

Abstract
Background
Resuscitation-induced intestinal edema is associated with early and profound mechanical changes in intestinal tissue. We hypothesize that the sodium hydrogen exchanger (NHE), a mechano-responsive ion channel, is a mediator of edema-induced intestinal contractile dysfunction.
Methods
An animal model of hydrostatic intestinal edema was utilized for all experiments. NHE isoforms 1-3 mRNA and protein were evaluated. Subsequently, the effects of NHE inhibition (with 5-(N-ethyl-N-isopropyl) amiloride (EIPA)) on wet to dry ratios, signal transduction and activator of transcription (STAT)-3, intestinal smooth muscle myosin light chain (MLC) phosphorylation, intestinal contractile activity, and intestinal transit were measured.
Results
NHE1-3 mRNA and protein levels were significantly increased in the small intestinal mucosa with the induction of intestinal edema. Administration of EIPA, an NHE inhibitor, attenuated validated markers of intestinal contractile dysfunction induced by edema as measured by decreased STAT-3 activation, increased MLC phosphorylation, improved intestinal contractile activity, and enhanced intestinal transit.
Conclusion
The mechano-responsive ion channel NHE may mediate edema-induced intestinal contractile dysfunction, possibly via a STAT-3 related mechanism.
MeSH Keywords: Edema, sodium hydrogen exchanger, intestine, ileus
Background
Interstitial edema is often a consequence of resuscitative measures, including high volume crystalloid administration and abdominal packing (as a component of damage control surgery). Most commonly, post-resuscitation intestinal edema is associated with ileus, leading to delays in initiation of enteral feeds, increased morbidity, prolonged hospital stay, and increased costs of hospitalization. (1–4)
Intestinal edema leads to ileus by decreasing intestinal contractile activity. The molecular pathway governing intestinal contractility converges on myosin light chain20 (MLC) phosphorylation, which is the obligatory step mediating intestinal contractility. (5, 6) Signal transducer and activator of transcription (STAT)-3 activity is increased in edematous intestinal smooth muscle and has been shown to at least partially mediate the decreased MLC phosphorylation seen with hydrostatic intestinal edema. (7) However, the mechanism by which edema induces increased STAT-3 activation and decreased MLC phosphorylation is not well understood.
A growing body of literature suggests that edema may mediate its downstream effects via early mechanical stimuli that trigger intracellular signaling cascades that culminate in decreased intestinal contractility and subsequent ileus. In previous work, we have demonstrated that the development of intestinal edema is rapid (within 30 minutes) and associated with early, concurrent mechanical changes as evidenced by increased strain and stress as well as increased interstitial pressure. (8) Additionally, cytoskeletal alterations, including alterations in filamentous to globular (F:G) actin, vimentin, and calponin are present. (9) The mechanism by which hypertonic saline alleviates edema induced intestinal dysfunction is believed to be due, in part, to reversing the mechanical changes induced by edema. (8–10)
The molecule(s) that transduces the mechanical stimulus of edema would have to react to alterations in cell stretch, external pressure, and/or shear stress. Both cell stretch and shear stress have been shown to modulate sodium-hydrogen exchanger (NHE) activity. (11–13) There are three main isoforms of NHE (1–3) expressed in the intestine. NHE1 is ubiquitously expressed, whereas NHE2 and 3 are predominantly expressed in epithelia. While NHE2 and 3 are expressed predominantly on the apical membrane, NHE1 is expressed on the basolateral membranes. (14) NHEs are integral membrane proteins that exchange an intracellular proton for an extracellular sodium ion. Studies in cardiomyocytes indicate that NHE might be partially involved in the stretch-induced phosphorylation of STAT3. (11) Furthermore, shear stress was shown to modulate NHE protein levels in cerebral microvascular endothelial cells. (13)
We hypothesize that NHE serves as a mediator of dysfunction in edema-induced intestinal contractile dysfunction. Using an established animal model of hydrostatic intestinal edema free of inflammatory or ischemic influences, we examined the effect of edema on NHE expression and protein levels. To further elucidate the role of NHE in edema-induced intestinal contractile dysfunction, we examined the effect of an NHE inhibitor with regards to validated endpoints of edema-induced intestinal contractile dysfunction.
Materials and Methods
All procedures were approved by the University of Texas Medical School at Houston’s Institutional Animal Care and Use Committee and were consistent with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals.
Animal Model
Male Sprague Dawley rats (250g–350g) were fasted 12–16 hours prior to surgery with free access to water. The rats were anesthetized with isoflurane, and a fluid filled external jugular vein catheter was placed under aseptic conditions. A midline laparotomy was subsequently performed and a silastic catheter was tunneled from the back of the neck, through the subcutaneous tissue and left abdominal wall musculature, and into the peritoneal cavity. The end of the silastic catheter was placed into the duodenum by needle puncture, advanced 1 centimeter distally, and secured in place using a 6-0 silk purse-string suture. The external end of the catheter was sealed using a rubber cap. Finally, the superior mesenteric vein (SMV) was dissected free from its mesenteric attachments. The small bowel was not manipulated.
Animals were divided into two groups, CONTROL and RESUS + VH (80mL/kg of 0.9% normal saline with induction of mesenteric venous hypertension, edema group, described below) (Figure 1a). We also included 4 additional groups to determine the effects of NHE inhibition (Figure 1b). For this subset of experiments, rats in the CONTROL and RESUS + VH groups were subdivided into two additional groups treated with either vehicle (VEH) (1 ml/kg bolus of 7% dimethyl sulfoxide (DMSO) in 0.9% normal saline) at the time of surgery and a continuous 10 μl/hr infusion of 50% DMSO (in 0.9% normal saline) via an ALZET osmotic pump) or with the NHE inhibitor, 5-(N-ethyl-N-isopropyl) amiloride (EIPA), (1 mg/kg (in 7% DMSO) bolus at time of surgery and a continuous infusion of 0.07 mg/hr (in 50% DMSO) via an ALZET osmotic pump). The bolus of VEH or EIPA was administered 30 minutes before surgery and the ALZET osmotic pump (Durect Corp., Cupertino, CA) was implanted subcutaneously over the shoulders at the time of surgery. The ALZET osmotic pump releases medications continuously into the subcutaneous tissue from which it is consequently absorbed into the systemic circulation. The dose for EIPA was chosen based on previous work by Wang et. al.. (15)
Figure 1.
