

Abstract
Mice lacking the Girk2 subunit of G protein-gated inwardly-rectifying K+ (Girk) channels exhibit dopamine-dependent hyperactivity and elevated responses to drugs that stimulate dopamine neurotransmission. The dopamine-dependent phenotypes seen in Girk2−/− mice could reflect increased intrinsic excitability of or diminished inhibitory feedback to midbrain dopamine neurons, or secondary adaptations triggered by Girk2 ablation. We addressed these possibilities by evaluating Girk−/− mice in behavioral, electrophysiological, and cell biological assays centered on the mesolimbic dopamine system. Despite differences in the contribution of Girk1 and Girk2 subunits to Girk signaling in midbrain dopamine neurons, Girk1−/− and Girk2−/− mice exhibited comparable baseline hyperactivities and enhanced responses to cocaine. Girk ablation also correlated with altered afferent input to dopamine neurons in the ventral tegmental area. Dopamine neurons from Girk1−/− and Girk2−/− mice exhibited elevated glutamatergic neurotransmission, paralleled by increased synaptic levels of AMPA glutamate receptors. In addition, synapse density, AMPA receptor levels, and glutamatergic neurotransmission were elevated in medium spiny neurons of the nucleus accumbens from Girk1−/− and Girk2−/− mice. We conclude that dopamine–dependent phenotypes in Girk2−/− mice are not solely attributable to a loss of Girk signaling in dopamine neurons, and likely involve secondary adaptations facilitating glutamatergic signaling in the mesolimbic reward system.
Keywords: Cocaine, dopamine, glutamate, plasticity, knockout
INTRODUCTION
The mesolimbic dopamine (DA) system consists of projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAcc) and mediates in part the motor-stimulatory and reinforcing effects of drugs of abuse (Nestler 2004; Wise 2004; Bjorklund and Dunnett 2007). Though drugs of abuse have distinct molecular targets, they share the ability to increase DA levels in the NAcc (Luscher and Ungless 2006). Cocaine, for example, elevates NAcc DA levels by blocking the re-uptake of released DA. The physiological relevance of this effect and location is underscored by the impact of destruction of the dopaminergic projection from the VTA to NAcc, which includes hypoactivity and insensitivity to the motor-stimulatory effects of cocaine (Koob et al. 1981).
In addition to potentiating DA signaling within the NAcc, cocaine also triggers “long-loop” and auto-inhibitory feedback to the VTA mediated by GABAB (GABABR) and D2 DA (D2R) receptors, respectively. G protein-gated inwardly-rectifying K+ (Girk/KIR3) channels are expressed in VTA DA neurons and mediate the postsynaptic inhibitory effect of GABABR and D2R activation (Beckstead et al. 2004; Cruz et al. 2004; Labouebe et al. 2007). The prototypical neuronal Girk channel is a heterotetramer formed by Girk1 and Girk2 subunits (Karschin et al. 1996; Liao et al. 1996; Luscher et al. 1997; Koyrakh et al. 2005). Midbrain DA neurons of the VTA and substantia nigra (SN), however, do not express Girk1 (Karschin et al. 1996; Inanobe et al. 1999; Cruz et al. 2004; Labouebe et al. 2007). Thus, genetic ablation of Girk2 correlates with blunted GABABR- and D2R-dependent postsynaptic inhibition in VTA and SN DA neurons, whereas loss of Girk1 has no effect (Beckstead et al. 2004; Cruz et al. 2004; Koyrakh et al. 2005; Labouebe et al. 2007).
Girk2−/− mice exhibit a basal hyperactivity that was normalized by the D1 DA receptor (D1R) antagonist SCH 23390 (Blednov et al. 2001; Blednov et al. 2002), indicating that Girk2−/− mice exhibit elevated DA signaling. Girk2−/− mice also exhibit enhanced responses to the acute motor-stimulatory effects of cocaine and the D1R partial agonist SKF 38393 (Blednov et al. 2002; Morgan et al. 2003). One interpretation of these data is that Girk2 ablation diminishes inhibitory feedback to VTA DA neurons and/or triggers elevated intrinsic excitability of VTA DA neurons, leading to elevated DA release in the NAcc and striatum. Secondary adaptations, perhaps linked to alterations in excitatory signaling, might also explain the DA-related phenotypes reported in Girk2−/− mice. Indeed, such adaptations are triggered by drugs of abuse and stress, and can promote persistent adaptations in the NAcc that are implicated in behavioral sensitization (Kauer and Malenka 2007; Engblom et al. 2008; Thomas et al. 2008; Zweifel et al. 2008; Heshmati 2009).
Here, we sought insight into the DA-related phenotypes reported in Girk2−/− mice. We reasoned that if these phenotypes reflect the loss of Girk signaling in VTA DA neurons, they would be observed in Girk2−/− but not Girk1−/− mice. We applied a combination of behavioral, electrophysiological, and cell biological approaches to examine the impact of Girk1 and Girk2 ablation on neurotransmission within the mesolimbic reward pathway. Our findings indicate that loss of Girk signaling in midbrain DA neurons is not the primary cause of the DA-related phenotypes reported in Girk2−/− mice, and suggest instead that secondary adaptations that facilitate glutamatergic neurotransmission may play a role.
MATERIALS AND METHODS
Drugs
Cocaine hydrochloride, baclofen, tetrodotoxin, picrotoxin, lidocaine hydrochloride and kynurenic acid were purchased from Sigma (St. Louis, MO). CGP54626 was purchased from Tocris (Ellisville, MO). rTertiapinQ was purchased from Alomone Labs, Inc. (Jerusalem, Israel).
Animals
All animal use was reviewed and approved by the Institutional Animal Care and Use Committee of the University of Minnesota. Efforts were made to minimize the pain and discomfort of the animals throughout the study. Adult mice were housed on a 12 h light/dark cycle, with food and water available ad libitum. The generation of Girk−/− was described previously (Signorini et al. 1997; Bettahi et al. 2002; Torrecilla et al. 2002). The Girk null mutations were backcrossed through >12 rounds against the C57BL/6 strain prior to experimentation.
Locomotor activity
Baseline and cocaine-induced motor activity were assessed in wild-type and Girk−/− mice (4-10 wk) using automated open field environments, as described (Pravetoni and Wickman 2008). Baseline activity data were obtained during 60-min daily sessions over a consecutive 3-d period. On day 3, subjects were given injections of saline (i.p.) and placed in the open field for 60-min. Subsequently, subjects were randomly assigned to groups that received saline or one of four cocaine doses (3, 15, 30, or 60 mg/kg, i.p.). Total distance traveled after the injection was monitored for 60 min.
