

Abstract
Loss of function mutations in the human RecQ helicase genes WRN and BLM respectively cause the genetic instability/cancer predisposition syndromes Werner syndrome and Bloom syndrome. In order to identify common and unique functions of WRN and BLM, we systematically analyzed cell proliferation, cell survival and genomic damage in isogenic cell lines depleted of WRN, BLM or both proteins. Cell proliferation and survival were assessed prior to and after treatment with camptothecin, cis-Pt, hydroxyurea or 5-fluorouracil. Genomic damage was assessed, prior to and after replication arrest, by γ-H2AX staining quantified at the single cell level by flow cytometry. Cell proliferation was strongly affected by the extent of WRN and/or BLM depletion, and more strongly by BLM than by WRN depletion (p=0.005). The proliferation of WRN/BLM co-depleted cells, in contrast, did not differ from BLM-depleted cells (p=0.34). BLM-depleted and WRN/BLM co-depleted cells had comparably impaired survivals after DNA damage, whereas WRN-depleted cells displayed a distinct pattern of sensitivity to DNA damage. BLM-depleted and WRN/BLM co-depleted cells had similar, significantly higher γ-H2AX induction levels than did WRN-depleted cells. Our results provide new information on the role of WRN and BLM in determining cell proliferation, cell survival and genomic damage after chemotherapeutic DNA damage or replication arrest. We also provide new information on functional redundancy between WRN and BLM. These results provide a strong rationale for further developing WRN and BLM as biomarkers of tumor chemotherapeutic responsiveness.
Keywords: RecQ helicase, Werner syndrome, Bloom syndrome, DNA damage, chemotherapy, therapeutic biomarker
Introduction
The human RecQ helicases are members of a deeply conserved protein family that plays important, albeit poorly understood, roles in DNA metabolism, genetic stability and the response to DNA damage (1;2). Germ line loss-of-function mutations in three human RecQ helicase genes, WRN, BLM and RECQL4, respectively cause Werner syndrome (WS), Bloom syndrome (BS) and the subset of Rothmund-Thomson syndrome (RTS) associated with a high risk of osteosarcoma. These genetic instability/cancer predisposition syndromes also have different developmental or acquired features. WS patients develop features resembling premature aging beginning in the second decade of life (3). BS patients, in contrast, are proportionately small from birth, display sun sensitivity and hypo- and hyper-pigmented skin lesions, are often immunodeficient; and have reduced fertility (4). RTS patients are typically short with sparse hair and eyebrows; have variable skeletal, dental and nail abnormalities; and develop a persistent skin rash in infancy together with a high risk of juvenile ocular cataracts (5). Epigenetic loss of expression of RecQ helicases may also be linked to human disease. For example, epigenetic silencing of WRN expression has been documented and is frequent in common adult epithelial malignancies such as colorectal cancer (6;7). No human disease has been linked thus far to mutation or epigenetic inactivation of the two other human RecQ helicase genes, RECQL or RECQL5 (1;2).
All five human RecQ proteins share a conserved helicase domain that encodes DNA dependent ATPase and 3′ to 5′ helicase activities. WRN also encodes a 3′ to 5′ exonuclease activity in an N-terminal domain. Purified human RecQ helicases preferentially bind, unwind, and in the case of WRN also degrade, partially double-stranded DNA molecules including model replication forks, D– and T–loops or synthetic Holliday junctions and highly structured DNAs such a G-quadruplexes. Several human RecQ helicases also possess DNA strand annealing activity (1;2;8). Functional correlates of these activities include a requirement for RecQ helicases in homology-dependent recombination, in replication initiation, in replication restart or fork elongation and in DNA repair (1;2;9–11).
In order to delineate redundant and unique in vivo functions of WRN and BLM, we systematically analyzed cell proliferation, genomic damage as assessed by γ-H2AX staining and cell survival in isogenic human cell lines depleted of WRN and/or BLM prior to and after treatment with DNA damaging chemotherapeutic drugs. Our results provide new information on the role of WRN and BLM in determining the response to chemotherapeutic damage, and on functional redundancy between WRN and BLM.