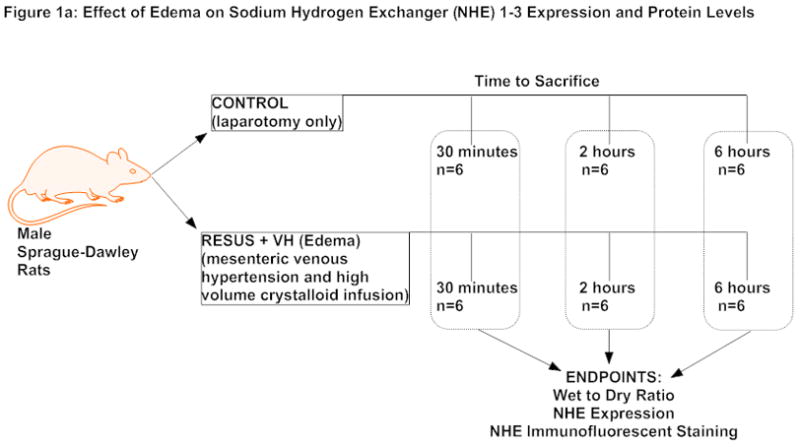
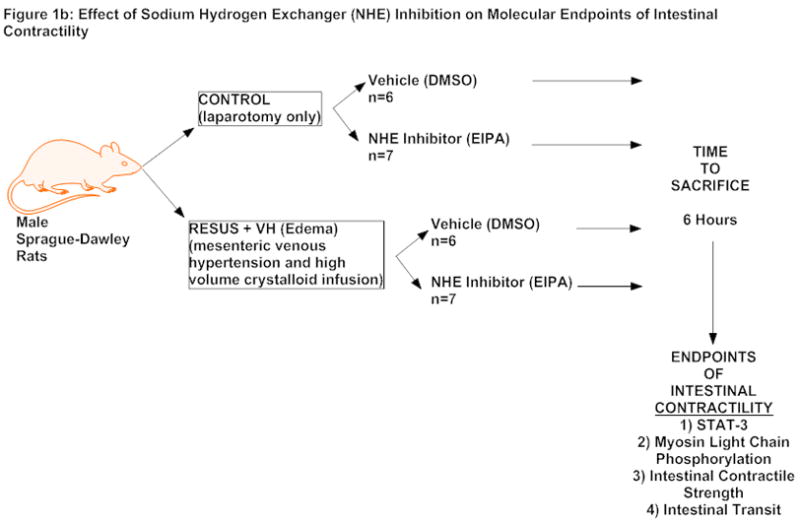
Figure 1a: Experimental design to determine effect of edema on NHE expression and protein levels
Figure 1b: Experimental design to determine effect of NHE inhibition on validated endpoints of intestinal contractility
The edema groups (RESUS + VH) had both administration of resuscitative fluids (80 mL/kg of 0.9% sodium chloride) and induction of mesenteric venous hypertension as described in previous publications. (5–7, 9, 10, 16, 17) Briefly, a 4-0 silk suture was placed around the superior mesenteric vein and subsequently tied over a PE-10 sized tube. The PE-10 sized tubing was then removed, creating a reproducible non-occlusive stenosis which causes a sustained elevation in mesenteric venous pressure. We have shown previously that this elevates the mesenteric venous pressure from 11 ± 1 to 20 ± 3 mmHg, which is in the clinically relevant range, corresponding to intra-abdominal pressures in patients undergoing high volume crystalloid resuscitation. (6) This was immediately followed by infusion of fluid (normal saline) slowly over five minutes. Control animals (CONTROL) had their laparotomy wounds closed without infusion of saline or induction of mesenteric venous hypertension. The animals were allowed to recover. Animals were sacrificed at either 30 minutes, 2 hours or 6 hours post-surgery.
Wet to Dry Ratio
Samples were weighed immediately after collection then dried in a 60°C oven until the weight did not change (approximately 2–3 days). The wet to dry ratio was calculated as: (wet weight − dry weight)/dry weight.
Quantitative Reverse Transcriptase Polymerase Chain Reaction
RNA was isolated from frozen tissue using RNA-Bee (Tel-Test, Inc., Friendswood, TX) following manufacturer’s suggested protocol. RNA samples were DNase treated immediately after RNA isolation to remove genomic DNA contamination. Samples were treated with an RNAse inhibitor and stored at −80°C until time of use.
1 μl RNA stock solution was reverse transcribed in triplicate at 50°C for 30 minutes using 1x reverse transcriptase (RT) buffer, 300nM specific reverse primer, 500 μM dNTPs and Superscript II (LTI, Bethesda, MD). After 10 minutes at 75°C, the RT reaction (10μl) was added to 40 μl of polymerase chain reaction (PCR) mix for a final concentration of 1x PCR buffer, 400nM primers, 100 nM fluorogenic probe, 4 mM MgCl2, and Taq Polymerase. Amplification and quantitation based on real-time monitoring of amplification was carried out using an ABI Prism 7700 (Applied Biosystems, Foster City, CA) performing 40 cycles of 95°C for 12 sec and 60°C for 1 min, following a 1 minute denaturation step at 95°C. Values of transcripts in unknown samples were obtained by interpolating their Ct (PCR cycles to threshold) values on a standard curve derived from known amounts of cognate, specific amplicons. Transcript levels were normalized to the level of 36b4 mRNA. All determinations were performed in triplicate and with a no RT control. Sequences of all primers and TaqMan probes used for amplification reaction assays are summarized in Table 1.
Table 1.
Primers and Probes used for qPCR Analysis
| Transcript | Accn. # | Forward Primer | Reverse Primer | Fluorogenic Probe |
|---|---|---|---|---|
| 36B4 | X15096 | AGAGGTGCTGGACATCACAG | CATTGCGGACACCCTCTAG | CAGGCCCTGCACACTCGCTT |
| NHE1 (Slc9A1) | NM_012652 | ACACAGTTCCTGGACCACCTT | TCCAGTGATGGTGGCCATA | TGACAGGCATCGAGGACATCTGTGG |
| NHE2 (Slc9A2) | NM_012653 | TGGTATCCTGCTGGGATTCA | GCTCAATGACCCGGATGTT | AGCAGCGTTCACCACCCGTTTCA |
| NHE3 (Slc9A3) | NM_012654 | ACGTGAAGGCCAACATCTCA | ACTTGCCAGCATCTTCATAGTGT | AGCAGTCGGCCACCACCGTG |
Sodium Hydrogen Exchanger Immunofluorescence
Tissue segments from the ileum were obtained from CONTROL and RESUS + VH animals sacrificed at the 6 hour time point (n=6/group). The tissues were fixed in buffered 10% formalin for at least 24 hours. After embedding in paraffin, 7 μm sections were placed onto slides. After deparaffinization, antigen recovery, and blocking, slides were incubated with an NHE-1 antibody (ab67313, 1:200 dilution, Abcam, Cambridge, MA), NHE-2 antibody (sc-30167, 1:50 dilution, Santa Cruz Biotechnology, Santa Cruz, CA), and NHE-3 antibody (sc-58636, 1:50 dilution, Santa Cruz Biotechnology, Santa Cruz, CA). After overnight incubation, slides were incubated with an appropriate phycoerythrin conjugated anti-rabbit fluorescent secondary antibody (ab7007, 1:200 dilution, Abcam, Cambridge, MA) or phycoerythrin conjugated anti-mouse fluorescent antibody (405307, 1:200 dilution, BioLegend, San Diego, CA). The slides were then stained with 4’,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA) for nuclear visualization and 2D deconvolution microscopy was utilized to determine NHE-1 localization. Negative control slides were prepared to exclude non-specific binding. Relative intensity of staining for NHE-1, 2, and 3 was determined by mean pixel count analysis (NIS-Elements, Nikon Instruments, Melville, NY).
