

Abstract
Ionophore-doped sensing membranes exhibit greater selectivities and wider measuring ranges if their membrane matrixes are noncoordinating and solvate interfering ions poorly. This is particularly true for fluorous phases, which are the least polar and polarizable condensed phases known. In this work, fluorous membrane matrixes were used to prepare silver ion-selective electrodes (ISEs). Sensing membranes composed of perfluoroperhydrophenanthrene, sodium tetrakis[3,5-bis(perfluorohexyl)phenyl]borate, and one of four fluorophilic Ag+-selective ionophores with one or two thioether groups were investigated. All electrodes exhibited Nernstian responses to Ag+ in a wide range of concentrations. Their selectivities for Ag+ over interfering ions were found to depend on host preorganization and the length of the –(CH2)n– spacers separating the coordinating thioether group from the strongly electron withdrawing perfluoroalkyl groups. ISEs based on the most selective of the four ionophores, i.e., 1,3-bis(perfluorodecylethylthiomethyl)benzene, provided much higher selectivities for Ag+ over many alkaline and heavy metal ions than most Ag+ ISEs reported in the literature (e.g., for K+, −11.6; Pb2+, −10.2; Cu2+, −13.0; Cd2+, −13.2). Moreover, the use of this ionophore with a linear perfluorooligoether as membrane matrix and solid contacts consisting of three-dimensionally ordered macroporous (3DOM) carbon resulted in a detection limit for Ag+ of 4.1 ppt (3.8×10−11 M).
Silver is utilized for a wide range of applications, such as in medicine, electronics, optics, photography, and the production of jewelry, coins, batteries, photovoltaic cells, bearings, and catalysts. Because of their antibacterial properties, silver salts and silver nanoparticles are used for the disinfection of drinking water and the preparation of topical gels, specialty bandages, implantable prostheses, and catheters. As a result, about 2500 tons of silver are released into the environment annually, and approximately 80 tons end up in surface waters.1 Silver is not as toxic to humans as many other heavy metals, but the US Environmental Protection Agency reported that a concentration higher than 0.17 μM is toxic to fish and microorganisms,2 and the maximum contaminant level for total silver in drinking water was set to 0.9 μM.3 While other methods for the determination of silver are available, atomic absorption spectrometry and the use of an inductively coupled plasma in combination with atomic emission or mass spectrometry are recommended methods.4,5 These and many other techniques often require quite extensive and time-consuming sample pretreatment, including preconcentration and matrix separation. In comparison, ion-selective electrodes (ISEs) require little sample preparation and manipulation while still permitting very wide ranges of linear response, low limits of detection, high selectivities, and the possibility to distinguish between the free metal ion and its complexes.6,7
Advances in the understanding of ion fluxes across ISE membranes made measurements with ISEs in the parts-per-billion (ppb) and parts-per-trillion (ppt) level possible.8–10 Both Ag+ ISEs with an inner filling solution in which the primary ion was buffered to a low activity and solid contact ISEs (SC-ISEs) without inner filling solution were reported. The use of Ag+ ISEs with inner filling solutions in contact with an ion-exchange resin were shown to minimize ion fluxes through the sensing membranes, resulting in detection limits as low as 3×10−10 M.11 In the first low-detection limit demonstration of Ag+ ISEs with a solid contact, polyoctylthiophene was used as the solid contact, and a detection limit of 2×10−9 M was achieved.12 More recently, we reported on ISEs with solid contacts made of three-dimensionally ordered macroporous (3DOM) carbon,13,14 which consists of a glassy carbon skeleton with a highly ordered array of uniformly sized macropores. The large interfacial area and the high capacitance of 3DOM carbon solid contacts was shown to result in excellent long-term stabilities.14 Ag+ ISEs with 3DOM carbon solid contacts exhibited a detection limit of 4.0×10−11 M.15
These low detection limits are not only of interest in view of silver measurements in environmental and biological samples, but enable new applications that were hindered so far by unsatisfactory detection limits. For example, the use of Ag+-selective electrodes for real-time monitoring of the growth dynamics of silver nanoparticles was described recently.16 Also, as an example of an assay with chemical signal amplification, a Ag+-selective electrode was used as a transducer for immunoassays. The electrodes were used to detect Ag+ ions released oxidatively from a nanoparticle label attached to an antibody. This permitted the detection of 12.5 pmol of the antigen immunoglobulin G (IgG) in samples as small as 50 μL.17
The existing needs for silver analysis and these emerging applications of Ag+ ISEs led us to wonder whether the selectivity of Ag+ ISEs could be increased by use of fluorous sensing membranes. Significant improvements in selectivity were recently achieved for ionophore-free cation18,19 and anion20,21 exchange electrodes with fluorous membranes as well as for ISEs based on fluorophilic H+ ionophores and fluorous liquid22 and perfluoropolymer membrane matrixes.23 The selectivity-enhancing effect of fluorous phases arises from the noncoordinating and poorly solvating properties of these materials, which are the least polar, least polarizable condensed phases known24–27 and are referred to as “fluorous” because of their high fluorine content and because they phase-separate both from aqueous solutions and from hydrocarbons.28 It is well known that weakly coordinating matrixes favor stronger binding between the ionophore and the ions for which the sensor is designed, and that weak solvation of interfering ions in the sensing membrane further increases the potentiometric selectivity.6,7 Moreover, weak solvation of counter ions inhibits the coextraction of target anions along with their counter ions into ISE membranes (Donnan failure), thereby widening the response range.29
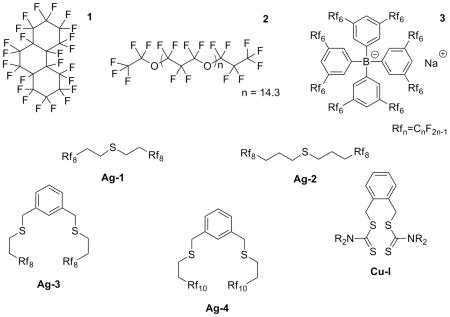
In order to prepare Ag+ ISEs with fluorous membrane matrixes, four fluorophilic ionophores with Ag+ coordinating thioether groups were used in this work in combination with perfluoroperhydrophenanthrene (1) or a linear perfluorooligoether (2) serving as the membrane matrix and sodium tetrakis[3,5-bis(perfluorohexyl)phenyl]borate (3) providing ionic sites. Compared to previously described ISEs, the selectivities for Ag+ over many alkaline and heavy metal ions were enhanced significantly. Moreover, using 3DOM carbon solid contacts, the detection limit for Ag+ was lowered to 3.8 × 10−11 M.
