

Summary
Background
Vascular smooth muscle cell (VSMC) migration is a critical process in arterial remodeling. Purified plasminogen activator inhibitor-1 (PAI-1) is reported to both promote and inhibit VSMC migration on 2-dimensional (D) surfaces.
Objective
To determine the effects of PAI-1 and vitronectin (VN) expressed by VSMC themselves on migration through physiological collagen matrices.
Methods
We studied migration of wild-type (WT), PAI-1-deficient, VN-deficient, PAI-1/VN doubly-deficient (DKO), and PAI-1-transgenic (Tg) VSMC through 3-D collagen gels.
Results
WT VSMC migrated significantly slower than PAI-1- and VN-deficient VSMC, but significantly faster than DKO VSMC. Experiments with recombinant PAI-1 suggested that basal VSMC PAI-1 expression inhibits migration by binding VN, which is secreted by VSMC and binds collagen. However, PAI-1-over-expressing Tg VSMC migrated faster than WT VSMC. Reconstitution experiments with recombinant PAI-1 mutants suggested that the pro-migratory effect of PAI-1 over-expression required its anti-plasminogen activator (PA) and LDL receptor-related protein (LRP) binding functions, but not VN binding. While promoting VSMC migration in the absence of PAI-1, VN inhibited the pro-migratory effect of active PAI-1.
Conclusions
In isolation, VN and PAI-1 are each pro-migratory. However, via formation of a high-affinity, non-motogenic complex, PAI-1 and VN each buffers the other's pro-migratory effect. The level of PAI-1 expression by VSMC and the concentration of VN in extracellular matrix are critical determinants of whether PAI-1 and VN promote or inhibit migration. These findings help to rectify previously conflicting reports and suggest that PAI-1/VN stoichiometry plays an important role in VSMC migration and vascular remodeling.
Keywords: Vascular smooth muscle cell, PAI-1, vitronectin, collagen
Intimal hyperplasia is a central process in acquired vascular diseases, such as atherosclerosis and restenosis after balloon angioplasty. A key step in intimal hyperplasia is the migration of vascular smooth muscle cells (VSMC) from the media, through extracellular matrix (ECM) composed of collagen, elastin, and multiple other components, into the intima. The plasminogen activation (PA) system plays a major role in the regulation of cell migration and the development of intimal hyperplasia [1;2]. Plasminogen activator inhibitor-1 (PAI-1) is the primary physiological inhibitor of tissue- and urinary-type plasminogen activators (t-PA and u-PA, respectively) and a major regulator of fibrinolysis [3]. PAI-1 is present in plasma, platelets, endothelial cells, VSMC, and the ECM. PAI-1 expression in the vascular wall is increased in several human vascular diseases characterized by neointima formation, suggesting that PAI-1 may regulate the development of intimal hyperplasia [4;5]. PAI-1 binds vitronectin (VN), an adhesive glycoprotein present in ECM that plays key roles in cell adhesion and migration [6;7]. Binding of PAI-1 inhibits VN's interactions with its receptors on VSMC, thereby inhibiting VSMC adhesion and migration [8;9]. However, PAI-1 has also been reported to promote VSMC migration by binding to low density lipoprotein receptor-related protein (LRP), which is expressed on the surface of VSMC [10]. When bound to VN, PAI-1's LRP binding site remains in an encrypted state that does not bind LRP [11;12]. Hence, PAI-1 and VN regulate each other's functions and are poised to play key roles in VSMC migration and intimal hyperplasia.
While PAI-1 can either promote or inhibit VSMC migration in vitro, depending on experimental conditions, the net effect of PAI-1 on VSMC migration in vivo is unknown. However, it is difficult to study VSMC migration in vivo. Intimal hyperplasia cannot be used as a surrogate because it depends not only on VSMC migration, but also on VSMC proliferation and apoptosis and other processes independent of VSMC. Prior in vitro studies of the roles of PAI-1 and VN in VSMC migration have involved 2-dimensional (D) culture systems in which cells are seeded on plastic surfaces coated with a purified matrix molecule and purified PAI-1 is added [8-10]. Two-D cell culture systems, such as the modified Boyden chamber (MBC) are useful, but do not adequately reproduce the complex, 3-D ECM in which VSMC migrate in vivo [13;14], nor does addition of recombinant PAI-1 to cells coated on VN or other purified matrix components give adequate insight into the functional significance of PAI-1 and VN produced by VSMC themselves. Within the vascular wall, an environment not directly accessible to plasma, the pool of PAI-1 produced by VSMC is likely to be a major determinant of the overall effect of PAI-1 on VSMC migration. However, the impact of PAI-1 expression by VSMC on their migration under physiological conditions has not been studied. To address these issues, we examined the migration of VSMC with variable levels of PAI-1 expression through 3-D collagen matrices. Given that VSMC express VN in vivo [15], as well as the key role of VN in determining PAI-1 function, we also studied the effects of VSMC VN expression and ECM VN concentration on PAI-1's migratory properties.
Materials and Methods
Proteins
Recombinant human PAI-1 was expressed and purified as described previously [16]. The following mutants were used: 1) PAI-1-14-1b (PAI-1 N150H, K154T, Q319L, M354I), which inhibits u-PA and t-PA and binds VN with WT activities [17]; however, unlike WT PAI-1 (which has a half-life of 1-2 hr under physiological conditions), PAI-1-14-1b is resistant to conversion to the inactive (i.e. latent) form (half-life >140 hrs), and hence is useful in experiments examining PAI-1 function over longer periods of time, as in the migration assay described below. Throughout this study PAI-1-14-1b is referred to as “PAI-1-WT.” 2) PAI-1-R (T333R, A335R), a reactive center mutant that binds VN normally, but has no detectable anti-proteolytic activity and cannot assume a latent conformation [16]; 3) PAI-1-AK (PAI-1 N150H, K154T, Q319L, M354I, R101A, Q123K), an active, stable mutant with no detectable VN binding [16;18]; and 4) PAI-1-R76E, I91L, an active, stable mutant that does not bind low density lipoprotein receptor-related protein (LRP) [19]. Latent human PAI-1 and mouse multimeric VN were from Molecular Innovations. Platelet-derived growth factor (PDGF)-BB and rat tail collagen type 1 were from Millipore.