Preparation of Cytoplasmic and Nuclear Extracts
All tissue samples were frozen in liquid nitrogen immediately after collection. Frozen tissue was ground with mortar and pestle over liquid nitrogen. Nuclear and cytoplasmic extracts were prepared using a nuclear extract kit (Active Motif, Carlsbad, CA). Briefly, cold hypotonic buffer containing protease inhibitors (Halt™ Kit Protease Inhibitor Cocktail, Pierce Biotechnology, Inc., Rockford, IL) and phosphatase inhibitors (2 mM orthovanadate and 2 mM sodium fluoride) was added to the ground tissue and the samples were homogenized. After a 15 minute incubation period at 4°C, the samples were centrifuged at 850 × g at 4°C for 10 minutes. The supernatant was saved. The pellet was resuspended in hypotonic buffer containing protease and phosphatase inhibitors and incubated on ice for 15 minutes. After addition of 0.05% detergent, the samples were vortexed and subsequently centrifuged for 10 minutes at 14000 × g at 4°C. The supernatant was saved and combined with the first supernatant and called the cytoplasmic extract. The pellet was resuspended in lysis buffer, vortexed, and incubated on ice for 30 minutes. The samples were then centrifuged at 14000 × g and the supernatant was called the nuclear extract. All samples were aliquotted and stored at −80°C until use.
STAT3 Phosphorylation
Phosphorylated STAT3 was measured in whole cell lysates from the intestinal muscularis layer of the ileum using an ELISA kit following manufacturer’s suggested protocol (PathScan Phospho-STAT3 Sandwich ELISA Kit, Cell Signaling Technology, Inc., Danvers, MA). Briefly, lysates were added to individual wells of a 96-well plate coated with a STAT3 rabbit monoclonal antibody. After overnight incubation at 4°C, the wells were washed and a primary phospho-STAT3 mouse monoclonal antibody was added. After a one hour incubation period, the wells were washed again and a secondary HRP conjugated anti-mouse IgG antibody was added. After 30 minutes of incubation, the wells were washed and an HRP substrate was added. After color development, a stop solution was added and the plate was read at 405 nm. Measurements were performed in duplicate. A standard curve was made using varying amounts of IL-6 activated HepG2 cells. Activity was normalized to total whole cell lysate protein added to assay. Activity is shown as units of activity/mg protein where one unit of activity equals one μl of activated HepG2 cells.
Myosin Light Chain Phosphorylation
MLC phosphorylation was determined as previously described. (5) Briefly, cytoplasmic extracts, prepared as described above, were separated using a 12.5 % SDS-PAGE gel. After transfer to polyvinyldiene fluoride (PVDF) membranes, Western blotting was performed with an primary MLC antibody that recognizes both the phosphorylated and unphosphorylated forms (sc-9449, 1:400 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) and corresponding secondary antibody obtained from Sigma-Aldrich (St. Louis, MO). Enhanced chemical luminescence (ECL) was used to visualize the proteins on the membranes (ECL Plus, Amersham Biosciences, Piscataway, NJ). Quantification of bands was performed with ImageJ software (National Institutes of Health, Bethesda, MD).
Intestinal Contractile Activity
Intestinal contractile activity was determined as previously described. (5) Briefly, 1 cm whole thickness strips from the ileum of each animal were mounted in duplicate in 25-ml organ baths filled with Krebs-Ringer solution (in mM: 103 NaCl, 4.7 KCl, 2.5 CaCl2, 25 NaHCO3, 15 glucose). The solution was buffered with albumin to avoid edema formation 1.1 NaH2PO4, force was during incubation in the tissue chamber and gassed with 5% CO2-95% O2. Isometric monitored by an external force displacement transducer (Quantametrics, Newtown, PA) connected to a PowerLab data acquisition system (AD Instruments, Colorado Springs, CO). Each strip was stretched to optimal length then allowed to equilibrate for at least 30 min. After equilibration, 30 min of basal contractile activity data was recorded. The strips were then treated with carbachol, 10−6 M, added to the bath and 5 minutes of data were recorded. This dose was based on previous dose response studies. (5) After recording contractile activity, the length of each strip was measured, removed, blotted lightly, and weighed. Contractile activity was calculated as the area under the curve over 10 minutes. Maximum stress was calculated as the area under the curve for the first 2 minutes after addition of carbachol. Contractile frequency (hertz (hz)) was measured during the same time periods. The cross-sectional area of each strip was calculated from length and weight data by assuming that the density of smooth muscle was 1.05 g/cm3. All force development was normalized to tissue cross-sectional area and expressed as stress (g/cm2/s). All contractility experiments were performed at 6 hours.
Intestinal Transit
After 5 ½ hours, the animals had 0.15 ml of a 5 mM solution of non-absorbable FITC-Dextran (MW=9400, FITC content 0.008 mol/mol glucose, Sigma-Aldrich, St. Louis, MO) injected into the duodenal catheter and flushed with 0.15 ml of normal saline. Approximately thirty minutes later, the animals were sacrificed and the entire small intestine was removed and divided into 10 equal segments. The intraluminal contents of each segment were then flushed using 3 ml of 5 mM Tris-buffer (pH 10.3) to recover the FITC-Dextran. The FITC-Dextran concentration was then measured using an optical scanner (Varioscan, Thermo Electron Corp., Waltham, MA) and expressed as a fraction of the total tracer recovered and presented as the geometric center of distribution as described in the literature and as we have previously published. (6, 10, 16–18)
Statistics
All data are reported as mean ± standard error of the mean (SEM). Values were compared using one and two way analysis of variance with a Newman-Keuls post-hoc analysis. A p value of ≤0.05 was considered significant.
