

Abstract
BLM helicase, the protein mutated in Bloom Syndrome, is involved in signal transduction cascades after DNA damage. BLM is phosphorylated on multiple residues by different kinases either after stress induction or during mitosis. Here we have provided evidence that both Chk1 and Chk2 phosphorylated the N-terminal 660 amino acids of BLM. An internal region within the DExH motif of BLM negatively regulated the Chk1/Chk2 dependent N-terminal phosphorylation event. Using in silico analysis involving the Chk1 structure and its known substrate specificity, we predicted that Chk1 should preferentially phosphorylate BLM on Serine 646 (Ser646). The prediction was validated in vitro by phosphopeptide analysis on BLM mutants and in vivo by usage of a newly generated phosphospecific polyclonal antibody. We demonstrated that the phosphorylation at Ser646 on BLM was constitutive and decreased rapidly after exposure to DNA damage. This resulted in diminished interaction of BLM with nucleolin and PML isoforms and consequent decreased BLM accumulation in nucleolus and PML nuclear bodies (PML NBs). Instead BLM relocalized to the sites of DNA damage and bound with the damage sensor protein, Nbs1. Mutant analysis confirmed that the binding to nucleolin and PML isoforms required Ser646 phosphorylation. These results indicated that Chk1-mediated phosphorylation on BLM at Ser646 maybe a determinant for regulating its subnuclear localization could act as a marker for the activation status of BLM in response to DNA damage.
Keywords: Bloom helicase, DExH motif, phosphopeptide mapping, nucleolin, PML isoforms, structural bioinformatics
INTRODUCTION
Signal transduction during DNA damage response is mediated via two proximal sensory kinases, ATM (ataxia telangiectasia-mutated) and ATR (ATM-Rad3-related) (1, 2). ATR and ATM initiate the signaling cascade via phosphorylation of its downstream checkpoint effecter kinases, Chk1 and Chk2 (3). While ATR/Chk1 predominantly sensed the damage in response to stalled replication, ATM/Chk2 were involved in response to double strand breaks. ATM/ATR along with Chk1/Chk2 are known to phosphorylate a variety of downstream targets involved in different cellular process including DNA damage response.
The highly conserved family of protein, RecQ helicases, is involved in DNA damage response in human (4, 5). Mutation in three members of the RecQ helicase family led to cancer predisposition syndromes: Bloom Syndrome (due to mutation in BLM), Werner Syndrome (due to mutation in WRN) and Rothmund Thomson Syndrome (due to mutation in RTS). Bloom Syndrome patients are prone to almost all forms of cancer, thereby possibly indicating that BLM is involved in the regulation of key DNA metabolism processes like DNA replication, recombination and repair (6). BLM is also an upstream sensory protein involved in DNA damage signaling cascade especially after stalled replication (7–9). Hence it could be hypothesized that phosphorylation of BLM by these kinases would be a prerequisite for the helicase to function effectively as a DNA damage sensory protein in vivo.
ATM was the first kinase demonstrated to be involved in the phosphorylation of BLM (10). ATM phosphorylated BLM on Thr99 and Thr122 in response to ionizing radiation, with Thr99 being the major site of phosphorylation. ATM-dependent phosphorylation on these two sites was responsible for correction of radiation induced damage in BS cells but had no role during SCE (11). BLM and ATR colocalized in nucleus and the percentage of colocalization increased following stalling of replication fork. ATR also phosphorylated BLM on Thr99 and Thr122 in response to HU (7). Loss of phosphorylation in Thr99/Thr122 of BLM led to a failure to recover from HU-induced replication blockade leading to a subsequent arrest in caffeine sensitive G2/M checkpoint. Phosphorylation at Thr99 was required for the colocalization of BLM with γ-H2AX (12) and 53BP1 (13). We have earlier reported that Chk1 phosphorylated BLM (8). Functional interaction of Chk1 also exists with Dna2, a known helicase cum nuclease, which can (like BLM) be recruited to the sites of replication origin and interact with sensory proteins involved in DNA damage response (like ATM and MRN) (14). Chk1 (and Chk2) induction and phosphorylation occurred even in absence of Dna2, indicating that the latter is not required for the induction or signaling of checkpoints.
Apart from the kinases activated after DNA damage (like ATM and ATR), cell cycle specific kinases are also known to use BLM as a substrate. MPS1 dependent phosphorylation of BLM at Ser144 was required to prevent early mitotic exit and resulted in binding of polo-like kinase 1 (PLK1) and subsequent phosphorylation of BLM (15). Phosphorylation of BLM by Cdc2 at Ser714 and Thr766 resulted in its exclusion from the chromatin and nuclear scaffold, thereby preventing BLM from interfering with mitotic processes such as chromosome condensation (16). Thus the studies till date have indicated that BLM phosphorylation occurred either only during mitosis or in response to exogenous stress.
However it is also possible that in addition to stimulus-induced phosphorylation, BLM could also be constitutively phosphorylated. Evidence does exist in literature regarding the constitutive phosphorylation of proteins involved in signal transduction. For example, Chk1-mediated constitutive phophorylation event had been observed for Cdc25B, one of the three human phosphatases that activate the CDK-cyclin complexes. Chk1 phosphorylated Cdc25B in vitro and in vivo on multiple residues, including Ser230 and Ser563. Chk1-dependent Ser230 phosphorylation was constitutively observed in absence of DNA damage. In vivo the mutation of Ser230 increased the mitotic-inducing activity of CDC25B leading to the speculation that Chk1 constitutively phosphorylated Cdc25B during interphase and thus prevented the premature initiation of mitosis by negatively regulating the activity of Cdc25B at the centrosome (17). Chk1 could phosphorylate its substrates constitutively because its essential function during cell cycle could be uncoupled from DNA damage response function and checkpoint control (18).
In this study we wanted to determine the regulatory mechanisms governing Chk1/Chk2-mediated phosphorylations. We found that an internal region within the DExH motif, till now thought to play a role in the helicase function of BLM, could also negatively regulate the N-terminal phosphorylation on the helicase by Chk1/Chk2. Using a structure based in silico approach we predicted the sites where Chk1 could phosphorylate BLM. Ser646 was predicted to be the site that would be most preferentially phosphorylated on BLM by Chk1. Using biochemical and cell biology techniques involving a newly generated phosphospecific polyclonal antibody we found that phosphorylation of BLM by Chk1 indeed occurred in vivo at Ser646. This phosphorylation on BLM by Chk1 was constitutive in nature and was diminished on exposure to multiple types of DNA damage. Loss of Chk1-dependent Ser646 phosphorylation resulted in decreased BLM binding to nucleolin and PML isoforms, reduced accumulation in nucleolus and PML NBs and correlated with its (i.e. BLM’s) relocalization to the sites of DNA damage and binding with damage sensor protein, Nbs1. These results indicated that Ser646 phosphorylation on BLM may one of the determinants that regulated its subnuclear localization and thereby act as a marker reflecting the activity status of the helicase.
