

Abstract
Ischemia-reperfusion injury (IRI) is a major cause of acute kidney injury (AKI) and both innate and adaptive immunity contribute to the pathogenesis. Kidney resident cells promote inflammation after IRI by increasing endothelial cell adhesion molecule expression and vascular permeability. Kidney epithelial cells bind complement and express tolllike receptors and resident and infiltrating cells produce cytokines/chemokines. Early activation of kidney dendritic cells (DCs) initiates a cascade of events leading to accumulation of interferon-γ-producing neutrophils, infiltrating macrophages, CD4+ T cells, B cells and invariant natural killer T (NKT) cells. Recent studies from our laboratory now implicate the IL23/IL17 pathway in kidney IRI. Following the initial early phase of inflammation, the late phase involves infiltration of anti-inflammatory cells including regulatory T cells, alternatively activated macrophages and stem cells leading to attenuation of inflammation and initiation of repair. Based upon these immune mechanisms of injury, recent studies hold promise for novel drug therapies. These pharmacological agents have been shown to reduce inflammation or cytotoxicity in rodent models of AKI and some show early promise in clinical trials. This review summarizes recent advances to further our understanding of the immune mechanisms of AKI and potential pharmacological therapies.
Keywords: Acute renal failure, ischemia-reperfusion injury, cisplatin, inflammation
Introduction
Effective therapy of acute kidney injury (AKI) especially in critically ill patients remains elusive. Mortality in these patients is alarmingly high despite substantial advances in techniques of resuscitation and renal replacement therapy. The incidence of AKI is increasing markedly [1, 2] as a result of the expansion of invasive medical and surgical procedures and the increasing expectation for aggressive medical management of critically ill patients. In critically ill patients, mortality is 40-60% [3-6] and traditionally attributed to comorbid conditions. Accumulating data suggests, however, that AKI has an independent negative impact on mortality [7, 8]. The success of therapeutic interventions for AKI necessitates a better understanding of its pathogenesis. Inflammation plays a critical role in AKI and the following review discusses immune mechanisms contributing to injury and repair associated with AKI as well a novel therapeutics.
Overview of Experimental Acute Kidney Injury
AKI is a consequence of vasospasm, alterations in ultrafiltration coefficient, tubular obstruction and/or back-leak [9, 10]. Renal ischemia-reperfusion (IR) is a major cause of AKI. The mechanisms involved in renal ischemia-reperfusion injury (IRI) are complex [9, 11], invoking both innate and adaptive immunity [12, 13]. Following IR, the cascade of events leading to endothelial cell dysfunction, tubular epithelial cell injury and activation of tissue-resident and infiltrating leukocytes consists of the coordinated action of cytokines/chemokines, reactive oxygen intermediates and adhesion molecules [11, 13]. The early phase of innate immune response to IR begins within minutes of reperfusion, whereas the late phase adaptive response requires days to manifest.
Early Phase Inflammation in Ischemia-Reperfusion Injury
Ischemia and/or reperfusion initiate changes in vascular endothelial cells, tubular epithelial cells (TEC) and resident renal dendritic cells (DCs) that cause the loss of immune system homeostasis in the kidney (see recent reviews [13-15]). As a result, numerous pro-inflammatory leukocytes are attracted to, and are activated within the post-ischemic kidney, potentiating the direct damage inflicted on kidney parenchymal cells by IR.
Endothelial Cells
Activation of the endothelium following kidney IRI leads to a loss of vascular endothelial cell integrity [16, 17] and up-regulation of adhesion molecules such as intracellular adhesion molecule 1 (ICAM-1) and P-selectin [18-20] that facilitate leukocyte-endothelial cell interactions. Macrophages expressing CX3CR1 and CCR2 are recruited by endothelial cells leading to the production of chemokines mediating their recruitment into the injured kidney [21, 22]. Therefore, the endothelium, which serves as the interface between immune cells and the renal parenchyma is a highly reactive tissue involved in the early phase of inflammation and kidney damage by promoting the accumulation of leukocytes.
Epithelial Cells
Epithelial cells contribute to inflammation and early kidney injury following IR. The basolateral membrane of proximal tubule cells express the complement inhibitor, Crry [23]. After renal IR, Crry is internalized allowing the deposition of complement component 3 (C3) on the tubular epithelium [23], complement activation and production of the pro-inflammatory chemokines macrophage inflammatory factor-2 (MIP-2) and keratinocyte-derived chemokine (KC) [24]. These chemokines attract neutrophils and macrophages to the post-ischemic kidney. In addition, toll-like receptors (TLR) 2 and 4 are up-regulated in epithelial cells after IR and deficiency of either TLR on kidney parenchymal cells was more effective at preventing kidney IRI than TLR deficiency on bone marrow derived cells [25, 26]. TLR2 or TLR4 deficiency blunted the IR-induced production of proinflammatory cytokines and chemokines and inhibited macrophage and neutrophil accumulation. These studies highlight the important role for renal epithelial cells in the inflammation of AKI.
Resident Kidney Dendritic Cells (DCs)
Immature DCs are present in virtually all tissues and participate in immune surveillance. DCs are an important link between innate and adaptive immunity and their role in AKI has only recently been investigated. CD11c+ and class II major histocompatibilty complex (MHC Class II)+ DCs are the most abundant leukocyte subset in the normal mouse kidney [22, 27] suggesting an important role in renal immunity and inflammation. In the normal C57BL/6 mouse kidney analyzed by 4 color FACS, all of the kidney F4/80high cells are CD11c+ and these cells also express CD86, MHC class IIhigh and CX3CR1, indicating that these cells have a DC phenotype [22]. Mature DCs are specialized for T cell activation. However, DCs can also initiate the innate immune response presenting endogenous glycolipids and stimulating NKT cells [28]. After IR, renal DCs produce the pro-inflammatory cytokines/chemokines TNF-α, IL-6, MCP-1 and RANTES, and depletion of DCs prior to IRI significantly reduced the kidney levels of TNF-α, [29]. IL-12 and IL-23 are mainly produced from activated DCs and macrophages, and their downstream cytokines, IFN-γ and IL-17, amplify the immune response following kidney reperfusion through macrophage activation and neutrophil recruitment (personal communication L Li and MD Okusa 2009).
Non-Resident Bone Marrow-Derived Cells
Non-resident bone marrow-derived cells, such as neutrophils [11, 13-15, 30, 31] macrophages [32, 33], natural killer cells [34], T cells [14, 35, 36] and natural killer T cells [28] rapidly infiltrate the kidney during reperfusion, in response to the signals from resident renal cells and in a distinct temporal profile [22]. A brief summary of the role of these leukocytes in the pathogenesis of renal IRI follows.
Neutrophils
Neutrophils rapidly respond to injury and are important mediators of innate immunity. Neutrophils release granules containing proteases and other enzymes, which generate reactive oxygen species. In inflammatory states, neutrophil degranulation can lead to the destruction of normal self cells in the inflamed tissue. One of the hallmarks of renal IRI is neutrophil accumulation in the post-ischemic kidney [18, 26, 28, 37] and depletion of neutrophils prevents AKI [18].