Electrophysiology
Coronal slices containing the VTA (200-300 μm) and sagittal slices containing the NAcc (240 μm) were prepared from drug-naïve wild-type and Girk−/− mice (4-6 weeks), and allowed to recover in oxygenated ACSF at room temperature for >1 h. Slices were transferred to recording chambers and perfused with oxygenated ACSF (+/− drugs) at a flow rate of approximately 2-2.5 mL/min during experiments. Currents, resistances, and potentials were measured using Multiclamp 700A amplifiers and pCLAMP v.9 software (Molecular Devices; Foster City CA) and stored on hard disk. All measured and command potentials factored in junction potentials predicted using JPCalc software (Molecular Devices).
Studies involving VTA DA neurons were performed at 32-34°C, with borosilicate (3-5 MΩ) electrodes containing (in mM): 140 K-gluconate, 2 MgCl2, 1.1 EGTA, 5 HEPES, 2 Na-ATP, 0.3 Na3GTP, and 5 phosphocreatine, pH 7.4. For sIPSC measurements, KCl replaced K-gluconate in the pipette solution. Upon achieving whole-cell access, the current response to a 1-s voltage-ramp (−60 to −120 mV) was measured to assess Ih presence and size. Following a 5-min stabilization period, spontaneous action potential frequency and duration were measured in whole-cell current clamp mode (I=0). Cells exhibiting relatively large capacitance (>50 pF), Ih current (>100 pA at −120 mV), moderate and stable spontaneous activity (1-5 Hz), and long action potential durations (>2.5 s) were considered to be putative DA neurons. Indeed, these properties correlate well with expression of tyrosine hydroxylase and Pitx3, selective markers of DA neurons (Grace and Onn 1989; Johnson and North 1992; Momiyama et al. 1996; Cameron et al. 1997; Klink et al. 2001; Neuhoff et al. 2002; Mathon et al. 2005; Labouebe et al. 2007).
Changes in whole-cell holding current evoked by baclofen were measured at a holding potential (Vhold) of −60 mV. The holding current, input resistance (Rin), and series resistance (RA) values were monitored throughout these experiments by tracking responses to periodic (0.2 Hz) voltage steps (−5 mV, 800 ms). Only experiments with stable (<20% variation) and low series resistances (<20 MΩ) were included in the final data set. The GABABR antagonist CGP54626 (2 μM) was used to verify the receptor-dependence of baclofen-evoked responses. sIPSCs were measured over a 2-3 min period at Vhold = −70 mV, in the presence of kynurenic acid (2 mM). sEPSCs were measured over 2-3 min period at Vhold = −80 mV, in the presence of picrotoxin (100 μM). All data were low-pass filtered at 2 kHz, digitized at 10 kHz, and analyzed using pCLAMP v.9 software (Molecular Devices).
Studies involving NAcc medium spiny neurons were conducted at room temperature, and recording electrodes contained the K-gluconate pipette solution described above or an alternative solution lacking K+ (in mM): 117 cesium gluconate, 2.8 NaCl, 20 HEPES, 0.4 EGTA, 5 tetraethylammonium-Cl, 2 MgATP, and 0.3 MgGTP, pH 7.2–7.4. mEPSC amplitudes and frequencies measured in wild-type NAcc neurons did not differ for these two pipette solutions and as such, data were pooled. Medium spiny neurons were identified by their morphology and hyperpolarized resting membrane potential (−75 to −85 mV). mEPSCs were measured at Vhold = −80 mV in the presence of picrotoxin (100 μM) and either lidocaine hydrochloride (0.6-0.8 mM) or TTX (1 μM). mEPSC data were filtered at 2 kHz, digitized at 5-10 kHz, and collected and analyzed using custom software (Igor Pro; Wavemetrics, Lake Oswego, OR).
AMPA receptor subunit quantification
Synaptic levels of GluR1 and GluR2/3 subunits were measured in the VTA and NAcc from wild-type and Girk−/− mice (8-12 wk) via post-embedding immunoelectron microscopy. Tissue was prepared as described (Lujan et al. 1996); sections containing of the VTA and NAcc were then cut at 500 μm incubated in a 1 M sucrose/PBS solution overnight, slammed onto copper blocks cooled in liquid nitrogen, and embedded in Lowicryl HM20 (TAAB Laboratories; Aldermaston, UK) after freeze substitution with methanol. Ultrathin sections (80 nm) from three Lowicryl-embedded blocks were incubated for 45 min on coated nickel grids with drops of blocking solution containing: 0.05 M TBS, 0.9% NaCl, 0.03% Triton X-100, and 2% albumin. Grids were then incubated at room temperature overnight in blocking solution containing rabbit polyclonal antibodies (10 μg/ml each) against GluR1 (AB1504, Millipore; Billerica, MA) or GluR2/3 (AB1506, Millipore). For VTA experiments, a mouse monoclonal antibody directed against TH (mAb 6D7, EMD Biosciences; San Diego, CA) was used to identify DA neurons. After washing in TBS, grids were incubated for 2 hr in drops of goat anti-rabbit IgG conjugated to 10 nm colloidal gold particles (BioCell International; Cardiff, UK) for experiments involving the NAcc, or a mixture of goat anti-rabbit IgG conjugated to 10 nm colloidal gold particles and goat anti-mouse IgG conjugated to 20 nm colloidal gold particles for experiments involving the VTA; in all cases, secondary antibodies were diluted 1:80 in a 0.05 M TBS solution containing 2% normal human serum and 0.5% polyethylene glycol. Grids were then washed in TBS for 30 min and counterstained for electron microscopy with saturated aqueous uranyl acetate and lead citrate.
Ultrastructural analyses were performed with a Jeol 1010 electron microscope (Peabody, MA). Electron photomicrographs were captured with a CCD camera (MegaView III Soft Imaging System; Munster, Germany). Digitized electron images were modified for brightness and contrast with Adobe Photoshop version 7.0. Quantification of GluR1 and GluR2/3 immunolabeling was performed in ultrathin sections obtained from 3 separate panels of wild-type, Girk1−/−, and Girk2−/− mice. Areas in the VTA and Nacc were chosen at random and images were captured at a magnification of 50,000X. In the VTA, only those synapses consisting of an axon terminal apposed to a PSD located on a dendritic shaft labelled for TH were included in the analysis. In the Nacc, only those synapses consisting of an axon terminal apposed to a PSD present on a dendritic spine were included in the analysis. In both locations, the length of the PSD was measured and the number of immunoparticles per PSD was tabulated. There was no impact of Girk ablation on PSD length (not shown).
