanna IS, Quon MJ, Shaul PW. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem. 2003;278:9142–9149. doi: 10.1074/jbc.M211394200. [DOI] [PubMed] [Google Scholar]
- 32.Gibbs TC, Rubio MV, Zhang Z, Xie Y, Kipp KR, Meier KE. Signal transduction responses to lysophosphatidic acid and sphingosine 1-phosphate in human prostate cancer cells. Prostate. 2009;69:1493–1506. doi: 10.1002/pros.20994. [DOI] [PubMed] [Google Scholar]
- 33.Okajima F, Sato K, Kimura T. Anti-atherogenic actions of high-density lipoprotein through sphingosine 1-phosphate receptors and scavenger receptor class B type I. Endocr J. 2009;56:317–334. doi: 10.1507/endocrj.k08e-228. [DOI] [PubMed] [Google Scholar]
- 34.Shannon J, Tewoderos S, Garzotto M, et al. Statins and prostate cancer risk: a case-control study. Am J Epidemiol. 2005;162:318–325. doi: 10.1093/aje/kwi203. [DOI] [PubMed] [Google Scholar]
- 35.Platz EA, Leitzmann MF, Visvanathan K, et al. Statin drugs and risk of advanced prostate cancer. J Natl Cancer Inst. 2006;98:1819–1825. doi: 10.1093/jnci/djj499. [DOI] [PubMed] [Google Scholar]
- 36.Cafforio P, Dammacco F, Gernone A, Silvestris F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis. 2005;26:883–891. doi: 10.1093/carcin/bgi036. [DOI] [PubMed] [Google Scholar]
- 37.Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyers CD, Kamanna VS, Kashyap ML. Niacin therapy in atherosclerosis. Curr Opin Lipidol. 2004;15:659–665. doi: 10.1097/00041433-200412000-00006. [DOI] [PubMed] [Google Scholar]
- 39.Langer C, Gansz B, Goepfert C, et al. Testosterone up-regulates scavenger receptor BI and stimulates cholesterol efflux from macrophages. Biochem Biophys Res Commun. 2002;296:1051–1057. doi: 10.1016/s0006-291x(02)02038-7. [DOI] [PubMed] [Google Scholar]
- 40.Redgrave TG, Roberts DC, West CE. Separation of plasma lipoproteins by density-gradient ultracentrifugation. Anal Biochem. 1975;65:42–49. doi: 10.1016/0003-2697(75)90488-1. [DOI] [PubMed] [Google Scholar]
- 41.Khovidhunkit W, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Cholesterol efflux by acute-phase high density lipoprotein: role of lecithin: cholesterol acyltransferase. J Lipid Res. 2001;42:967–975. [PubMed] [Google Scholar]
- 42.Rothblat GH, Bamberger M, Phillips MC. Reverse cholesterol transport. Methods Enzymol. 1986;129:628–644. doi: 10.1016/0076-6879(86)29095-3. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki K, Koike H, Matsui H, et al. Genistein, a soy isoflavone, induces glutathione peroxidase in the human prostate cancer cell lines LNCaP and PC-3. Int J Cancer. 2002;99:846–852. doi: 10.1002/ijc.10428. [DOI] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.