Materials and Methods
Cells and cell culture
The SV40-transformed GM639 human fibroblast cell line developed from a normal donor was originally obtained from the Coriell Institute Cell Repositories (Camden, NJ) in 1990. GM639-cc1 is a clonal derivative of GM639 that carries an integrated copy of the pNeoA direct repeat homologous recombination reporter plasmid (12). The human osteosarcoma cell line U-2 OS (13) was obtained from the American Type Culture Collection (Manassas, VA) in 2008. GM639 cells are functionally TP53(−), whereas U-2 OS cells express TP53 protein and are functionally TP53(+). Cell lines were initially DNA fingerprinted and screened to verify the absence of Mycoplasma infection using PCR kits obtained from the Coriell Institute Cell Repositories. Subsequent fingerprinting and Mycoplasma screening verifications have been performed by the University of Missouri Research Animal Diagnostic Laboratory (RADIL; www.radil.missouri.edu/). Recently thawed aliquots of both lines were used for all experiments. GM639-cc1 cells were grown in Dulbecco-modified Eagle’s medium, and U-2 OS cells in McCoy’s 5A medium (MediaTech CellGro, Manassas, VA) in a humidified 37°C, 7% incubator. Both growth media were supplemented with 4,500 mg/L glucose, 10% (v/v) fetal bovine serum (Hyclone, Logan, UT) and penicillin (100U/ml) and streptomycin sulfate (100 mg/ml; Invitrogen, Carlsbad, CA).
Drugs and dyes
Stock solutions of cis-diamminedichloroplatinum(II) (CDDP or cis-Pt; 2 mM in 0.9% NaCl), camptothecin (CPT; 1 mM in DMSO), hydroxyurea (HU; 1M in phosphate-buffered saline), 5-fluorouracil (5-FU; 1 mg/ml in DMSO), and 5-bromodeoxyuridine (BrdU; 10 mM in sterile water) were stored at −20°C and diluted just prior to use. Propidium iodide (10 mg/ml in PBS) was stored at 4 °C in the dark, and 4,6′-diamidino-2-phenylindole (DAPI, 1 mg/ml) −20°C. DAPI was obtained from Accurate Chemical and Scientific Corp (Westbury, NY). All other chemicals and drugs were obtained from Sigma/Aldrich (St. Louis, MO).
Cell proliferation and survival assays
Population-based cell proliferation assays were performed by plating 104 cells per well in six-well plates (9.1 cm2/well). Duplicate wells were trypsinized and counted every three days. Cell survival was quantified by colony-forming efficiency (CFE) determined as previously described (12). In brief 100 – 500 control cells, or 4,000 – 20,000 RecQ-depleted cells/well were plated in six-well plates 24 hr prior to drug treatment, then treated for 24 hrs followed by 8 days growth in the absence of drug prior to crystal violet staining to identify colonies containing ≥6 cells.
shRNA-mediated depletion of WRN and BLM
We screened WRN and BLM-specific shRNAs designed by The RNAi Consortium (TRC; http://www.broad.mit.edu/rnai/trc/lib) or by Rosetta Inpharmatics, Inc. (Seattle, WA) to identify shRNAs that reproducibly depleted WRN or BLM when expressed from pLKO.1, a lentiviral expression vector containing a human U6 promoter (http://www.broad.mit.edu/genome_bio/trc/protocols/pLKO1.noStuffer.pdf; (14). pLKO.1 shRNA vectors (Figure 1A) were packaged by co-transfecting pLKO.1 DNA with packaging plasmid pCMV-dR8.2 dvpr and envelope plasmid pCMV-VSVG (kindly provided by Robert Weinberg, Whitehead/MIT) into human 293T cells as previously described (15). Viral supernatants were filtered through a 45 micron filter and stored at −80°C until use. shRNA-mediated depletions were performed by transducing cells with shRNA lentivirus for 48 hrs, followed by an additional 96 hrs of puromycin selection (1.5–2.0 μg/ml). Depletions were quantified by Western blot analysis (Figure 1B). Controls included cells transduced with pLKO.1 vector DNA alone, or with pLKO.1 expressing a scrambled shRNA with no known target sequence in the human genome (plasmid 1864, ‘scramble shRNA’; Addgene, Cambridge, MA). Co-depletions were achieved by simultaneously transducing cells with WRN- and BLM-specific shRNA lentiviruses.
FIGURE 1. RNA-interference-mediated depletion of WRN and BLM.