Synapse quantification
Mice (8-10 weeks) were anesthetized and perfused with a solution containing 2% paraformaldehyde and 1.5% glutaraldehyde, as described (Marker et al. 2005). Coronal sections (60 μm) were cut through the level of the NAcc using a vibrating microtome. After several washes in PB, sections were post-fixed with 1% osmium tetraoxide in PB and block-stained with 1% uranyl acetate in distilled water. Sections were then dehydrated in ascending series of ethanol (to 100%) followed by propylene oxide and flat-embedded on glass slides in Durcupan (Sigma; St. Louis, MO). For quantitative analysis of the density of spines/synapses in the NAcc, sampling fields were chosen by using the systematic random sampling method (Geinisman et al. 1996). After collection of semi-thin sections and determination of sample sites, serial ultrathin sections (80 nm thick) of the neuropil containing the sampling fields were collected from three different parts of the NAcc of three different animals. Sections were placed on slot grids and stained with Reynolds’ lead citrate.
Ultrastructural analyses were performed on a Jeol-1010 electron microscope from sections at a magnification of 40,000X. Spine/synapse density was estimated by using the unbiased physical dissector method. In brief, data were collected from pairs of serial sections (dissectors). Synapses were identified on the reference section by the presence of: 1) a postsynaptic density (PSD), 2) synaptic vesicles at the presynaptic terminal, and 3) opposing membranes between the pre- and the postsynaptic terminals. Only PSDs found on dendritic spines and only one PSD per spine were analyzed. An unbiased counting frame was superimposed over each of the two micrographs, and the PSD was used as a counting unit. Synapses were labeled on the reference section micrograph if their PSD profiles were located either entirely or partly within the frame and did not intersect the forbidden edges of the frame and its extensions. Finally, only synapses that had a PSD profile in the reference, but not in the look-up section, were counted. At least seven neuropil fields (each 163.65 μm2) were photographed from each animal, corresponding to a total section area of 3.437 μm2. Synapse density was calculated as the number of synapses per counting divided by the product of area of the counting frame and the height of the dissector.
Data analysis
Data are presented throughout as the mean±SEM. Statistical analyses were performed using GraphPad Prism (GraphPad Software; La Jolla, CA). Quantal events were analyzed using Minianalysis software (Synaptosoft, Decatur, GA). The impact of genotype on VTA DA neuron characteristics, baclofen-induced current amplitude, EPSC and IPSC frequency and amplitude, synaptic level of AMPA subunits, synapse density, and cocaine-induced motor activity were assessed using one-way ANOVA, followed by Tukey’s Multiple Comparison post hoc test when appropriate. For all statistical comparisons, P values of less than 0.05 were considered to reflect significant differences between groups.
RESULTS
We first measured basal activity levels and the motor-stimulatory effect of systemic cocaine (0-60 mg/kg) in wild-type and Girk−/− mice in a standard open-field environment (Fig. 1). Girk1−/− and Girk2−/− mice were similarly hyperactive during their initial and subsequent exposures to the novel environment (Fig. 1A). Cocaine triggered dose-dependent increases in motor activity in all groups (Fig. 1B). While none of the genotypes responded significantly to 3 mg/kg cocaine, Girk1−/− and Girk2−/− mice showed comparable and enhanced responses to 15 mg/kg cocaine. Maximal responses did not differ across genotypes, though they were observed at 15 mg/kg for Girk2−/− mice and at 30 mg/kg for the other three groups. Responses of Girk2−/− mice to 30 mg/kg were notably, though not significantly, smaller than those seen at the 15 mg/kg. Similarly, 60 mg/kg cocaine evoked sub-maximal responses in wild-type, Girk1−/−, and Girk3−/− mice.
Figure 1. Baseline and cocaine-induced motor activity in wild-type and Girk−/− mice.
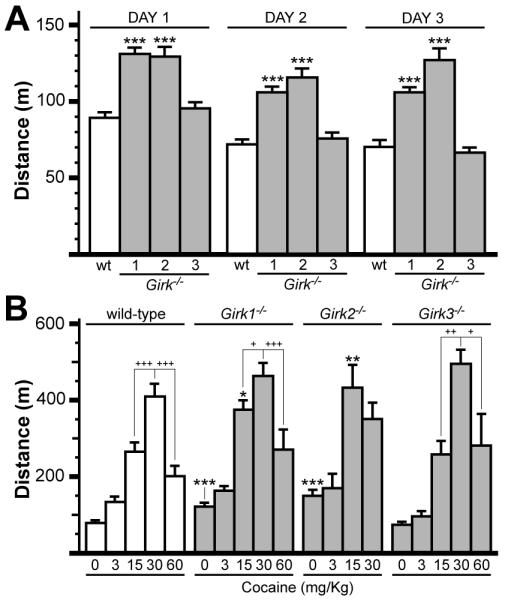
A) Total distance traveled (m) for wild-type (wt), Girk1−/−, Girk2−/−, and Girk3−/− mice in open-field environments during 1-h sessions on 3 consecutive days (n=36-81 per genotype). Statistical symbols: *** − P<0.001, vs. wild-type (within day). B) Cocaine-induced motor activity in wild-type and Girk−/− mice. Subjects were challenged with saline or one of 4 cocaine doses (3, 15, 30, 60 mg/kg i.p.) and total distance traveled (m) was measured during the 60-min post-injection window. Cocaine triggered dose-dependent increases in motor activity in wild-type (F4,125=54.6; P<0.001), Girk1−/− (F4,130=65.3; P<0.001), Girk2−/− (F3,52=14.4; P<0.001), and Girk3−/− (F4,81=65.3; P<0.001) mice. Genotype-dependent differences in cocaine-induced responses were observed following injection of 0 (F3,192=23.0; P<0.001) and 15 mg/kg cocaine (F3,87=6.1; P<0.001), but not after the 3 (F3,41=2.0; P=0.1), 30 (F3,43=1.8; P=0.2), or 60 (F3,41=2.0; P=0.1) mg/kg injections (n=6-30 per genotype and dose). Statistical symbols: *, **, *** − P<0.05, 0.01, and 0.001, respectively, vs. wild-type (within dose); within-genotype comparisons are displayed to convey differences observed at the higher cocaine doses: +, ++, +++ − P<0.05, 0.01, and 0.001, respectively, relative to the response at 30 mg/kg cocaine.
Given the differential expression patterns of Girk1 and Girk2 in midbrain DA neurons, the comparable profiles for Girk1−/− and Girk2−/− mice with respect to basal and cocaine-induced motor activity argue that the loss of Girk signaling in these neurons is not the primary cause. Thus, we asked next whether Girk ablation altered other aspects of midbrain DA neuron physiology. We first examined the impact of Girk subunit ablation on the spontaneous activity and afferent input to putative DA neurons in acutely-isolated slices of the VTA. VTA DA neurons were identified using morphological and electrophysiological criteria (Materials and Methods) that we demonstrated previously correlated well with expression of Pitx3 (Labouebe et al. 2007), a DA neuron-specific transcription factor. These criteria also correlated well with robust GABABR-dependent somatodendritic outward currents in slices from wild-type mice (Fig. 2A). Peak outward currents evoked by the GABABR agonist baclofen measured in slices from Girk1−/− and Girk3−/− mice were indistinguishable from wild-type, whereas currents in slices from Girk2−/− mice were dramatically attenuated (Fig. 2A,B).