MATERIALS AND METHODS
Antibodies
A polyclonal antibody against phosphorylated Ser646 in BLM was raised in rabbits (Abexome Biosciences, Bangalore, India). Crude serum from inoculated rabbits was double-affinity purified using a phosphor-peptide and non-phosphor-peptide-conjugated Sepharose columns and measured for antibody concentration using an ELISA assay. Anti-BLM: rabbit polyclonal A300-110A (Bethyl) for westerns, goal polyclonal A-300-120A (Bethyl) for immunoprecipitations and immunofluorescence, Anti-hsp90: sc-7947 (Santa Cruz Biotechnology), Anti-nucleolin (C23): sc-8031 (Santa Cruz Biotechnology), Anti-PML: sc-966 (Santa Cruz Biotechnology), Anti-Nbs1: NB100-143 (Novus Biologicals), Anti-Lamin A/C: 612163 (BD Biosciences). Anti-Flag antibody and beads: F1804, A2220 (Sigma). Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories.
Recombinants
pGEX4T-1 BLM (1–212), pcDNA3 Flag BLM (gifted by Ian Hickson), pHook Chk1 (WT) (gifted by Carol Prives), GST Chk1 (WT) and GST Chk1 (D130A, kinase dead mutant) (gifted by Steve Elldege), pCDZF Chk2 and GST Chk2 (gifted by Thanos Halazonetis). pGEX4T-1 BLM (191–660), pGEX4T-1 BLM (621–1041), pGEX4T-1 BLM (1001–1417) (13). pGEX4T-1 BLM (1–1417) (9). pGEX4T-1 BLM (1–660), pGEX4T-1 BLM (1–800), pGEX4T-1 BLM (1–900), pGEX4T-1 BLM (1–1006), pGEX4T-1 BLM (1–1041) and pGEX4T-1 BLM (661–800) were obtained by cloning the respective PCR products into the BamH1/XhoI sites of the vector. pGEX4T-1 BLM (1–1211) and pGEX4T-1 BLM (1–1292) were obtained by cloning the respective PCR products into the BamH1 site of the vector and checking the orientation. pGEX4T-1 BLM (1–115), pGEX4T-1 BLM (109–212), pGEX4T-1 BLM (191–320), pGEX4T-1 BLM (321–530) and pGEX4T-1 BLM (531–660) were obtained by cloning the respective PCR products into the EcoR1/XhoI sites of the vector. GST Chk2 D347A (kinase dead) and pcDNA3 Flag BLM (S646A) mutants were obtained by site directed mutagenesis kit (Stratagene).
Kinase and peptide binding assays
Kinase assays with wild type or kinase dead (KD) Chk1 or Chk2 were carried out as described earlier (8). 5ng (for Chk1) or 10ng (for Chk2) of were used at 30°C for 20 minutes. Amounts of BLM and its derivative used in individual experiments, as described in the respective figure legends were obtained by quantitating the respective Coomassie visible bands in ImageJ software (NIH). A modified kinase assay used to determine the regions in BLM undergoing phosphorylation in presence of nuclear extracts had been earlier described (13). For peptide binding assays, the wild type or the mutant peptide (200 µg) were phosphorylated by the above method. The reactions were stopped with 10%TCA, samples were spotted on P81 phosphocellulose paper (Whatman), extensively washed and incorporation assessed by scintillation counting. UCN-01 (NCI, NIH) was used at 100nM in both in vitro and cell based assays. Time of incubation of UCN-01 on cells was 2 h.
Phosphopeptide analysis and phosphoamino analysis
For two-dimensional phosphopeptide maps or phosphoamino acid analysis, radiolabelled bands corresponding to the protein(s) of interest were excised from the nitrocellulose membrane and digested with mass spectrometry grade trypsin gold (Promega). The phosphopeptides were analyzed by two-dimensional resolution on thin-layer cellulose plates (19). In cases where the extent was phosphorylation was low, multiple kinase reactions were pooled together so that an equal amount of counts (25,000 cpm) were available for the two-dimensional resolution. Aliquots of the tryptic peptide mixtures were further processed and phosphoamino acid analysis was carried out as described (19).
Expression, purification and interaction of proteins
GST-tagged proteins were expressed according to standard protocols in E. coli at 16°C and subsequently purified by binding to Glutathione S-Sepharose (GE Healthcare) for use in interaction studies. Soluble proteins were obtained by eluting the bound proteins with reduced glutathione. pHook Chk1 (WT) and pCDZF Chk2 were used for coupled in vitro transcription/translation reactions of Chk1 and Chk2 respectively using T7 Quick coupled Transcription/Translation (TNT) System kit (Promega). GST-bound target proteins, whose expressions were visualized by Coomassie and quantitated by ImageJ software (NIH), were incubated with the in vitro translated interacting partner (one fifth of an entire TNT reaction) for 4 h at 4°C with constant inversion. Interaction was assayed by autoradiography. BLM was produced in S. cerevisiae using the yeast strain JEL1 (20) (gifted by Ian Hickson).
Cell culture conditions and treatments
hTERT-immortalized Bloom Syndrome fibroblasts (referred as BS), chromosome 15 minochromosome corrected BS fibroblasts (referred as A-15), hTERT immortalized Normal Human Fibroblasts (referred as NHF) were maintained as described (21). For HU experiments during IPs and IFs, cells were either left untreated (−HU) or treated (+HU) for 12 h. The cells were washed and allowed to grow for a further 6 h (PW). For neocarzinostatin (NCS) treatment, the cells were either left untreated (−NCS) or exposed to the drug for 1 h and 6 h (+NCS). After the 6 h exposure, the drug was washed off and treatment continued for a further 6 h (PW). Transfections were carried out with Lipofectamine2000 (Invitrogen). Whole cell lysates were made 36 h post-transfection in RIPA buffer.
Immunoprecipitations, confocal microscopy and siRNA
Cytoplasmic and nuclear extracts from cells were made using NE-PER Nuclear and Cytoplasmic Extraction reagent (Pierce). IPs were done as described previously (9) using 1 mg of the nuclear extracts. IFs was carried out as described previously (9). For confocal microscopy, the slides were analyzed on a Zeiss 510 Meta system with 63x/1.4 oil immersion or 40x/0.95 Corr objective. The laser lines used were Argon 458/477/488/514 nm (for FITC), DPSS 561 nm (for Texas Red) and a Chameleon Ultra autotunable femtosecond laser with a tuning range 690–1050 nm (for DAPI). LSM5 software was used for image acquisition. Quantitation was carried out after visualization of atleast 200 cells over three experiments. siRNA transfection for Chk1 (synthesized by Dharmacon, USA) was carried out as previously described (8).
Flow cytometry analysis
Cells, at different stages of the cell cycle or after different treatments, were subjected to cell cycle analysis in BD FACS Calibur. The data was analyzed either by FloJo or in WinMDI software.
