

Abstract
Background
Highly pathogenic avian influenza viruses of the H5N1 subtype continue to cross the species barrier to infect humans and cause severe disease. It has been suggested that an exaggerated immune response contributes to the pathogenesis of H5N1 virus infection in mammals. In particular, H5N1 virus infections are associated with a high expression of the proinflammatory cytokines, including interleukin-1 (IL-1) and tumor necrosis factor- alpha (TNF-α).
Methods
We investigated the compounding affects of both cytokines on the outcome of H5N1 virus disease by using triple mutant (TM) mice deficient in three signaling receptors, TNF-R1, TNF-R2, and IL-1-RI.
Results
TM mice exhibited reduced morbidity and a significant delay in mortality following lethal challenge with a lethal H5N1 virus, whereas no such differences were observed with the less virulent A/PR/8/34 (H1N1) virus. H5N1 infected TM mice displayed diminished cytokine production in lung tissue and a quantifiable decline of macrophages and neutrophils in the lungs post-infection. Moreover, morphometric analysis of airway sections revealed less extensive inflammation in H5N1-infected TM mice compared with infected wild type mice.
Conclusions
The combined signaling from the TNF or IL-1 receptors promotes maximal lung inflammation that may contribute to the severity of disease caused by H5N1 virus infection.
The emergence of the highly pathogenic H5N1 avian influenza virus and its associated high lethality still remains a public health concern. Since the re-emergence of the H5N1 subtype in 2003, there have been over 495 human cases worldwide, of which approximately 60% have been fatal [1]. H5N1 virus infected patients initially present with fever, coughing, dyspnea and in some cases radiological evidence of bronchopneumonia [2, 3]. In severe cases, bilateral pneumonia and the development of acute respiratory distress syndrome (ARDS) is followed by progressive respiratory failure [4, 5]. ARDS is a clinical syndrome usually secondary to an intense host cytokine and inflammatory response that includes increased levels of tumor necrosis factor alpha (TNF-α) [5-9]. Thus, it has been suggested that elevated levels of cytokines in H5N1 infected patients may contribute to the overall pathogenesis of these viral infections further complicating recovery [10-13].
The hypothesis that an aberrant cytokine and subsequent cellular immune responses to H5N1 virus detrimentally affect severity and patient outcome has been understudied. In comparison to seasonal influenza strains, H5N1 viruses are potent inducers of TNF and IL-1 cytokines both in vivo and in vitro studies [3, 10-15]. In mice, studies demonstrated that no individual pro-inflammatory cytokine was responsible for the pathogenesis associated with virus or bacterial infection [16-19]. Thus, the lack of these individual cytokine receptors ultimately did not lead to an improvement in host survival following infection with H5N1 virus. However, because of the functional redundancy of some cytokine products, the effect of deleting only one cytokine might be masked by compensatory host responses.
To determine the combined contributions of TNF and IL-1 signaling to the inflammatory responses to H5N1 virus infection, we utilized triple mutant (TM) mice deficient in TNF (R1 and R2 receptors) and IL-1 (type I receptor) and measured their host responses to a virulent H5N1 virus. We found that TM mice survived longer, exhibited less morbidity, and displayed a reduction in lung inflammation and cytokine response compared to wild-type (WT) mice. The specific immune cell subpopulations contributing to the differences in inflammation observed between infected TM and WT mice are also presented here. This study shows an important role of the combined effects of TNF and IL-1 in contributing to the pathogenic outcome of H5N1 virus infection.
Methods
Virus
Influenza A virus, A/Hong Kong/483/97 (HK/483), isolated from a fatal case [20-22], and A/PR/8/34 (PR8; H1N1) were grown and titered in embryonating hen's eggs and 50% egg infectious dose (EID50) was calculated as described previously [23, 24]. All experiments were performed in biosafety level 3 laboratories with enhancements as outlined in the Biomedical Microbiological and Biomedical Laboratory [25].
Mice
Mouse strains (6-8 weeks old) utilized in these studies included: TNF-R1, TNF-R2, and IL-1-RI triple-mutant (TM) C57BL/6J × 129S background [18, 19, 26, 27]. IL-1-RI single cytokine receptor knock-out mice (IL-1-R KO), C57BL/6J × 129Sv -Il1r1tm1Roml/J), and single cytokine receptor TNFR1 KO mice ((TNF-R KO), C57BL/6J × 129Sv-Tnfrsf1atm1ImxTnfrsf1btm1Imx/J), were purchased from The Jackson Laboratory (Bar Harbor, ME). Age-matched C57B6J × 129 WT mice were used as genetic controls and utilized for 50% mouse infectious dose (MID50) determination [23].