Macrophages
Macrophages are derived from monocytes [38-40] in the blood and have heterogeneous functions [13, 40-42]. Macrophages also produce pro-inflammatory cytokines that can stimulate the activity of other leukocytes. Macrophages infiltrate the injured kidney early within 1 hour of reperfusion, and this infiltration is mediated by CCR2 [22] and CX3CR1 signaling pathways [21, 22]. These macrophages have a distinct F4/80lowLy6ChighGR1+CX3CR1low “inflamed” phenotype [22] and through depletion and transfer studies macrophages have been documented to contribute to kidney IRI [32]. Analysis of kidney infiltrating macrophages by flow cytometry demonstrated that these leukocytes are significant producers of the cytokines IL-1α, IL-6, IL-12p40/70 and TNF-α [22].
Natural Killer (NK) Cells
NK cells have recently been reported to infiltrate the post-ischemic kidney by 4 hours of reperfusion [34]. IR induced the expression of an NK cell-activating ligand (Rae-1) on TECs and in vitro studies demonstrated that the interaction of the NKG2D receptor on NK cells with Rae-1 on TECs causes perforin-dependent lysis of cultured kidney cells. Antibody-mediated depletion of NK cells inhibited IRI in wild-type (WT) mice and adoptive transfer of WT, but not perforin KO, NK cells into a T, B and NK cell-deficient mouse enhanced IRI.
T Lymphocytes
A role for T cells in the pathogenesis of kidney IRI has been established in different mouse models lacking certain types of lymphocytes [35, 36]. In nu/nu mice (which lack CD4+ and CD8+ T cells), IRI measured by serum creatinine levels and renal histology, was significantly reduced compared to WT controls [36]. Reconstitution of nu/nu mice with CD4+ T cells alone, but not CD8+ T cells alone, restored kidney injury after IR. Additionally, Rag-1 KO mice (lacking both B and T cells) suffer less severe injury from IR and adoptive transfer of CD4+ T cells from WT mice reconstitutes injury [35]. Importantly, transfer of CD4+ T cells from IFN-γ KO mice failed to re-establish injury in this model [35]. These results suggest that CD4+ T cells, and specifically IFN-γ produced by these cells, mediate the early phase of IRI.
Invariant Natural Killer T (iNKT) Cells
Conventional CD4+ T cells are thought to play a role in antigen-specific, adaptive immunity that requires several days, a time course that cannot explain the rapid, innate immune response following IRI. NKT cells are a unique subset of T lymphocytes with surface receptors and functional properties shared with conventional T cells and NK cells. Invariant NKT cells posses a conserved invariant T cell receptor (TCR) together with the NK cell marker NK1.1. In contrast to conventional T cells, iNKT cells are activated by endogenously released glycolipid antigens. The most remarkable property of iNKT cells is their ability to rapidly produce large quantities of cytokines, including Th1-type (IFN-γ, TNF-α) and Th2-type (IL-4, IL-13), at the same time, within 1-2 hours of activation. A recent finding from our laboratory is that the number of IFN-γ producing iNKT cells in the kidney is significantly increased by 3 hours of reperfusion compared to sham-operated mice [28]. Blockade of NKT cell activation with the anti-CD1d mAb, NKT cell depletion with an anti-NK1.1 mAb in WT mice, or use of iNKT cell deficient mice (Jα18-/-) inhibited the accumulation of IFN-γ producing neutrophils after IRI and prevented AKI [28]. These results demonstrate the important contribution of iNKT cells in kidney IRI.
Late Phase Inflammation and Repair in Ischemia-Reperfusion Injury
Compared to the early/innate response to kidney IR, less is known about the adaptive immune response. The late or adaptive immune response to specific antigens (from pathogens or dead self cells) occurs over the course of several days and includes DC maturation and antigen presentation and CD4+ and CD8+ T lymphocyte proliferation and activation.
Antigen Presenting Cells
Leukocytes such as DCs and macrophages play a key role in adaptive immunity by producing pro-inflammatory cytokines and in antigen presentation, although this latter process has received little attention until recently [43]. As discussed above, DCs are the most abundant leukocyte in the kidney. Upon stimulation, DCs can convert to a mature cell type characterized by high levels of MHC class II and costimulatory molecules and low phagocytic capacity. After kidney IR, DCs undergo maturational processing, migrate to the renal draining lymph nodes (LNs) and induce T cell proliferation in an antigen-specific fashion [44], implicating renal DCs in the adaptive immune response to IRI.
T Cells
T lymphocytes are major mediators of adaptive immunity. Antigen presentation by antigen presenting cells, in the presence of sufficient co-stimulation, causes expansion and activation of T cells with a T cell receptor (TCR) specific for the presented antigen. Kidney-derived DCs traffic to draining lymph nodes and activate T cells, a process that might result in local delayed kidney injury [44, 45]. Recent studies demonstrate that T cells with diverse T cell repertoire induced greater IRI than T cells with restricted TCR repertoire providing evidence for activation of T cell receptors (TCR), through an antigen-dependent mechanism, in the pathogenesis of kidney IRI [43]. TCR α/β- and TCR γ/δ-deficient mice were mildly protected from IRI [46], an effect not observed by others [47]. A recent study by Ascon et al. demonstrated the accumulation of activated (CD4+CD69+ and CD8+CD69+) and effector-memory (CD4+CD44hiCD62L- and CD8+CD44hiCD62L-) T cells in the kidney 2 and 6 weeks after ischemia [48]. However, global depletion of T lymphocytes, starting 3 days after ischemia, had no protective effect on cortex or medulla injury scores, measured 6 weeks after IRI [48]. So, at this time, a long-term pathogenic role of TCR activation has not been demonstrated conclusively.
Stem Cells
Following AKI, tissue repair may result from the proliferation of adjacent surviving dedifferentiated epithelial cells, mobilization of kidney specific stem cells which migrate to the site of regeneration, or from bone marrow derived mesenchymal or hematopoietic stem cells that gain access to the injured epithelium and differentiate into mature cells Fig. (1) [49]. There is evidence that kidneys possess adult stem cells [50-52] that have pluripotent potential. After tissue injury, intrinsic tissue stem cells may replace damaged tissue. Other studies have demonstrated that BM mesenchymal stem cells reduce inflammation and protect against IRI [53]. The effects were likely not due to differentiation of mesenchymal stem cells into tubular or endothelial cells, but to a paracrine effect that reduced expression of proinflammatory cytokines (e.g. IL-1β, TNF-α and IFN-γ) and inducible nitric oxide synthase and increased anti-inflammatory IL-10, basic-FGF, TGF-α and Bcl-2 in affected kidneys. BM mesenchymal stem cells limit activation of natural killer cells [54], which could attenuate IRI during the early phase. Other studies, in which antigen presentation or maturation of dendritic cells were blocked by BM mesenchymal stem cells, show that these cells might also be effective at inhibiting the late phase activation of the adaptive immune response [55]. In an elegant study, Humphreys and Bonventre generated transgenic mice in which tubular epithelial cells were labeled with beta-galactosidase or red fluorescent protein to determine the source of reparative cells [56]. Two days following IRI no “dilution” of label was observed indicating that surviving tubular epithelial cells were the predominant mechanism of repair. Thus although there is controversy as to mechanism of tissue repair, there is much excitement that the use of the appropriate progenitor cells may eventually lead to new therapies in limiting injury and repairing acutely injured kidneys.