Animals
C57BL/6J mice were from Jackson Labs. C57BL/6J-congenic PAI-1-deficient (Pai1-/-) mice were a gift from Dr. Peter Carmeliet, University of Leuven [20]. C57BL/6J-congenic VN-deficient (Vn-/-) mice and PAI-1-transgenic (Tg) mice that over-express PAI-1 under the control of the CMV promoter were from Dr. David Ginsburg, University of Michigan [21;22]. Mice received standard chow. All experiments were approved by the University of Missouri Office of Animal Resources.
Cell culture
VSMC were isolated from mouse aortas and grown in culture as described previously [23]. VSMC (passage 2-3) were cultured in DMEM supplemented with 20% fetal bovine serum (FBS) and gentamycin/amphotericin B. After achieving 70-90% confluency, cells were serum-starved (0.2% FBS) overnight. Cells were harvested by trypsin digestion, washed, and resuspended in 0.2% FBS for use in the 3-D cell migration assay. To determine if genetic deletion of PAI-1, VN, or both affected the general migration properties of cultured VSMC, we performed control experiments in which WT, PAI-1-deficient, VN-deficient, and PAI-1/VN doubly-deficient VSMC were seeded on collagen-coated porous membranes in a modified Boyden chamber and VSMC migration through pores into the lower chamber was stimulated with PDGF-BB [24]. At 6 hrs after seeding of VSMC on collagen there were no significant differences in migration between the 4 groups of cells (data not shown), suggesting that genetic deletion of PAI-1 and/or VN did not significantly alter the general migration properties of VSMC.
3-D cell migration assay
Collagen gels were prepared as described previously [25], except that final collagen concentration was 2.2 mg/mL. Polymerizing collagen mixture (30 μL) was pipetted into the upper chambers of 24-well Transwell inserts (Corning) whose bottoms consisted of porous (8 μm pore diameter) membranes. After gels polymerized, DMEM medium (200 μL) containing 0.2% fetal bovine serum (FBS) and murine VSMC (105) were added to the upper chamber. Inserts were placed into lower chamber wells filled with DMEM (600 μL) containing 2.5% FBS and PDGF-BB (20 ng/mL), which stimulate VSMC migration through the collagen gel into the lower chamber. After 72 hr of standard cell culture conditions, inserts were removed, collagen gels were scraped away, and membranes were stained with Diff-Quick (Siemens Healthcare Diagnostics). The lower-chamber side of the membrane, to which cells that migrate through the collagen gel and pores adhere, was visualized en-face with a microscope and cells were counted. In some experiments VN or recombinant PAI-1 was added to polymerizing collagen solutions and upper chamber culture media.
Western blotting and ELISA
VSMC were grown to confluency. Conditioned media (CM) was removed and cells were washed twice with phosphate-buffered saline and lysed by addition of SDS solution [26]. The lysate was centrifuged to remove insoluble material. Total protein concentration of the supernatant was measured with the BCA reagent. Samples (30 μg total protein) were subjected to SDS-PAGE and Western blotting with rabbit antibodies raised against murine PAI-1 (Santa Cruz Biotechnology) and beta-actin (Cell Signaling Technology), as described [27]. PAI-1 concentration in CM was measured using an ELISA for murine PAI-1 (Innovative Research). Results were normalized to total protein concentration of CM.
Data analyses
Duplicate or triplicate wells were performed for each set of experimental conditions in each experiment and mean results were calculated. A control group consisting of WT or untreated cells was included in each experiment. As indicated, results are reported as % control. All experiments were performed at least in triplicate, with results reported as mean ± standard error of mean. Experimental groups were compared by two-tailed Student's t test or one-way analysis of variance.
Results
Isolated deficiency PAI-1 or VN increases VSMC migration through collagen, while combined deficiency of PAI-1 and VN inhibits migration
To study the functions of PAI-1 and VN expressed by VSMC during migration through collagen, we compared the migration of VSMC isolated from WT, Pai1-/-, and Vn-/- mice. PAI-1-deficient VSMC migrated significantly faster than WT VSMC (Fig. 1), suggesting that under physiological conditions the dominant effect of basal PAI-1 expression by VSMC is anti-migratory. VN-deficient VSMC also migrated significantly faster than WT VSMC, though significantly slower than PAI-1-deficient VSMC (Fig. 1). To examine the impact of combined PAI-1 and VN deficiency on VSMC migration, we crossed Pai1-/- mice and Vn-/- mice to generate double-knock-out mice, from which we established cultured lines of VSMC. While VSMC lacking only PAI-1 or VN each migrated faster than WT VSMC, VSMC lacking both PAI-1 and VN migrated significantly slower than WT VSMC (Fig. 1). These results suggested that a basal level of PAI-1 expression by VSMC inhibited migration only if VSMC VN expression is preserved. However, genetic deletion of VN converted the effect of VSMC PAI-1 expression to pro-migratory, as evidenced by comparing the migratory properties of VN-deficient VSMC (which express PAI-1) to those of VSMC lacking both PAI-1 and VN. These results also suggested that basal VN expression inhibited VSMC migration when PAI-1 expression was preserved, but promoted migration when PAI-1 was genetically deleted, as evidenced by comparing the migratory properties of PAI-1-deficient VSMC (which express VN) to those of VSMC lacking both PAI-1 and VN.