Inoculations
Mice were anesthetized with Avertin (Sigma-Aldrich, St Louis, MO) and infected intranasally (i.n.) (50μl) with 1,000 MID50 of HK/483 or PR8 virus [23]. Mice were weighed daily and euthanatized when total weight loss exceeded 25% of starting body weight. Survival data and mean days to death (MDD) were analyzed using SPSS software and Kaplan-Meier survival analysis.
Virus titration and cytokine analysis
Mouse lung tissues were removed on days 3 and 5 p.i. and homogenized in 1 ml of PBS. Clarified homogenates were titrated for virus in hens' eggs, as described [23] and expressed as the mean log10 EID50/ml. Statistical differences in virus titers between infection groups were measured by Student's t test. Cytokines were measured in homogenates in duplicate using the Bioplex Protein Array system (Bio-Rad, Hercules, CA), according to manufacturer's instructions [14]. The mean cytokine values from six mock-infected WT and TM mice were determined.
Pulmonary histopathology
Lungs were collected from WT and TM mice 3 days after i.n. infection with HK/483 virus and processed for routine histology. Dimensions from a stage micrometer and square grid coherent test systems were reflected onto histology slides using a drawing arm microscope. All airway sections with an aspect ratio less than 2 (i.e., cut perpendicular to the longitudinal dimension of the airway) and a diameter of >50 μm from all lung lobes from all mice were included in analyses, totaling 31-81 distinct airway sections per mouse. The fraction of the airway surface area that was inflamed (defined by the presence of extravasated leukocytes) or denuded (defined by exposed basement membrane) was calculated using standard stereological approaches [19].
Flow cytometry and lung immune cell quantification
WT, IL-1-R KO, TNFR KO and TM mice were infected i.n. with HK/483 virus as described above. On days 3 and 5 p.i., mice were euthanatized, exsanguinated and lungs were removed from individual mice without PBS perfusion. Individual whole lung cell suspensions were prepared as described [14] and total viable lung cell numbers were determined by trypan blue exclusion. Lung cell suspensions were incubated with Fc blocking antibody (CD16/32) followed by antibodies to cell sub-populations: CD11b- APC (pan-macrophage), CD11c-PE (pan-dendritic), Ly6G/C- FITC (neutrophil), CD3-FITC (pan- T cell), CD4- PE (T cell) and CD8β- APC (T cell) (BD Biosciences, San Diego, CA). Cells were incubated with antibodies, fixed with 2% paraformaldehyde and quantified on a LSR II flow cytometer (BD Biosciences). Percentages of cell types were determined by appropriate gating on positively labeled cells. The numbers of cells in each sub-population were calculated using the percent populations determined by the cytometer statistics and the total lung cell counts. Statistically significant differences in immune cell numbers between experimental groups were determined by Analysis of Variance.
Results
Triple-mutant (TM) mice exhibit a delay in mortality following lethal H5N1 virus challenge
TM mice lacking the receptors for TNF and IL-1 were inoculated with 1,000 MID50 of HK/483 or PR8 virus and mortality was compared to infected WT (C57B6 × 129) mice. As shown in Figure 1A, all infected TM and WT mice eventually succumbed to H5N1 virus infection; however the TM mice showed a significant delay in mean time to death compared to WT mice. HK/483-infected TM mice lived an average of 2 days longer (mean days until death (MDD) = 8) compared to virus infected WT mice (MDD= 6) (P < 0.05). Moreover, TM-infected mice lost significantly less weight at earlier time points than WT-infected mice (Figure 1B), whereas mock-infected controls that received PBS in place of virus gained approximately 6% body weight over the observation period (not shown). In contrast, differences in morbidity and mortality between TM and WT mouse strains were not observed among PR8-inoculated mice. These results suggest that the lack of both IL-1-R and TNF-R signaling affects the outcome of H5N1 virus disease in mice.
Figure 1. Morbidity and mortality in triple-mutant (TM) mice.
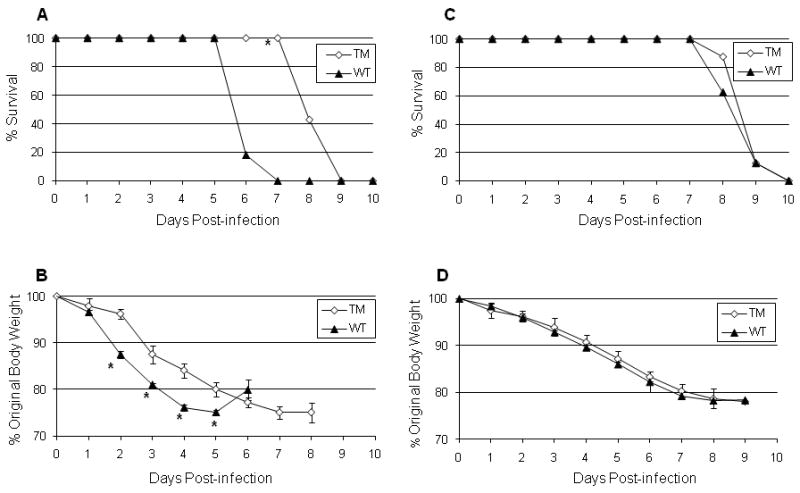
Wild type C57B6 × 129 (WT) (n = 11) and TM mice (n = 7) were inoculated intranasally (i.n.) with 1,000 MID50 of A/Hong Kong/483/97 (A and B) or A/PR/8/34 (C and D) virus and monitored daily for survival (A and C) and weight loss (B and D). Kaplan-Meier survival analysis was performed and significant differences in average time to death was observed between infected WT and TM mice (* p < 0.05). Lines represent average percent weight loss over time for each group and error bars show standard errors of the means.