Results
Wet to Dry Ratios
Mesenteric venous hypertension and high volume crystalloid administration leads to development of significant intestinal edema as early as 30 minutes after surgery. Wet to dry ratios were significantly elevated in the RESUS + VH groups compared to CONTROL groups at 30 minutes (4.4 ± 0.2 versus 3.7 ± 0.1), 2 hours (4.5 ± 0.2 versus 3.4 ± 0), and 6 hours (4.0 ± 0.1 versus 3.5 ± 0.1) after surgery. Values are expressed as a unitless ratio.
Treatment with the NHE inhibitor EIPA did not significantly affect edema formation in the RESUS + VH group (RESUS + VH + VEH (4.2 ± 0.1) versus RESUS + VH + EIPA (3.8 ± 0.1)); wet to dry ratios in both groups were significantly elevated compared to CONTROL + VEH (3.5 ± 0) or CONTROL + EIPA (3.4 ± 0). Of note, there was no significant difference between CONTROL groups. (Figure 2) Values are expressed as a unitless ratio.
Figure 2.
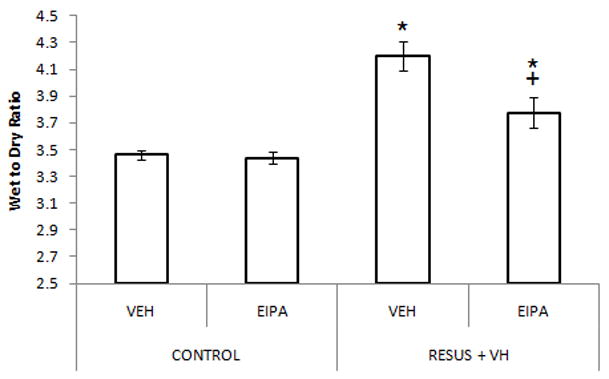
Wet to dry ratios, indicating edema development, as measured 6 hours after surgery in CONTROL and RESUS + VH rats treated with VEH or EIPA. Values are represented as mean ± SEM. (n=6 per VEH treatment group; n=7 per EIPA treatment group; *, p<0.05 versus CONTROL groups; +, p<0.05 versus RESUS + VH + VEH)
NHE Expression
Changes in expression of NHE isoforms 1-3 in both the intestinal smooth muscle and mucosa were measured from 30 minutes to 6 hours after surgery as demonstrated in Figure 3. In general, NHE expression was higher in the intestinal mucosa compared to intestinal muscular layers. NHE1 expression in the mucosa and the smooth muscle layers were similar, with expression in the mucosa approximately 2 fold higher than in the muscle. However, NHE2 and 3 mRNA levels were much higher, approximately 10–30 fold higher, in the intestinal mucosa as compared to intestinal muscle.
Figure 3.
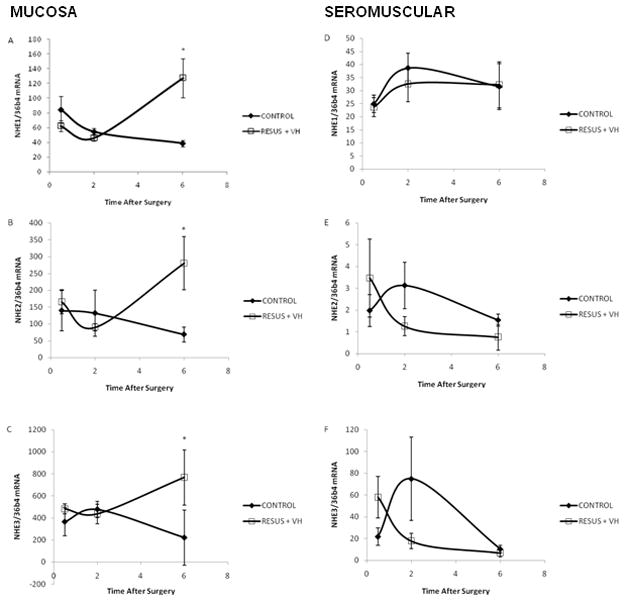
Figure 3a–f: The ratio of NHE mRNA to 36B4 mRNA from 0–6 hours after surgery in CONTROL and RESUS + VH groups. Panels A–C, intestinal mucosa mRNA levels; Panels D–F, intestinal smooth muscle mRNA levels; Panels A and D, NHE1 mRNA; Panels B and E, NHE2 mRNA; Panels C and F, NHE3 mRNA. Values are represented as mean ± SEM. (n=6 per group; *, p<0.05 versus CONTROL)
In the mucosal layer of the ileum, NHE1 (3.3 fold), NHE2 (4.1 fold), and NHE3 (3.5 fold) expression was significantly elevated in the RESUS + VH group 6 hours after surgery to induce edema as compared to CONTROL (genes Slc9a1-3). (Figure 3a–c) There were no significant changes in mRNA levels for NHE isoforms 1-3 in the smooth muscle of the ileum with edema development. (Figure 3d–f, RESUS + VH versus CONTROL)
NHE Immunofluorescence
Edema caused a significant upregulation of NHE1 (403.39 ± 23.18 versus 696.74 ± 90.08 in CONTROL and RESUS + VH groups, respectively), NHE2 (437.21 ± 26.43 versus 1015.62 ± 233.58 in CONTROL and RESUS + VH groups, respectively), and NHE3 (237.71 ± 12.29 versus 391.90 ± 45.01 in CONTROL and RESUS + VH groups, respectively) as determined by mean pixel count analysis. The staining appears to be localized to the mucosal layers, corroborating the expression data. Representative images (20x) are shown in Figure 4a–f.
Figure 4.