Figure 1.
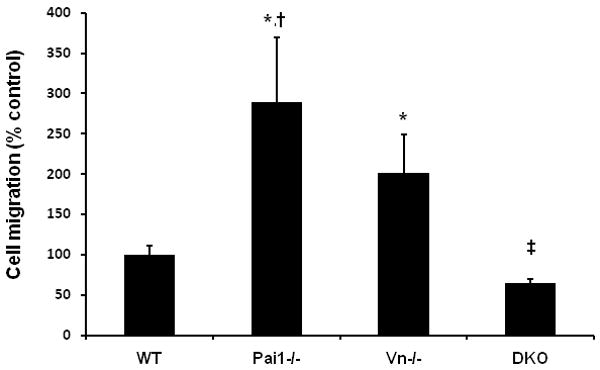
Deficiency of PAI-1 or VN enhances VSMC migration, while combined PAI-1/VN deficiency inhibits migration. The number of VSMC migrating through 3-D collagen matrix in 72 hr was determined, with results expressed as % control (i.e. WT VSMC). *P<0.001 vs. WT; †P<0.01 vs. Vn-/- VSMC; ‡P<0.001 vs. all other groups. DKO, double knockout genotype.
Recombinant PAI-1 mutants promote and inhibit VSMC migration through collagen by distinct mechanisms
We added several forms of recombinant PAI-1 (each at 1 μg/mL) to 3-D migration assays containing PAI-1-deficient VSMC to enable us to study the impact of specific functional defects in PAI-1 on VSMC migration through collagen. PAI-1-WT significantly increased migration of PAI-1-deficient VSMC (Fig. 2). PAI-1-AK (which inhibits PAs, but does not bind VN) also significantly increased migration of PAI-1-deficient VSMC. However, PAI-1-R76E, I91L (which inhibits PAs and binds VN, but does not bind LRP) did not increase PAI-1-deficient VSMC migration, rather producing a significant inhibitory effect (Fig. 2). These results suggested that active PAI-1 can stimulate VSMC migration through collagen by a mechanism that requires LRP binding, but does not require binding to VN, which is secreted by VSMC and binds collagen [28]. PAI-1-R (which does not inhibit PAs, but binds VN with normal affinity) significantly inhibited migration of PAI-1-deficient VSMC (Fig. 2). Latent PAI-1, which does not inhibit PAs and has low binding affinity for VN, had no significant effect on migration of PAI-1-deficient VSMC. Together, these results suggested that PAI-1 can inhibit VSMC migration through collagen under physiological conditions by a VN-dependent mechanism that does not require protease inhibition or LRP binding.
Figure 2.
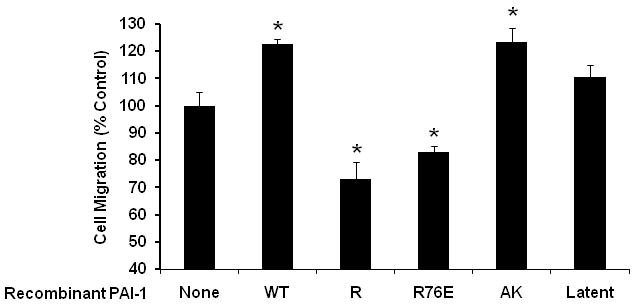
Effects of recombinant PAI-1 on VSMC migration. All cells used in this experiment were PAI-1-deficient. *P<0.002 vs. control PAI-1-deficient VSMC that were not treated with recombinant PAI-1 (“None” group). “R76E” designates PAI-1-R76E, I91L.
We also studied the effect of selected forms of recombinant PAI-1 on migration of VN-deficient VSMC. PAI-1-WT significantly increased migration of VN-deficient VSMC (i.e. 1.4±0.1-fold compared to control matrices lacking PAI-1-WT, P<0.001), further suggesting that enhancement of VSMC migration by PAI-1-WT does not require VN binding. PAI-1-R had no significant effect on migration of VN-deficient VSMC (data not shown), further suggesting that inhibition of VSMC migration by PAI-1-R requires VN binding.
ECM PAI-1 and VN increase migration of WT VSMC, but each factor inhibits the other's pro-migratory effect
Concentrations of PAI-1 and VN in the vascular wall are increased in some human disease states [4;29]. We supplemented collagen matrices with PAI-1-WT (1 μg/mL=23.3 nM), multimeric VN (10 μg/mL=133 nM), or both PAI-1-WT and VN to examine the effects of increased ECM concentrations of these factors on VSMC migration. PAI-1-WT significantly increased migration of WT VSMC through collagen (Fig. 3, bar 1 vs. 2). VN also significantly increased migration of WT VSMC through collagen (Fig. 3, bar 1 vs. 3). These results suggested that increased ECM concentration of either active PAI-1 or VN promotes VSMC migration. However, addition of both PAI-1-WT and VN to collagen matrices resulted in no significant change in VSMC migration compared to collagen matrices to which neither factor was added (Fig. 3, bar 1 vs. 4), suggesting that active PAI-1 and VN can each inhibit the pro-migratory effect of the other. Consistent with this hypothesis, addition of VN (10 μg/mL) to collagen matrices reduced the migration of VN-deficient VSMC (which express PAI-1) by 57.2±6.6% (n=3, P<0.001). We also examined the effect of adding a higher concentration of PAI-1-WT (5 μg/mL=116 nM) along with VN (10 μg/mL) on migration of WT VSMC. Under these conditions VSMC migration was significantly increased compared to control conditions lacking addition of either factor (Fig. 3, bar 1 vs. 5). These results suggested that a stimulatory effect of active PAI-1 on VSMC migration can be observed in the presence of VN if PAI-1 concentration is sufficiently high.