In silico studies
In order to model peptides in complex with the Chk1 complex structure (11A8), the peptide bound crystal structure, 2PHK (22) was chosen as a template, as it was known to be structurally and functionally similar to Chk1 (23). Chk1 was superimposed onto 2PHK-MC peptide bound structure using the program ProFit (http://www.bioinf.org.uk/software/profit/) with a rmsd value of 1.372 Å and then MC peptide coordinates were transferred to Chk1, which led to the generation of substrate bound Chk1. Following standard nomenclature, the site of phosphorylation on the substrate peptide was referred as P0, while the three residues flanking the phosphorylation site on the N- and C-terminus were referred as P(−3), P(−2), P(−1) and P(+1), P(+2) and P(+3) respectively. For modeling the 22 known substrates of Chk1, backbone dependent rotamer library approach (24) was used for generating sidechains corresponding to the known substrates at sites P(−3) to P(+3). The 10 predicted peptides of BLM protein were also modeled onto Chk1 kinase using the same approach (24).
The resulting Chk1-peptide complexes were minimized by using CVFF forcefield and InsightII. Minimizations were carried out using steepest decent algorithm for initial iterations followed by 5000 iterations of conjugate gradient algorithm. The convergence criterion was set to rms gradient of 0.001 kcal mol−1Å−1. The models of Chk1 with 22 known substrates were analyzed for any conserved structural complementarities. Contacting residue pairs between the kinase and the peptide were identified using the criteria of any two atoms of the residue pair being at a distance, less than or equal to 4 Å. Another web based tool - WHAT IF (25), was used calculate the interactions. The interaction energy (van der Waals attraction/repulsion and electrostatic forces) between the 10 predicted BLM peptides and Chk1 were calculated by using the docking module of InsightII. The contribution of −3 and +1 position to the total binding energy of the peptide was also evaluated. Apart from all atom interactions, values for 10 BLM peptides were also calculated. The binding energy, based on the total number of contacts between kinase and the peptide, was evaluated using residue based statistical pair potential (26).
RESULTS
Chk1 and Chk2 phosphorylated BLM on serine and threonine residues in the first 660 amino acids
We had earlier demonstrated that Chk1 phosphorylated BLM in vitro (8). It is also known that Chk1 and Chk2 have overlapping substrate specificities (3). Indeed like Chk1, wild type Chk2, but not its kinase-dead counterpart, was able to phosphorylate recombinant wild type BLM (Figure 1A). In vitro translated Chk1 and Chk2 could bind to wild type BLM and its fragments (Figure 1B-1D). Chk1 preferably interacted with the N-terminal (1–212) region of BLM. However the central region of BLM, encompassing the two fragments (191–660) and (621–1024), also interacted with the kinase (Figure 1C).
Figure 1. Chk1 and Chk2 phosphorylate BLM in the first 660 amino acids.

A. Chk2 phosphorylated wildtype BLM. Recombinant Chk1 (WT or KD) was incubated with BLM (1–1417) (400 ng on left and 100 ng, 400 ng on right) in the presence of γ-32P ATP. The proteins were resolved by SDS-PAGE and detected by autoradiography.
B. (Left) Schematic diagram of full-length BLM and its fragments (1–212), (191–660), (621–1041) and (1001–1417). (Right) Levels of BLM (1–212), BLM (191–660), BLM (621–1041) and BLM (1001–1417) as detected by Coomassie. Approximately 1 µg of protein was leaded in each lane.
C. Interaction between in vitro translated S35-radiolabelled Chk1 and equal amounts (5 µg) of the glutathione-sepharose bound BLM fragments or GST alone. The amount of bound radioactivity was detected by autoradiography.
D. Same as (C), except S35-radiolabelled Chk2 was used for interaction with BLM (1–1417).
E. Chk1 phosphorylated BLM (1–212) and (1–660). Chk1-dependent kinase assays were carried out with 400 ng of BLM (1–212), BLM (191–660), BLM (621–1041) and BLM (1001–417). Arrows indicated the phosphorylated products.
F. Same as (E), except Chk2 was used as the kinase to phosphorylate BLM fragments.
Since BLM is a protein consisting of 1417 amino acids, we wanted to narrow down the region(s) where the Chk1/Chk2-mediated phosphorylations on BLM possibly occurred. With recombinant BLM fragments and the two kinases we carried out in vitro phosphorylation on each of the four fragments in presence of γ32P-ATP. Both BLM (1–212) and BLM (191–660) underwent robust Chk1/Chk2-dependent phosphorylation (Figure 1E, 1F). To fine map the regions of BLM that were highly phosphorylated by Chk1, smaller regions of BLM within the first 660 amino acids were cloned, expressed and purified (Supplementary Figure 1A). While both Chk1 and Chk2 phosphorylated BLM (109–212) and BLM (531–660) to a high extent, Chk2 phosphorylated BLM exclusively between residues (191–320) (Supplementary Figure 1B, 1C)
Next we wanted to determine the region(s) of BLM that were phosphorylated by Chk1 in presence of nuclear extracts prepared from asynchronously growing hTERT immortalized Normal Human Fibroblasts (NHF). Equal amounts of BLM fragments, BLM (1–212), BLM (191–660), BLM (621–1041) and BLM (1001–1417) were subjected to a modified kinase assay where nuclear extract was used as the source of kinase. BLM (1–212) and (191–660) were phosphoryated by the kinase present in the nuclear extract of BLM (Supplementary Figure 1D). The phosphorylation of BLM (1–212) and BLM (191–660) with the nuclear kinase was decreased when Chk1 inhibitor, UCN-01 was included in the reaction mixture (Supplementary Figure 1E), indicating that the predominant fraction of BLM phosphorylation in the N-terminus was dependent on Chk1.
Region within the DExH motif of the helicase negatively regulated Chk1 and Chk2-mediated N-terminal phosphorylation on BLM
We hypothesized that an internal stretch of amino acids in BLM may regulate its Chk1/Chk2-mediated N-terminal phosphorylation. To test this hypothesis we cloned, expressed and purified in E.coli full-length BLM and its seven C-terminal fragments (Figure 2A), and subsequently carried out phosphorylation with Chk1 or Chk2. BLM (1–660) was highly phosphorylated by both Chk1 and Chk2 (Figure 2B and Supplementary Figure 2A), which drastically decreased in BLM (1–800), thereby indicating the presence of a region between amino acids 660–800 of the helicase, which negatively regulated Chk1/Chk2-mediated BLM phosphorylation in the first 660 amino acids.
Figure 2. Amino acids within DExH motif negatively regulated the Chk1-mediated N-terminal phosphorylation of BLM.

A. Schematic diagram and relative expression levels of wild type BLM and its derivatives. (Left) Full length BLM (1–1417) and its various fragments (1–660), (1–800), (1–900), (1–1006), (1–1041), (1–1211), (1–1292) and (661–800). The helicase, RQC and HRDC domains are indicated. (Right, top) The expression of the BLM (1–660), (1–800), (1–900), (1–1006), (1–41041), (1–1211), (1–31292) and (1–1417) fragments were determined by Coomassie. Approximately 1 µg of protein was leaded in each lane.
B. In vitro phosphorylation of full-length BLM and C-terminal deletion fragments [i.e. BLM (1–660), (1–800), (1–900), (1–1006), (1–1041), (1–1211) and (1–1292)] (400 ng each) in presence of γ32P-ATP and recombinant Chk1.