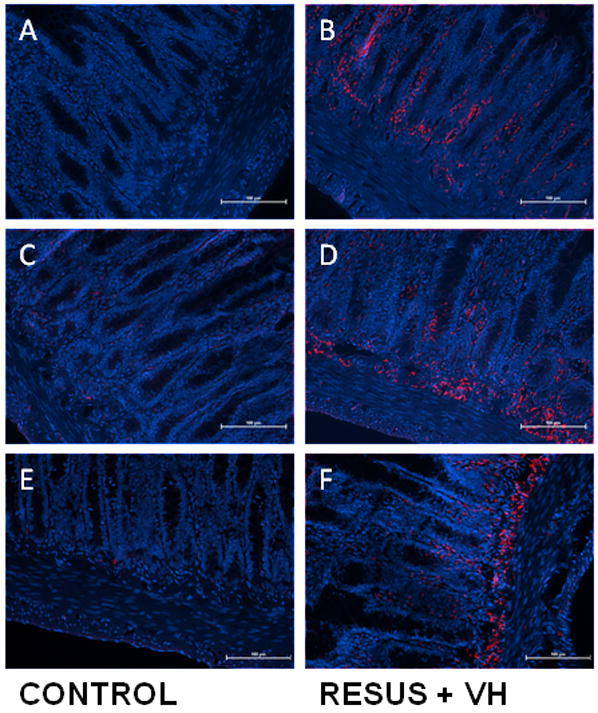
Figure 4a–f: Representative images demonstrating immunofluorescent staining of NHE-1 (A, B), NHE-2 (C, D), and NHE-3 (E, F) in ileum of CONTROL and RESUS + VH animals. Note the significant upregulation of NHE-1, 2, and 3 in the RESUS + VH (edema) group as compared to CONTROL, with a predominance of staining at the submucosa/muscularis interface. All images are 20x. (blue – DAPI; red – NHE-1, 2, and 3, respectively)
Effects of NHE Inhibition
STAT-3 Phosphorylation
Edema caused an increase in STAT-3 phosphorylation (146.6 ± 20.7 versus 46.2 ± 8.7, RESUS + VH + VEH versus CONTROL + VEH, respectively). EIPA treatment prevented the increase in STAT-3 phosphorylation in the edema groups (75.7 ± 8.7 versus 146.6 ± 20.7, RESUS + VH + EIPA versus RESUS + VH + VEH, respectively). (Figure 5) Activity is shown as units of activity/mg protein where one unit of activity equals one μl of activated HepG2 cells.
Figure 5.
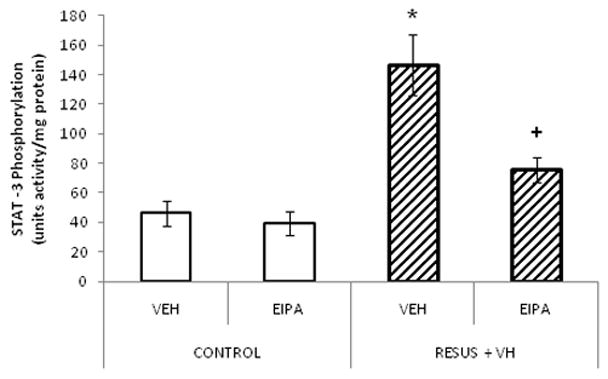
STAT3 phosphorylation measured in ileum 6 hours after surgery in CONTROL and RESUS + VH groups treated with VEH or EIPA. Values are represented as mean ± SEM. (n=6 per VEH treatment group; n=7 per EIPA treatment group; *, p<0.05 versus CONTROL; +, p<0.05 versus RESUS + VH + VEH)
Myosin Light Chain Phosphorylation
As shown in Figure 6, MLC phosphorylation measured 6 hours after surgery was significantly decreased in the RESUS + VH + VEH group as compared to CONTROL + VEH (0.6 ± 0.1 versus 3.6 ± 0.4, respectively). EIPA treatment in the RESUS+VH group partially attenuated the decrease in MLC phosphorylation (1.9 ± 0.5 versus 5.2 ± 1.4 and 3.6 ± 0.4 in RESUS + VH + EIPA versus CONTROL + EIPA and CONTROL + VEH respectively). Values are expressed as the ratio of phosphorylated to un-phosphorylated MLC.
Figure 6.
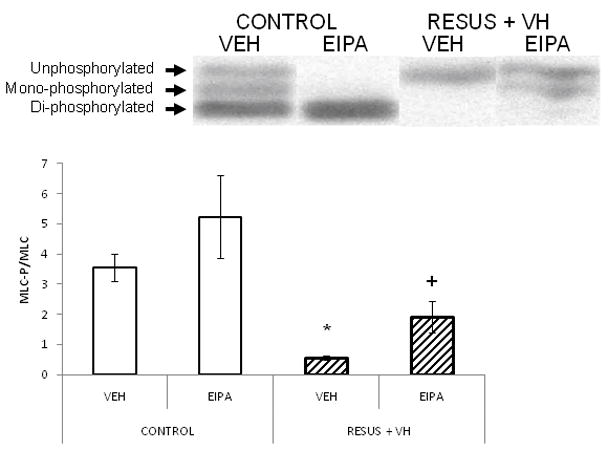
The ratio of phosphorylated myosin light chain to total myosin light chain measured in the ileum 6 hours after surgery in CONTROL and RESUS + VH groups treated with VEH or EIPA. Values are represented as mean ± SEM. (n=6 per VEH treatment group; n=7 per EIPA treatment group; *, p<0.05 versus CONTROL; +, p<0.05 versus RESUS + VH + VEH)
Contractile Activity
Figures 7a and 7b demonstrate basal and carbachol stimulated contractile activity with and without EIPA treatment, respectively, measured 6 hours after surgery. Both basal contractile activity (27.2 ± 10.4 versus 69.3 ± 4.7) and carbachol stimulated contractile activity (17.2 ± 5.4 versus 56.6 ± 8.5) decreased significantly in the edematous RESUS + VH + VEH group as compared to CONTROL + VEH. Treatment with EIPA attenuated the changes in basal (53.3 ± 7.0 versus 27.2 ± 10.4) and carbachol stimulated (48.6 ± 8.1 versus 17.2 ± 5.4) contractile activity (RESUS +VH + EIPA compared to RESUS + VH + VEH). EIPA had no effect in the CONTROL groups with regards to basal (81.3 ± 8.7 versus 69.3 ± 4.7, EIPA versus VEH, respectively) and carbachol stimulated contractile activity (55.7 ± 7.5 versus 56.6 ± 8.5, EIPA versus VEH, respectively). There were no significant changes in contractile frequency (0.6 ± 0, 0.5 ± 0, 0.5 ± 0, and 0.5 ± 0 in CONTROL + VEH, CONTROL + EIPA, RESUS + VH + VEH, and RESUS + VH + EIPA groups, respectively). (Figure 7c) Contractile activity is expressed as stress (g/cm2/s) and frequency as Hz.
Figure 7.