Figure 3.
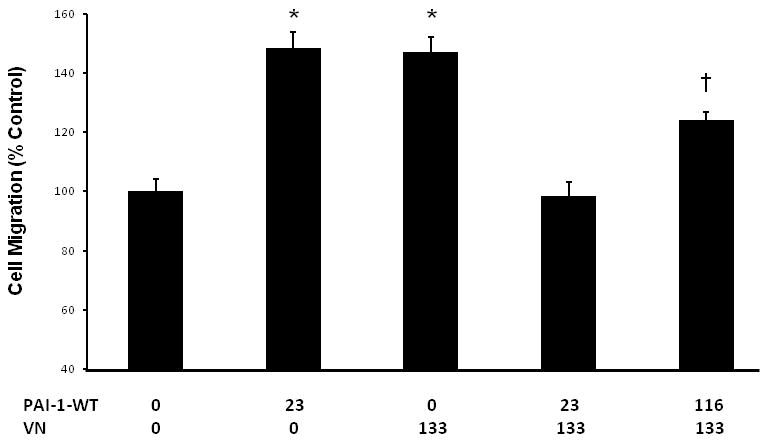
ECM PAI-1 and VN increase migration of WT VSMC, but inhibit each other's pro-migratory effect. X-axis values indicate concentrations of purified PAI-1 and VN (in nM) added in each experiment. *P<0.001 vs. control (i.e. no added PAI-1 or VN); †P<0.002 vs. control.
PAI-1 over-expression increases VSMC through collagen, but VN blunts this effect
Based on our findings that addition of active PAI-1 to collagen matrix promoted VSMC migration, we hypothesized that genetic over-expression of PAI-1 would also enhance VSMC migration. To test this hypothesis, we isolated aortic VSMC from PAI-1-Tg mice. SDS-PAGE/Western blot analysis of cultured PAI-1-Tg VSMC confirmed that they expressed significantly more PAI-1 than WT VSMC (Fig. 4). Consistent with these data, PAI-1 concentration in CM of confluent PAI-1-Tg VSMC was significantly higher than that of confluent WT VSMC (430±88.1 vs. 154±13.3 ng PAI-1/mg total protein, respectively, n=4/group, P=0.02), whereas PAI-1 was undetectable in CM of confluent PAI-1-deficient VSMC. PAI-1-Tg VSMC migrated through collagen significantly faster than WT VSMC (Fig. 5), suggesting that over-expression of PAI-1 by VSMC, as occurs in human vascular diseases [4], promotes migration through collagen. We also compared the migration of WT and PAI-1-Tg VSMC in collagen matrices supplemented with VN (10 μg/mL). Under these conditions, the mean number of VSMC migrating through collagen+VN matrix in 72 hr was higher for PAI-1-Tg VSMC than WT VSMC, but the difference between cell types did not achieve statistical significance (Fig. 5). Furthermore, the ratio of the number of VSMC (i.e. PAI-1-Tg/WT) that migrated through a collagen-only matrix in 72 hr (4.7±0.8, n=3) was significantly higher (P<0.04) than the ratio of PAI-1-Tg/WT VSMC that migrated through a collagen+VN matrix (2.4±0.5, n=3). Together, these results suggested that the pro-migratory effect of PAI-1 over-expression is blunted by VN.
Figure 4.
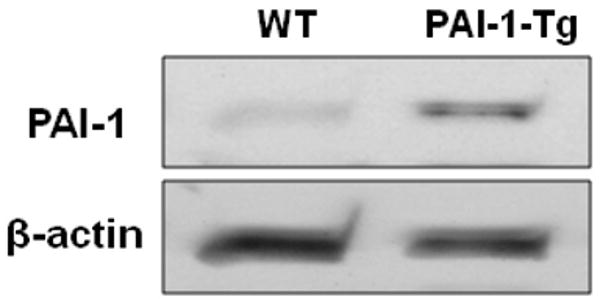
Western blot analysis of PAI-1 expression by cultured VSMC. Blots were stripped and re-probed with anti-beta-actin antibody. Results shown are representative of 3 independent experiments.
Figure 5.
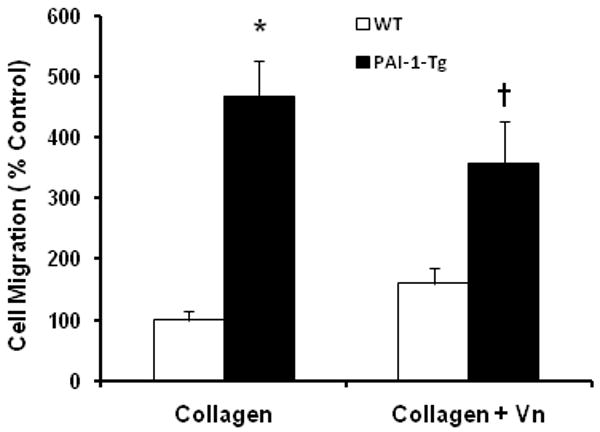
PAI-1 over-expression promotes VSMC migration through collagen matrix, but VN blunts this effect. WT and PAI-1-Tg VSMC were seeded on collagen matrices and collagen matrices containing VN (collagen + VN). Migration was assessed and expressed as % control (WT VSMC migrating through collagen). *P<0.03 vs. WT VSMC/collagen matrix. †P=0.09 vs. WT VSMC/collagen+VN matrix.