Macrophages
Given their diverse phenotype [13, 38-42], macrophages appear to participate in early stages of injury, and in the late stage repair process, following IR. Upon activation, alternative macrophages (M2) produce anti-inflammatory cytokines including IL-10 and TGF-β and also increase matrix production [57], suggesting the potential for this population of macrophages to participate in tissue repair [58]. A causal link between macrophages and repair processes was reported by Duffield et al. [59]. A conditional ablation system was used that takes advantage of transgenic mice expressing human diptheria toxin (DT) receptor in CD11b+ cells (i.e. macrophages), which confers sensitivity to DT and permits macrophage depletion in vivo when DT is injected. DT treatment leads to failure in the liver repair process following liver injury [59]. Subsequently, Jang et al. [60] found impaired recovery from kidney IRI when macrophages were depleted following kidney IRI using liposomal clodronate. The anti-inflammatory phenotype of alternatively activated macrophages discussed above provides evidence for a role of macrophages in tissue repair processes.
Regulatory T Cells
Regulatory T (Treg) cells are lymphocytes with immunosuppressive properties. One important subset of Treg cells express CD4 and CD25 on the cell surface and the transcription factor, FoxP3 [61]. The mechanisms of suppression by Treg cells are diverse and include: production of antiinflammatory cytokines such as IL-10 or TGF-β, direct cell-cell contact or CTLA-4 mediated inhibition and production of extracellular adenosine [62]. Recently, Treg cells have been identified in normal mouse kidneys [63, 64]. In WT mice, treatment with an anti-CD25 monoclonal antibody (PC61) selectively decreased kidney, spleen and blood CD4+ FoxP3+ Treg cell numbers by approximately 50%, five days after PC61 treatment [64]. At that time point, Treg cell deficiency potentiated kidney IRI, measured by plasma creatinine, acute tubular necrosis (ATN), neutrophil and macrophage accumulation and pro-inflammatory cytokine transcription in the kidney after 24 hr of reperfusion [64]. In lymphocyte-deficient Rag-1 KO mice, adoptive transfer of WT, but not IL-10 KO, Treg cells blocked IR-induced inflammation and kidney injury [64]. These findings demonstrate that Treg cells can directly suppress the early innate inflammation, induced by IR, in an IL-10 dependent manner. In a different study, PC61 was administered 1 day prior to IRI, and while BUN levels and ATN scores were no different than control antibody-treated mice at 24 hr of reperfusion, the necrosis failed to resolve by 72 hr in the PC61-treated mice [65]. These results strongly support an important role of regulatory T cells during IRI and in kidney repair after IRI.
In summary, the early/innate immune response to kidney IR is well-characterized and robust, involving resident kidney cells and circulating pro-inflammatory leukocytes. While an adaptive immune response occurs, as evidenced by DC migration to kidney draining LNs and antigen presentation followed by kidney accumulation of activated and effector-memory T cells, direct evidence for antigen-dependent long-term kidney damage after IRI is currently lacking. Finally, BM mesenchymal stem cells, alternatively activated macrophages and Treg cells show promise for protecting the kidney during injury and/or facilitating kidney repair after IRI.
Drugs That Block Inflammation and Reduce Cytotoxicity in Acute Kidney Injury
Pharmacological therapy in the prevention and treatment of AKI has been largely unsuccessful despite proven benefits seen in preclinical studies. Prevention and treatment of AKI is indeed an important clinical issue as mortality in patients with AKI especially in critically ill patients remain alarmingly high despite substantial advances in techniques of resuscitation and renal replacement therapy. A number of drugs and investigational compounds appear promising in preclinical studies and promising investigational compounds are in use in clinical trials for a variety of indications including AKI. The success of these compound will necessitate an understanding of the pathogenesis of AKI so that appropriate rational therapies and combination therapies can be implemented in well designed clinical trials [66].
Spingosine 1 Phosphate (S1P) Analogs
S1P is a specific ligand for a family of G protein coupled endothelial differentiation gene (Edg) receptors (also referred to as S1PRs1-5) that evoke diverse cellular signaling responses. S1PRs regulate different biological processes depending on their pattern of expression. S1P binds to receptors or acts as a second messenger to stimulate cell survival, inhibit cellular apoptosis, and is involved in cell adhesion and movement [67]. An S1P analog, FTY720, acts as an agonist at four S1PRs, with exception of S1P2R. FTY720 binding to S1P1Rs lead to sequestration of lymphocytes in secondary lymphatic tissue [68]. In studies of kidney IRI, FTY720 or similar compounds induce lymphopenia and protect renal tissue from IRI [69, 70]. With discovery of new more potent and selective S1PR analogs, like SEW2871, these selective agents will soon be available for preclinical and clinical studies [71]. A comprehensive list of S1PR analogs can be found in a review by Jo SK et al. [72]. Recently, in a phase II study, FTY720 reduced the number of lesions detected on magnetic resonance imaging and clinical disease activity in patients with multiple sclerosis [73].
A2A Agonists and Other Adenosine Analogs
Locally produced adenosine in the kidney controls renal circulation and metabolic cellular activity [74]. Adenosine binds to receptor subtypes, which are members of the G-protein coupled receptor family that includes four subtypes: A1-, A2A-, A2B- and A3Rs [75]. Adenosine acts on these receptors in organs such as brain, heart and skeletal muscle and induces vasodilation to allow matching of oxygen delivery and work [74]. Based upon these findings, theophylline, an adenosine A1R antagonist, has been used successfully in several randomized controlled studies to prevent radiocontrast-induced AKI (as reviewed in [76-78]). Accumulating data demonstrates that selective activation of A2ARs reduces parenchymal injury in non-renal tissues including heart, liver, spinal cord, lung, and brain [79-81]. The selective A2AR-agonist, ATL146e, is highly protective against kidney IRI by 70-80% [13, 82, 83]. Following administration either before or immediately at the onset of reperfusion, ATL146e alone or in combination with a phosphodiesterase inhibitor reduced renal injury [84]. ATL146e is in human clinical studies for cardiac imaging and current efforts are directed toward human clinical studies in AKI. Additional studies demonstrate that strategies using A1 agonists or A3 blockers maybe effective in AKI [85, 86].
Statins
3-Hydroxy-3-methylglutaryl coenzyme (HMG-CoA) reductase inhibitors (statins) have been clinically approved for the reduction of cholesterol. Additional studies demonstrate that statins have anti-inflammatory and anti-oxidant activities that improve endothelial function, decrease platelet aggregation and procoagulant factors [87-89]. Pravastatin decreases the rise in plasma creatinine when compared to vehicle treatment without a change in plasma cholesterol levels [90]. Co-administraton of mevalonate, a product of HMG-CoA reductase, in renal IRI reversed the protective effect; demonstrating that the tissue protective effect of pravastatin was through inhibition of the mevalonate pathway. Similarly, when simvastatin was administered prior to cecal ligation puncture (CLP) induced sepsis, the rise in plasma creatinine, TNF-α and vascular permeability was attenuated [91]. In human studies, retrospective case controlled studies and registry studies found that statins reduced radiocontrast-induced AKI [92, 93] whereas a recent prospective study did not find a benefit [94]. Several concerns regarding the latter studies preclude any firm conclusions based upon this study. Thus at this time statins continue to have potential for the prevention of radiocontrast nephropathy and perhaps other forms of AKI.