C. BLM (661–800) negatively regulated the phosphorylation of BLM (1–660). (Left) BLM (661–800) was expressed, purified and checked by Coomassie along with BLM (1–660) and GST. Approximately 1 µg of protein was leaded in each lane. (Middle) Chk1-dependent phosphorylation of BLM (1–660) (400 ng) was carried out either alone or in presence of increasing amounts (50 ng, 100 ng, 200 ng, 400 ng) of BLM (661–800). As control, phosphorylation was done for GST (400 ng), BLM (661–800) (400 ng) and BLM (1–660) (400 ng) in presence of GST (400 ng). (Right) Graph indicates the extent of inhibition of the phosphorylation of BLM (1–660) by GST, Chk1 or Chk2. The values are represented by mean with the standard deviation.
D. BLM (661–800) inhibited BLM (1–600) phosphorylation even after preincubation with AMP-PNP. Chk1-dependent phosphorylation of BLM (1–660) (400 ng), alone or in presence of BLM (661–800) (400 ng) was carried out either without or after preincubation of BLM (661–800) with AMP-PNP (5 µM).
In BLM, the amino acids 683–833 encodes for DExH motif (27). Our results (Figure 2B and Supplementary Figure 2A) indicated that an aminoacid sequence within the DExH motif in BLM might negatively regulate the Chk1/Chk2-mediated phosphorylation in the first 660 amino acids of the helicase. To test this hypothesis we cloned, expressed and purified GST-BLM (661–800) (Figure 2C, left). GST-BLM (661–800), like GST itself, was not itself phosphorylated by Chk1. However addition of GST-BLM (661–800), but not GST alone, in trans, decreased Chk1/Chk2-mediated BLM (1–660) phosphorylation in a concentration dependent manner (Figure 2C, middle and right and Supplementary Figure 2B). Since BLM (660–800) lies within the ATP binding/helicase domain, it is possible that the decrease in Chk1/Chk2-mediated phosphorylation was a reflection of sequestration of ATP by this stretch of amino acids. Hence we carried out the kinase reactions in parallel, either without or after preincubation of BLM (661–800) with AMP-PNP, a competitive inhibitor of most ATP-dependent systems. BLM (661–800) could inhibit the Chk1/Chk2-mediated phosphorylation on BLM (1–660), irrespective of AMP-PNP preincubation (Figure 2D and Supplementary Figure 2C).
Chk1 phosphorylated BLM at Ser646 in vitro
BLM has been implicated as an early responder to replication stress (8, 9). Since Chk1 and its upstream kinase, ATR, are known to be the key determinants in the signal transduction pathway activated in response to stalled replication forks, we decided to determine the sites on BLM which were phosphorylated by Chk1. To have an idea about the number of sites on BLM, which were phosphorylated by Chk1 in vitro, we carried out two-dimensional phosphopeptide map analysis. For this assay we used recombinant full-length BLM, produced either in E.coli (9) or S. cerevisiae (20). Phosphopeptide map analysis with BLM (1–1417) (Figure 3A) indicated the presence of 25–30 phosphopeptides [as verified by color coding (Supplementary Figure 3A)], thereby indicating the presence of atleast that many sites at which Chk1 phosphorylated full-length BLM in vitro. Moreover the similar pattern obtained in the maps derived from recombinant BLM produced in two different hosts, indicated a similar three-dimensional structure and folding for both sources of recombinant human BLM. Interestingly comparison of the phosphopeptide maps of BLM (1–1417), BLM (1–1041) and BLM (1–660) indicated that all the phosphopeptides seen in full-length BLM [except one phosphopeptide present in BLM (1–1417) and BLM (1–1041) as indicated by a red arrow in Figure 3A] were also present within the first 660 amino acids of the helicase (compare the color codes across peptide maps in Supplementary Figure 3A), confirming the earlier data (Figure 1E) that this region of BLM was preferentially phosphorylated by Chk1.
Figure 3. Chk1 phosphorylated BLM at Ser646 in vitro.

A. Phosphopeptide maps of human BLM (1–1417) (produced either in S. cerevisiae or in E. coli) and BLM fragments (1–1041) and (1–660) phosphorylated in vitro by Chk1. Red arrow indicates a phosphopeptide present in all except BLM (1–660). The black arrows indicate the directions in which the phosphopeptides were separated by electrophoresis and chromatography in the first and second dimensions, respectively.
B. Same as (A) except the following BLM derivatives were used: BLM (1–212), BLM (109–212), BLM (191–660), BLM (531–660), BLM (1–660) and BLM (1–1417).
C. Phosphoaminoacid analysis of BLM (1417). The arrows indicate the directions during the chromatographic runs. The broken circles indicate the positions of co-migrating cold phosphoaminoacid standards. The position of the origin and products of the partial hydrolysis have been indicated.
D. Phosphopeptide analysis of BLM (1–1041) and the two mutants BLM (1–1041) S646A and BLM (1–660) S646A. Arrow indicates the position of the phosphopeptide decreased in the mutants.
E. Peptides (containing either wild type or mutant Ser646 residue) were phosphorylated with either the wild type or kinase dead recombinant Chk1. Bound radioactivity was quantitated by scintillation counting.
Next we wanted to determine whether the similarity in the phosphopeptides was conserved within smaller BLM fragments when they were phosphorylated by Chk1. Hence peptide maps were carried out for the smaller fragments of BLM known to be highly phosphorylated by Chk1 - i.e. BLM (1–212), BLM (109–212), BLM (191–660), BLM (531–660) and BLM (1–660). Almost all the phosphopeptides seen in BLM (1–212), BLM (109–212), BLM (191–660) and BLM (531–660) were also observed within BLM (1–660) and BLM (1–1417) (Figure 3B, Supplementary Figure 3B by comparison of color codes). The fragment analysis of Chk1-phosphorylation on BLM thus indicated a possible way by which each phosphorylation site on the helicase could be authentically mapped on BLM.
Chk1 is known to phosphorylate its substrates on serine and threonine residues, both in vitro and in vivo (3). To check whether Chk1 phosphorylated BLM on serine and/or threonine residues, we carried out phosphoamino acid analysis using purified full-length BLM (1–1417) (Figure 3C). Phosphoaminoacid analysis indicated that both serine(s) and to a much lesser extent threonine(s) were phosphorylated on BLM by Chk1.