A. pLKO.1 lentiviral vector used for shRNA expression. key: LTRs: 5′ RSV and 3′ self-inactivating HIV long terminal repeats; Hs U6: human U6 promoter; Hs PGK: human phosphoglycerate kinase promoter; puro: puromycin resistance gene. B. Experimental protocol for generation/use of WRN and/or BLM-depleted cells. Depletion of WRN and BLM were maximal from 5–6 d post-transduction, and persisted for >14 days (15;16; additional results not shown).
Western blot analyses
Solubilized cell pellets (~1 × 106 cells) prepared as previously described (16;17) were resolved by SDS-PAGE electrophoresis (Invitrogen, Nu-PAGE) prior to transfer onto PVDF membrane (Bio-Rad, Richmond, CA, USA). WRN protein was detected with mouse monoclonal anti-WRN primary antibody 195C (kindly provided by Dr. Patricia Opresko, University of Pittsburgh; 18). BLM protein was detected using an affinity-purified rabbit polyclonal anti-BLM antisera directed against the BLM C-terminal peptide KPINRPFLKPSYAFS (additional results not shown). Human CHK1 protein was detected with an anti-Chk1 mouse monoclonal primary antibody (Santa Cruz #sc-8408; Santa Cruz, CA). Bound antibodies were detected ECL detection (GE Healthcare, UK; Figure 2A). Blots were scanned and quantified using a Storm Phosphorimager and ImageQuant software (Molecular Dynamics; Sunnyvale, CA) as previously described (16).
FIGURE 2. Lentiviral shRNAs selectively deplete or co-deplete WRN and BLM.
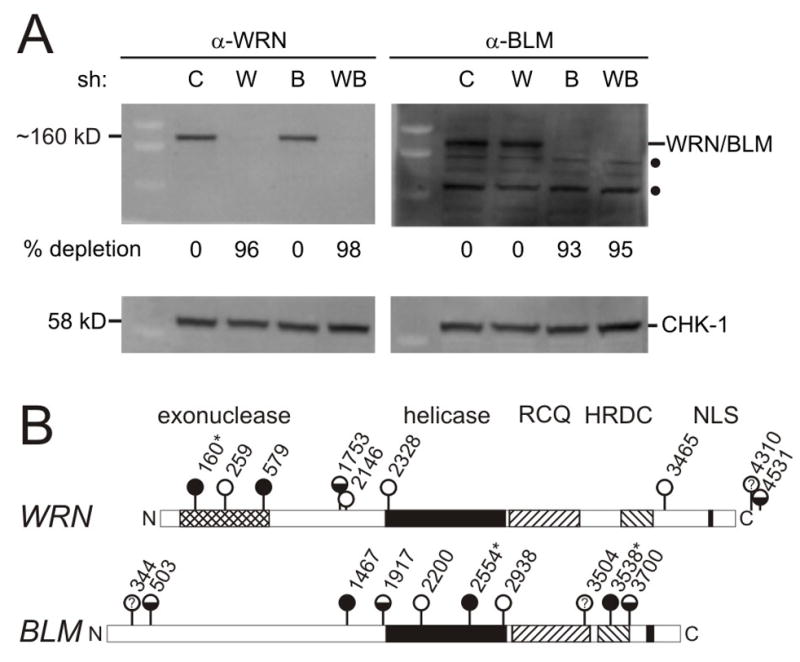
A. Western blot analysis of GM639 cells after WRN- and/or BLM-specific shRNA expression. key: C, pLKO.1 vector control; W, shWRN579; B, shBLM2554; W+B, shWRN579 and shBLM2554, where individual shRNAs are numbered from their 5′ end in the corresponding WRN or BLM cDNA sequence (see below, panel B). Predicted molecular weight of WRN is 162 kDa, and BLM 159 kDa. Filled circles (●) indicate nonspecific cross-reacting bands detected by our polyclonal BLM antisera. CHK1 protein was used as a loading control. B. Summary of shRNAs screened for ability to deplete WRN or BLM proteins, where symbols indicate extent of depletion: filled circles, >90% depletion; half-fill, partial (20–90% depletion); open circles, <20% or no detectable depletion; ?, inconclusive data. (*) shRNAs verified in both retroviral and lentiviral vectors.