Fibrates
Peroxisome proliferator-activated receptors (PPARs) are transcription factors that regulate glucose and lipid metabolism. PPAR-α is expressed in the renal proximal tubule which upon activation, hetero-dimerizes with the retinoic X receptor (RXR) and binds to PPAR response elements (PPREs) to regulate gene transcription involved in lipid metabolism [95-97]. IRI and cisplatin-induced AKI reduces kidney PPAR-α activity and inhibits peroxisomal and mitochondrial fatty acid oxidation [98-100] leading to the accumulation and cellular toxicity of oxidized long-chain fatty acids and long-chain acylcarnitines [101]. Fibrates have been shown to reduce cisplatin induced AKI [97, 102]. Liver fatty acid binding protein (L-FABP) belongs to a super family of lipid-binding proteins with low molecular weight (14-15 kDa) whose transcription rate is regulated by fibrates through a PPRE located in its promoter region [103, 104]. Most recently, fibrates have been shown to increase L-FAPB and decrease cisplatin-induced AKI [105]. When proximal tubule epithelial cells were exposed to cisplatin, the increase apoptosis was suppressed with bezafibrate [102]. The effect of bezafibrate to reduce apoptosis was associated with attenuation of cisplatin-induced translocation of proapoptotic Bax from the cytosol to the mitochondria, and increase in the expression of anti-apoptotic molecule Bcl-2 [102]. Furthermore, recent studies have indicated PPARs play an important role in inflammation and immunity [106]. Pretreatment of animals with WY-14, 643 (WY), a fibrate class of PPAR-α ligand ameliorated cisplatin-induced renal dysfunction and this was accompanied by suppression of NF-kB activation, cytokine/chemokine expression and neutrophil infiltration, suggesting that the protective effect of fibrates is mediated through its anti-inflammatory effect [107]. The current use of fibrates in lipid management may facilitate clinical trials in AKI.
Inducible Nitic Oxide Synthase (iNOS) Inhibitors
The role of nitric oxide and nitric oxide synthases has been extensively studied. Both in vivo and in vitro studies point toward the important role of iiNOS in mediating injury to proximal tubules [108]. Selective iNOS inhibitors are currently used in human investigation for a variety of indications.
Antioxidants and Antioxidant Enzymes
Excessive reactive oxygen species (ROS) generation, decreases in antioxidant defense, or both, are known to contribute to IRI and antioxidants that effectively remove ROS have been found to be effective against IRI. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a potent free radical scavenger, improved survival and renal function in rats subjected to renal IRI [109]. Recently, stobadine, a novel synthetic pyridoindole antioxidant, which diminishes lipid peroxidation and protein impairment by free radical scavenging and anti-oxidant activity, has been shown to provide significant protection from IRI in rat kidneys [110].
Thrombomodulin and Activated Protein C (APC)
Proteolytic activation of protein C occurs on the endothelial cell by two membrane receptors, thrombomodulin and endothelial protein C receptors (EPCRs). Binding of thrombin to thrombomodulin on the endothelial surface promotes its anticoagulant properties by APC by the thrombin-thrombomodulin complex and is enhanced by binding of protein C to EPCR [111]. In addition, soluble thrombomodulin, independent of its ability to generate APC, reduced ischemia-reperfusion injury [112]. In this study an aortic clamp model was used; soluble thrombomodulin (sTM) not only attenuated the rise in creatinine following reperfusion, but it also improved microvascular erythrocyte flow, reduced microvascular endothelial leukocyte adhesion, and minimized endothelial permeability. A mutant, F376L, in which a point mutation was made in sTM, reduced ischemia-reperfusion injury suggesting that the protective effect of sTM is independent of its ability to generate APC.
APC, in addition to its effect on coagulation, has been shown to have direct cellular effects via EPCRs including: anti-inflammatory and anti-apoptotic activities leukocyte activation and stability of barrier function [111, 113-118]. Through genetic engineering of wild type APC, mutants have been created that have the cytoprotective and anticoagulant activity of APC [119]. Following endotoxemia, these molecules with preserved cytoprotective properties are effective in preserving renal blood flow and attenuating acute kidney injury [119] and reducing mortality [120]. On the other hand an APC mutant with potent antithrombotic activity but minimal cytoprotection was less effective in reducing endotoxin-induced murine mortality [121]. Thus it is the hope that genetically- engineered APC mutants and thrombomodulin might yield specific agents that take advantage of selective anticoagulant and cytoprotective properties in future clinical studies of AKI from sepsis or in critically ill patients.
Erythropoietin (EPO) and EPO Derivatives
The haematopoietic factor erythropoietin (EPO) has recently been recognized to play a physiological role in the brain and other tissues. The EPO receptor is present in the glomerulus and TECs in the kidney [122]. EPO attenuates the dysfunction and histological changes associated with IRI [123] and in animal models of systemic shock and cisplatin-induced nephrotoxicity [124]. Non-hematopoietic erythropoietin analogs have shown similar benefit in experimental contrast-induced nephropathy [125]. In a pilot clinical trial, the efficacy of EPO to prevent AKI after coronary artery bypass grafting (CABG) was examined [126]. Seventy one patients scheduled for elective CABG randomly received either 300 U/kg of EPO or saline intravenously before surgery. EPO administration resulted in an incidence of AKI of 8% compared with an incidence of 29% in the placebo group (p = 0.035). These data are preliminary and provide promise for this mode of treatment for AKI.
In summary the complexity of AKI is due in part to activation of multiple overlapping and distinct temporal pathways. Inflammation (cellular and humoral) is a key mediator of AKI. Recent studies have highlighted novel immune mechanisms contributing to AKI that provide the foundation for newer classes of pharmacological agents that block inflammation. It is likely that new strategies to treat or prevent AKI will require drugs that target multiple pathways, or combination of drugs that targets several areas, rather than one.
Fig. (1).