An analysis of BLM full-length protein sequence indicated the presence of 157 serine and 90 threonine sites. Within the first 660 amino acids, where BLM is supposed to be phosphorylated by Chk1, there were 83 serine and 52 threonine residues. Hence an in silico analysis was carried out which could potentially help us to understand in molecular details the interactions between Chk1 and short peptide motifs within BLM which may allow a particular site to be preferentially phosphorylated by this kinase. Initially we used web based prediction tools such as KinasePhos (28), GPS (29), PPSP (30) and Scansite (31) to identify potential sites on BLM, which could be phosphorylated by Chk1 (Supplementary Table 1). However these tools are based purely on motif or amino acid sequences. The number of substrates of Chk1 in literature was relatively few and hence the motif derived from these substrates and used in the sequence-based approaches by the web-based tools may not give robust results. Our recent study (32) found structure-based methods to be superior to sequence based methods for identification of substrates for CHK family of kinases. Therefore we used a structure-based method to predict the phosphorylation sites in BLM by Chk1. The method extracted the information from the three dimensional structure of the protein–peptide complex and revealed important physico-chemical interactions between the kinase and peptide (32). Initially all the 22 known substrates of Chk1 present in vertebrates (Supplementary Table 2) and reported in PhosphoELM database (33) were modeled in complex with Chk1 using Insight II software. The modeled complexes were then analyzed in detail to identify crucial contact residues, which governed the recognition of the peptide by Chk1. The analysis showed us that two positions namely arginine at P (−3) and hydrophobic residue at P (+1) position in the peptide were more preferred due to its favorable interactions with the Chk1 binding pockets. There were 10 peptides within fulllength BLM that matched to the motif [R/K] x x [S/T][hydrophobic] x x (where x indicated any amino acid) which could be potential binders to Chk1. Interestingly the analysis of the complete BLM sequence by a structure based kinase substrate prediction tool, MODPROPEP (34), indicated that the 9 out of 10 [R/K] x x [S/T][hydrophobic] x x motif also lied within 30% of the total number of possible Ser/Thr sites on BLM.
Each of these peptides was modeled onto Chk1 and interactions were scored and ranked using residue-based statistical energy potentials by Betancourt and Thirumalai (26). Apart from statistical potentials, binding energy calculations were performed for these 10 peptides using all atom force field. Out of these 10 structural motifs, 6 were within BLM (1–660). Both residue-based statistical pair potentials and all atom binding energy values indicated that Ser646 had the highest probability to be a Chk1 substrate (Table 1). Other sites listed in Table 1 could also be potential substrates for Chk1.
Table 1. Ranking of BLM peptides potentially phosphorylated by Chk1 as obtained by in silico analysis.
The residue based statistical pair potential scores, and all atom binding energies for the peptides are listed. The peptides are ranked based on their residue based statistical pair potential scores. The rows for the 6 peptides that lie in 1 to 660 amino acid region of BLM protein are highlighted in pink. VDW represents the contribution due to van der Waals interaction while ELE represents the contribution due to electrostatic interaction.
| Serial number |
Ser/Thr position |
Peptide sequence |
Residue based statistical pair potential scores (BT) |
Binding energy calculated using Insight II (Kcal mol−1) |
||
|---|---|---|---|---|---|---|
| VDW | ELE | TOTAL | ||||
| 1 | Ser646 | −7.72 | −68.724 | −36.337 | −105.061 | |
| 2 | Ser729 | −5.26 | −56.895 | −26.337 | −83.232 | |
| 3 | Thr581 | −5.03 | −55.567 | −34.583 | −90.15 | |
| 4 | Ser1361 | −4.81 | −43.757 | −26.804 | −70.561 | |
| 5 | Ser1375 | −3.79 | −53.314 | −35.054 | −88.368 | |
| 6 | Ser434 | −3.22 | −57.886 | −39.371 | −97.257 | |
| 7 | Thr1350 | −3.01 | −55.531 | −41.978 | −97.509 | |
| 8 | Ser367 | −2.72 | −66.311 | −43.226 | −109.537 | |
| 9 | Ser602 | −2.4 | −52.816 | −41.17 | −93.986 | |
| 10 | Thr182 | −2.08 | −53.744 | −29.651 | −83.395 | |
We mutated Ser646 in the context of both BLM (1–1041) and BLM (1–660). The mutant proteins were expressed in E. coli, purified, phosphorylated by Chk1 and two-dimensional phosphopeptide map analysis was carried out alongside BLM (1–1417) as control. We found that compared to BLM (1–1417) peptide map, the intensity of one specific phosphopeptide (indicated by red arrow) was reproducibly decreased in the mutant maps obtained with two different BLM substrates (Figure 3D). The residual radiolabel in the mutant peptide map was probably due to the low level of Chk1 phosphorylation on another serine residue present within the specific tryptic fragment. Interestingly the phosphopeptide in which Ser646 was present was also observed in the phosphopeptide map analysis of BLM (191–660) and BLM (531–660) (Figure 3B, Supplementary Figure 3B), thereby validating the robustness of our assay system. Together these results indicated that Chk1 phosphorylated BLM at Ser646 in vitro.
To verify whether Chk1 could phosphorylate BLM at Ser646 in the context of a specific peptide, wild type and mutant peptides spanning BLM (641–651), containing a mismatch at Ser646 were generated. Kinase assays were carried out with Chk1, either wild type or mutant, and the extent of phosphorylation on the two peptides was determined using the peptide-binding assay (Figure 3E). Wild type Chk1 phosphorylated the wild type peptide but not the mutant one, while mutant Chk1 phosphorylated the peptides to a basal level. The above results indicated that in vitro Chk1 phosphorylation on BLMSer646 was a specific phenomenon.
BLM Ser646 phosphorylation was lost after DNA damage
Next we wanted to determine whether Ser646 phosphorylation on BLM was observed in vivo. Immortalized cells obtained from BS patients were used along with chromosome 15 complemented BS cells (A-15). The cells were either left untreated or treated with Chk1 inhibitor UCN-01 for 2 h which did not lead to any drastic change in the cell cycle profile (Figure 4A, left) or change in the expression level of endogenous BLM in the nucleus (Figure 4A, middle). We generated a new phosphospecific polyclonal antibody against a BLM peptide phosphorylated at Ser646. Since the levels of pSer646BLM after direct western analysis were not equivocally apparent, we enriched the phosphorylated moiety by immunoprecipitating endogenous BLM from nuclear extracts of A-15 cells grown in absence or presence of UCN-01. Phosphorylation on BLM at Ser646 was decreased when A-15 was treated for 2 h with UCN-01 (Figure 4A, right), indicating that Ser646 was phosphorylated in a Chk1-dependent manner on asynchronously growing cells. A similar decrease of phosphorylation on BLM at Ser646 was observed when asynchronously growing A-15 cells were depleted of Chk1 by Chk1 siRNA transfection (Supplementary Figure 4A).
Figure 4. BLM phosphorylation at Ser646 decreased after DNAdamage.

A. (Left) Cell cycle profile of A-15 and BS, both grown asynchronously, and A-15 treated with UCN-01 for 2 h. (Middle) Nuclear extracts (50 µg) from A-15, grown in the above two conditions, and BS (grown asynchronously) were subjected to SDS-PAGE and western blotting to determine the level of BLM and hsp90. (Right) Nuclear extracts (1 mg) from A-15 and BS were immunoprecipitated with BLM antibody and the immoprecipitates subjected to western blotting with either BLM or pSer646BLM antibody.
B. BLM was immunoprecipitated from nuclear extracts (1 mg) obtained from BS and A-15 cells (−HU, +HU, PW) with anti-BLM antibody. The immunoprecipitates were probed with either BLM (top) or pSer646BLM antibody (bottom). Immunoprecipitates from A-15 cells were additionally incubated with Calf Intestinal Alkaline Phosphatase (CIAP, 10 units) for 30 min before western blotting to check for the specificity of pSer646BLM signal under-HU condition. (*) indicated a cross-reactive band.