EXPERIMENTAL SECTION
Reagents
All chemicals were of the highest commercially available purity and were used as received, unless noted otherwise. Perfluoroperhydrophenanthrene (1) was purchased from Alfa Aesar (Ward Hill, MA) and the linear perfluorooligoether α-(heptafluoropropyl)-ω-(pentafluoroethoxy)-poly[oxy(1,1,2,2,3,3-hexafluoro-1,3-propanediyl)] (2) was purchased from Daikin Industries (Osaka, Japan). Sodium tetrakis[3,5-bis(perfluorohexyl)phenyl]borate (3), bis[perfluorooctylethyl]sulfane (Ag-1), bis[perfluorooctylpropyl]sulfane (Ag-2), 1,3-bis(perfluorooctylethylthiomethyl)benzene (Ag-3) and 1,3-bis(perfluorodecylethylthiomethyl)benzene (Ag-4), were prepared according to previously described procedures.18,19,30,31 All metal ion salts were obtained from Sigma-Aldrich (St. Louis, MO). Deionized and charcoal-treated water (18.2 MΩ· cm specific resistance) obtained with a Milli-Q PLUS reagent-grade water system (Millipore, Bedford, MA) was used for all sample solutions.
Sensing Membranes
To prepare sensing membranes, ionic sites and ionophore were added to the fluorous matrix material (see below for exact concentrations of membrane components), and the resulting mixture was stirred for at least 24 h to ensure complete dissolution. The fluorous sensing phases were then applied with a micropipette onto a stack of 1–6 porous filter disks used to mechanically support the fluorous membrane. Full penetration of the fluorous phase into the porous supports was confirmed by a translucent appearance of the thus prepared sensing membranes.
For all selectivity measurements, Fluoropore™ filters (porous poly(tetrafluoroethylene) without backing, 47 mm diameter, 0.45 μm pore size, 50 μm thick, 85% porosity) from Millipore were used as membrane supports. As described previously,18–23 the Fluoropore filters were sandwiched between two note cards, and appropriate disks of 13 mm diameter were cut out with a hole punch. Six layers of filter disks were used for each electrode when selectivity measurements involved direct exposure to Ag+; four layers were sufficient when selectivities for one interfering ion relative to another interfering ion were determined with the fixed interference method.32 The sensing phases consisted of 1 doped with 3 and one of the four ionophores. For each filter disk in the stack, 5 μL of fluorous sensing phase was used.
For low detection limit measurements, Fluoropore™ membrane disks made of a porous poly(tetrafluoroethylene) layer (47 mm diameter, 0.22 μm pore size, 175 μm thickness, 70% porosity) with a polyethylene backing were used. Membrane disks of 16 mm diameter were cut with a hole punch as described above but only one layer was used for each electrode. The sensing membranes were prepared with 10 μL of the linear perfluorooligoether 2, containing ionic sites (0.5 mM) and ionophore (1.5 mM). Ionophore Ag-4 was used for all low detection limit experiments.
Electrode Assembly
For selectivity measurements, the thus prepared fluorous membranes were mounted into custom-machined electrode bodies made from poly(chlorotrifluoroethylene), as described previously.19 In brief, a screw cap with a hole (8.3 mm diameter) in the center was screwed onto the electrode body, securing the sensing membrane in between the electrode body and the cap but leaving the center of the membrane exposed (see Figure 1 in ref. 19). To measure the response to Ag+, a 0.1 mM AgNO3 solution was added into the electrode body, and a Ag/AgCl wire was inserted as reference electrode. Prior to measurements, all electrodes were conditioned in a 10 mM AgNO3 solution for 5 h. To determine selectivities directly with respect to Ag+, the inner filling solution contained a 1.0 mM nitrate and a 0.1 mM chloride salt of the interfering ion. Before measurements, the electrodes were conditioned in 10 mM solutions of the nitrate salt of the interfering ion, and the responses to interfering ions were determined before the electrodes were brought into contact with Ag+ solutions (see ref. 33 for similar procedures).