TM mice exhibited similar lung virus titers but displayed increased systemic spread of virus to brain tissue
In an effort to identify biological mechanisms contributing to the enhanced survival of H5N1 virus infected TM mice, we first assessed virus replication in the lungs of infected mice on days 3 and 5 p.i., time points that correlated with reductions in weight loss as shown in Figure 1. TM and WT inoculated mice exhibited similar lung virus titers at both time periods (Figure 2). While virus titers in brain tissue were similar between the infected TM and WT groups on day 3 p.i., virus titers were significantly higher in TM mice compared to WT mice on day 5 p.i. (P < 0.05), suggesting a role for TNF and/or IL-1 in controlling extrapulmonary spread of virus.
Figure 2. Infectious virus in lung and brain tissues.

WT and TM mice were inoculated i.n. with 1,000 MID50 of HK/483 virus and whole lungs were harvested (n= 4 mice per time point) for virus titration on days 3 (A) and 5 p.i. (B). The limit of detection was 101.5 EID50/ml. A significant increase in titers was observed from day 3 to 5 in the brains of both WT and TM infected mice (*p < 0.05, Students t test).
Diminished cytokine production in the lungs of H5N1 infected TM mice
We next determined whether cytokine responses were altered in the lungs of TM mice. A panel of cytokines critical for the recruitment and activation of immune cells, shown previously to be secreted at high concentrations in lungs of H5N1 infected mice [14, 17] were measured. Beginning on day 3 p.i., compared to WT mice, the TM infected mice exhibited significantly lower levels of MCP-1 and KC in the lungs, shown previously to be important for recruitment of monocytes [28] and neutrophils [29], respectively (Figure 3). Conversely, the lung levels of MIP-1α and β, IL-3, IL-12 (p40), IL-12 (p70), and RANTES were generally similar or only moderately higher on day 3 p.i. among TM mice compared to WT mice. However, by day 5 p.i., all cytokines measured were lower in the lungs of infected TM mice compared to WT mice, with significant (P < 0.05) differences observed in the levels of KC, MIP-1-β, MIP-1-α, IL-3, IFN-γ, G-CSF, IL-12 (p40).
Figure 3. Lung cytokine levels.

Lung cytokine levels from HK/483 infected mice (days 3 and 5 p.i.) were measured individually (n = 4 mice per group) by Bioplex Protein Array, in duplicate. Bars represent means from each infection group ± standard deviation (SD). The mean lung cytokine values among mock-infected WT mice are as follows; TNF-α (10.8 pg/ml) and RANTES (15.6 pg/ml) and the mean cytokine values among mock-infected TM mice are as follows; TNF-α (11.8 pg/ml) and RANTES (18.2 pg/ml). The following cytokines were not detectable in the mock-infected lungs of either mouse strain; G-CSF, IL-1-α, IL-3, IL-6, IL-12 (p70), IL-12 (p40), MIP-1-α, MCP-1, IL-17, KC, MIP-1-β, and IFN-γ. The minimum cytokine detection was <10 pg/ml. * p < 0.05 between TM and WT mouse groups for each day post-infection (Students t test).
Reduced inflammation in the lungs of TM mice
To examine the contribution of TNF and IL-1 signaling to lung inflammation, we performed histopathological analyses of H5N1 infected mouse lung sections (n = 10 WT and 7 TM mice). Airway inflammation and injury was examined using morphometry, including all conducting airway sections perpendicular to the longitudinal axis (>30 airway sections per mouse, ranging from 60-620 microns in diameter). There was no detectable inflammation or necrosis among uninfected TM or WT mice that received PBS in place in virus (not shown). The overall type of inflammation, necrotizing bronchitis and bronchiolitis, was qualitatively similar in the 2 genotypes (Figure 4A). However, when the fraction of total airway surface area that was inflamed was quantified, the extent of airway inflammation was lower in the lungs of TM mice compared to WT mice (Figure 4B). The localization of inflammation was similar in both genotypes, with the larger and more central airways exhibiting more inflammation than the smaller and more distal airways (Figure 4C), but TM mice showed less inflammation (across airway sizes) throughout the infected lungs (Figure 4C). Similar fractions of the inflamed regions were necrotic or denuded of epithelial cells regardless of mouse genotype (Figure 4D), suggesting that these cytokine receptors were not essential for progression to epithelial necrosis during airway infection with this H5N1 virus.