C. Immunoprecipitaion with anti-BLM antibody was carried out on nuclear extracts isolated from A-15 cells obtained from different conditions. The immunoprecipitates were probed for the presence of BLM, pSer646BLM, nucleolin, PML isoforms and Nbs1.
D. Immunoprecipitaion with anti-pSer646BLM antibody was carried out on nuclear extracts isolated from A-15 cells obtained from different conditions. The immunoprecipitates were probed for the presence of pSer646BLM, BLM and nucleolin.
The presence of BLM phosphorylation at Ser646 in asynchronous culture led us to investigate its status after stalled replication forks. A-15 and BS cells were grown either asynchronously (−HU) or in presence of hydroxyurea (+HU) for 12 h. The cells were also kept for 6 h post-removal of HU (referred to as post-wash, PW), which allowed the cells to proceed to G2/M phase (Supplementary Figure 4B). The levels of BLM increased after stalling of the replication forks and in G2/M phase (Supplementary Figure 4C, top). It has been reported that replication arrest lead to the generation of double stand breaks (DSBs) (35). It was found that BLM was phosphorylated at Ser646 only under asynchronous conditions and this phosphorylation was lost when the cells were treated with HU (Figure 4B, bottom). Loss of Ser646 phosphorylation after calf intestinal alkaline phosphatase (CIAP) treatment, acted as a specificity control for the Ser646BLM phosphorylation under asynchronous conditions.
Since DSBs are the end result of stalled replication (35), we hypothesized that loss of Ser646 phosphorylation on BLM should also be observed after exposure of cells to DSB inducers like neocarzinostatin (NCS). Hence we treated A-15 cells with NCS for 1 h or 6 h and also allowed a subsequent recovery for 6 h after wash-off. Such a treatment regime did not lead to much alteration in the levels of endogenous BLM, but led to the accumulation of γ-H2AX (Supplementary Figure 4D). Immunoprecipitation of endogenous BLM from the nuclear extracts of A-15 cells revealed that Ser646 phosphorylation was much reduced within 1 h after NCS treatment and was no longer detectable after 6 h (Figure 4C).
Since Ser646 phosphorylation on BLM was present only in the asynchronous cultures, we wanted to investigate whether the loss of this specific phosphorylation coordinated with the cellular relocalization of BLM. For this purpose, A-15 cells were either left untreated or treated with HU for 12 h and co-immunostained with BLM and pSer646BLM antibody. Under asynchronous conditions, both BLM and pSer646BLM colocalized (Figure 5A, a) within the PML NBs and nucleolus (Figure 5A, b and c). After HU-treatment, BLM localization was decreased in the PML NBs and nucleolus and increased at the sites of stalled replication where the helicase colocalized with proteins involving in sensing and resolution of DNA damage like RAD51, 53BP1 (8, 21) and Nbs1 (Figure 5A, e and data not shown). Very little staining of pSer646BLM was observed in cells treated with HU (Figure 5A, d). Similar loss of pSer646BLM staining within the PML NBs and nucleolus was also observed after treatment of A-15 cells with NCS (data not shown). Incidentally robust Ser646 phosphorylation on BLM was observed within 6 h after NCS wash-off (Figure 4C), which coincided with the co-localization of BLM within PML NBs and nucleolus (data not shown). The lack of pSer646BLM signal in PW condition after HU treatment (Figure 4B, bottom), possibly points to the differences in the dynamics of BLM relocalization post-NCS and -HU treatments. The above results indicated that the relocalization of BLM post DNA damage correlated with the loss of Ser646 phosphorylation. The relocalization of BLM was verified by its immunoprecipitation from A-15 cells. While the binding of BLM to both nucleolin and PML isoforms decreased after DNA damage, its in vivo association with Nbs1 increased within 1 h and persisted upto 6 h after DNA damage (Figure 4C). To further determine whether Ser646 phosphorylation was one of the prerequisite for the relocalization of BLM from nucleolus after DNA damage, we carried out co-immunoprecipitation with pSer646BLM antibody. Loss of BLM Ser646 phosphorylation increased after DNA damage and correlated with decreased binding to nucleolin (Figure 4D).
Figure 5. Ser646 phosphorylation of BLM regulates its localization in PML NBs and nucleolin.
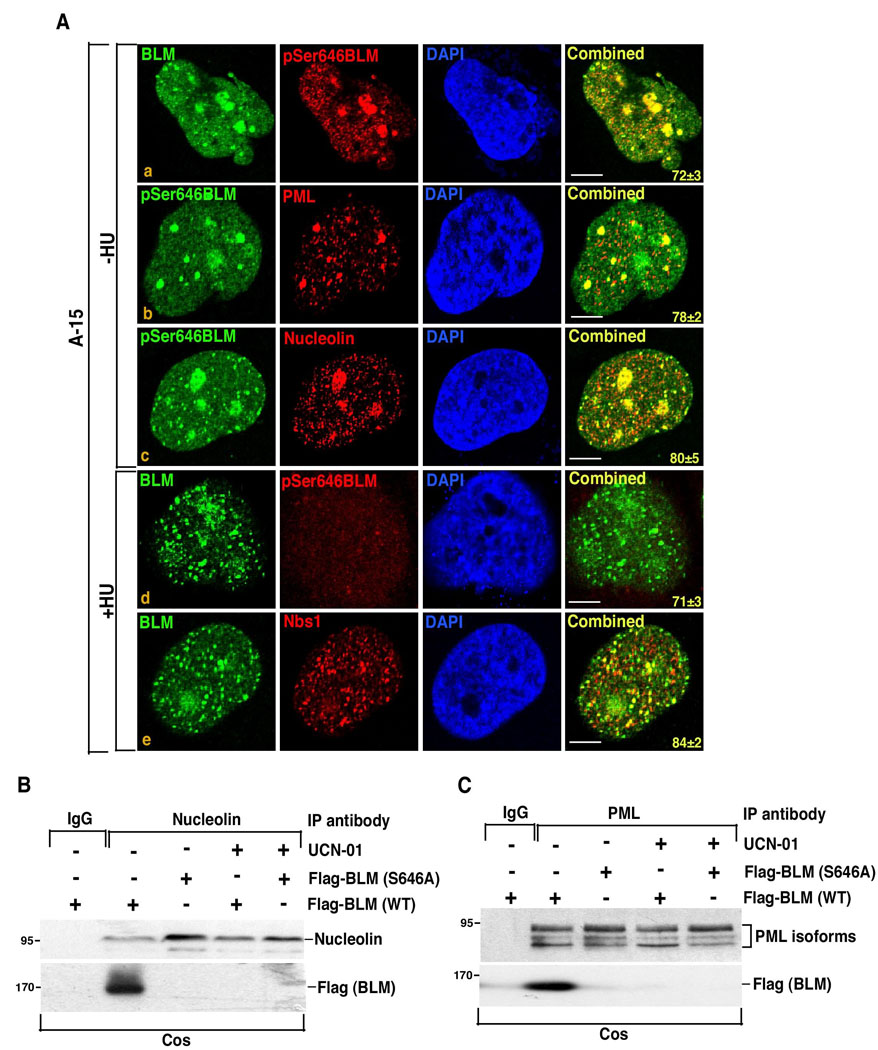
A. A-15 cells were either left untreated (−HU) or treated with HU (+HU) for 12 h. Immunofluorescence was carried out with antibodies against (a, d) BLM/pSer646BLM, (b) pSer646BLM/PML, (c) pSer646BLM/nucleolin (e) BLM/Nbs1. Nucleus is stained by DAPI. Combined indicates the merged image from the red and green channel. The numbers indicate the percentage of cells having similar colocalization.