Figure 2. Functional characterization of VTA DA neurons from wild-type and Girk−/− mice.
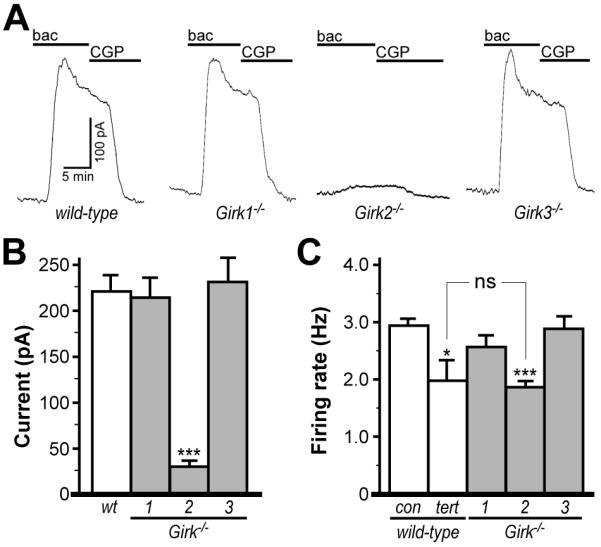
A) Outward currents evoked by baclofen (bac, 200 μM) in VTA DA neurons from wild-type, Girk1−/−, Girk2−/−, and Girk3−/− mice. Outward currents corresponded with a decrease in input resistance and were sensitive to 0.3 external Ba2+ (not shown), and were reversed by the GABABR antagonist CGP54626 (CGP, 2 μM). B) Summary of baclofen-induced outward currents in VTA DA neurons from wild-type (219±17 pA), Girk1−/− (212±21 pA), Girk2−/− mice (31±4 pA), and Girk3−/− (229±37 pA) mice (n=6-22 per genotype). Genotype-dependent differences in peak baclofen-induced current amplitude were evident (F3,39=11.9, P<0.001). Symbols: ***, P<0.001 vs. wild-type. C) A significant effect of group on firing rate was observed (F4,147=10.5, P<0.001) with respect to ex vivo firing frequencies of VTA DA neurons from wild-type mice at baseline (con, 2.93±0.12 Hz) and following application of the Girk channel blocker tertiapin (tert, 1.98±0.34 Hz), and at baseline in slices from Girk1−/− (2.55±0.20 Hz), Girk2−/− mice (1.84±0.11 Hz) and Girk3−/− (2.87±0.22 Hz) mice (n=9-49 per group). Note that there was no significant (ns) difference in firing rates of VTA DA neurons from Girk2−/− mice and wild-type neurons treated with tertiapin. Symbols: *, ***, P<0.05 and 0.001, respectively, vs. wild-type.
Girk ablation had no impact on apparent capacitance, input resistance, Ih current amplitude, or action potential duration in putative VTA DA neurons (not shown). Genotype-dependent differences in spontaneous rates, however, were evident. Surprisingly, VTA DA neurons from Girk2−/− mice exhibited lower rates than wild-type, Girk1−/−, and Girk3−/− counterparts (Fig. 2C). Firing rates of VTA DA neurons from wild-type slices were consistently and reversibly lowered in the presence of the Girk channel blocker tertiapin, suggesting that the lower firing rates observed in DA neurons from Girk2−/− mice were largely attributable to the loss of Girk channel function (Fig. 2C). Indeed, tertiapin application had no effect on firing rates of VTA DA neurons in slices from Girk2−/− mice (not shown).
We hypothesized that VTA DA neurons in Girk2−/− mice receive elevated inhibitory input from local GABA interneurons. To test this hypothesis, we first recorded spontaneous inhibitory postsynaptic currents (sIPSCs) in VTA DA neurons from wild-type and Girk−/− mice (Fig. 3). The frequency and amplitude of sIPSCs measured in wild-type mice were consistent with previous reports (Mathon et al. 2005). Consistent with our prediction, sIPSC frequency was elevated in DA neurons from Girk2−/− mice, relative to frequencies measured in wild-type, Girk1−/−, and Girk3−/− mice (Fig. 3A,B). sIPSC amplitudes did not differ across genotypes (Fig. 3C). Second, we examined the impact of picrotoxin on the spontaneous activity of VTA DA neurons in slices from wild-type and Girk−/− mice, reasoning that GABAA receptor blockade should normalize firing rates if the lower basal firing rates seen in Girk2−/− mice were attributable to elevated GABAergic input. Indeed, VTA DA neuron firing rates did not differ across genotypes in the presence of picrotoxin application (Fig. 3D).
Figure 3. Inhibitory input to VTA DA neurons in wild-type and Girk−/− mice.

A) Representative traces of sIPSCs measured in VTA DA neurons from wild-type (upper) and Girk2−/− mice (middle); sIPSCs were blocked by the GABA receptor antagonist picrotoxin (100 μM, lower). Scale bars: 40 pA/5s. Summary histograms of sIPSC frequency and amplitude in VTA DA neurons from wild-type (wt, 1.2±0.2 Hz, 34.4±5.7 pA), Girk1−/− (1.7±0.2 Hz, 26.9±3.1 pA), Girk2−/− (2.9±0.3 Hz, 29.2±2.2 pA), and Girk3−/− (1.2±0.3 Hz, 36.6±5.5 pA), mice are shown in panels B and C, respectively (n=7-10 per genotype). A significant effect of genotype was observed for sIPSC frequency (F3,29=10.1: P<0.001) but not amplitude (F3,29=1.1: P=0.4). Symbols: *** P<0.001 vs. wild-type and Girk3−/−; + P<0.05, Girk1−/− vs. Girk2−/−. D) Firing frequency of VTA DA neurons from wild-type and Girk−/− mice (n=8-21 per genotype) were measured at baseline (left) and following administration of 100 μM picrotoxin (right). An effect of genotype on firing rates was observed at baseline (F3,56=10.0; P<0.001) but not after picrotoxin application (F3,56=1.0: P=0.4). Symbols: ** P<0.01 vs. wild-type and Girk3−/−; + P<0.05, Girk1−/− vs. Girk2−/−.
Elevated inhibitory input to, and lower ex vivo firing rates of, VTA DA neurons in Girk2−/− mice is inconsistent with the DA-related phenotypes shown in Fig. 1 and reported previously (Blednov et al. 2001; Blednov et al. 2002; Morgan et al. 2003). We reasoned that Girk2 ablation may also trigger adaptations facilitating excitatory neurotransmission in VTA DA neurons, and that the influence of these adaptations would be muted due to the transection of excitatory afferents that occurs during slice preparation. To address this possibility, we first measured spontaneous excitatory postsynaptic currents (sEPSCs) in VTA DA neurons from wild-type and Girk−/− mice (Fig. 4). sEPSCs were readily observed in VTA DA neurons from wild-type mice (Fig. 4A), with amplitudes and frequencies consistent with previous reports using similar conditions (Engblom et al. 2008; Zweifel et al. 2008). sEPSC frequency was elevated significantly and similarly in VTA DA neurons from Girk1−/− and Girk2−/− mice, but not Girk3−/− mice (Fig. 4B). Interestingly, sEPSC amplitudes were elevated significantly in Girk2−/− mice (Fig. 4C).