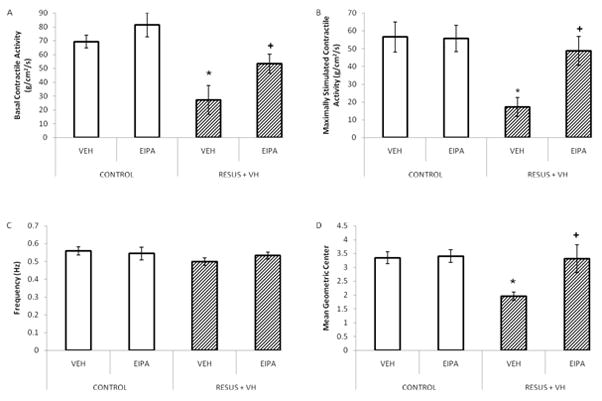
Figure 7a–d: Intestinal function measured in the ileum 6 hours after surgery in CONTROL and RESUS + VH groups treated with VEH or EIPA. Panel A, Basal (un-stimulated) contractile activity; Panel B, Carbachol stimulated contractile activity; Panel C, Contractile frequency; Panel D, Intestinal transit. Values represent mean ± SEM. (Panels A–C, n=6 per VEH treatment group; n=7 per EIPA treatment group; Panel D, n=6 per group; *, p<0.05 versus CONTROL; +, p<0.05 versus RESUS + VH + VEH)
Intestinal Transit
Decreased contractile activity resulted in significantly slower intestinal transit in the RESUS + VH + VEH group (2.0 ± 0.1) compared to CONTROL + VEH (3.3 ± 0.2). (Figure 7d) As with intestinal contractile activity, EIPA treatment significantly attenuated edema-induced slowed intestinal transit (3.3 ± 0.5 versus 2.0 ± 0.1 in RESUS + VH + VEH versus RESUS + VH + EIPA, respectively). (Figure 7d) Values are expressed as mean geometric center.
Discussion
Our data demonstrate that hydrostatic edema is associated with an increase in expression of NHE1-3 and pharmacologic inhibition of NHE with EIPA attenuates intestinal contractile dysfunction induced by edema by modulating STAT-3 phosphorylation, MLC phosphorylation, intestinal contractile activity, and intestinal transit. In previously published data, we have established that intestinal edema causes ileus by decreasing intestinal contractility and MLC phosphorylation. (5–7, 19) The data presented currently builds on our previous work and further demonstrates the potential role of mechanical changes and mechanosensitive channels in edema induced contractile dysfunction. (8, 20)
The increase in NHE expression observed with edema may be secondary to a mechanotransduction related phenomenon. In our animal model, development of edema is rapid (i.e., within 30 minutes) and associated with concurrent early (increased stress, strain, and interstitial pressure) and sustained mechanical changes (altered F:G actin ratios, calponin, and vimentin as well as altered stiffness and residual stress). These changes may serve as a stimulus for alterations in gene expression. (8, 9, 17) NHE has been shown to interact with the actin cytoskeleton. In fibroblasts, NHE1 is linked to the actin cytoskeleton and can affect cytoskeletal organization (21). Conversely, NHE3 activity was shown to be regulated by interaction with the actin cytoskeleton (22). As mentioned above, in our intestinal edema model, the F:G actin ratio changes in response to edema, suggesting cytoskeletal alterations that may serve as a precursor to alteration in NHE expression. (9)
Changes in cytoskeletal elements, such as NHE, may represent the response of the host to injurious stimuli. While the initial response may be protective, changes in these elements result in downstream deleterious effects resulting in ileus. We are therefore interested in both the responsible aspect of the initiating stimuli as well as the downstream effects of the resultant alterations in signaling pathways leading to end organ dysfunction.
In previous studies, we have measured intestinal transit as a functional endpoint of edema induced contractile dysfunction. We have typically measured transit at 6 hours to exclude any confounding effects of anesthesia or bowel manipulation. (6, 9, 10, 16) Therefore, in this study, we measured intestinal transit as well as validated molecular and functional regulators of intestinal transit (i.e., STAT-3, MLC phosphorylation, and intestinal contractility and frequency of contraction) at the 6 hour time point. The ileum of the animals was used for all studies, as we have done in all previous experiments.
The molecular pathway governing smooth muscle contractile activity converges at the level of MLC phosphorylation, and edema-induced intestinal contractile dysfunction has been shown to be associated with decreases in MLC phosphorylation. (5) In previous work, we have demonstrated that MLC phosphorylation is at least partially mediated by STAT-3 activation and that STAT-3 activation is a precursor to decreased MLC phosphorylation. (7) We therefore measured STAT-3 activation and the ratio of phosphorylated to un-phosphorylated MLC in EIPA treated RESUS + VH animals as definable endpoints of pharmacologic inhibition. Other investigators have also shown that NHE inhibition can block STAT-3 activation as well as improve contractility. Pan et. al. showed that the NHE inhibitor, HOE642, blocked stretch-induced activation of STAT-3 in cardiomyocytes. (11) Khan et. al demonstrated that amiloride improved intestinal contractile activity in an experimental colitis model. (23) In this model, NHE1 acting via Interleukin 1-β and mitogen-activated protein (MAP) kinase was implicated. (23) NHE2 inhibition has also been shown to restore intestinal barrier function in ischemia induced intestinal injury. (24) This study further contributes by demonstrating that intestinal edema, in the absence of inflammatory, ischemic, or reperfusion injury, is associated with increased NHE expression, and pharmacologic inhibition of NHE with EIPA results in improved contractile function. The effects of NHE on MLC phosphorylation may be mediated by its effect on STAT-3 activation.
There are several limitations of our study. One of the primary weaknesses of studies involving pharmacologic inhibition is that inhibitors frequently have non-specific effects apart from the ones desired. EIPA was chosen for NHE inhibition for three main reasons. It has inhibitory activity against NHE3 in addition to NHE1 and NHE2. It is more selective than amiloride and has significantly less inhibitory activity against sodium and sodium/calcium channels as compared to other NHE inhibitors. (25) Another limitation was that NHE activity was not measured. It is ideal to confirm that increased expression and/or increased protein levels of NHE correlates with increased activity of the ion channel. We recognize that changes in expression are not necessarily a pre-requisite to changes in activity in ion channels and that activity may be affected without a change in expression. Indeed, in edematous animals, EIPA administration did not decrease NHE1-3 expression or NHE1-3 protein levels (as determined by immunofluorescence), suggesting that pharmacologic inhibition may have affected activity independent of a change in expression or protein levels. (DATA NOT SHOWN) Additionally, the fact that a functional effect of NHE inhibition was seen at 6 hours suggests a change in NHE activity prior to that timepoint, despite a lack of change in expression until 6 hours. The measurement of ion channel activity in tissue, however, poses a unique set of challenges. The majority of the published literature on NHE activity in intestinal tissue involves the use of radioactive sodium (22Na). (24, 26–31) The use of radioactive sodium poses a distinctive set of issues. Extensive lead shielding is necessary to protect investigators secondary to the gamma emission properties. Additionally, and more importantly, a half life of over 2.5 years poses significant issues for the safe storage and disposal of radioactive waste and equipment. NHE activity is widely measured in cells using probes that reflect intracellular pH (most commonly 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF)). The recovery of intracellular pH in response to an acid load is generally considered representative of NHE activity. We are not aware of any published reports of using BCECF as a measure of NHE activity in intestinal tissue. We did attempt to measure NHE activity in the ileum by using BCECF. Although we were able to load BCECF into the tissues with minimal autofluorescence, we were unable to detect a change in fluorescence as determined by ratiometric methods in the presence of media of varying pH’s and nigericin (a K+/H+ ionophore frequently utilized for pH calibration). We do note, however, that multiple investigators have noted that an increase in NHE expression was associated with an increase in NHE activity. (32–34)
In our intestinal edema model, NHE expression changed in the mucosa only while functional changes in intestinal contractile activity (with NHE inhibition) were demonstrated. Conceivably, NHE inhibition can indirectly affect intestinal contractile activity via changes in intestinal barrier function. Alternatively, post-translational changes resulting in increased NHE activity may have occurred in the intestinal smooth muscle with intestinal edema development. In this case, NHE inhibition may directly affect intestinal smooth muscle NHE activity and consequently intestinal contractile activity.