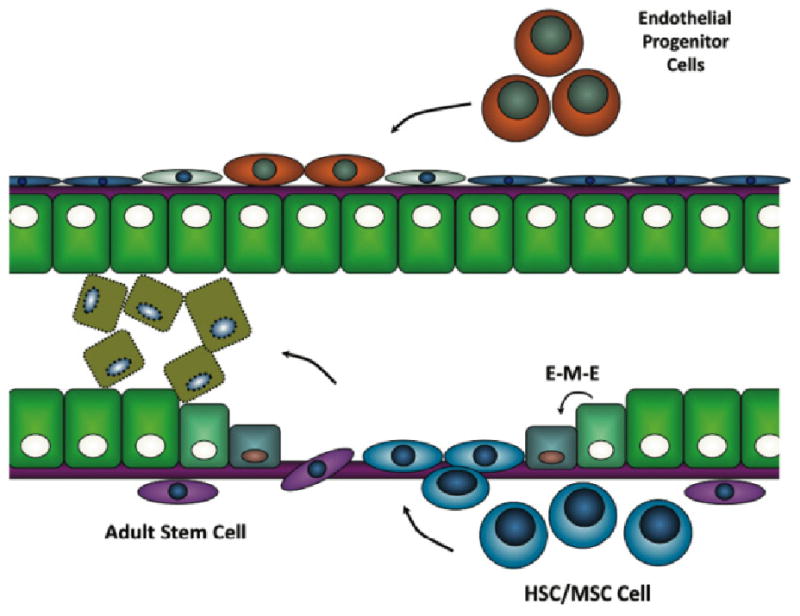
Progenitor cells in tissue repair following AKI. A) Adjacent surviving epithelial cells dedifferentiate undergo epithelial-mesenchymal-epithelial (EME) transition into new epithelial cells, B) kidney specific adult stem cells may migrate to the site of injury or C) bone marrow derived mesenchymal or hematopoietic stem cells gain access to the injured epithelium and differentiate into mature epithelial cells.
Acknowledgments
We would like to acknowledge members of the Okusa laboratory whose data formed the basis for much of this review. This work was supported by grants from the National Institutes of Health RO1 DK56223, RO1 DK62324, RO1 DK065957, T32 DK072922, F32 DK083185, Genzyme (Genzyme Renal Innovations Program) and the National Kidney Foundation Research Fellowship Award.
References
- 1.Xue JL, Daniels F, Star RA, Kimmel PL, Eggers PW, Molitoris BA, et al. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol. 2006;17:1135–42. doi: 10.1681/ASN.2005060668. [DOI] [PubMed] [Google Scholar]
- 2.Waikar SS, Curhan GC, Wald R, McCarthy EP, Chertow GM. Declining mortality in patients with acute renal failure, 1988 to 2002. J Am Soc Nephrol. 2006;17:1143–50. doi: 10.1681/ASN.2005091017. [DOI] [PubMed] [Google Scholar]
- 3.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–8. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 4.Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8:R204–12. doi: 10.1186/cc2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liano F, Pascual J. Outcomes in acute renal failure. Semin Nephrol. 1998;18:541–50. [PubMed] [Google Scholar]
- 6.Hoste EA, Schurgers M. Epidemiology of acute kidney injury: how big is the problem? Crit Care Med. 2008;36:S146–51. doi: 10.1097/CCM.0b013e318168c590. [DOI] [PubMed] [Google Scholar]
- 7.Chertow GM, Levy EM, Hammermeister KE, Grover F, Daley J. Independent association between acute renal failure and mortality following cardiac surgery. Am J Med. 1998;104:343–8. doi: 10.1016/s0002-9343(98)00058-8. [DOI] [PubMed] [Google Scholar]
- 8.Levy EM, Viscoli CM, Horwitz RI. The effect of acute renal failure on mortality. A cohort analysis. JAMA. 1996;275:1489–94. [PubMed] [Google Scholar]
- 9.Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;114:5–14. doi: 10.1172/JCI22353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–60. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 11.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–210. doi: 10.1097/01.asn.0000079785.13922.f6. [DOI] [PubMed] [Google Scholar]
- 12.Rabb H. The T cell as a bridge between innate and adaptive immune systems: implications for the kidney. Kidney Int. 2002;61:1935–46. doi: 10.1046/j.1523-1755.2002.00378.x. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Okusa MD. Blocking the Immune respone in ischemic acute kidney injury: the role of adenosine 2A agonists. Nat Clin Pract Nephrology. 2006;2:432–44. doi: 10.1038/ncpneph0238. [DOI] [PubMed] [Google Scholar]
- 14.Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 2004;66:486–91. doi: 10.1111/j.1523-1755.2004.761_3.x. [DOI] [PubMed] [Google Scholar]
- 15.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. 2006;17:1503–20. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- 16.Sutton TA, Mang HE, Campos SB, Sandoval RM, Yoder MC, Molitoris BA. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Renal Physiol. 2003;285:F191–8. doi: 10.1152/ajprenal.00042.2003. [DOI] [PubMed] [Google Scholar]
- 17.Brodsky SV, Yamamoto T, Tada T, Kim B, Chen J, Kajiya F, et al. Endothelial dysfunction in ischemic acute renal failure: rescue by transplanted endothelial cells. Am J Physiol Renal Physiol. 2002 Jun;282:F1140–9. doi: 10.1152/ajprenal.00329.2001. [DOI] [PubMed] [Google Scholar]
- 18.Kelly KJ, Williams WW, Colvin RB, Meehan SM, Springer TA, Gutierrez-Ramos J, et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97:1056–63. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singbartl K, Green SA, Ley K. Blocking P-selectin protects from ischemia/reperfusion-induced acute renal failure. FASEB J. 2000;14:48–54. doi: 10.1096/fasebj.14.1.48. [DOI] [PubMed] [Google Scholar]
- 20.Okusa MD, Linden J, Huang L, Rieger JM, Macdonald TL, Huynh LP. A2a-Adenosine receptor mediated inhibition of renal injury and neutrophil adhesion. Am J Physiol. 2000;279:F809–18. doi: 10.1152/ajprenal.2000.279.5.F809. [DOI] [PubMed] [Google Scholar]
- 21.Oh DJ, Dursun B, He Z, Lu L, Hoke TS, Ljubanovic D, et al. Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am J Physiol Renal Physiol. 2008;294:F264–71. doi: 10.1152/ajprenal.00204.2007. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Huang L, Sung SS, Vergis AL, Rosin DL, Rose CE, Jr, et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008;74:1526–37. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thurman JM, Ljubanovic D, Royer PA, Kraus DM, Molina H, Barry NP, et al. Altered renal tubular expression of the complement inhibitor Crry permits complement activation after ischemia/reperfusion. J Clin Invest. 2006;116:357–68. doi: 10.1172/JCI24521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thurman JM, Lenderink AM, Royer PA, Coleman KE, Zhou J, Lambris JD, et al. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ishemia/reperfusion. J Immunol. 2007;178:1819–28. doi: 10.4049/jimmunol.178.3.1819. [DOI] [PubMed] [Google Scholar]
- 25.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115:2894–903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–59. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soos TJ, Sims TN, Barisoni L, Lin K, Littman DR, Dustin ML, et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006;70:591–6. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Huang L, Sung SJ, Lobo PI, Brown MG, Gregg RK, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;178:5899–911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 29.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007;71:619–28. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- 30.Okusa MD. The inflammatory cascade in acute ischemic renal failure. Nephron. 2002;90:133–8. doi: 10.1159/000049032. [DOI] [PubMed] [Google Scholar]
- 31.Koo DD, Welsh KI, Roake JA, Morris PJ, Fuggle SV. Ischemia/reperfusion injury in human kidney transplantation: an immunohistochemical analysis of changes after reperfusion. Am J Pathol. 1998;153:557–66. doi: 10.1016/S0002-9440(10)65598-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005;288:F722–31. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- 33.Jo SK, Sung SA, Cho WY, Go KJ, Kim HK. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol Dial Transplant. 2006;21:1231–9. doi: 10.1093/ndt/gfk047. [DOI] [PubMed] [Google Scholar]