Figure 4. Excitatory input to VTA DA neurons in wild-type and Girk−/− mice.
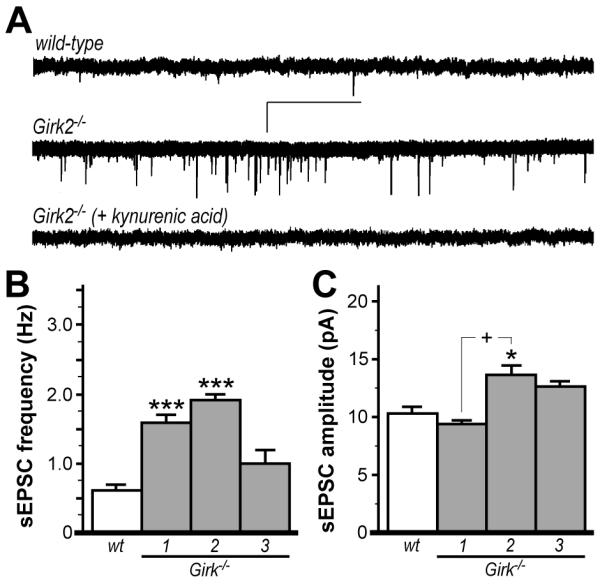
A) Representative traces of sEPSCs measured in VTA DA neurons from wild-type (upper) and Girk2−/− mice (middle); sEPSCs were blocked by the non-selective ionotropic glutamate receptor antagonist kynurenic acid (2 mM; lower). Scale bars: 20 pA/5 s. Summary histograms of sEPSC frequency and amplitude in VTA DA neurons from wild-type (wt, 0.6±0.1 Hz, 10.4±0.8 pA), Girk1−/− (1.6±0.1 Hz, 8.9±0.4 pA), Girk2−/− (1.9±0.1 Hz, 13.5±0.8 pA), and Girk3−/− (1.0±0.2 Hz, 12.7±0.4 pA), mice are shown in panels B and C, respectively (n=7-12 per genotype). A significant effect of genotype was observed for both sEPSC frequency (F3,31=18.4; P<0.001) and amplitude (F3,31=8.8; P<0.001). Symbols: *,*** P<0.05 and 0.001, respectively, vs. wild-type; + P<0.001, Girk1−/− vs. Girk2−/−.
We next examined the synaptic levels of AMPA receptor subunits in VTA DA neurons using double-labeling post-embedding immunoelectron microscopy (Fig. 5). Specifically, the relative amounts of the AMPA receptor subunits GluR1 (Fig. 5A-D) and GluR2/3 (Fig. 5E-H) in the postsynaptic density (PSD) in VTA DA neurons (identified by tyrosine hydroxylase labeling) were compared in wild-type, Girk1−/−, and Girk2−/− mice. Synaptic densities of GluR1 and GluR2/3 levels were significantly elevated (~2-fold) in VTA DA neurons from Girk1−/− and Girk2−/− mice, suggesting that the elevated frequency of sEPSCs measured in VTA DA neurons from Girk1−/− and Girk2−/− mice is due to the increased synaptic distribution of AMPA receptors.
Figure 5. AMPA subunit labeling in VTA DA neurons from wild-type and Girk−/− mice.

Electron micrographs showing GluR1 (A-C) and GluR2/3 (E-G) immunoreactivities in VTA DA neurons from wild-type, Girk1−/−, and Girk2−/− mice, as detected using double-labeling, post-embedding immunogold electron microscopy. In sections from wild-type mice, immunoparticles for GluR1 (A) and GluR2/3 (E) subunits (arrows, 10 nm gold particles) were found along the PSDs of dendritic shafts (Den) of DA neurons (TH-positive cells, arrowheads, 20 nm gold particles) establishing contact with axon terminals (b). In sections from Girk1−/− mice (B,F) and Girk2−/− mice (C,G), an increase in the number of immunoparticles for both GluR1 and GluR2/3 subunits along the PSD was observed. These images are representative of data from three separate panels of wild-type and Girk−/− mice. Scale bar: 0.2 μm. D,H) Quantification of GluR1 and GluR2/3 synaptic density in VTA DA neurons from wild-type, Girk1−/− (G1−/−), and Girk2−/− (G2−/−) mice. Synaptic density is expressed as number of immunoparticles per 1 μm length of PSD. Symbols: *** P<0.001 vs. wild-type.
Persistent elevation in glutamatergic signaling in VTA DA neurons can trigger adaptations in the NAcc, including increased glutamatergic input, dendritic spine density, and expression of the AMPA receptor subunit GluR1 (Robinson and Kolb 2004; Boudreau and Wolf 2005; Pulipparacharuvil et al. 2008; Mameli et al. 2009). Given the evidence for increased glutamatergic neurotransmission in VTA DA neurons from Girk1−/− and Girk2−/− mice, we next probed for adaptations related to excitatory neurotransmission in the NAcc of these mice. Specifically, we measured miniature EPSCs (mEPSCs) in NAcc shell neurons from wild-type and Girk−/− mice (Fig. 6). We found that mEPSC frequency and amplitude were elevated in NAcc neurons from Girk1−/− and Girk2−/− mice, relative to wild-type controls. Interestingly, mEPSC frequency and amplitude were significantly higher in NAcc neurons from Girk2−/− mice as compared to neurons from Girk1−/− mice.
Figure 6. Excitatory input to NAcc medium spiny neurons in wild-type and Girk−/− mice.