To improve the detection limit for Ag+, ISEs with 3DOM carbon solid contacts were utilized. 3DOM carbon monoliths were prepared by colloidal crystal templating with monodisperse poly(methyl methacrylate) spheres, as reported previously.13 Before use, the 3DOM carbon monoliths were polished with sandpaper (600 grit, 3M, St. Paul, MN) to remove the untemplated crust on top of the monolith and to produce the desired size (5×5×0.5 mm3). The monolith was then attached with colloidal silver paste (Ted Pella, Redding, CA) to the end of a copper wire, which was shaped into a loop at that end for improved mechanical integrity. (No negative effects due to copper surface oxidation were observed in this work, but use of a noble metal wire may be preferable to obtain particularly long sensor lifetimes.) Then about 30 μL of the fluorous sensing phase was pipetted onto the monolith, which wicked up the sensing phase. The pores of the monolith were considered to be saturated with the fluorous sensing phase when no more air bubbles came out of the monolith and the surface of the monolith had a wet appearance. The monolith was then mounted along with a support disk (loaded with 10 μL fluorous sensing phase, as described above) into a custom-machined electrode body of the same construction as for the selectivity measurements (see Figure 1 for a schematic of the electrode setup). The conditioning process used for low detection limit experiments is explained below.
Figure 1.

Schematic setup of 3DOM carbon-contacted Ag+-selective electrode with a fluorous membrane, as used for the low detection limit experiments (see also Figure S2, Supplementary Information).
EMF Measurements
Potentials were monitored with an EMF 16 potentiometer (Lawson Labs, Malvern, PA) controlled with EMF Suite 1.02 software (Lawson Labs) at room temperature (25 °C) in stirred solutions. The external reference electrode consisted of a double-junction Ag/AgCl electrode with a 1 M LiOAc bridge electrolyte and a 3 M KCl reference electrolyte. All emf values were corrected for liquid-junction potentials with the Henderson equation.34 Selectivity coefficients over K+ and Na+ were determined with the separate solution method, and for all other ions the fixed interference method was used with respect to K+ for ease of measurements.32 For all interfering ions Nernstian responses were confirmed by successive dilution of stock solutions in the concentration range where selectivities were measured. The reported selectivities are averages for 6 to 8 electrodes. Activity coefficients were calculated with a two-parameter Debye–Hückel approximation.35 All measurements were performed with polypropylene beakers, which were cleaned overnight in 0.1 M HNO3 before use.
RESULTS AND DISCUSSION
Since the use of a dithia crown ether as the first electrically neutral ionophore for Ag+ was reported in 1986,36 many new ionophores have been applied for the construction of Ag+-ISEs.7,37 In most cases, sulfur was the key atom of the coordinating group(s). For example, ionophores with thioether,38–40 thiophosphate,41 and thiocarbamate groups were reported.33 The silver ion binds to these functional groups with high selectivity. This can be rationalized on the basis of the hard–soft acid base (HSAB) theory, which predicts that sulfur as a soft Lewis base has a high affinity for the silver ion, which is a soft Lewis acid.42 Interestingly, remarkably high potentiometric selectivities have been achieved with thioether derivatives as ionophores, while use of thiocarbonyl groups often appears to result in more pronounced interferences from other heavy metal ions such as Pb2+ and Cd2+.43
Based on this prior work, we anticipated that fluorophilic thioether derivatives would be promising ionophores for the selective recognition of Ag+ in fluorous phases. Two fluorophilic dialkyl sulfides of the structure Rf8(CH2)nS (where Rf8=CF3(CF2)7; n=2 for Ag-1, and n=3 for Ag-2) were obtained by reaction of Rf8(CH2)nI with Li2S.30 The pincer ligands 1,3-C6H4(CH2SCHCH2Rfn)2 (n=8 for Ag-3, and n=10 for Ag 4) were synthesized from 1,3-C6H4(CH2Br)2 and the thiols HSCH2CH2Rfn, or from the dithiols 1,3-C6H4(CH2SH)2 and ICH2CH2Rfn.31 These compounds were first introduced as ligands for fluorophilic palladium complexes, which were shown to be catalyst precursors for Suzuki and Heck reactions.
Potentiometric Responses and Selectivities
For initial experiments, fluorous sensing membranes consisting of perfluoroperhydrophenanthrene doped with 1.0 mM sodium tetrakis[3,5-bis(perfluorohexyl)phenyl]borate and one of the four Ag+ ionophores (3.0 mM) were used. The potentiometric Ag+ responses of electrodes with these four types of membranes were measured in AgNO3 solutions (for the calibration curves, see Figure S1 in the Supporting Information). Independent of the type of ionophore, every electrode exhibited a Nernstian response to Ag+, and without further optimization the Ag+ concentration range for which a linear response was observed was between 10−7 M and 10−2 M. The potentiometric responses were fast (≤5 s) and, therefore, likely limited by the speed of sample change.