- 34.Zhang ZX, Wang S, Huang X, Min WP, Sun H, Liu W, et al. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J Immunol. 2008;181:7489–98. doi: 10.4049/jimmunol.181.11.7489. [DOI] [PubMed] [Google Scholar]
- 35.Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD. Renal ischemia-reperfusion and adenosine 2A receptor-mediated tissue protection: The role of CD4+ T cells and inteferon gamma. J Immunol. 2006;176:3108–14. doi: 10.4049/jimmunol.176.5.3108. [DOI] [PubMed] [Google Scholar]
- 36.Burne MJ, Daniels F, El Ghandour A, Mauiyyedi S, Colvin RB, O'Donnell MP, et al. Identification of the CD4+ T cell as a major pathogenic factor in ischemic acute renal failure. J Clin Invest. 2001;108:1283–90. doi: 10.1172/JCI12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiao H, Kohda Y, McLeroy P, Craig L, Housini I, Star RA. Alpha-melanocyte-stimulating hormone protects against renal injury after Ischemia in mice and rats. J Clin Invest. 1997;99:1165–72. doi: 10.1172/JCI119272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 39.Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol. 2002;72:101–6. [PubMed] [Google Scholar]
- 40.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 41.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118:3522–30. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferenbach D, Hughes J. Macrophages and dendritic cells: what is the difference? Kidney Int. 2008;74:5–7. doi: 10.1038/ki.2008.189. [DOI] [PubMed] [Google Scholar]
- 43.Satpute SR, Park JM, Jang HR, Agreda P, Liu M, Gandolfo MT, et al. The role for T cell repertoire/antigen-specific interactions in experimental kidney ischemia reperfusion injury. J Immunol. 2009;183:984–92. doi: 10.4049/jimmunol.0801928. [DOI] [PubMed] [Google Scholar]
- 44.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. 2005;68:1096–108. doi: 10.1111/j.1523-1755.2005.00502.x. [DOI] [PubMed] [Google Scholar]
- 45.Jang MH, Sougawa N, Tanaka T, Hirata T, Hiroi T, Tohya K, et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J Immunol. 2006;176:803–10. doi: 10.4049/jimmunol.176.2.803. [DOI] [PubMed] [Google Scholar]
- 46.Savransky V, Molls RR, Burne-Taney MJ, Chien CC, Racusen L, Rabb H. Role of T cell receptor in kidney ischemia reperfusion injury. Kidney Int. 2006;69:233–8. doi: 10.1038/sj.ki.5000038. [DOI] [PubMed] [Google Scholar]
- 47.Faubel S, Ljubanovic D, Poole B, Dursun B, He Z, Cushing S, et al. Peripheral CD4 T-cell depletion is not sufficient to prevent ischemic acute renal failure. Transplantation. 2005;80:643–9. doi: 10.1097/01.tp.0000173396.07368.55. [DOI] [PubMed] [Google Scholar]
- 48.Ascon M, Ascon DB, Liu M, Cheadle C, Sarkar C, Racusen L, et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009;75:526–35. doi: 10.1038/ki.2008.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krause D, Cantley LG. Bone marrow plasticity revisited: protection or differentiation in the kidney tubule? J Clin Invest. 2005;115:1705–8. doi: 10.1172/JCI25540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupta S, Verfaillie C, Chmielewski D, Kren S, Eidman K, Connaire J, et al. Isolation and characterization of kidney-derived stem cells. J Am Soc Nephrol. 2006;17:3028–40. doi: 10.1681/ASN.2006030275. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, Park HC, Addabbo F, Ni J, Pelger E, Li H, et al. Kidney-derived mesenchymal stem cells contribute to vasculogenesis, angiogenesis and endothelial repair. Kidney Int. 2008;74:879–89. doi: 10.1038/ki.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin F. Stem cells in kidney regeneration following acute renal injury. Pediatr Res. 2006;59:74R–8R. doi: 10.1203/01.pdr.0000205156.85990.12. [DOI] [PubMed] [Google Scholar]
- 53.Togel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol. 2005;289:F31–42. doi: 10.1152/ajprenal.00007.2005. [DOI] [PubMed] [Google Scholar]
- 54.Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells. 2006;24(1):74–85. doi: 10.1634/stemcells.2004-0359. [DOI] [PubMed] [Google Scholar]
- 55.Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD, et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105:4120–6. doi: 10.1182/blood-2004-02-0586. [DOI] [PubMed] [Google Scholar]
- 56.Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2:284–91. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 57.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 58.Kerjaschki D. The crucial role of macrophages in lymphangiogenesis. J Clin Invest. 2005;115:2316–9. doi: 10.1172/JCI26354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jang HS, Kim J, Park YK, Park KM. Infiltrated macrophages contribute to recovery after ischemic injury but not to ischemic preconditioning in kidneys. Transplantation. 2008;85:447–55. doi: 10.1097/TP.0b013e318160f0d1. [DOI] [PubMed] [Google Scholar]
- 61.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 62.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–45. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 63.Ascon DB, Ascon M, Satpute S, Lopez-Briones S, Racusen L, Colvin RB, et al. Normal mouse kidneys contain activated and CD3+CD4- CD8- double-negative T lymphocytes with a distinct TCR repertoire. J Leukoc Biol. 2008;84:1400–9. doi: 10.1189/jlb.0907651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, et al. Regulatory T Cells Suppress Innate Immunity in Kidney Ischemia-Reperfusion Injury. J Am Soc Nephrol. 2009;20:1744–53. doi: 10.1681/ASN.2008111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Monteiro RM, Camara NO, Rodrigues MM, Tzelepis F, Damiao MJ, Cenedeze MA, et al. A role for regulatory T cells in renal acute kidney injury. Transpl Immunol. 2009;21:50–5. doi: 10.1016/j.trim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 66.Jo SK, Rosner MH, Okusa MD. Pharmacologic Treatment of Acute Kidney Injury. Why Drugs Haven't: Why Drugs Haven't Worked and What is on the Horizon. Clin J Am Soc Nephrol. 2007;2:256–365. doi: 10.2215/CJN.03280906. [DOI] [PubMed] [Google Scholar]