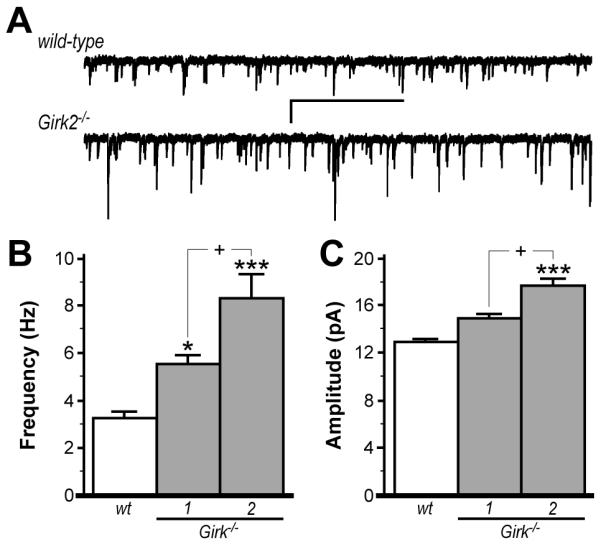
A) Representative traces of mEPSCs in NAcc DA neurons from wild-type (upper) and Girk2−/− (lower) mice. Scale bars: 2 s/5 pA. mEPSCs were blocked completely by the non-selective ionotropic glutamate receptor antagonist kynurenic acid (2 mM, not shown). Summary of mEPSC frequency and amplitude for medium spiny neurons in the NAcc shell from wild-type (wt, 3.3±0.3 Hz, 12.9±0.4 pA), Girk1−/− (5.5±0.4 Hz, 14.6±0.7 pA), and Girk2−/− (8.3±1.0 Hz, 17.1±0.8 pA) mice are shown in panels B and C, respectively (n=11-23 per genotype). Genotype-dependent differences were observed for both mEPSC frequency (F2,44=21.8; P<0.001) and amplitude (F2,44=14.6; P<0.001). Symbols: *,*** P<0.05 and 0.001, respectively, vs. wild-type; + P<0.05, Girk1−/− vs. Girk2−/−.
We next examined and quantified key cellular and molecular markers of excitatory neurotransmission in the NAcc of wild-type and Girk−/− mice. First, we quantified excitatory synapse density in medium spiny neurons of the NAcc using an optical dissector approach (Sterio 1984). Synapse density in the NAcc from Girk1−/− (1.31 synapses/μm3) and Girk2−/− (1.35 synapses/μm3) mice was ~15% higher as compared to wild-type control (1.12 synapses/μm3) (Fig. 7A-D). Moreover, synaptic densities of GluR1 (Fig. 7E-H) & GluR2/3 (Fig. 7I-L) were significantly elevated (~2-fold) in medium spiny neurons in the NAcc from Girk1−/− and Girk2−/− mice. Similar increases in synapse density and AMPA subunit labeling were observed in both the NAcc shell and core.
Figure 7. Markers of excitatory neurotransmission in the NAcc of wild-type and Girk−/− mice.

Electron micrographs taken from the NAcc core of wild-type (A), Girk1−/− (B), and Girk2−/− (C) mice, showing excitatory synapses (numbered) in the neuropil. Synapses between dendritic spines and axon terminals that exhibited a clear PSD were counted. Scale bar: 0.2 μm. D) Quantification of synapses from wild-type, Girk1−/− (G1−/−), and Girk2−/− (G2−/−) mice. The number of synapses per frame (#/frame) is plotted as a function of genotype. Synapses were tabulated in 21 frames per genotype, from 3 different animals per genotype. A significant effect of genotype on synapse density was observed (F2,60=5.4; P<0.01). Symbols: *,** P<0.05 and 0.01, respectively, vs. wild-type. E-L) AMPA subunit labeling in NAcc medium spiny neurons from wild-type and Girk−/− mice. Electron micrographs showing GluR1 (E-G) and GluR2/3 (I-K) immunoreactivity in medium spiny neurons from wild-type, Girk1−/−, and Girk2−/− mice, as detected using post-embedding immunogold electron microscopy. In sections from wild-type mice, immunoparticles for GluR1 (E) and GluR2/3 (I) subunits (arrows, 10 nm gold particles) were found along PSDs on individual spines (s) in contact with axon terminals (b). In sections from Girk1−/− (F,J) and Girk2−/− (G,K) mice, an increase in the number of immunoparticles for both GluR1 and GluR2/3 subunits along the PSD was observed. Scale bar: 0.2 μm. H,L) Quantification of GluR1 and GluR2/3 synaptic density in Nacc medium spiny neurons from wild-type, Girk1−/− (G1−/−), and Girk2−/− (G2−/−) mice. Synaptic density is expressed as number of immunoparticles per 1 μm length of PSD. Symbols: *** P<0.001 vs. wild-type.
DISCUSSION
Long-loop (GABABR-dependent) and autocrine (D2R-dependent) inhibitory feedback to VTA DA neurons is triggered by cocaine-induced increases in DA levels in the NAcc and VTA, respectively, and lead to tempered VTA DA neuron output and motor stimulation (Wolf et al. 1978; Einhorn et al. 1988; Napier and Potter 1989; Chen and Reith 1994). Simultaneous pharmacological inhibition of both feedback pathways evokes hyperactivity and exaggerated responses to psychostimulants, including cocaine (Steketee et al. 1991, 1992; Narayanan et al. 1996). Since Girk channels mediate the postsynaptic inhibitory effects linked to GABABR and D2R activation, Girk ablation in midbrain DA neurons should promote hyperactivity and exaggerated motor-stimulatory responses to cocaine due to the weakening of these inhibitory feedback mechanisms. Moreover, given that the Girk channel in midbrain DA neurons contains Girk2 but not Girk1, one would predict that hyperactivity and enhanced responses to cocaine would be seen in Girk2−/− but not Girk1−/− mice. To the contrary, Girk1−/− and Girk2−/− mice showed comparable baseline hyperactivities and enhanced responses to acute cocaine. Thus, the loss of inhibitory feedback to VTA DA neurons cannot explain the DA-dependent phenotypes manifest in Girk2−/− mice.
By the same logic, one can conclude that alterations in intrinsic excitability of VTA DA neurons are not the primary cause of the DA-dependent phenotypes in Girk2−/− mice. Indeed, ablation of Girk2 but not Girk1 should enhance the intrinsic excitability of VTA DA neurons, particularly if these neurons exhibit a high degree of basal Girk channel activity. While intrinsic excitability of VTA DA neurons may be elevated in Girk2−/− mice, these neurons exhibited lower spontaneous activity ex vivo. Firing rates were normalized by picrotoxin, indicating that VTA DA neurons from Girk2−/− mice receive elevated inhibitory input from local GABA neurons (Sugita et al. 1992). While this elevated inhibitory tone may be offset in vivo by elevated excitatory input (as our data suggest), these data do not support the contention that the DA-dependent hyperactivity in Girk2−/− mice reflects the enhanced intrinsic excitability of VTA DA neurons.
Chronic elevation of glutamatergic signaling in VTA DA neurons promotes persistent adaptations in glutamatergic signaling in the NAcc (Mameli et al. 2009). These adaptations are thought to underlie drug-seeking behavior and behavioral sensitization (Kalivas 2004; Self 2004). Interestingly, Girk1−/− and Girk2−/− mice show enhanced responses to acute cocaine administration, and the NAcc adaptations seen in Girk1−/− and Girk2−/− mice, including increased synapse density and elevated excitatory signaling, overlap with those in animals treated chronically with psychostimulants. Enhanced excitatory signaling in the NAcc, however, is thought to be a response to cocaine withdrawal rather than a consequence of repeated cocaine exposure (Kourrich et al. 2007). As such, adaptations in drug-naïve, constitutive Girk1−/− and Girk2−/− mice may be distinct from those linked to drug exposure.