To screen the selectivities of these electrode membranes, the selectivities for Ag+ over the monocations K+ and Na+ and the dication Cu2+ were determined. Figure 2 shows the selectivity coefficients in logarithmic format ( ; see Table S1 of the Supporting Information for numerical values), illustrating a steep increase in selectivity from ionophore Ag-1 to Ag-4. This trend is the result of host preorganization6 and electronic effects, as will be discussed in the following.
Figure 2.

Selectivity coefficients, , for ISE membranes with perfluoroperhydrophenanthrene doped with 1.0 mM borate salt 3 and one of the four ionophores Ag-1, Ag-2, Ag-3, or Ag-4 (3.0 mM). For comparison, the selectivities for a fluorous ionophore-free ion-exchanger membrane made of 1.0 mM borate salt 3, in perfluoroperhydrophenanthrene are shown on the left.
The perfluoroalkyl chains of all four ionophores ensure that these compounds are sufficiently soluble in the fluorous matrix. However, since perfluoroalkyl groups are strongly electron withdrawing, the sulfur atoms must be separated from these perfluoroalkyl groups by –(CH2)n– spacers in order for the thioethers to bind strongly to Ag+. While the coordinative properties of perfluoroalkyl thioethers (Rf–S–Rf) have not been studied, it is well known that the direct substitution of ether oxygens,19,44,45 amino nitrogens,19,22 and phosphorus atoms of phosphines46 with perfluoroalkyl groups results in only extremely weak interactions with protons and metal cations. However, –(CH2)n– spacers can be used to separate the perfluoroalkyl groups from the coordinating group, shielding the latter with increasing efficiency as more methylene units are added.24 Indeed, as shown in Figure 2, membranes doped with ionophore Ag-2 have a higher selectivity for Ag+ than electrodes doped with ionophore Ag-1. For example, the K+ discrimination by Ag-2, which has two –CH2CH2CH2– spacers, is 15.5 times higher than that of Ag-1, which has two –CH2CH2– spacers. Because Ag-1 and Ag-2 have otherwise identical structures, the different selectivities arise from the different lengths of the –(CH2)n– spacers.
In spite of the electron-withdrawing perfluoroalkyl groups, the selectivities for Ag+ of membranes doped with Ag-1 and Ag-2 are 2 to 3 orders of magnitude higher than those of conventional membranes with nonfluorinated monothioether ionophores (such as diethyl sulfide, ethyl phenyl sulfide and diphenyl sulfide).38,39 This appears to be the result of the fluorous matrix and not an electronic effect. Quite to the contrary, while the –CH2CH2CH2– spacers of Ag-2 clearly reduce the electron withdrawing effect of the perfluorooctyl groups on the thioether group, it is well documented that even this type of spacer cannot completely shield tertiary amines and phosphines from perfluoroalkyl groups.19,24 Therefore, the Lewis basicities of Ag-1 and Ag-2 are almost certainly lower than those of nonfluorinated ones, suggesting that the higher selectivities of membranes doped with Ag-1 and Ag-2 do not result from stronger binding of Ag+ but poorer solvation of interfering ions in the fluorous sensing membranes.
Subsequently, the effect of the ratio of ionophore to ionic sites on the potentiometric selectivities was investigated. As is well documented, the optimum ratio in view of selectivity is controlled by the complex stoichiometry and, therefore, the coordination number, of the primary and interfering ions.47 For Ag+, the most common coordination number in complexes with inorganic and organic ligands is four.48,49 However, tri- and dicoordinate complexes make up roughly half of all known crystal structures, demonstrating that Ag+ has a preference for low coordination numbers. Indeed, while there are some examples of penta- and hexacoordinated Ag+ complexes with multidentate receptors such as thia crown ethers, several tetracoordinated hexathio crown ether complexes of Ag+ were reported.49,50 Evidently, the gain in free energy from ligation of the fifth and sixth thioether group is small. While similar data for monodentate thioether ligands are not available, the preference of Ag+ for lower coordination numbers is also exemplified well by the stability of its complexes with the monodentate model ligand NH3. In aqueous solution dicoordination predominates (K1=103.15 M−1, K2=103.75 M−1) and tricoordination is extremely weak (K3=0.025 M−1),51 unusual environments such as liquid ammonia or a water-free NH3 gas are needed for tetracoordination,52 and computational studies confirm that the penta- and hexacoordinated species are thermodynamically unstable.