Discussion
In some studies PAI-1 has been reported to inhibit VSMC migration [8], while in others PAI-1 has been reported to promote VSMC migration [10]. These experiments involved cell culture on 2-D surfaces coated with purified VN or another ECM molecule and the addition of purified PAI-1 into the experimental system. These discordant results beg the questions, what is the net effect of PAI-1 on VSMC migration under physiological conditions reflective of the arterial wall in vivo, and how does PAI-1 produced by VSMC themselves (which are likely to be the dominant source of PAI-1 within the vascular media [30]) affect migration? To address these questions, we studied the migration of VSMC with variable levels of PAI-1 expression through 3-D type I collagen matrices, as type I collagen is a major component of the ECM of the arterial media and intima [31]. We found that a basal level of PAI-1 expression by VSMC inhibits migration through collagen. It has been proposed that an important mechanism underlying VSMC migration is activation of plasminogen by uPA bound to its cell surface receptor, uPAR, resulting in plasmin formation and digestion of ECM by plasmin or metalloproteinases activated by plasmin. Under such a scenario, PAI-1 would be expected to inhibit cell migration by inhibiting uPA. However, we found no data to support this mechanism in our experimental system (which contained plasminogen because serum was added to culture media), as reconstitution of PAI-1-deficient VSMC with recombinant PAI-1-WT promoted, rather than inhibited, migration, while PAI-1-R, a mutant devoid of anti-proteolytic activity, mimicked the anti-migratory effect of endogenous PAI-1 expression. The capacity of PAI-1 to inhibit VSMC migration through collagen was critically dependent on VN expression, as demonstrated by the inability of PAI-1-R to inhibit migration of VN-deficient VSMC, and by the contrasting effects of PAI-1 deficiency in VN-expressing vs. VN-deficient VSMC (Fig. 1). VN engages integrins and uPAR on VSMC to promote adhesion and migration [32]. Given that VN binds collagen [28], our data suggest that VN secreted by VSMC bridges collagen and cell surface VN receptors to promote migration, and that PAI-1 can inhibit the migration of VSMC through collagen by blocking the bridging function of VN.
Conversely, we found that PAI-1-Tg VSMC migrate through collagen faster than WT VSMC do. The pro-migratory effect of enhanced PAI-1 expression appeared to depend on its anti-protease and LRP binding properties, as addition of latent PAI-1, PAI-1-R, or PAI-1-R76E, I91L to PAI-1-deficient VSMC did not stimulate migration, while addition of PAI-1-WT or PAI-1-AK did. Previous studies demonstrated that PAI-1 binds to uPA and other proteases, which results in exposure of a cryptic LRP binding site in PAI-1 [11]. Engagement of LRP by PAI-1 leads to internalization of PAI-1-protease complex, LRP, and associated integrin, thereby enabling cell detachment, which is necessary for migration [19;33]. Our results demonstrate that VSMC PAI-1 expression can stimulate migration under physiological conditions, but apparently only if PAI-1 expression level is increased, as a pro-migratory effect of PAI-1 was observed in PAI-1-Tg VSMC, but not in WT cells. PAI-1 over-expression within the vascular wall occurs in disease states characterized by migration of VSMC from the media to the intima, including diabetes mellitus and atherosclerosis [4;5]. Based upon our results we hypothesize that basal PAI-1 expression by VSMC, as occurs in a healthy artery, may inhibit intimal hyperplasia, while enhanced PAI-1 expression by VSMC, as occurs in diabetes mellitus and other disease states, may promote intimal hyperplasia. Consistent with this hypothesis, Otsuka et al. observed less intimal hyperplasia in WT mice than in PAI-1-deficient mice, whereas up-regulation of arterial PAI-1 expression by TGF-beta-1 promoted intimal hyperplasia [34]. Active PAI-1 spontaneously converts to a latent form, a process that is delayed by binding of VN to PAI-1 [35]. While Degryse et al. observed that latent PAI-1 produced a motogenic effect in VSMC cultured on a 2-D surface [10], we did not find any apparent effect of latent PAI-1 on VSMC migration through 3-D collagen, consistent with the low affinity of latent PAI-1 for both VN and LRP [11].
Our experiments involving VN-deficient VSMC provide important insights into the role of de novo VN expression by VSMC in migration. While VN is generally considered to promote VSMC migration though interactions with integrin and non-integrin receptors on VSMC [6], we demonstrated that VN can also exert an anti-migratory effect, as WT VSMC migrated through collagen slower than VN-deficient VSMC did, addition of purified VN to collagen inhibited migration of VN-deficient VSMC, and addition of VN to collagen matrices blunted the pro-migratory effects of recombinant PAI-1-WT and transgenic over-expression of PAI-1. However, we showed that PAI-1-deficient VSMC (which express VN) migrate faster than PAI-1/VN doubly-deficient VSMC did. Together these experiments demonstrate that the anti-migratory effect of VN depends on concomitant PAI-1 expression, while in the absence of PAI-1, only a pro-migratory effect of VN is observed. These results are consistent with the hypothesis that when VN expression is low, up-regulation of PAI-1 expression increases the pool of unbound, motogenic PAI-1, fostering VSMC migration. However, when VN expression is high, up-regulation of PAI-1 expression does not necessarily produce a motogenic pool of PAI-1, as PAI-1 binds VN, blocking its motogenic interactions with VSMC VN receptors, while at the same time sequestering PAI-1 from LRP [11;12]. Nevertheless, if PAI-1 expression is sufficiently high to saturate even increased levels of VN, a motogenic pool of free PAI-1 can be created. Some in vivo experiments involving PAI-1-knockout mice concluded that PAI-1 promotes intimal hyperplasia [36;37], others concluded that PAI-1 inhibits intimal hyperplasia [38;39], and one study found no effect [40]. While multiple factors are likely to have contributed to these variable results, it is possible that variability in VN expression between different vascular injury models [29] and mouse strains could have accounted for the variable effects of PAI-1 on neointima formation in these studies. Conversely, differences in PAI-1 expression level between experimental models may have contributed to the generation of conflicting reports that VN promotes [36] and inhibits [39] intimal hyperplasia after vascular injury.