Figure 4. Airway inflammation is reduced in mice infected with H5N1 virus.

TM and WT mice were infected with HK/483 virus and 3 days later lungs were filled with fixative and sections were prepared for histopathological analysis. A) Inflamed airways in TM and WT mice. B) Inflammation throughout the conducting airways was quantified and percent surface area calculated as a measure of airway inflammation (n = 7-10 mice per group). C) Pulmonary inflammation was most prominent in larger and more central airways, but less in TM mice throughout the range of airway sizes. Airway diameter was measured throughout the lung sections and airway sizes binned within each genotype. Epithelial necrosis is shown in D). Inflamed airway sections were screened for areas of cellular necrosis and exposed basement membranes, and the fraction of inflamed airways that was denuded was calculated.
TNF receptor deficient mice exhibit diminished numbers of macrophages and neutrophils following virus challenge
To discriminate cellular contributions to the differences in inflammation observed between TM and WT mice, we measured key immune cell subpopulations in lungs following H5N1 virus or mock (PBS) infection by flow cytometry. For such studies, we included single cytokine knock-out mice, deficient in the IL-1 receptor (IL-1-R KO) or TNF receptor 1 (TNF-R KO) (17), in attempts to determine if changes to specific cell populations is due to the lack of IL-1R or TNF-R signaling. First, a comprehensive view of the total lung cell profile for each of the KO mice is shown in Figure 5A. The mock WT and both single cytokine KO mice exhibited similar total lung cell numbers (1.5 × 108 mean cells per lung). However, mock TM mice had significantly lower (P < 0.05) total lung cell numbers (5.0 × 107 mean cells per lung) at either time point examined. Following H5N1 virus inoculation, all mice exhibited a marked increase (*P < 0.05) in total lung cellularity compared to their respective uninfected controls, regardless of genetic background. However, TM mice failed to demonstrate an increase in total lung cellularity from days 3 to 5 p.i. (Figure 5A). Thus, infected TNF-R KO, IL-1-R KO, and WT mice exhibited an increase of 3.1 × 107 to 1.1 × 108 total cells from days 3 to 5 p.i, whereas a decrease in total lung cell number (by 1.0 × 107) was measured among H5N1 infected TM mice during the same time period. On day 5 p.i., infected TM mice had significantly fewer (P = 0.0012) total lung cells than infected single cytokine knock-out mice (Figure 5A, right panel).
Figure 5. Effect of cytokine deficiency on lung immune cell populations following H5N1 virus infection.

Mice were infected i.n. with 1,000 MID50 of HK/483 virus (WT n= 8, TM n = 7, IL-1-R KO n = 5, TNF-R KO n = 4) or given PBS (WT n= 5, TM n = 8, IL-1-R KO n = 3, TNF-R KO n = 4). Lungs were removed on days 3 and 5 p.i. and single cell suspensions prepared from individual lungs. Total viable cells from individual lungs were counted on a hemocytometer by trypan blue exclusion prior to antibody labeling and cytometric analysis (A). Bars represent the average total number of live lung cells (× 107 cells) per treatment group ± SD. Statistical differences between treatment groups were measured using the analysis of variance test (ANOVA). * P <0.05 between PBS inoculated mice and H5N1 infected mice in the same genetic group. (x002C6) P <0.05 between WT-H5N1 infected mice and cytokine deficient-H5N1 infected mice. † P <0.05 between WT-PBS inoculated mice and cytokine deficient-PBS inoculated mice. ‡ P <0.05 between TM-H5N1 infected mice and single cytokine deficient-H5N1 infected mice. Lung suspensions from individual mice were stained with fluorescent antibodies and analyzed by flow cytometry to determine immune cell composition following infection B). Numbers of macrophages (CD11b+, CD11c-, Ly6G/C-, CD3-, CD4-, CD8-)(a), neutrophils (CD11b+, CD11c-, Ly6G/C+, CD3-, CD4-, CD8-) (b), dendritic cells (CD11b-, CD11c+, Ly6G/C-, CD3-, CD4-, CD8-) (c) and T cell numbers (d and e) (CD11b-, CD11c-, Ly6G/C-, CD3+ and CD4+ or CD8+) were determined by appropriate gating within the total lung cell population. Bars represent the average number of immune cells (× 106 cells) per treatment group ± SD.