Tetracoordination of Ag+ by the fluorophilic ionophores used in this study is indeed consistent with the selectivities shown in Table 1.53 When the molar ratio of ionophore to ionic sites was increased from 3:1 to 5:1 for electrodes with the monodentate ionophore Ag-2, the selectivity over K+, Na+ and Cu2+ increased by at least 1.5 orders of magnitude. This significant increase in selectivity cannot be explained by a modest increase in the free ionophore concentration from 1.0 to 3.0 mM, as it would be expected if 2:1 complexation were dominant. This suggests that in the membranes with the 3:1 ionophore-to-site ratio, and thereby a 3:1 ratio of total ionophore and Ag+, there is 1:3 complexation and only a low concentration of free ionophore. The 5:1 ionophore-to-site ratio permits formation of tetracoordinated complexes with an excess of free ionophore, which is crucial for high selectivity.47 An analogous effect is observed for ionophore Ag-4, which has two thioether groups and can, therefore, achieve tetracoordination already at a 2:1 ratio of ionophore to Ag+. A ratio of Ag-4 to ionic sites of 2:1 permits tetracoordinate binding of all Ag+, but this would leave the free ionophore concentration in the sensing membrane at the very low level determined by the dissociation constant of this complex. In contrast, an ionophore–ionic site ratio of 3:1 and tetrahedral Ag+ coordination as a result of the formation of 2:1 complexes leaves one third of the ionophore in its free form (1.0 mM), resulting in high selectivity. Indeed, the selectivity of the 3:1 membranes doped with ionophore Ag-4 is approximately 2.5 orders of magnitude higher than for membranes with the 2:1 ratio.
Table 1.
Silver Ion Selectivities ( ) of Fluorous Ionophore-Doped Electrode Membranes Based on Perfluoroperhydrophenanthrene with Different Ratios of Ionophore and Ionic Sitesa
| Ionophore | Ionophore/Ionic Site Ratio | Ag+ selectivity ( ) | ||
|---|---|---|---|---|
| K+ | Na+ | Cu2+ | ||
| Ag-2 | 3:1 | −5.97±0.07 | −7.60±0.09 | −8.09±0.19 |
| 5:1 | −8.12±0.08 | −9.06±0.15 | −10.09±0.18 | |
| Ag-3 | 3:1 | −9.45±0.15 | −10.93±0.21 | −11.22±0.21 |
| Ag-4 | 2:1 | −9.11±0.21 | −10.35±0.22 | −10.50±0.25 |
| 3:1 | −11.60±0.13 | −12.94±0.14 | −13.04±0.22 | |
The concentration of ionic sites, 3, for all electrode membranes was 1.0 mM.
For a meaningful comparison of the performance of different ionophores, selectivities of membranes with an excess of free ionophore should be considered. Here, selectivities from membranes containing Ag-2 in a molar ratio of 5:1 to the ionic sites and membranes with Ag-3 or Ag-4 in a ratio of 3:1 to the ionic sites may be compared. As Table 1 shows, the monodentate Ag-2 provides 1.5 and 3 orders of magnitude lower selectivity than the bidentate ionophores Ag-3 and Ag-4, respectively. While the superior performance of Ag-3 and Ag-4 in comparison to Ag-2 is easily understood as an effect of host preorganization, the higher selectivity of Ag-4 in comparison to Ag-3 is puzzling. These two ionophores share the same spacer between the two thioether groups, and they have the same number of methylene groups between the thioether groups and the perfluoroalkyl substituents. The only difference in their structure is that the perfluoroalkyl chains of Ag-4 (perfluorodecyl) are longer than in Ag-3 (perfluorooctyl). While the higher selectivity of Ag-4 is not currently understood, one may wonder if the higher selectivity is related to intramolecular interactions between the two perfluoroalkyl chains of one ionophore molecule, or alternatively interactions between the perfluoroalkyl groups of two ionophores within a Ag+ complex. Fluorophilic compounds with very long straight-chain perfluoroalkyl groups are often not very soluble even in fluorous solvents because they crystallize quite readily as the result of the relative stiffness of perfluoroalkyl groups24 and the concomitant low loss of entropy upon crystallization. Thereby, the perfluorodecyl groups may contribute in an unusual way to host preorganization.
As Ag-4 is the most selective ionophore among these four ionophores, the selectivities for Ag+ over more interfering ions were determined (see Table 2). In comparison to the selectivities for the so far most Ag+ selective ISE (based on a conventional nonfluorous membrane and ionophore Cu-I),11 the ISE with Ag-4 as fluorophilic ionophore shows a Ag+ selectivity enhanced by about two orders of magnitude over many interfering ions. For example, Pb2+ and Cu2+, which coexist with Ag+ in many environmental samples, are highly discriminated with −10.2 and −13.0, while the nonfluorous membrane ISE listed in Table 2 exhibits a of −11.1 and the majority of other ionophore-based ISEs described in the literature were reported to exhibit and values in a range of −1.0 to −6.0.
Table 2.
Silver Ion Selectivities ( ) of Fluorous Ionophore-Doped Electrode Membranes Based on Perfluoroperhydrophenanthrene and Ionophore Ag-4a
Detection Limit
It is well known that in the presence of interfering ions ISEs with higher selectivities can detect primary ions at lower concentration. Moreover, strong binding of the primary ion to the ionophore reduces co-extraction of the primary ion and the ionic sites into the sample, an effect that can improve the detection limit when the concentration of interfering ions is low. Since the best selectivity in this work was obtained with ionophore Ag-4, further experiments to improve the detection limit for Ag+ were performed with this ionophore in a molar ratio of 3:1 to the ionic sites. Because perfluoroperhydrophenanthrene, 1, which was used in the selectivity evaluation described above, has a fairly high vapor pressure and is, therefore, not suitable for long-term use, the linear perfluorooligoether 2 was utilized as membrane matrix. Earlier research with 2 and H+ ionophores showed that similar and even slightly better selectivities were obtained with ISE membranes based on 2 as compared to ISEs based on 1, which is consistent with the low Lewis basicity of the oxygens in perfluoroethers.23 Moreover, the linear perfluorooligoether 2 is more viscous than 1 (0.099 Pa•s and 0.028 Pa•s, respectively),54 which is expected to favor low detection limits as this decreases the diffusion coefficients of ions in sensing membranes and thereby diminishes ion fluxes.