In summary, we examined the roles of PAI-1 and VN expression by VSMC in their migration through physiological collagen matrices. Based on our results we propose the following paradigm. In isolation, VN and PAI-1 are each pro-migratory, the former by fostering cell adhesion, the latter by fostering cell detachment—both of which are necessary in coordinated fashion for VSMC migration. However, via formation of a high-affinity, non-motogenic complex, PAI-1 and VN each buffers the other's pro-migratory effect (Fig. 6). Binding of PAI-1 to VN blocks VN's interactions with its VSMC receptors, thereby inhibiting cell adhesion. Binding of VN to PAI-1 can inhibit exposure of PAI-1's LRP binding site and may sequester PAI-1 from interacting with VSMC, thereby inhibiting PAI-1's pro-migratory properties [11,12]. Hence, PAI-1 and VN exhibit a distinct functional interdependence in regulating VSMC migration [41]. The level of PAI-1 expression by VSMC and the concentration of VN in ECM are critical determinants of whether PAI-1 (or VN) promotes or inhibits migration. If VN expression is low, free PAI-1 is more likely to accumulate and stimulate VSMC migration after exposing its LRP binding site. However, if VN expression is high, PAI-1 is more likely to bind VN, thereby producing an anti-migratory effect by blocking VN's interactions with cell surface receptors. Conversely, if PAI-1 expression is low, unbound VN can promote VSMC migration, while if PAI-1 expression is high, VN is more likely to be consumed into PAI-1-VN complex, thereby inhibiting PAI-1's pro-migratory function. This paradigm establishes the important role of PAI-1/VN stoichiometry in determining each factor's vascular effects. While our in vitro assay allowed us to study VSMC migration under physiological conditions, it did not reproduce several important factors present in vivo, including complex, multi-component ECM, cell types other than VSMC, and other factors that regulate cell migration, including thrombin, metalloproteinases, and cytokines. Therefore, animal studies will be necessary to further test the hypothesis that PAI-1/VN stoichiometry is an important regulator of arterial remodeling.
Figure 6.
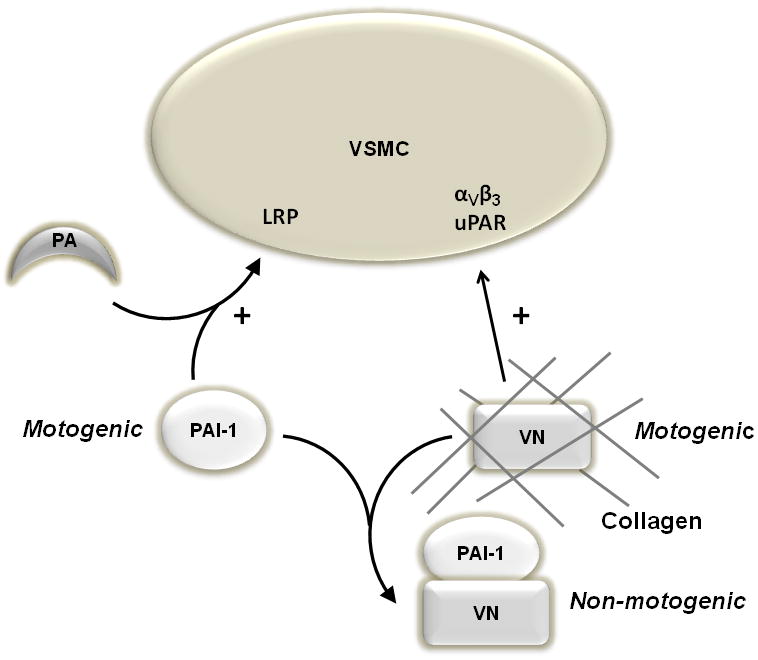
Pro- and anti-migratory effects of PAI-1 and VN on VSMC. VN binds collagen and stimulates (+) VSMC migration by interacting with αVβ3 and uPA receptor (uPAR). PAI-1 stimulates VSMC migration by binding to a plasminogen activator (PA), which exposes an LRP binding site on PAI-1, leading to engagement of the complex with LRP, a motogenic receptor. Binding of PAI-1 to VN yields a complex that is not motogenic, providing a mechanism for each factor to inhibit the pro-migratory effect of the other.
Acknowledgments
This work was supported by a research grant from the Missouri Life Sciences Research Board (WPF), the Department of Veterans Affairs (WPF), and NIH grants HL57346 (WPF) and HL55374, HL54710, HL57346, and HL89407 (DAL).
Footnotes
Disclosure of Conflict of Interest: The authors state that they have no conflicts of interest.