B–C. Immunoprecipitaions were carried out with either anti-nucleolin (B) or anti-PML (C) antibody on cell extracts (1 mg) obtained after transfecting Cos cells with either pcDNA3 Flag BLM (wildtype) or pcDNA3 Flag BLM (S646A). The immunoprecipitates were probed for the presence of nucleolin and Flag i.e BLM (for B) or PML isoforms and Flag i.e. BLM (for C).
To further validate the above results, mutational analysis was carried out with overexpressed Flag-tagged wildtype or BLM (S646A) variant in Cos cells (Supplementary Figure 4E). UCN-01 treatment for 2 h did not cause any change in the expression levels of the transfected BLM, PML isoforms or nucleolin. Immunoprecipitation was carried out with either nucleolin (Figure 5B) or PML (Figure 5C). Wildtype BLM but not the S646A mutant interacted with both nucleolin and PML isoforms in asynchronous conditions (Figure 5B, 5C). However both nucleolin-BLM and PML-BLM interactions were much decreased after UCN-01 treatment, indicating that Ser646 constitutive phosphorylation maybe one of the post-translational events that regulated BLM localization under asynchronous conditions.
DISCUSSION
In this communication we have provided evidence that apart from Chk1, Chk2 also phosphorylated BLM in vitro in the N-terminal 660 amino acids (Figure 1 and Supplementary Figure 1). Chk1/Chk2 mediated phosphorylation of BLM was regulated by an internal stretch of amino acids present within the DExH motif of the helicase (Figure 2, Supplementary Figure 2). Using biochemical, in silico and cell biology techniques we have demonstrated that Chk1 could phosphorylate BLM at Ser646 in a constitutive manner in vivo (Figure 4, 5). Phosphorylation on BLM at Ser646 was only present in asynchronous cultures and decreased rapidly after DNA damage. Loss of this phosphorylation event led to a decrease of BLM binding to nucleolin and PML isoforms, diminished accumulation in the nucleolus and PML NBs, coinciding with its (i.e. BLM’s) simultaneous relocalization to the sites of the DNA damage and binding with DNA sensor protein, Nbs1 (Figure 4C, 4D, 5). Thus the results indicated that Ser646 phosphorylation could act as a marker to determine the activity status of the helicase. Constitutive phosphorylation of H2AX, ATM and Chk2 had been found in human cancerous tissues and shown to be associated with precancerous lesions (36, 37). Hence it will be of interest to know the status of BLMSer646 phosphorylation in different stages and grades of tissues obtained from cancer patients.
Events involving phosphorylation cascades are spatially and temporarily regulated. SMC3 (Structural maintenance of chromosomes subunit 3) controls the activity of chromosomes during cell division and also plays important roles in stabilizing cells' genetic information and repairing damaged DNA. SMC3 is phosphorylated at Ser1067 and Ser1083 in vivo. Phosphorylation at Ser1083 was IR induced, depended on ATM and NBS1, and was required for intra-S-phase checkpoint. ATM-dependent phosphorylation at Ser1083 was in turn dependent on the constitutive phosphorylation of Ser1067 by CK2 (38). Similarly it is possible that phosphorylation at Ser646 may either coordinate or even regulate other post-translational events on BLM, which may happen after DNA damage. These post-translational events may involve other Chk1-dependent phosphorylation events on BLM. Indeed around 25–30 phosphopeptides were present in the peptide map of Chk1-phosphorylated wild type BLM (Figure 3A, Supplementary Figure 3A), indicating that Chk1 possibly phosphorylated BLM on multiple residues. The Chk1-mediated phoshorylation events on BLM may mutually regulate each other temporarily and thereby modulate BLM functions during signal transduction cascades.
DExH motif containing proteins are more common among the RNA helicases. Mutations in the conserved residues of DExH motif revealed its role in ATPase and RNA helicase activities (39). DExH motif is also present in the DNA helicases of the SF2 superfamily, to which BLM belongs (27). DExH motif in BLM, which extended from amino acids 683–832, was contained within the ATP-binding region in BLM. During this study we have provided evidence that amino acids (661–800) within the DExH motif negatively regulated the Chk1/Chk2 mediated N-terminal phosphorylation of BLM (Figure 2, Supplementary Figure 2). Addition of BLM (661–800) in trans inhibited Chk1/Chk2 phosphorylation (Figure 2D, Supplementary Figure 2C), thereby indicating that in the native conformation the DExH motif and the N-terminal region of the protein were probably in close proximity which allowed the former to regulate the phosphorylation event in the latter. Hence a regulatory circuitry probably exist in vivo which may coordinate and temporarily control vital biochemical processes mediated by the helicase domain of BLM and the phosphorylation events mediated by Chk1/Chk2. We have recently shown that BLM could enhance the ATPase function of RAD54 and thereby stimulated RAD54-mediated chromatin remodeling (9). Since DExH RNA helicases has been shown to remodel ribonucleoprotein complexes (40), it will be tempting to speculate that Chk1/Chk2 phosphorylation in general and Ser646 phosphorylation on BLM in particular could also regulate the stimulation by BLM on RAD54-mediated chromatin remodeling. BLM functions during the latter stages of recombination like disruption of RAD51 nucleofilaments (41, 42) could depend on the DExH motif of BLM, as members of the DExH group processively translocate along single-stranded RNA (or DNA) and displace paired strands (or proteins) in their path (43). Hence Chk1/Chk2 phosphorylation on BLM may regulate the effect of the helicase during disruption of RAD51 nucleofilaments. Future research will determine how Chk1/Chk2 mediated phosphorylations in the N-terminus of BLM indeed affected the functions of the helicase during homologous recombination.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge Ian Hickson, Carol Prives, Steve Elldege and Thanos Halazonetis for plasmids and Ian Hickson for the yeast strain JEL1. This work is supported by National Institute of Immunology core funds, Department of Biotechnology, India (BT/PR9598/Med/30/33/2007, BT/PR11258/BRB/10/645/2008), Council of Scientific and Industrial Research [37(1348)/08/EMR-II], India and National Institutes of Health, USA (1 R01 TW007302-05).