The adaptations observed in the mesolimbic DA system of Girk1−/− and Girk2−/− mice may reflect synaptic scaling, a form of homeostatic plasticity triggered by persistent alterations in factors that influence neuronal excitability (Turrigiano and Nelson 2004; Kato et al. 2007; Thiagarajan et al. 2007). Synaptic scaling, manifest as an increase in the synaptic expression of GluR1 and GluR2-containing AMPA receptors, has been reported in the NAcc during cocaine withdrawal (Boudreau and Wolf 2005; Boudreau et al. 2007; Kourrich et al. 2007; Boudreau et al. 2009). Similarly, we observed elevated synaptic levels of both GluR1 and GluR2/3 in VTA DA and NAcc medium spiny neurons in Girk1−/− and Girk2−/− mice. It is possible, therefore, that the enhanced synaptic AMPA receptor distribution and elevated glutamatergic neurotransmission in VTA DA neurons and NAcc medium spiny neurons from Girk1−/− and Girk2−/− mice reflects compensatory responses of these neurons to persistent inhibition.
While the similarities between Girk1−/− and Girk2−/− mice with respect to adaptations relevant to excitatory neurotransmission are striking, some differences were evident. Notably, sEPSC amplitudes measured in VTA DA neurons were greater in Girk2−/− mice as compared to Girk1−/− mice. Similarly, mEPSC frequency and amplitude were both significantly elevated in NAcc medium spiny neurons in Girk2−/− mice relative to Girk1−/− mice. These differences may result directly from the loss of Girk signaling in VTA DA neurons, or may reflect more global differences linked to the relative impact of Girk1 and Girk2 ablation on neuronal physiology. While the genetic ablation of Girk1 and Girk2 yields similar outcomes in terms of somatodendritic currents in hippocampal pyramidal, locus ceruleus, and spinal cord dorsal horn neurons (Koyrakh et al. 2005; Marker et al. 2006; Cruz et al. 2008), Girk2 ablation may have a more dramatic impact than Girk1 ablation on synaptic Girk currents, which likely account for a small fraction of the somatodendritic currents measured in most studies. In support of this contention, a significant fraction of Girk2 protein, but not Girk1, is found within the postsynaptic specialization at excitatory synapses (Koyrakh et al. 2005; Marker et al. 2006). Clearly, understanding the more subtle differences between Girk1−/− and Girk2−/− mice reported in this study and other studies will require more selective genetic and/or pharmacologic approaches.
In summary, we demonstrate that genetic ablation of Girk1 and Girk2 promotes adaptations in the mesolimbic DA system that facilitate excitatory glutamatergic neurotransmission. The adaptations observed are reminiscent of those reported for synaptic scaling and following drug administration, suggesting that Girk channels may normally serve as a barrier to such adaptations. Accordingly, the regulation of Girk signaling strength, perhaps in VTA DA neurons and/or in neurons that provide input to the VTA, may constitute in part the mechanism by which drugs of abuse evoke adaptations that promote chronic drug intake, or provide a target for therapeutic interventions aimed at preventing such adaptations.
Acknowledgements
This work was supported by NIH grants MH061933 (KW) and DA011806 (KW) and by grants from the Spanish Ministry Science and Innovation (BFU2009-08404/BFI and CONSOLIDER-Ingenio CSD2008-00005; RL).
Abbreviations used
- VTA
ventral tegmental area
- NAcc
nucleus accumbens
- DA
dopamine
- Girk
G protein-gated inwardly-rectifying K+ channel
- SN
substantia nigra
- GABABR
GABAB receptor
- D1R
D1 dopamine receptor
- D2R
D2 dopamine receptor
- AMPA
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate
- EPSC
excitatory postsynaptic current
- IPSC
inhibitory postsynaptic current
- PSD
postsynaptic density
Footnotes
The authors have no conflicts of interest to declare.
REFERENCES
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Bettahi I, Marker CL, Roman MI, Wickman K. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem. 2002;277:48282–48288. doi: 10.1074/jbc.M209599200. [DOI] [PubMed] [Google Scholar]
- Bjorklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. doi: 10.1016/j.tins.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Stoffel M, Chang SR, Harris RA. GIRK2 deficient mice. Evidence for hyperactivity and reduced anxiety. Physiol Behav. 2001;74:109–117. doi: 10.1016/s0031-9384(01)00555-8. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Stoffel M, Cooper R, Wallace D, Mane N, Harris RA. Hyperactivity and dopamine D1 receptor activation in mice lacking girk2 channels. Psychopharmacology (Berl) 2002;159:370–378. doi: 10.1007/s00213-001-0937-6. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Ferrario CR, Glucksman MJ, Wolf ME. Signaling pathway adaptations and novel protein kinase A substrates related to behavioral sensitization to cocaine. J Neurochem. 2009;110:363–377. doi: 10.1111/j.1471-4159.2009.06140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Wessendorf MW, Williams JT. A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience. 1997;77:155–166. doi: 10.1016/s0306-4522(96)00444-7. [DOI] [PubMed] [Google Scholar]
- Chen NH, Reith ME. Autoregulation and monoamine interactions in the ventral tegmental area in the absence and presence of cocaine: a microdialysis study in freely moving rats. J Pharmacol Exp Ther. 1994;271:1597–1610. [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Luscher C. Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci. 2004;7:153–159. doi: 10.1038/nn1181. [DOI] [PubMed] [Google Scholar]
- Cruz HG, Berton F, Sollini M, Blanchet C, Pravetoni M, Wickman K, Luscher C. Absence and rescue of morphine withdrawal in GIRK/Kir3 knock-out mice. J Neurosci. 2008;28:4069–4077. doi: 10.1523/JNEUROSCI.0267-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einhorn LC, Johansen PA, White FJ. Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: studies in the ventral tegmental area. J Neurosci. 1988;8:100–112. doi: 10.1523/JNEUROSCI.08-01-00100.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, Parkitna JR, Lujan R, Halbout B, Mameli M, Parlato R, Sprengel R, Luscher C, Schutz G, Spanagel R. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron. 2008;59:497–508. doi: 10.1016/j.neuron.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Gundersen HJ, van der Zee E, West MJ. Unbiased stereological estimation of the total number of synapses in a brain region. J Neurocytol. 1996;25:805–819. doi: 10.1007/BF02284843. [DOI] [PubMed] [Google Scholar]
- Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heshmati M. Cocaine-induced LTP in the ventral tegmental area: new insights into mechanism and time course illuminate the cellular substrates of addiction. J Neurophysiol. 2009;101:2735–2737. doi: 10.1152/jn.00127.2009. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Yoshimoto Y, Horio Y, Morishige KI, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y, Kurachi Y. Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J Neurosci. 1999;19:1006–1017. doi: 10.1523/JNEUROSCI.19-03-01006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. Glutamate systems in cocaine addiction. Curr Opin Pharmacol. 2004;4:23–29. doi: 10.1016/j.coph.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Karschin C, Dissmann E, Stuhmer W, Karschin A. IRK(1-3) and GIRK(1-4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J Neurosci. 1996;16:3559–3570. doi: 10.1523/JNEUROSCI.16-11-03559.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Sekino Y, Takahashi H, Yasuda H, Shirao T. Increase in AMPA receptor-mediated miniature EPSC amplitude after chronic NMDA receptor blockade in cultured hippocampal neurons. Neurosci Lett. 2007;418:4–8. doi: 10.1016/j.neulet.2007.02.058. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Klink R, de Kerchove d’Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–1463. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Stinus L, Le Moal M. Hyperactivity and hypoactivity produced by lesions to the mesolimbic dopamine system. Behav Brain Res. 1981;3:341–359. doi: 10.1016/0166-4328(81)90004-8. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyrakh L, Lujan R, Colon J, Karschin C, Kurachi Y, Karschin A, Wickman K. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci. 2005;25:11468–11478. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouebe G, Lomazzi M, Cruz HG, Creton C, Lujan R, Li M, Yanagawa Y, Obata K, Watanabe M, Wickman K, Boyer SB, Slesinger PA, Luscher C. RGS2 modulates coupling between GABA(B) receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci. 2007 doi: 10.1038/nn2006. [DOI] [PubMed] [Google Scholar]
- Liao YJ, Jan YN, Jan LY. Heteromultimerization of G-protein-gated inwardly rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in weaver brain. J Neurosci. 1996;16:7137–7150. doi: 10.1523/JNEUROSCI.16-22-07137.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Luscher C, Ungless MA. The mechanistic classification of addictive drugs. PLoS Med. 2006;3:e437. doi: 10.1371/journal.pmed.0030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Luscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Marker CL, Lujan R, Loh HH, Wickman K. Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of mu- and delta- but not kappa-opioids. J Neurosci. 2005;25:3551–3559. doi: 10.1523/JNEUROSCI.4899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker CL, Lujan R, Colon J, Wickman K. Distinct populations of spinal cord lamina II interneurons expressing G-protein-gated potassium channels. J Neurosci. 2006;26:12251–12259. doi: 10.1523/JNEUROSCI.3693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathon DS, Lesscher HM, Gerrits MA, Kamal A, Pintar JE, Schuller AG, Spruijt BM, Burbach JP, Smidt MP, van Ree JM, Ramakers GM. Increased gabaergic input to ventral tegmental area dopaminergic neurons associated with decreased cocaine reinforcement in mu-opioid receptor knockout mice. Neuroscience. 2005;130:359–367. doi: 10.1016/j.neuroscience.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Momiyama T, Amano T, Todo N, Sasa M. Inhibition by a putative antipsychotic quinolinone derivative (OPC-14597) of dopaminergic neurons in the ventral tegmental area. Eur J Pharmacol. 1996;310:1–8. doi: 10.1016/0014-2999(96)00350-0. [DOI] [PubMed] [Google Scholar]
- Morgan AD, Carroll ME, Loth AK, Stoffel M, Wickman K. Decreased cocaine self-administration in Kir3 potassium channel subunit knockout mice. Neuropsychopharmacology. 2003;28:932–938. doi: 10.1038/sj.npp.1300100. [DOI] [PubMed] [Google Scholar]
- Napier TC, Potter PE. Dopamine in the rat ventral pallidum/substantia innominata: biochemical and electrophysiological studies. Neuropharmacology. 1989;28:757–760. doi: 10.1016/0028-3908(89)90163-9. [DOI] [PubMed] [Google Scholar]
- Narayanan S, Wallace L, Uretsky N. Spontaneous and drug-stimulated locomotor activity after the administration of pertussis toxin into the ventral tegmental area. J Psychiatry Neurosci. 1996;21:172–180. [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacology. 2004;47(Suppl 1):24–32. doi: 10.1016/j.neuropharm.2004.06.031. [DOI] [PubMed] [Google Scholar]
- Neuhoff H, Neu A, Liss B, Roeper J. I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J Neurosci. 2002;22:1290–1302. doi: 10.1523/JNEUROSCI.22-04-01290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravetoni M, Wickman K. Behavioral characterization of mice lacking GIRK/Kir3 channel subunits. Genes Brain Behav. 2008;7:523–531. doi: 10.1111/j.1601-183X.2008.00388.x. [DOI] [PubMed] [Google Scholar]
- Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, Greengard P, Nairn AC, Nestler EJ, Cowan CW. Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron. 2008;59:621–633. doi: 10.1016/j.neuron.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Self DW. Regulation of drug-taking and -seeking behaviors by neuroadaptations in the mesolimbic dopamine system. Neuropharmacology. 2004;47(Suppl 1):242–255. doi: 10.1016/j.neuropharm.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M. Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci U S A. 1997;94:923–927. doi: 10.1073/pnas.94.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steketee JD, Striplin CD, Murray TF, Kalivas PW. Possible role for G-proteins in behavioral sensitization to cocaine. Brain Res. 1991;545:287–291. doi: 10.1016/0006-8993(91)91299-g. [DOI] [PubMed] [Google Scholar]
- Steketee JD, Striplin CD, Murray TF, Kalivas PW. Pertussis toxin in the A10 region increases dopamine synthesis and metabolism. J Neurochem. 1992;58:811–816. doi: 10.1111/j.1471-4159.1992.tb09329.x. [DOI] [PubMed] [Google Scholar]
- Sterio DC. The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc. 1984;134:127–136. doi: 10.1111/j.1365-2818.1984.tb02501.x. [DOI] [PubMed] [Google Scholar]
- Sugita S, Johnson SW, North RA. Synaptic inputs to GABAA and GABAB receptors originate from discrete afferent neurons. Neurosci Lett. 1992;134:207–211. doi: 10.1016/0304-3940(92)90518-c. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Malgaroli A, Tsien RW. LTP and adaptation to inactivity: overlapping mechanisms and implications for metaplasticity. Neuropharmacology. 2007;52:156–175. doi: 10.1016/j.neuropharm.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, Wickman K. G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J Neurosci. 2002;22:4328–4334. doi: 10.1523/JNEUROSCI.22-11-04328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Wolf P, Olpe HR, Avrith D, Haas HL. GABAergic inhibition of neurons in the ventral tegmental area. Experientia. 1978;34:73–74. doi: 10.1007/BF01921910. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Argilli E, Bonci A, Palmiter RD. Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron. 2008;59:486–496. doi: 10.1016/j.neuron.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]