- 67.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 68.Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–7. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 69.Awad AS, Ye H, Huang L, Li L, Foss FW, Jr, Macdonald TL, et al. Selective Sphingosine 1-Phosphate 1 (S1P1) Receptor Activation Reduces Ischemia-Reperfusion Injury in Mouse Kidney. Am J Physiol Renal Physiol. 2006;290:F1516–24. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 70.Lien YH, Yong KC, Cho C, Igarashi S, Lai LW. S1P(1)-selective agonist, SEW2871, ameliorates ischemic acute renal failure. Kidney Int. 2006;69:1601–8. doi: 10.1038/sj.ki.5000360. [DOI] [PubMed] [Google Scholar]
- 71.Foss FW, Jr, Clemens JJ, Davis MD, Snyder AH, Zigler MA, Lynch KR, et al. Synthesis, stability, and implications of phosphothioate agonists of sphingosine-1-phosphate receptors. Bioorg Med Chem Lett. 2005;15:4470–4. doi: 10.1016/j.bmcl.2005.07.057. [DOI] [PubMed] [Google Scholar]
- 72.Jo SK, Bajwa A, Awad AS, Lynch KR, Okusa MD. Sphingosine-1-phosphate receptors: biology and therapeutic potential in kidney disease. Kidney Int. 2008;73:1220–30. doi: 10.1038/ki.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kappos L, Antel J, Comi G, Montalban X, O'Connor P, Polman CH, et al. Radue EW Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–40. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 74.Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev. 2006;86:901–40. doi: 10.1152/physrev.00031.2005. [DOI] [PubMed] [Google Scholar]
- 75.Linden J. Molecular approach to adenosine receptors: receptor mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–87. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 76.Bagshaw SM, Ghali WA. Theophylline for prevention of contrast-induced nephropathy: a systematic review and meta-analysis. Arch Intern Med. 2005;165:1087–93. doi: 10.1001/archinte.165.10.1087. [DOI] [PubMed] [Google Scholar]
- 77.Kapoor A, Kumar S, Gulati S, Gambhir S, Sethi RS, Sinha N. The role of theophylline in contrast-induced nephropathy: a case-control study. Nephrol Dial Transplant. 2002;17:1936–41. doi: 10.1093/ndt/17.11.1936. [DOI] [PubMed] [Google Scholar]
- 78.Ix JH, McCulloch CE, Chertow GM. Theophylline for the prevention of radiocontrast nephropathy: a meta-analysis. Nephrol Dial Transplant. 2004;19:2747–53. doi: 10.1093/ndt/gfh468. [DOI] [PubMed] [Google Scholar]
- 79.Jordan JE, Zhao Z, Sato H, Taft S, Vinten-Johansen J. Adenosine A2 receptor activation attenuates reperfusion injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J Pharmacol Exp Ther. 1997;280:301–9. [PubMed] [Google Scholar]
- 80.Lasley RDJM, Mentzer RM., Jr Beneficial effects of adenosine (2a) agonist CGS-21680 in infarcted and stunned porcine myocardium. Am J Physiol Heart Circ Physiol. 2001;280:H1660–6. doi: 10.1152/ajpheart.2001.280.4.H1660. [DOI] [PubMed] [Google Scholar]
- 81.Day YJ, Marshall MA, Huang L, McDuffie MJ, Okusa MD, Linden J. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: inhibition of chemokine induction. Am J Physiol Gastrointest Liver Physiol. 2004;286:G285–93. doi: 10.1152/ajpgi.00348.2003. [DOI] [PubMed] [Google Scholar]
- 82.Day YJ, Huang L, McDuffie MJ, Rosin DL, Ye H, Chen JF, et al. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J Clin Invest. 2003;112:883–91. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okusa MD, Linden J, Macdonald T, Huang L. Selective A2A-adenosine receptor activation during reperfusion reduces ischemia-reperfusion injury in rat kidney. Am J Physiol. 1999;277:F404–F12. doi: 10.1152/ajprenal.1999.277.3.F404. [DOI] [PubMed] [Google Scholar]
- 84.Okusa MD, Linden J, Huang L, Rosin DL, Smith DF, Sullivan G. Enhanced protection from renal ischemia-reperfusion injury with A2A-adenosine receptor activation and PDE 4 inhibition. Kidney Int. 2001;59:2114–25. doi: 10.1046/j.1523-1755.2001.00726.x. [DOI] [PubMed] [Google Scholar]
- 85.Lee HT, Ota-Setlik A, Xu H, D'Agati VD, Jacobson MA, Emala CW. A3 adenosine receptor knockout mice are protected against ischemia- and myoglobinuria-induced renal failure. Am J Physiol Renal Physiol. 2003;284:F267–73. doi: 10.1152/ajprenal.00271.2002. [DOI] [PubMed] [Google Scholar]
- 86.Lee HT, Gallos G, Nasr SH, Emala CW. A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J Am Soc Nephrol. 2004;15:102–11. doi: 10.1097/01.asn.0000102474.68613.ae. [DOI] [PubMed] [Google Scholar]
- 87.Wheeler DC. Are there potential non-lipid-lowering uses of statins? Drugs. 1998;56:517–22. doi: 10.2165/00003495-199856040-00001. [DOI] [PubMed] [Google Scholar]
- 88.Haslinger-Loffler B. Multiple effects of HMG-CoA reductase inhibitors (statins) besides their lipid-lowering function. Kidney Int. 2008;74:553–5. doi: 10.1038/ki.2008.323. [DOI] [PubMed] [Google Scholar]
- 89.Schonbeck U, Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation. 2004;109:1118–26. doi: 10.1161/01.CIR.0000129505.34151.23. [DOI] [PubMed] [Google Scholar]
- 90.Sharyo S, Yokota-Ikeda N, Mori M, Kumagai K, Uchida K, Ito K, et al. Pravastatin improves renal ischemia-reperfusion injury by inhibiting the mevalonate pathway. Kidney Int. 2008;74:577–84. doi: 10.1038/ki.2008.210. [DOI] [PubMed] [Google Scholar]
- 91.Yasuda H, Yuen PS, Hu X, Zhou H, Star RA. Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney Int. 2006;69:1535–42. doi: 10.1038/sj.ki.5000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Attallah N, Yassine L, Musial J, Yee J, Fisher K. The potential role of statins in contrast nephropathy. Clin Nephrol. 2004;62:273–8. doi: 10.5414/cnp62273. [DOI] [PubMed] [Google Scholar]
- 93.Khanal S, Attallah N, Smith DE, Kline-Rogers E, Share D, O'Donnell MJ, et al. Statin therapy reduces contrast-induced nephropathy: an analysis of contemporary percutaneous interventions. Am J Med. 2005;118:843–9. doi: 10.1016/j.amjmed.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 94.Jo SH, Koo BK, Park JS, Kang HJ, Cho YS, Kim YJ, et al. Prevention of radiocontrast medium-induced nephropathy using short-term high-dose simvastatin in patients with renal insufficiency undergoing coronary angiography (PROMISS) trial--a randomized controlled study. Am Heart J. 2008;155:499, e1–8. doi: 10.1016/j.ahj.2007.11.042. [DOI] [PubMed] [Google Scholar]
- 95.Smirnov AN. Nuclear receptors: nomenclature, ligands, mechanisms of their effects on gene expression. Biochemistry (Mosc) 2002;67:957–77. doi: 10.1023/a:1020545200302. [DOI] [PubMed] [Google Scholar]
- 96.Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Evans RM Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 1994;91:7355–9. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li S, Basnakian A, Bhatt R, Megyesi J, Gokden N, Shah SV, et al. PPAR-alpha ligand ameliorates acute renal failure by reducing cisplatin-induced increased expression of renal endonuclease G. Am J Physiol Renal Physiol. 2004;287:F990–8. doi: 10.1152/ajprenal.00206.2004. [DOI] [PubMed] [Google Scholar]