Figure 8 shows a proposed model of the effects of intestinal edema development on intestinal contractile activity. We demonstrated that intestinal interstitial edema induces changes in NHE mRNA and protein levels. Pharmacological inhibition of NHE attenuated the changes in intestinal contractile activity induced by intestinal edema suggesting that NHE may adversely affect intestinal contractile function in edema, possibly through a STAT-3 mediated pathway.
Figure 8.
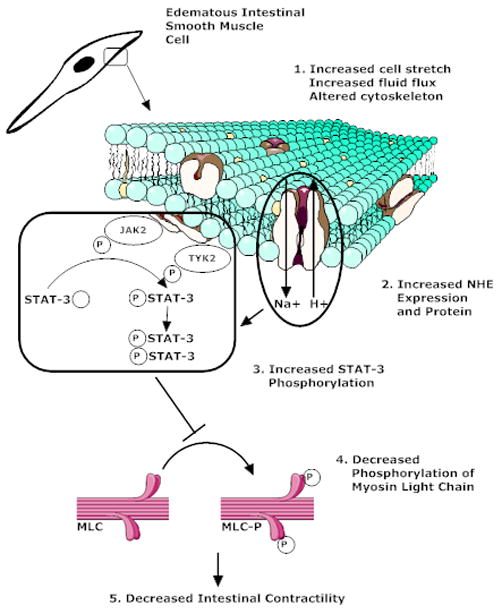
Suggested model of the effects of intestinal interstitial edema development on intestinal contractile activity. Edema alters both the mechanical properties of the tissue and the cytoskeletal properties in the mucosal and muscle layers of the small intestine. These changes may induce increased NHE activity in the mucosa that eventually lead to sequential increases in intestinal smooth muscle STAT3 phosphorylation, decreased myosin light chain phosphorylation and, finally, decreased intestinal contractile activity.
Acknowledgments
SOURCES OF SUPPORT: NIH Grants KO1 DK 070758, RO1 HL 092916, T32 GM08792 and P50 GM 38529; Texas Higher Education Coordinating Board
The authors would like to acknowledge the technical expertise of Karina Kislitsyna, BS and Hasen Xue, MD.
Footnotes
Conflict Statement: There are no known conflicts between the authors and the information presented in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schuster TG, Montie JE. Postoperative ileus after abdominal surgery. Urology. 2002;59(4):465–471. doi: 10.1016/s0090-4295(01)01561-8. [DOI] [PubMed] [Google Scholar]
- 2.Kehlet H, Holte K. Review of postoperative ileus. Am J Surg. 2001;182(5A Suppl):3S–10S. doi: 10.1016/s0002-9610(01)00781-4. [DOI] [PubMed] [Google Scholar]
- 3.Bauer AJ, Schwarz NT, Moore BA, et al. Ileus in critical illness: mechanisms and management. Curr Opin Crit Care. 2002;8(2):152–157. doi: 10.1097/00075198-200204000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Balogh Z, McKinley BA, Cox CS, Jr, et al. Abdominal compartment syndrome: the cause or effect of postinjury multiple organ failure. Shock. 2003;20(6):483–492. doi: 10.1097/01.shk.0000093346.68755.43. [DOI] [PubMed] [Google Scholar]
- 5.Uray KS, Laine GA, Xue H, et al. Intestinal edema decreases intestinal contractile activity via decreased myosin light chain phosphorylation. Crit Care Med. 2006;34(10):2630–2637. doi: 10.1097/01.CCM.0000239195.06781.8C. [DOI] [PubMed] [Google Scholar]
- 6.Moore-Olufemi SD, Xue H, Attuwaybi BO, et al. Resuscitation-induced gut edema and intestinal dysfunction. J Trauma. 2005;58(2):264–270. doi: 10.1097/01.ta.0000133571.64393.d2. [DOI] [PubMed] [Google Scholar]
- 7.Uray KS, Laine GA, Xue H, et al. Edema-induced intestinal dysfunction is mediated by STAT3 activation. Shock. 2007;28(2):239–244. doi: 10.1097/shk.0b013e318033eaae. [DOI] [PubMed] [Google Scholar]
- 8.Cox CS, Jr, Radhakrishnan R, Villarrubia L, et al. Hypertonic saline modulation of intestinal tissue stress and fluid balance. Shock. 2008;29(5):598–602. doi: 10.1097/SHK.0b013e318157eba7. [DOI] [PubMed] [Google Scholar]
- 9.Radhakrishnan RS, Radhakrishnan HR, Xue H, et al. Hypertonic saline reverses stiffness in a Sprague-Dawley rat model of acute intestinal edema, leading to improved intestinal function. Crit Care Med. 2007;35(2):538–543. doi: 10.1097/01.CCM.0000254330.39804.9C. [DOI] [PubMed] [Google Scholar]
- 10.Radhakrishnan RS, Xue H, Moore-Olufemi SD, et al. Hypertonic saline resuscitation prevents hydrostatically induced intestinal edema and ileus. Crit Care Med. 2006;34(6):1713–1718. doi: 10.1097/01.CCM.0000218811.39686.3D. [DOI] [PubMed] [Google Scholar]
- 11.Pan J, Fukuda K, Saito M, et al. Mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes. Circ Res. 1999;84(10):1127–1136. doi: 10.1161/01.res.84.10.1127. [DOI] [PubMed] [Google Scholar]
- 12.Cingolani HE, Perez NG, Pieske B, et al. Stretch-elicited Na+/H+ exchanger activation: the autocrine/paracrine loop and its mechanical counterpart. Cardiovasc Res. 2003;57(4):953–960. doi: 10.1016/s0008-6363(02)00768-x. [DOI] [PubMed] [Google Scholar]
- 13.Chang E, O’Donnell ME, Barakat AI. Shear stress and 17beta-estradiol modulate cerebral microvascular endothelial Na-K-Cl cotransporter and Na/H exchanger protein levels. Am J Physiol Cell Physiol. 2008;294(1):C363–371. doi: 10.1152/ajpcell.00045.2007. [DOI] [PubMed] [Google Scholar]
- 14.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Dou K, Song Z, et al. The Na+/H+ exchange inhibitor: a new therapeutic approach for hepatic ischemia injury in rats. Transplant Proc. 2003;35(8):3134–3135. doi: 10.1016/j.transproceed.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 16.Moore-Olufemi SD, Xue H, Allen SJ, et al. Inhibition of intestinal transit by resuscitation-induced gut edema is reversed by L-NIL. J Surg Res. 2005;129(1):1–5. doi: 10.1016/j.jss.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 17.Radhakrishnan RS, Xue H, Weisbrodt N, et al. Resuscitation-induced intestinal edema decreases the stiffness and residual stress of the intestine. Shock. 2005;24(2):165–170. doi: 10.1097/01.shk.0000168873.45283.4c. [DOI] [PubMed] [Google Scholar]