References
- 1.Carmeliet P, Collen D. Evaluation of the plasminogen/plasmin system in transgenic mice. Fibrinolysis. 1994;8:269–276. [Google Scholar]
- 2.Fay WP, Garg N, Sunkar M. Vascular functions of the plasminogen activation system. Arterioscler Thromb Vasc Biol. 2007;27:1231–1237. doi: 10.1161/ATVBAHA.107.140046. [DOI] [PubMed] [Google Scholar]
- 3.Loskutoff DJ, Sawdey M, Mimuro J. Type 1 plasminogen activator inhibitor. Prog Hemost Thromb. 1989;9:87–115. [PubMed] [Google Scholar]
- 4.Pandolfi A, Cetrullo D, Polishuck R, Alberta MM, Calafiore A, Pellegrini G, Vitacolonna E, Capani F, Consoli A. Plasminogen activator inhibitor type 1 is increased in the arterial wall of type II diabetic subjects. Arterioscler Thromb Vasc Biol. 2001;21:1378–1382. doi: 10.1161/hq0801.093667. [DOI] [PubMed] [Google Scholar]
- 5.Schneiderman J, Sawdey MS, Keeton MR, Bordin GM, Bernstein EF, Dilley RB, Loskutoff DJ. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc Natl Acad Sci. 1992;89:6998–7002. doi: 10.1073/pnas.89.15.6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown SL, Lundgren CH, Nordt T, Fujii S. Stimulation of migration of human aortic smooth muscle cells by vitronectin: Implications for atherosclerosis. Cardiovasc Res. 1994;28:1815–1820. doi: 10.1093/cvr/28.12.1815. [DOI] [PubMed] [Google Scholar]
- 7.Preissner K. Structure and biological role of Vitronectin. Annu Rev Cell Biol. 1991;7:275–310. doi: 10.1146/annurev.cb.07.110191.001423. [DOI] [PubMed] [Google Scholar]
- 8.Stefansson S, Lawrence DA. The serpin PAI-1 inhibits cell migration by blocking integrin alpha-V beta-3 binding to vitronectin. Nature. 1996;383:441–443. doi: 10.1038/383441a0. [DOI] [PubMed] [Google Scholar]
- 9.Deng G, Curriden SA, Hu G, Czekay RP, Loskutoff DJ. Plasminogen activator inhibitor-1 regulates cell adhesion by binding to the somatomedin B domain of vitronectin. J Cell Physiol. 2001;189:23–33. doi: 10.1002/jcp.1133. [DOI] [PubMed] [Google Scholar]
- 10.Degryse B, Neels JG, Czekay RP, Aertgeerts K, Kamikubo Y, Loskutoff DJ. The Low Density Lipoprotein Receptor-related Protein Is a Motogenic Receptor for Plasminogen Activator Inhibitor-1. J Biol Chem. 2004;279:22595–22604. doi: 10.1074/jbc.M313004200. [DOI] [PubMed] [Google Scholar]
- 11.Stefansson S, Muhammad S, Cheng XF, Battey F, Strickland DK, Lawrence DA. Plasminogen activator inhibitor-1 contains a cryptic high afinity binding site for the low density lipoprotein receptor-related protein. J Biol Chem. 1998;273:6358–6366. doi: 10.1074/jbc.273.11.6358. [DOI] [PubMed] [Google Scholar]
- 12.Kamikubo Y, Neels JG, Degryse B. Vitronectin inhibits plasminogen activator inhibitor-1-induced signalling and chemotaxis by blocking plasminogen activator inhibitor-1 binding to the low-density lipoprotein receptor-related protein. Int J Biochem Cell Biol. 2009;41:578–585. doi: 10.1016/j.biocel.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Li S, Lao J, Chen BPC, Li Y, Zhao Y, Chu J, Chen KD, Tsou TC, Peck K, Chien S. Genomic analysis of smooth muscle cells in three-dimensional collagen matrix. FASEB J. 2003;17:97–99. doi: 10.1096/fj.02-0256fje. [DOI] [PubMed] [Google Scholar]
- 14.Stegemann JP, Nerem RM. Altered response of vascular smooth muscle cells to exogenous biochemical stimulation in two- and three-dimensional culture. Exper Cell Res. 2003;283:146–155. doi: 10.1016/s0014-4827(02)00041-1. [DOI] [PubMed] [Google Scholar]
- 15.Dufourcq P, Louis H, Moreau C, Daret D, Boisseau MR, Lamazière JMD, Bonnet J. Vitronectin expression and interaction with receptors in smooth muscle cells from human atheromatous plaque. Arterioscler Thromb Vasc Biol. 1998;18:168–176. doi: 10.1161/01.atv.18.2.168. [DOI] [PubMed] [Google Scholar]
- 16.Stefansson S, Petitclerc E, Wong MKK, McMahon GA, Brooks PC, Lawrence DA. Inhibition of angiogenesis in vivo by plasminogen activator inhibitor-1. J Biol Chem. 2001;276:8135–8141. doi: 10.1074/jbc.M007609200. [DOI] [PubMed] [Google Scholar]
- 17.Berkenpas MB, Lawrence DA, Ginsburg D. Molecular evolution of plasminogen activator inhibitor-1 functional stability. EMBO J. 1995;14:2969–2977. doi: 10.1002/j.1460-2075.1995.tb07299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li SH, Gorlatova NV, Lawrence DA, Schwartz BS. Structural differences between active forms of plasminogen activator inhibitor type 1 revealed by conformationally-sensitive ligands. J Biol Chem. 2008;283:18147–18157. doi: 10.1074/jbc.M709455200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao C, Lawrence DA, Li Y, Von Arnim CA, Herz J, Su EJ, Makarova A, Hyman BT, Strickland DK, Zhang L. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO Journal. 2006;25:1860–1870. doi: 10.1038/sj.emboj.7601082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carmeliet P, Kieckens L, Schoonjans L, Ream B, Nuffelen A, Prendergast G, Cole M, Bronson R, Collen D, Mulligan R. Plasminogen activator inhibitor-1 gene deficient mice. I. Generation by homologous recombination and characterization. J Clin Invest. 1993;92:2746–2755. doi: 10.1172/JCI116892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng X, Saunders T, Camper S, Samuelson L, Ginsburg D. Vitronectin is not essential for normal mammalian development and fertility. Proc Natl Acad Sci. 1995;92:12426–12430. doi: 10.1073/pnas.92.26.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest. 1996;97:232–237. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray JL, Leach R, Herbert JM, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Meth Cell Sci. 2002;23:185–188. doi: 10.1023/a:1016357510143. [DOI] [PubMed] [Google Scholar]