Low detection limits for Ag+ with membranes based on 2 were achieved by use of solid contacted membranes. As in our earlier work with PVC-based Ag+ ISE membranes,15 three-dimensionally ordered macroporous (3DOM) carbon was used as a solid contact. Initial experiments using Ag-4 and 3DOM carbon attempted the direct attachment of fluorous polymeric sensing membranes onto 3DOM carbon monoliths to give an electrode setup identical to the one previously used in our work with PVC-based membranes (see Figure 1 in ref. 13 and Figure 1 in ref. 15). For this purpose, the sensing membranes were composed of Teflon AF2400 (poly[4,5-difluoro-2,2,-bis(trifluoromethyl)-1,3-dioxole]-co-poly(tetrafluoroethylene)), plasticized with 2 and doped with the borate salt 3 and ionophore Ag-4. However, due to the low polarity of the fluorous sensing phase, the fluorous sensing membranes readily peeled off from the supporting poly(vinyl chloride) (PVC) substrate. Therefore, the electrode setup shown in Figure 1 was used instead. The same electrode bodies were used as for the ISEs with inner filling solutions, and the screw cap permitted a tight contact between the 3DOM carbon and the sensing membrane. The use of porous poly(tetrafluoroethylene) disks with a backing provided additional mechanical stability and did not affect selectivities. Following the conditioning protocol already used in our previous work for the detection of Ag+ at very low concentrations,15 the electrodes were first exposed to 1 μM AgNO3 solution for 2 days to replace the Na+ introduced into the fluorous membranes in the form of the salt of the ionic site, 3, with Ag+ from the conditioning solution. This was followed by conditioning of the electrodes in 1 nM AgNO3 solution for another 2 days to remove extra Ag+ from the sensing membranes. The electrodes were then moved to freshly prepared 1 nM AgNO3 solutions to determine the detection limit. A typical calibration curve is shown in Figure 3. The response to Ag+ was Nernstian in the range of 10−10 to 10−9 M, and started to level off at lower concentrations. The detection limit achieved was 3.8 ×10−11 M, or 4.1 ppt, which is very close to the one reported earlier by us for PVC-based 3DOM carbon-contacted membranes, which had a 4.0 ×10−11 M (4.3 ppt) detection limit for Ag+.15 Whether the extremely favorable detection limits of these sensors are simply the result of the combination of the use of solid-contacts, very selective ionophores and membrane formulations, and ideal conditioning procedures, or whether 3DOM carbon solid contacts generally provide somewhat lower detection limits than solid contacts based on conductive polymers such as polypyrroles or polythiophenes remains to be seen.
Figure 3.

Potentiometric Ag+ response with the lowest detection limit achieved in this work, as recorded after conditioning of the electrode in 1 μM AgNO3 for 2 days and 1 nM AgNO3 for another 2 days. The fluorous membrane was composed of the linear perfluorooligoether 2, doped with 0.5 mM borate salt 3, and 1.5 mM ionophore Ag-4. The solid line is a fit based on the Nicolskii–Eisenman Formalism6 and the interrupted lines show how the detection limit was determined according to IUPAC recommendations.55
CONCLUSIONS
This paper describes the first use of a fluorous membrane matrix along with 3DOM carbon solid-contacts, resulting in a detection limit as low as 4.1 ppt for Ag+. This opens a venue to combine the advantages of fluorous membranes and 3DOM carbon solid contacts for many other ionophores and analytes. Following previous work with H+ selective ionophores, this work is also the second example of how fluorous membranes can greatly improve the selectivity of ionophore-based ISEs. The strong interaction between Ag+ and the ditopic receptors in the fluorous membrane matrix results in exceptional selectivities. Attempts to optimize the preorganization of the ditopic receptors have not been made yet, and one may wonder what further improvements in Ag+ selectivities may be achieved if, for example, the linker between the two thioether groups were varied or fluorophilic macrocyclic thia crown ethers were used to dope fluorous ISE membranes.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (CTS-0428046, EXP-SA 0730437), the National Institute of Health (R01 EB005225-01), the Office of Naval Research (ONR, Grant N00014-07-1-0608), the MRSEC program of the NSF (DMR-0819885), and the Welch Foundation (Grant A-1656). C.-Z. L. thanks the University of Minnesota for a Louise T. Dosdall fellowship. M. A. F. thanks 3M for a Science and Technology Fellowship and the University of Minnesota Graduate School for a Graduate School Fellowship. The authors thank Yun Jiang for his assistance in making Figure 1.