- 18.Hassoun HT, Weisbrodt NW, Mercer DW, et al. Inducible nitric oxide synthase mediates gut ischemia/reperfusion-induced ileus only after severe insults. J Surg Res. 2001;97(2):150–154. doi: 10.1006/jsre.2001.6140. [DOI] [PubMed] [Google Scholar]
- 19.Wehner S, Schwarz NT, Hundsdoerfer R, et al. Induction of IL-6 within the rodent intestinal muscularis after intestinal surgical stress. Surgery. 2005;137(4):436–446. doi: 10.1016/j.surg.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Shah SK, Fogle LN, Aroom KR, Gill BS, Uray KS, Laine GA, Stewart RH, Cox CS. Mechanotransduction as a mechanistic explanation for edema induced intestinal dysfunction. J Am Coll Surg. 2009;209(3S):S43. [Google Scholar]
- 21.Denker SP, Huang DC, Orlowski J, et al. Direct binding of the Na--H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H(+) translocation. Mol Cell. 2000;6(6):1425–1436. doi: 10.1016/s1097-2765(00)00139-8. [DOI] [PubMed] [Google Scholar]
- 22.Kurashima K, D’Souza S, Szaszi K, et al. The apical Na(+)/H(+) exchanger isoform NHE3 is regulated by the actin cytoskeleton. J Biol Chem. 1999;274(42):29843–29849. doi: 10.1074/jbc.274.42.29843. [DOI] [PubMed] [Google Scholar]
- 23.Khan I, Oriowo MA, Anim JT. Amelioration of experimental colitis by Na-H exchanger-1 inhibitor amiloride is associated with reversal of IL-1ss and ERK mitogen-activated protein kinase. Scand J Gastroenterol. 2005;40(5):578–585. doi: 10.1080/00365520510012352. [DOI] [PubMed] [Google Scholar]
- 24.Moeser AJ, Nighot PK, Ryan KA, et al. Prostaglandin-mediated inhibition of Na+/H+ exchanger isoform 2 stimulates recovery of barrier function in ischemia-injured intestine. Am J Physiol Gastrointest Liver Physiol. 2006;291(5):G885–894. doi: 10.1152/ajpgi.00380.2005. [DOI] [PubMed] [Google Scholar]
- 25.Masereel B, Pochet L, Laeckmann D. An overview of inhibitors of Na(+)/H(+) exchanger. Eur J Med Chem. 2003;38(6):547–554. doi: 10.1016/s0223-5234(03)00100-4. [DOI] [PubMed] [Google Scholar]
- 26.Subramanya SB, Rajendran VM, Srinivasan P, et al. Differential regulation of cholera toxin-inhibited Na-H exchange isoforms by butyrate in rat ileum. Am J Physiol Gastrointest Liver Physiol. 2007;293(4):G857–863. doi: 10.1152/ajpgi.00462.2006. [DOI] [PubMed] [Google Scholar]
- 27.Narins SC, Ramakrishnan R, Park EH, et al. Protein kinase C-alpha regulation of gallbladder Na+ transport becomes progressively more dysfunctional during gallstone formation. J Lab Clin Med. 2005;146(4):227–237. doi: 10.1016/j.lab.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 28.Broere N, Chen M, Cinar A, et al. Defective jejunal and colonic salt absorption and alteredNa(+)/H (+) exchanger 3 (NHE3) activity in NHE regulatory factor 1 (NHERF1) adaptor protein-deficient mice. Pflugers Arch. 2009;457(5):1079–1091. doi: 10.1007/s00424-008-0579-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musch MW, Bookstein C, Rocha F, et al. Region-specific adaptation of apical Na/H exchangers after extensive proximal small bowel resection. Am J Physiol Gastrointest Liver Physiol. 2002;283(4):G975–985. doi: 10.1152/ajpgi.00528.2001. [DOI] [PubMed] [Google Scholar]
- 30.Cho JH, Musch MW, DePaoli AM, et al. Glucocorticoids regulate Na+/H+ exchange expression and activity in region- and tissue-specific manner. Am J Physiol. 1994;267(3 Pt 1):C796–803. doi: 10.1152/ajpcell.1994.267.3.C796. [DOI] [PubMed] [Google Scholar]
- 31.Lucioni A, Womack C, Musch MW, et al. Metabolic acidosis in rats increases intestinal NHE2 and NHE3 expression and function. Am J Physiol Gastrointest Liver Physiol. 2002;283(1):G51–56. doi: 10.1152/ajpgi.00529.2001. [DOI] [PubMed] [Google Scholar]
- 32.Damkier HH, Prasad V, Hubner CA, et al. Nhe1 is a luminal Na+/H+ exchanger in mouse choroid plexus and is targeted to the basolateral membrane in Ncbe/Nbcn2-null mice. Am J Physiol Cell Physiol. 2009;296(6):C1291–1300. doi: 10.1152/ajpcell.00062.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu H, Collins JF, Bai L, et al. Epidermal growth factor regulation of rat NHE2 gene expression. Am J Physiol Cell Physiol. 2001;281(2):C504–513. doi: 10.1152/ajpcell.2001.281.2.C504. [DOI] [PubMed] [Google Scholar]
- 34.Rocha F, Musch MW, Lishanskiy L, et al. IFN-gamma downregulates expression of Na(+)/H(+) exchangers NHE2 and NHE3 in rat intestine and human Caco-2/bbe cells. Am J Physiol Cell Physiol. 2001;280(5):C1224–1232. doi: 10.1152/ajpcell.2001.280.5.C1224. [DOI] [PubMed] [Google Scholar]