- 98.Li S, Wu P, Yarlagadda P, Vadjunec NM, Proia AD, Harris RA, et al. PPAR alpha ligand protects during cisplatin-induced acute renal failure by preventing inhibition of renal FAO and PDC activity. Am J Physiol Renal Physiol. 2004;286:F572–80. doi: 10.1152/ajprenal.00190.2003. [DOI] [PubMed] [Google Scholar]
- 99.Portilla D. Energy metabolism and cytotoxicity. Semin Nephrol. 2003;23:432–8. doi: 10.1016/s0270-9295(03)00088-3. [DOI] [PubMed] [Google Scholar]
- 100.Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol. 2000;278:F667–75. doi: 10.1152/ajprenal.2000.278.4.F667. [DOI] [PubMed] [Google Scholar]
- 101.Feldkamp T, Kribben A, Roeser NF, Senter RA, Weinberg JM. Accumulation of nonesterified fatty acids causes the sustained energetic deficit in kidney proximal tubules after hypoxia-reoxygenation. Am J Physiol Renal Physiol. 2006;290:F465–77. doi: 10.1152/ajprenal.00305.2005. [DOI] [PubMed] [Google Scholar]
- 102.Nagothu KK, Bhatt R, Kaushal GP, Portilla D. Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int. 2005;68:2680–93. doi: 10.1111/j.1523-1755.2005.00739.x. [DOI] [PubMed] [Google Scholar]
- 103.Kaikaus RM, Chan WK, Ortiz de Montellano PR, Bass NM. Mechanisms of regulation of liver fatty acid-binding protein. Mol Cell Biochem. 1993;123:93–100. doi: 10.1007/BF01076479. [DOI] [PubMed] [Google Scholar]
- 104.Reddy JK. Peroxisome proliferators and peroxisome proliferator-activated receptor alpha: biotic and xenobiotic sensing. Am J Pathol. 2004;164:2305–21. doi: 10.1016/s0002-9440(10)63787-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Negishi K, Noiri E, Maeda R, Portilla D, Sugaya T, Fujita T. Renal L-type fatty acid-binding protein mediates the bezafibrate reduction of cisplatin-induced acute kidney injury. Kidney Int. 2008;73:1374–84. doi: 10.1038/ki.2008.106. [DOI] [PubMed] [Google Scholar]
- 106.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–59. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 107.Li S, Gokden N, Okusa MD, Bhatt R, Portilla D. Antiinflammatory effect of fibrate protects from cisplatin-induced ARF. Am J Physiol Renal Physiol. 2005;289:F469–80. doi: 10.1152/ajprenal.00038.2005. [DOI] [PubMed] [Google Scholar]
- 108.Noiri E, Peresleni T, Miller F, Goligorsky MS. In vivo targeting of inducible NO synthase wtih oligodeoxynucleotides protects rat kidneys against ischemia. J Clin Invest. 1996;97:2377–83. doi: 10.1172/JCI118681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Doi K, Suzuki Y, Nakao A, Fujita T, Noiri E. Radical scavenger edaravone developed for clinical use ameliorates ischemia/reperfusion injury in rat kidney. Kidney Int. 2004;65:1714–23. doi: 10.1111/j.1523-1755.2004.00567.x. [DOI] [PubMed] [Google Scholar]
- 110.Guz G, Demirogullari B, Ulusu NN, Dogu C, Demirtola A, Kavutcu M, et al. Stobadine protects rat kidney against ischaemia/reperfusion injury. Clin Exp Pharmacol Physiol. 2007;34:210–6. doi: 10.1111/j.1440-1681.2007.04574.x. [DOI] [PubMed] [Google Scholar]
- 111.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–72. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 112.Sharfuddin AA, Sandoval RM, Berg DT, McDougal GE, Campos SB, Phillips CL, et al. Soluble thrombomodulin protects ischemic kidneys. J Am Soc Nephrol. 2009;20:524–34. doi: 10.1681/ASN.2008060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grey S, Hau H, Salem HH, Hancock WW. Selective effects of protein C on activation of human monocytes by lipopolysaccharide, interferon-gamma, or PMA: modulation of effects on CD11b and CD14 but not CD25 or CD54 induction. Transplant Proc. 1993;25:2913–4. [PubMed] [Google Scholar]
- 114.Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–42. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 115.Domotor E, Benzakour O, Griffin JH, Yule D, Fukudome K, Zlokovic BV. Activated protein C alters cytosolic calcium flux in human brain endothelium via binding to endothelial protein C receptor and activation of protease activated receptor-1. Blood. 2003;101:4797–801. doi: 10.1182/blood-2002-12-3680. [DOI] [PubMed] [Google Scholar]
- 116.Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. 2003;373:65–70. doi: 10.1042/BJ20030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–2. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 118.Mizutani A, Okajima K, Uchiba M, Noguchi T. Activated protein C reduces ischemia/reperfusion-induced renal injury in rats by inhibiting leukocyte activation. Blood. 2000;95:3781–7. [PubMed] [Google Scholar]
- 119.Gupta A, Gerlitz B, Richardson MA, Bull C, Berg DT, Syed S, et al. Grinnell BW Distinct functions of activated protein C differentially attenuate acute kidney injury. J Am Soc Nephrol. 2009;20:267–77. doi: 10.1681/ASN.2008030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–48. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mosnier LO, Zampolli A, Kerschen EJ, Schuepbach RA, Banerjee Y, Fernandez JA, et al. Hyper-antithrombotic, non-cytoprotective Glu149Ala-activated protein C mutant. Blood. 2009;113:5970–8. doi: 10.1182/blood-2008-10-183327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Westenfelder C, Biddle DL, Baranowski RL. Human, rat, and mouse kidney cells express functional erythropoietin receptors. Kidney Int. 1999;55:808–20. doi: 10.1046/j.1523-1755.1999.055003808.x. [DOI] [PubMed] [Google Scholar]
- 123.Sharples EJ, Patel N, Brown P, Stewart K, Mota-Philipe H, Sheaff M, et al. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J Am Soc Nephrol. 2004;15:2115–24. doi: 10.1097/01.ASN.0000135059.67385.5D. [DOI] [PubMed] [Google Scholar]
- 124.Vaziri ND, Zhou XJ, Liao SY. Erythropoietin enhances recovery from cisplatin-induced acute renal failure. Am J Physiol. 1994;266:F360–6. doi: 10.1152/ajprenal.1994.266.3.F360. [DOI] [PubMed] [Google Scholar]
- 125.Yokomaku Y, Sugimoto T, Kume S, Araki S, Isshiki K, Chin-Kanasaki M, et al. Asialoerythropoietin prevents contrast-induced nephropathy. J Am Soc Nephrol. 2008;19:321–8. doi: 10.1681/ASN.2007040481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Song YR, Lee T, You SJ, Chin HJ, Chae DW, Lim C, Park KH, et al. Prevention of Acute Kidney Injury by Erythropoietin in Patients Undergoing Coronary Artery Bypass Grafting: A Pilot Study. Am J Nephrol. 2009;30:253–60. doi: 10.1159/000223229. [DOI] [PubMed] [Google Scholar]