Footnotes
Supporting Information Available
Ag+ response curves of ISEs with inner filling solutions. Full-page color version of Figure 1. Selectivity coefficients of electrodes based on membranes containing ionophore and ionic sites in a molar ratio of 3:1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ratte HT. Environ Toxicol Chem. 1999;18:89–108. [Google Scholar]
- 2.EPA Drinking Water Criteria Document for Silver. Environmental Protection Agency; Washington DC: 1989. EPA CARSRN 7440-7422-7444. [Google Scholar]
- 3.National Primary Drinking Water Regulations: Final Rule. Environment Protection Agency; Washington DC: 1991. Fed Reg 56:3526. [Google Scholar]
- 4.Henneth H, editor. Official Methods of Analysis of the Association of Official Analytical Chemists. Association of Official Analytical Chemists; Arlington, Virginia: 1990. p. 324. [Google Scholar]
- 5.EPA Method 200.7, “Inductively Coupled Plasma Atomic Emission Spectrometric Method for Trace Element Analysis of Water and Wastes,” and EPA Method 200.8, “Determination of Trace Elements in Waters and Wastes by Inductively Coupled Plasma-Mass Spectrometry,” from “Methods for Determination of Metals in Environmental Samples—Supplement I”, Environmental Protection Agency, EPA–600/R–94–111, May 1994.
- 6.Bakker E, Bühlmann P, Pretsch E. Chem Rev. 1997;97:3083–3132. doi: 10.1021/cr940394a. [DOI] [PubMed] [Google Scholar]
- 7.Bühlmann P, Pretsch E, Bakker E. Chem Rev. 1998;98:1593–1687. doi: 10.1021/cr970113+. [DOI] [PubMed] [Google Scholar]
- 8.Sokalski T, Ceresa A, Zwickl T, Pretsch E. J Am Chem Soc. 1997;119:11347–11348. [Google Scholar]
- 9.Sokalski T, Zwickl T, Bakker E, Pretsch E. Anal Chem. 1999;71:1204–1209. [Google Scholar]
- 10.Sokalski T, Ceresa A, Fibbioli M, Zwickl T, Bakker E, Pretsch E. Anal Chem. 1999;71:1210–1214. [Google Scholar]
- 11.Wygladacz K, Radu A, Xu C, Qin Y, Bakker E. Anal Chem. 2005;77:4706–4712. doi: 10.1021/ac050856s. [DOI] [PubMed] [Google Scholar]
- 12.Chumbimuni-Torres KY, Rubinova N, Radu A, Kubota LT, Bakker E. Anal Chem. 2006;78:1318–1322. doi: 10.1021/ac050749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai CZ, Fierke MA, Stein A, Bühlmann P. Anal Chem. 2007;79:4621–4626. doi: 10.1021/ac070132b. [DOI] [PubMed] [Google Scholar]
- 14.Fierke MA, Lai CZ, Bühlmann P, Stein A. Anal Chem. 2010;82:680–688. doi: 10.1021/ac902222n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai CZ, Joyer MM, Fierke MA, Petkovich ND, Stein A, Bühlmann P. J Solid State Electrochem. 2009;13:123–128. doi: 10.1007/s10008-008-0579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chumbimuni-Torres KY, Bakker E, Wang J. Electrochem Commun. 2009;11:1964–1967. doi: 10.1016/j.elecom.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chumbimuni-Torres KY, Dai Z, Rubinova N, Xiang Y, Pretsch E, Wang J, Bakker E. J Am Chem Soc. 2006;128:13676–13677. doi: 10.1021/ja065899k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boswell PG, Bühlmann P. J Am Chem Soc. 2005;127:8958–8959. doi: 10.1021/ja052403a. [DOI] [PubMed] [Google Scholar]
- 19.Boswell PG, Lugert EC, Rábai J, Amin EA, Bühlmann P. J Am Chem Soc. 2005;127:16976–16984. doi: 10.1021/ja055816k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boswell PG, Anfang AC, Bühlmann P. J Fluorine Chem. 2008;129:961–967. doi: 10.1016/j.jfluchem.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen LD, Mandal D, Gladysz JA, Bühlmann P. New J Chem. doi: 10.1039/b9nj00696f. in press. published on-line 5 May 2010. [DOI] [Google Scholar]
- 22.Boswell PG, Szíjjártó C, Jurisch M, Gladysz JA, Rábai J, Bühlmann P. Anal Chem. 2008;80:2084–2090. doi: 10.1021/ac702161c. [DOI] [PubMed] [Google Scholar]