- 24.Poon M, Marx SO, Gallo R, Badimon JJ, Taubman MB, Marks AR. Rapamycin inhibits vascular smooth muscle cell migration. J Clin Invest. 1996;98:2277–2283. doi: 10.1172/JCI119038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotary K, Allen E, Punturieri A, Yana I, Weiss SJ. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J Cell Biol. 2000;149:1309–1323. doi: 10.1083/jcb.149.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim TJ, Kim JH, Jin YR, Yun YP. The inhibitory effect and mechanism of luteolin 7-glucoside on rat aortic vascular smooth muscle cell proliferation. Arch Pharm Res. 2006;29:67–72. doi: 10.1007/BF02977471. [DOI] [PubMed] [Google Scholar]
- 27.Wu J, Stevenson MJ, Brown JM, Grunz EA, Strawn TL, Fay WP. C-reactive protein enhances tissue factor expression by vascular smooth muscle cells: Mechanisms and in vivo significance. Arterioscler Thromb Vasc Biol. 2008;28:698–704. doi: 10.1161/ATVBAHA.107.160903. [DOI] [PubMed] [Google Scholar]
- 28.Izumi M, Shimo-Oka T, Morishita N, Ii I, Hayashi I. Identification of the collagen-binding domain of vitronectin using monoclonal antibodies. Cell Struct Funct. 1988;13:217–25. doi: 10.1247/csf.13.217. [DOI] [PubMed] [Google Scholar]
- 29.Dufourcq P, Couffinhal T, Alzieu P, Daret D, Moreau C, Duplaa C, Bonnet J. Vitronectin is up-regulated after vascular injury and vitronectin blockage prevents neointima formation. Cardiovasc Res. 2002;53:952–962. doi: 10.1016/s0008-6363(01)00547-8. [DOI] [PubMed] [Google Scholar]
- 30.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89:957–989. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voss B, Rauterberg J. Localization of collagen types I, III, IV and V, fibronectin and laminin in human arteries by the indirect immunofluorescence method. Pathol Res Pract. 1986;181:568–575. doi: 10.1016/S0344-0338(86)80151-0. [DOI] [PubMed] [Google Scholar]
- 32.Blasi F. uPA, uPAR, PAI-1: key intersection of proteolytic, adhesive and chemotactic highways? Immunol Today. 1997;18:415–417. doi: 10.1016/s0167-5699(97)01121-3. [DOI] [PubMed] [Google Scholar]
- 33.Czekay RP, Aertgeerts K, Curriden SA, Loskutoff DJ. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol. 2003;160:781–791. [Google Scholar]
- 34.Otsuka G, Agah R, Frutkin AD, Wight TN, Dichek DA. Transforming growth factor beta-1 induces neointima formation through plasminogen activator inhibitor-1-dependent pathways. Arterioscler Thromb Vasc Biol. 2006;26:737–743. doi: 10.1161/01.ATV.0000201087.23877.e1. [DOI] [PubMed] [Google Scholar]
- 35.Mimuro J, Loskutoff DJ. Purification of a protein from bovine plasma that binds to type 1 plasminogen activator inhibitor and prevents its interaction with extracellular matrix. J Biol Chem. 1989;264:936–939. [PubMed] [Google Scholar]
- 36.Peng L, Bhatia N, Parker AC, Zhu Y, Fay WP. Endogenous vitronectin and plasminogen activator inhibitor-1 promote neointima formation in murine carotid arteries. Arterioscler Thromb Vasc Biol. 2002;22:934–939. doi: 10.1161/01.atv.0000019360.14554.53. [DOI] [PubMed] [Google Scholar]
- 37.Ploplis VA, Cornelissen I, Sandoval-Cooper MJ, Weeks L, Noria FA, Castellino FJ. Remodeling of the vessel wall after copper-induced injury is highly attenuated in mice with a total deficiency of plasminogen activator inhibitor-1. Am J Pathol. 2001;158:107–117. doi: 10.1016/S0002-9440(10)63949-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carmeliet P, Moons L, Lijnen HR, Janssens S, Lupu F, Collen D, Gerard RD. Inhibitory role of plasminogen activator inhibitor-1 in arterial wound healing and neointima formation--A gene targeting and gene transfer study in mice. Circulation. 1997;96:3180–3191. doi: 10.1161/01.cir.96.9.3180. [DOI] [PubMed] [Google Scholar]
- 39.de Waard V, Arkenbout EK, Carmeliet P, Lindner V, Pannekoek H. Plasminogen activator inhibitor 1 and vitronectin protect against stenosis in a murine carotid artery ligation model. Arterioscler Thromb Vasc Biol. 2002;22:1978–1983. doi: 10.1161/01.atv.0000042231.04318.e6. [DOI] [PubMed] [Google Scholar]
- 40.Lijnen HR, Van Hoef B, Umans K, Collen D. Neointima formation and thrombosis after vascular injury in transgenic mice overexpressing plasminogen activator inhibitor-1 (PAI-1) J Thromb Haemost. 2004;2:16–22. doi: 10.1111/j.1538-7836.2003.00533.x. [DOI] [PubMed] [Google Scholar]
- 41.Stefansson S, Haudenschild CC, Lawrence DA. Beyond fibrinolysis: The role of plasminogen activator inhibitor-1 and vitronectin in vascular wound healing. Trends Cardiovasc Med. 1998;8:175–180. doi: 10.1016/S1050-1738(98)00003-6. [DOI] [PubMed] [Google Scholar]