Flow cytometry was used to determine the specific immune cell composition in the infected lungs, targeting macrophages, neutrophils, dendritic cells (DC) and T cells. With respect to macrophages, the baseline numbers of these cells were over 106 cells per lung (range 1.5-3.0 × 106 mean cells/lung) among all PBS-inoculated mouse strains except for PBS-control TM mice which exhibited 7.2-8.2 × 105 mean macrophages per lung on days 3 and 5 p.i (Figure 5B, panel a). Following H5N1 virus inoculation, the numbers of macrophages increased in the lung tissue in all mouse strains compared to their respective PBS controls, however the intensity of macrophage infiltration differed between mouse strains. In IL-1-R KO and WT mice on day 3 p.i., the number of macrophages was approximately 20-fold higher among infected mice compared to PBS inoculated mice. Infected TM mice exhibited a macrophage increase of 15-fold compared to PBS inoculated TM mice, however no increase in the number of macrophages was observed among infected TNF-R KO mice on day 3 p.i., and only a 1.8-fold increase of lung macrophages was observed on day 5 p.i. Importantly, infected WT and IL-1-R KO mice exhibited a significantly (P = 0.0016) greater infiltration of macrophages compared to the other mouse strains lacking TNF receptors.
Differences in the neutrophil response between WT and TM or TNF-R KO mice also became apparent following H5N1 infection (Figure 5B, panel b). In general, WT and IL-1-R KO infected mice exhibited greater infiltration of neutrophils compared to TNF-R KO and TM mice. On day 3 p.i. infected WT and IL-1-R KO mice had significantly more neutrophils in their lungs compared to PBS controls, whereas infected TM and TNF-R KO mice exhibited a more modest increase. Differences in neutrophil numbers between infected WT and the other PBS-inoculated groups was most apparent on day 5 p.i. where infected WT mice had 1.5 × 107 (P = 0.0064) and 6.7 × 106 more neutrophils than H5N1 infected TM and IL-1-R KO mice, respectively. As was observed with the macrophage dynamics following H5N1 infection, both WT and IL-1-R KO mice had more lung neutrophils on both days than either mouse strain lacking TNF receptors.
DCs were included in this analysis because of their importance regulating the immune response by bridging innate and adaptive immunity [30]. In general, infection caused an increase in the numbers of DCs, compared to their respective PBS controls, however the increase among IL-1-R and TNF-R KO mice was only observed on day 5 p.i (Figure 6B, panel c). In contrast to what was observed for macrophages and neutrophils, DC's were not significantly reduced among infected TM mice, and actually exhibited slightly higher numbers in their lungs than all other infected mouse strains on both days examined.
No significant change in total numbers of lung associated CD4+ T cells was observed among infected WT mice on either day compared to PBS controls, with the exception of IL-1-R KO (P = 0.014) mice on day 3 p.i. (Figure 5B, panel d). With respect to lung associated CD8+ T cells, the baseline numbers of this T cell subset was significantly lower among naïve TM lungs at both time periods examined (Figure 5B, panel e). In general CD8+ T cells did not dramatically increase after infection, although infected WT mice exhibited slightly higher numbers of CD8+ T cells on day 3 p.i. than any of the cytokine receptor deficient mice.
Discussion
ARDS is characterized by diffuse alveolar capillary damage, usually following an intense inflammatory response to infection. Fatal outcome in patients with H5N1 virus infection may be attributed to ARDS characterized by elevated levels of inflammatory mediators [5, 8, 31]. This has prompted the hypothesis that an exacerbated proinflammatory response may contribute to the severity of H5N1 disease [10-15]. TNF and IL-1 are of particular interest in ARDS because they are early response cytokines produced by a variety of pulmonary cells and can instigate a cascade of physiological changes including recruitment of neutrophils leading to alveolar capillary damage [5-8]. In a bacterial pneumonia model, inhibiting the actions of TNF or IL-1 individually results in little improvement in lung inflammation compared to blocking both proinflammatory pathways simultaneously [18, 19, 32], suggesting functional redundancy among these cytokines. Thus, studying the effect of deleting individual cytokines might be of limited significance. In this study, we investigated the effects of inhibiting the actions of both TNF (R1 and R2 receptors) and IL-1 (type I receptor) in mice challenged with a lethal H5N1 virus. We used TM mice to address the hypercytokinemia hypothesis of host pathogenesis and to measure affects of combined cytokine depletion on overall lung inflammation. We observed that H5N1-, but not H1N1-infected TM mice exhibited reduced morbidity and survived longer compared to infected WT mice. The substantial reduction in lung inflammation and significant delay in mortality were not observed in previous H5N1 infection studies testing cytokine knockout mice deficient in either TNFR-1 or IL-1 signaling alone [16, 17].