- 23.Lai CZ, Koseoglu SS, Lugert EC, Boswell PG, Rábai J, Lodge TP, Bühlmann P. J Am Chem Soc. 2009;131:1598–1606. doi: 10.1021/ja808047x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gladysz JA, Curran DP, Horváth IT. Handbook of Fluorous Chemistry. Wiley & Sons; New York: 2005. [Google Scholar]
- 25.Chambers RD. Fluorine in Organic Chemistry. Blackwell; Oxford, U.K.: 2004. [Google Scholar]
- 26.Grec JJ, Riess JG, Devallez B. Nouv J Chim. 1985;9:109–117. [Google Scholar]
- 27.Vincent JM. J Fluorine Chem. 2008;129:903–909. [Google Scholar]
- 28.Horváth IT, Rábai J. Science. 1994;266:72–75. doi: 10.1126/science.266.5182.72. [DOI] [PubMed] [Google Scholar]
- 29.Bakker E, Xu A, Pretsch E. Anal Chim Acta. 1994;295:253–262. [Google Scholar]
- 30.Rocaboy C, Gladysz JA. Tetrahedron. 2002;58:4007–4014. [Google Scholar]
- 31.Corrêa da Costa R, Jurisch M, Gladysz JA. Inorg Chim Acta. 2008;361:3205–3214. [Google Scholar]
- 32.Bakker E, Pretsch E, Bühlmann P. Anal Chem. 2000;72:1127–1133. doi: 10.1021/ac991146n. [DOI] [PubMed] [Google Scholar]
- 33.Bakker E. Anal Chem. 1997;69:1061–1069. [Google Scholar]
- 34.Morf WE. The Principles of Ion-Selective Electrodes and of Membrane Transport. Elsevier; New York: 1981. [Google Scholar]
- 35.Meier PC. Anal Chim Acta. 1982;136:363–368. [Google Scholar]
- 36.Lai MT, Shih JS. Analyst. 1986;111:891–895. [Google Scholar]
- 37.Bakker E, Bühlmann P, Pretsch E. Electroanalysis. 1999;11:915–933. [Google Scholar]
- 38.Teixidor F, Flores MA, Escriche L, Viñas C, Casabó J. J Chem Soc, Chem Commun. 1994:963–964. [Google Scholar]
- 39.Errachid A, Bausells J, Merlos A, Esteve J, Teixidor F, Pérez-Jiménez C, Casabó J, Jiménez C, Bartrolí J. Sens Actuators B. 1995;26–27:321–324. [Google Scholar]
- 40.Mashhadizadeh MH, Shamsipur M. Anal Chim Acta. 1999;381:111–116. [Google Scholar]
- 41.Liu D, Liu J, Tian D, Hong W, Zhou X, Yu JC. Anal Chim Acta. 2000;416:139–144. [Google Scholar]
- 42.Pearson RG. J Am Chem Soc. 1963;85:3533–3539. [Google Scholar]
- 43.Brzozka Z, Cobben PLHM, Reinhoudt DN, Edema JJH, Buter J, Kellogg RM. Anal Chim Acta. 1993;273:139–144. [Google Scholar]
- 44.Lai CZ, Reardon ME, Boswell PG, Bühlmann P. J Fluorine Chem. 2010;131:42–46. doi: 10.1016/j.jfluchem.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szlávik Z, Tárkányi G, Gömöry A, Tarczay G, Rábai J. J Fluorine Chem. 2001;108:7–14. [Google Scholar]
- 46.Jiao H, Le Stang S, Soos T, Meier R, Kowski K, Rademacher P, Jafarpour L, Hamard JB, Nolan SP, Gladysz JA. J Am Chem Soc. 2002;124:1516–1523. doi: 10.1021/ja011877g. [DOI] [PubMed] [Google Scholar]
- 47.Amemiya S, Bühlmann P, Pretsch E, Rusterholz B, Umezawa Y. Anal Chem. 2000;72:1618–1631. doi: 10.1021/ac991167h. [DOI] [PubMed] [Google Scholar]
- 48.Carvajal MA, Novoa JJ, Alvarez S. J Am Chem Soc. 2004;126:1465–1477. doi: 10.1021/ja038416a. [DOI] [PubMed] [Google Scholar]
- 49.Alberto R, Angst D, Ortner K, Abram U, Schubiger PA, Kaden TA. New J Chem. 2007;31:409–417. [Google Scholar]
- 50.Alberto R, Nef W, Smith A, Kaden TA, Neuburger M, Zehnder M, Frey A, Abram U, Schubiger PA. Inorg Chem. 1996;35:3420–3427. doi: 10.1021/ic951421y. [DOI] [PubMed] [Google Scholar]
- 51.Bjerrum J. Acta Chem Scand Ser A. 1986;40:392–395. [Google Scholar]
- 52.Inoue K, Ohashi K, Iino T, Sasaki J, Judai K, Nishi N, Sekiya H. Phys Chem Chem Phys. 2008;10:3052–3062. doi: 10.1039/b802050g. [DOI] [PubMed] [Google Scholar]
- 53.Shoeib T, Milburn RK, Koyanagi GK, Lavrov VV, Bohme DK, Siu KWM, Hopkinson AC. Int J Mass Spectrom. 2000;201:87–100. [Google Scholar]
- 54.Demnum data sheet. Daikin Industries; Osaka, Japan: May, 2009. [Google Scholar]
- 55.Guilbault GG, Durst RA, Frant MS, Freiser H, Hansen EH, Light TS, Pungor E, Rechnitz G, Rice NM, Rohm TJ, Simon W, Thomas JDR. Pure Appl Chem. 1976;48:127. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.