REFERENCES
- 1.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hurley PJ, Bunz F. ATM and ATR: components of an integrated circuit. Cell Cycle. 2007;6:414–417. doi: 10.4161/cc.6.4.3886. [DOI] [PubMed] [Google Scholar]
- 3.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 4.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 5.Sharma S, Doherty KM, Brosh RM., Jr Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J. 2006;398:319–337. doi: 10.1042/BJ20060450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009;9:644–654. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 7.Davies SL, North PS, Dart A, Lakin ND, Hickson ID. Phosphorylation of the Bloom's syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004;24:1279–1291. doi: 10.1128/MCB.24.3.1279-1291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sengupta S, Robles AI, Linke SP, et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol. 2004;166:801–813. doi: 10.1083/jcb.200405128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivastava V, Modi P, Tripathi V, Mudgal R, De S, Sengupta S. BLM helicase stimulates the ATPase and chromatin-remodeling activities of RAD54. J Cell Sci. 2009;122:3093–3103. doi: 10.1242/jcs.051813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ababou M, Dutertre S, Lecluse Y, Onclercq R, Chatton B, Amor-Gueret M. ATM-dependent phosphorylation and accumulation of endogenous BLM protein in response to ionizing radiation. Oncogene. 2000;19:5955–5963. doi: 10.1038/sj.onc.1204003. [DOI] [PubMed] [Google Scholar]
- 11.Beamish H, Kedar P, Kaneko H, et al. Functional link between BLM defective in Bloom's syndrome and the ataxia-telangiectasia-mutated protein, ATM. J Biol Chem. 2002;277:30515–30523. doi: 10.1074/jbc.M203801200. [DOI] [PubMed] [Google Scholar]
- 12.Rao VA, Fan AM, Meng L, et al. Phosphorylation of BLM, dissociation from topoisomerase IIIalpha, and colocalization with gamma-H2AX after topoisomerase I-induced replication damage. Mol Cell Biol. 2005;25:8925–8937. doi: 10.1128/MCB.25.20.8925-8937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tripathi V, Kaur S, Sengupta S. Phosphorylation-dependent interactions of BLM and 53BP1 are required for their anti-recombinogenic roles during homologous recombination. Carcinogenesis. 2008;29:52–61. doi: 10.1093/carcin/bgm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wawrousek KE, Fortini BK, Polaczek P, et al. Xenopus DNA2 is a helicase/nuclease that is found in complexes with replication proteins And-1/Ctf4 and Mcm10 and DSB response proteins Nbs1 and ATM. Cell Cycle. 2010:9. doi: 10.4161/cc.9.6.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leng M, Chan DW, Luo H, Zhu C, Qin J, Wang Y. MPS1-dependent mitotic BLM phosphorylation is important for chromosome stability. Proc Natl Acad Sci U S A. 2006;103:11485–11490. doi: 10.1073/pnas.0601828103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bayart E, Dutertre S, Jaulin C, Guo RB, Xi XG, Amor-Gueret M. The Bloom syndrome helicase is a substrate of the mitotic Cdc2 kinase. Cell Cycle. 2006;5:1681–1686. doi: 10.4161/cc.5.15.3122. [DOI] [PubMed] [Google Scholar]
- 17.Schmitt E, Boutros R, Froment C, Monsarrat B, Ducommun B, Dozier C. CHK1 phosphorylates CDC25B during the cell cycle in the absence of DNA damage. J Cell Sci. 2006;119:4269–4275. doi: 10.1242/jcs.03200. [DOI] [PubMed] [Google Scholar]
- 18.Wilsker D, Petermann E, Helleday T, Bunz F. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc Natl Acad Sci U S A. 2008;105:20752–20757. doi: 10.1073/pnas.0806917106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyle WJ, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- 20.Karow JK, Chakraverty RK, Hickson ID. The Bloom's syndrome gene product is a 3'-5' DNA helicase. J Biol Chem. 1997;272:30611–30614. doi: 10.1074/jbc.272.49.30611. [DOI] [PubMed] [Google Scholar]
- 21.Sengupta S, Linke SP, Pedeux R, et al. BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. Embo J. 2003;22:1210–1222. doi: 10.1093/emboj/cdg114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowe ED, Noble ME, Skamnaki VT, Oikonomakos NG, Owen DJ, Johnson LN. The crystal structure of a phosphorylase kinase peptide substrate complex: kinase substrate recognition. EMBO J. 1997;16:6646–6658. doi: 10.1093/emboj/16.22.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen P, Luo C, Deng Y, et al. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell. 2000;100:681–692. doi: 10.1016/s0092-8674(00)80704-7. [DOI] [PubMed] [Google Scholar]
- 24.Canutescu AA, Shelenkov AA, Dunbrack RL., Jr A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. 29. [DOI] [PubMed] [Google Scholar]
- 26.Betancourt MR, Thirumalai D. Pair potentials for protein folding: choice of reference states and sensitivity of predicted native states to variations in the interaction schemes. Protein Sci. 1999;8:361–369. doi: 10.1110/ps.8.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis NA, Groden J, Ye TZ, et al. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 28.Huang HD, Lee TY, Tzeng SW, Horng JT. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 2005;33:W226–W229. doi: 10.1093/nar/gki471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou FF, Xue Y, Chen GL, Yao X. GPS: a novel group-based phosphorylation predicting and scoring method. Biochem Biophys Res Commun. 2004;325:1443–1448. doi: 10.1016/j.bbrc.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Xue Y, Li A, Wang L, Feng H, Yao X. PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics. 2006;7:163. doi: 10.1186/1471-2105-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar N, Mohanty D. Identification of substrates for Ser/Thr kinases using residue-based statistical pair potentials. Bioinformatics. 2010;26:189–197. doi: 10.1093/bioinformatics/btp633. [DOI] [PubMed] [Google Scholar]
- 33.Diella F, Gould CM, Chica C, Via A, Gibson TJ. Phospho.ELM: a database of phosphorylation sites--update 2008. Nucleic Acids Res. 2008;36:D240–D244. doi: 10.1093/nar/gkm772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar N, Mohanty D. MODPROPEP: a program for knowledge-based modeling of protein-peptide complexes. Nucleic Acids Res. 2007;35:W549–W555. doi: 10.1093/nar/gkm266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saintigny Y, Delacote F, Vares G, et al. Characterization of homologous recombination induced by replication inhibition in mammalian cells. Embo J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 37.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 38.Luo H, Li Y, Mu JJ, et al. Regulation of intra-S phase checkpoint by ionizing radiation (IR)-dependent and IR-independent phosphorylation of SMC3. J Biol Chem. 2008;283:19176–19183. doi: 10.1074/jbc.M802299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Utama A, Shimizu H, Hasebe F, et al. Role of the DExH motif of the Japanese encephalitis virus and hepatitis C virus NS3 proteins in the ATPase and RNA helicase activities. Virology. 2000;273:316–324. doi: 10.1006/viro.2000.0417. [DOI] [PubMed] [Google Scholar]
- 40.Jankowsky E, Bowers H. Remodeling of ribonucleoprotein complexes with DExH/D RNA helicases. Nucleic Acids Res. 2006;34:4181–4188. doi: 10.1093/nar/gkl410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev. 2007;21:3085–3094. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tripathi V, Nagarjuna T, Sengupta S. BLM helicase-dependent and -independent roles of 53BP1 during replication stress-mediated homologous recombination. J Cell Biol. 2007;178:9–14. doi: 10.1083/jcb.200610051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pyle AM. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys. 2008;37:317–336. doi: 10.1146/annurev.biophys.37.032807.125908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.