Histologically, the extent of airway inflammation was lower in the lungs of TM mice compared to WT mice, suggesting that TNF and IL-1 signaling pathways contribute to H5N1 induced inflammation of lung tissue. Within the inflamed areas, similar proportions of necrotic or denuded tissue were observed, regardless of mouse genotype, indicating that these cytokine receptors are not essential for progression to epithelial necrosis during H5N1 virus infection. These histopathological observations were supported further by cytometric quantification of immune cell populations in the lung following H5N1 infection. The reduced inflammation observed in H5N1 virus infected TM mice was largely due to the significantly reduced numbers of macrophages and neutrophils observed in lung tissue compared to WT mice. Macrophages and neutrophils accounted for the majority of increased cellularity in the lungs of infected WT mice (figure 5) and in the lungs of H5N1 infected BALB/c mice [14]. Macrophage and neutrophil cell function was not addressed in this study but there is evidence that TNF-α and IL-1 can activate macrophages and neutrophils [33] and interrupting TNF-α and IL-1 signaling affects bacterial burdens in pneumonia models [34].
Unlike H5N1, PR8 virus is not a potent inducer of TNF-α and IL-1 cytokines [35], which may partially explain the lack of reduced morbidity and mortality observed among H1N1-infected TM mice. Conversely, the virulent HK/483 virus induced relatively high lung levels of IL-1α and TNF-α that persisted up until the death of WT (C57B6 × 129) mice, and observed previously in BALB/c mice [17, 35]. We found here that among the ten cytokines measured in the lungs on day 5 p.i., seven were significantly lower in TM mice compared to WT mice. A few cytokines, such as IL-12 (p70) and RANTES were not reduced in TM mice, suggesting that these proteins are can be produced independent of IL-1 and TNF receptor signaling. TM mice had lower amounts of two key chemokines, MCP-1 and KC. The potent chemoattractant properties of MCP-1 for monocytes and KC for neutrophils are well documented [8, 28]. The question is how these chemokines contribute to H5N1-induced pathogenesis. Normally, IL-1 and TNF receptors are present on the pulmonary epithelium and on neutrophils passing through the interstitium to the alveolar airways [8]. Overall, the inhibition of the TNF receptors has been shown to decrease vascular permeability through the destabilization of endothelial cell cytoskeleton [36, 37] and affects subsequent immune cell transmigration across the endothelium [38, 39]. It has been suggested that alveolar macrophage-produced TNF-α further activates alveolar type II pneumocytes to release KC and MCP-1, which in turn contributes to a pro-inflammatory cascade and neutrophil sequestration during pulmonary ischemia-reperfusion injury [40-41]. It is interesting to note that type II pneumocytes, not columnar tracheal epithelial cells, are the major site of H5N1 viral replication in patients with fatal H5N1 disease [42-44]. Conceivably, infection of type II pneumocytes and subsequent release of cytokines/chemokines could result in excessive accumulation of neutrophils and macrophages and contribute to tissue damage by causing vascular injury and destruction of the parenchymal cells [45, 46]. The resulting loss of functional alveolar surface area could result in inadequate gas exchange, lower respiration, and ultimately death. Deterioration of pulmonary function has been indicated by the need for hospital ventilator support in H5N1 infected patients [47].
We found that TM mice had no detectable defects in virus clearance in their lungs; however systemic spread of HK/483, which is a common feature of this H5N1 strain [17], was isolated at higher titers in brain tissues of TM mice. This suggests that together, TNF and IL-1 are important in controlling extrapulmonary viral spread and should be considered when designing potential prophylactic approaches targeting these cytokines for the treatment of inflammatory conditions induced by H5N1 infection. Treatment with anti-inflammatory drugs have been proposed as a therapeutic option for patients infected with H5N1 viruses however pre-clinical testing in H5N1 virus infection models shows little benefit of these treatments [16, 48, 49]. Moreover, treatments with anti-inflammatory drugs, such as corticosteroids to combat an exaggerated immune response are far too broad in their affects on the immune system. The development of new, more targeted therapies for H5N1 disease along with combination antiviral drug treatment could be effective in reducing acute lung injury and mortality caused by H5N1 virus.
Acknowledgments
The authors wish to thank the Hong Kong SAR Ministry of Health for providing the H5N1 virus. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the funding agency.
Sources of financial support: L.A.P was supported by fellowships from the American Society for Microbiology/CDC Coordinating Center for Infectious Diseases and the Oak Ridge Institute for Science and Education. J.P.M was supported by the National Institutes of Health, R01 HL-068153 and the Centers for Disease Control and Prevention.
Footnotes
Competing interests statement: The authors L.A.P., K.J.S., J.M.K., J.P.M., and T.M.T., declare that they have no competing financial interests.
This work in part has been presented by J.P.M. at the IX International Symposium on Respiratory Viral Infections, February 2009, Bangkok, Thailand (Abstract title: Mechanism of Lung Injuries and Pulmonary Inflammation)
References
- 1.World Health Organization. http://www.who.int/csr/disease/avian_influenza/country/en/index.html.
- 2.Tran TH, Nguyen TL, Nguyen TD, et al. Avian influenza A (H5N1) in 10 patients in Vietnam. N Engl J Med. 2004;350:1179–88. doi: 10.1056/NEJMoa040419. [DOI] [PubMed] [Google Scholar]
- 3.Yuen KY, Chan PK, Peiris M, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet. 1998;351:467–71. doi: 10.1016/s0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- 4.Gambotto A, Barratt-Boyes SM, de Jong MD, Neumann G, Kawaoka Y. Human infection with highly pathogenic H5N1 influenza virus. Lancet. 2008;371:1464–75. doi: 10.1016/S0140-6736(08)60627-3. [DOI] [PubMed] [Google Scholar]
- 5.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27:337–49. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 6.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol. 2009;77:1763–72. doi: 10.1016/j.bcp.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFalpha in pulmonary pathophysiology. Respir Res. 2006;7:125. doi: 10.1186/1465-9921-7-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2005;288:L3–15. doi: 10.1152/ajplung.00405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ware CF. The TNF Superfamily-2008. Cytokine Growth Factor Rev. 2008;19:183–6. doi: 10.1016/j.cytogfr.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan MC, Cheung CY, Chui WH, et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res. 2005;6:135. doi: 10.1186/1465-9921-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung CY, Poon LL, Lau AS, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360:1831–7. doi: 10.1016/s0140-6736(02)11772-7. [DOI] [PubMed] [Google Scholar]
- 12.de Jong MD, Simmons CP, Thanh T, et al. Fatal outcome of human influenza A H5N1 is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–7. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peiris JS, Yu WC, Leung CW, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet. 2004;363:617–9. doi: 10.1016/S0140-6736(04)15595-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.To KF, Chan PK, Chan KF, et al. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J Med Virol. 2001;63:242–6. doi: 10.1002/1096-9071(200103)63:3<242::aid-jmv1007>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 16.Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci U S A. 2007;104:12479–81. doi: 10.1073/pnas.0705289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szretter KJ, Gangappa S, Lu X, et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol. 2007;81:2736–44. doi: 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Lung NF-kappaB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J Immunol. 2005;175:7530–5. doi: 10.4049/jimmunol.175.11.7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizgerd JP, Lupa MM, Hjoberg J, et al. Roles for early response cytokines during Escherichia coli pneumonia revealed by mice with combined deficiencies of all signaling receptors for TNF and IL-1. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1302–10. doi: 10.1152/ajplung.00353.2003. [DOI] [PubMed] [Google Scholar]
- 20.Bender C, Hall H, Huang J, et al. Characterization of the surface proteins of influenza A (H5N1) viruses isolated from humans in 1997-1998. Virology. 1999;254:115–23. doi: 10.1006/viro.1998.9529. [DOI] [PubMed] [Google Scholar]
- 21.Claas EC, Osterhaus AD, van Beek R, et al. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet. 1998;351:472–7. doi: 10.1016/S0140-6736(97)11212-0. [DOI] [PubMed] [Google Scholar]
- 22.Suarez DL, Perdue ML, Cox N, et al. Comparisons of highly virulent H5N1 influenza A viruses isolated from humans and chickens from Hong Kong. J Virol. 1998;72:6678–88. doi: 10.1128/jvi.72.8.6678-6688.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu X, Tumpey TM, Morken T, Zaki SR, Cox NJ, Katz JM. A mouse model for the evaluation of pathogenesis and immunity to influenza A (H5N1) viruses isolated from humans. J Virol. 1999;73:5903–11. doi: 10.1128/jvi.73.7.5903-5911.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reed LJ, M H. A simple method for estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 25.Wilson DE, Chosewood LC, editors. US Department of Health and Human Services, Centers for Disease Control and Prevention and National Institutes of Health. 5th. U.S. Government Printing Office; Washington, D.C.: 2007. Biosafety in Microbiological and Biomedical Laboratories. http://www.cdc.gov/od/ohs/biosfty/bmbl5/bmbl5toc.htm. [Google Scholar]
- 26.Mizgerd JP. Molecular mechanisms of neutrophil recruitment elicited by bacteria in the lungs. Semin Immunol. 2002;14:123–32. doi: 10.1006/smim.2001.0349. [DOI] [PubMed] [Google Scholar]
- 27.Peschon JJ, Torrance DS, Stocking KL, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–52. [PubMed] [Google Scholar]
- 28.Melgarejo E, Medina MA, Sanchez-Jimenez F, Urdiales JL. Monocyte chemoattractant protein-1: a key mediator in inflammatory processes. Int J Biochem Cell Biol. 2009;41:998–1001. doi: 10.1016/j.biocel.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008;13:2400–7. doi: 10.2741/2853. [DOI] [PubMed] [Google Scholar]
- 30.Hammad H, Lambrecht BN. Lung dendritic cell migration. Adv Immunol. 2007;93:265–78. doi: 10.1016/S0065-2776(06)93007-7. [DOI] [PubMed] [Google Scholar]
- 31.de Jong MD. H5N1 transmission and disease: observations from the frontlines. Pediatr Infect Dis J. 2008;27:S54–6. doi: 10.1097/INF.0b013e3181684d2d. [DOI] [PubMed] [Google Scholar]
- 32.Mizgerd JP, Spieker MR, Doerschuk CM. Early response cytokines and innate immunity: essential roles for TNF receptor 1 and type I IL-1 receptor during Escherichia coli pneumonia in mice. J Immunol. 2001;166:4042–8. doi: 10.4049/jimmunol.166.6.4042. [DOI] [PubMed] [Google Scholar]
- 33.Simms HH, D'Amico R. Studies on polymorphonuclear leukocyte bactericidal function: II. The role of oxidative stress. Shock. 1997;7:339–44. [PubMed] [Google Scholar]
- 34.Lee JH, Del Sorbo L, Khine AA, et al. Modulation of bacterial growth by tumor necrosis factor-alpha in vitro and in vivo. Am J Respir Crit Care Med. 2003;168:1462–70. doi: 10.1164/rccm.200302-303OC. [DOI] [PubMed] [Google Scholar]
- 35.Tumpey TM, Lu X, Morken T, Zaki SR, Katz JM. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J Virol. 2000;74:6105–16. doi: 10.1128/jvi.74.13.6105-6116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28:574–81. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- 37.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123:134–45. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 38.Kusters S, Tiegs G, Alexopoulou L, et al. In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur J Immunol. 1997;27:2870–5. doi: 10.1002/eji.1830271119. [DOI] [PubMed] [Google Scholar]
- 39.Piguet PF, Kan CD, Vesin C. Role of the tumor necrosis factor receptor 2 (TNFR2) in cerebral malaria in mice. Lab Invest. 2002;82:1155–66. doi: 10.1097/01.lab.0000028822.94883.8a. [DOI] [PubMed] [Google Scholar]
- 40.Fudala R, Krupa A, Stankowska D, Allen TC, Kurdowska AK. Anti-interleukin-8 autoantibody:interleukin-8 immune complexes in acute lung injury/acute respiratory distress syndrome. Clin Sci (Lond) 2008;114:403–12. doi: 10.1042/CS20070272. [DOI] [PubMed] [Google Scholar]
- 41.Sharma AK, Fernandez LG, Awad AS, Kron IL, Laubach VE. Proinflammatory response of alveolar epithelial cells is enhanced by alveolar macrophage-produced TNF-alpha during pulmonary ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol. 2007;293:L105–13. doi: 10.1152/ajplung.00470.2006. [DOI] [PubMed] [Google Scholar]
- 42.Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440:435–6. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 43.Uiprasertkul M, Puthavathana P, Sangsiriwut K, et al. Influenza A H5N1 replication sites in humans. Emerg Infect Dis. 2005;11:1036–41. doi: 10.3201/eid1107.041313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Riel D, Munster VJ, de Wit E, et al. H5N1 Virus Attachment to Lower Respiratory Tract. Science. 2006;312:399. doi: 10.1126/science.1125548. [DOI] [PubMed] [Google Scholar]
- 45.Azoulay E, Darmon M, Delclaux C, et al. Deterioration of previous acute lung injury during neutropenia recovery. Crit Care Med. 2002;30:781–6. doi: 10.1097/00003246-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 46.La Gruta NL, Kedzierska K, Stambas J, Doherty PC. A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol. 2007;85:85–92. doi: 10.1038/sj.icb.7100026. [DOI] [PubMed] [Google Scholar]
- 47.Uyeki TM. Human infection with highly pathogenic avian influenza A (H5N1) virus: review of clinical issues. Clin Infect Dis. 2009;49:279–90. doi: 10.1086/600035. [DOI] [PubMed] [Google Scholar]
- 48.Carter MJ. A rationale for using steroids in the treatment of severe cases of H5N1 avian influenza. J Med Microbiol. 2007;56:875–83. doi: 10.1099/jmm.0.47124-0. [DOI] [PubMed] [Google Scholar]
- 49.Xu T, Qiao J, Zhao L, et al. Effect of dexamethasone on acute respiratory distress syndrome induced by the H5N1 virus in mice. Eur Respir J. 2009;33:852–60. doi: 10.1183/09031936.00130507. [DOI] [PubMed] [Google Scholar]