Flow cytometric analysis of γ-H2AX-stained cells
Genomic damage was quantified by flow cytometric analysis of cellular γ-H2AX staining as previously described (19). In brief, 1–2 × 105 cells/well were plated in 6 well plates prior to treatment with hydroxyurea (HU) for 2–8 hrs. Cells were scrape-harvested, washed twice in 1X PBS, fixed with 1 ml cold 66% ethanol and stored at 4 °C until analyzed. Fixed cells were resuspended in TBS (25 mM Tris-HCl [pH 7.4], 137 mM NaCl, 5 mM KCl) supplemented with 4% fetal bovine serum and 0.1% Triton X-100, incubated on ice for 10 min, and stained for 1 hr with a mouse monoclonal anti-phospho-histone H2AX (Ser139) primary antibody (Millipore-clone JBW301; Billerica, MA) for 1 hr. Bound antibody was detected by staining for 1 hr with a goat-anti-mouse Alexa 488-conjugated-secondary antibody (Molecular Probes A1100110C; Carlsbad, CA). Cells resuspended in TBS were stained with DAPI (10 μg/ml) prior to flow cytometric analysis on an inFlux® flow cytometer (Cytopeia Inc., Seattle, WA). Control cells (pLKO.1 vector only or pLKO.1-scramble sh) were used to define baseline staining set to include 1% of the cell population in the positive cell fraction (Gate 1). A second gate included all cells while excluding cell debris (Gate 2). The same gating was used for all samples within an experiment. Gate 1 vs. Gate 2 ratios defined the ‘% γ-H2AX positive’ cells, and ‘fold inductions’ were calculated by dividing experimental by control, γ-H2AX positive, cell frequencies.
Cell cycle distribution determined by BrdU labeling and flow cytometry
RecQ-depleted and control cells were labeled with 50 mM BrdU for 2 hrs, then harvested as previously described (15). BrdU content was determined by fixing cells in cold 66% ethanol/PBS, denaturing in 2N HCl/0.5% Triton X100 for 30 min each, and then neutralizing samples with 100 mM Na borate (pH8.5). Immunostaining to detect incorporated BrdU was for 1 hr each at 4°C in the dark with mouse anti-BrdU primary antibody (347580, BD Biosciences, San Jose, CA), followed by Alexa 488-conjugated anti-mouse secondary antibody (Molecular Probes A1100110C). Cells were strained with propidium iodide (10 μg/mL) in PBS containing 100 μg/mL RNAse A prior to analysis on an inFlux® flow cytometer. Data analyses were done using Summit software (Dako, Carpinteria, CA). Cell cycle fractions were estimated using FCS Express (De Novo Software, Los Angeles, CA) or M-cycle (Phoenix Flow Systems, San Diego, CA).
Statistical analysis of cell proliferation, survival and γ-H2AX straining
Regression modeling was used as the most rigorous way to analyze outcomes as a function of RecQ depletion type while controlling for time, within-experiment correlations, extent of depletion, drug and drug dose. This approach allowed us to analyze primary proliferation, survival or staining data to identify significant differences, while correcting for interactions between variable and for multiple testing. Differences were considered significant if they met a p value that was Bonferroni-corrected for multiple-testing by experiment type in order to preserve a family-wise type I error rate of 0.05. These corrected p values were: cell proliferation, p = 0.012; cell survival, p = 0.00096; and γ-H2AX induction, p = 0.007. Analysis of log cell counts and γ-H2AX straining were fitted as a linear function of time. Data for survival of drug-treated U-2 OS cells with fewer than 500 cells plated were excluded from regression analysis in order to avoid generating artificially high co-linearity within the treated survival design matrix. CFE outcomes were normalized to zero-dose CFE by including the latter in the regression model. This approach requires fewer modeling assumptions than using a ‘ratio of ratios’ approach (20), and thus avoids the high variability generated when dividing by small numbers. Differences between depletion states (control, scrambled sh, WRN or BLM-depleted and WRN/BLM co-depleted) were tested after adjusting for dose and experiment.
Results
Depletion of WRN and BLM from human fibroblasts
We identified 2 WRN-specific and 3 BLM-specific shRNAs that reproducibly depleted their respective target proteins in different cell types by ≥90%. We also identified 2 additional WRN-specific, and 3 additional BLM-specific, shRNAs that partially depleted WRN or BLM by 30 to 70% (Figures 1 and 2; additional results not shown). Western blot analyses indicated that both WRN and BLM were maximally depleted by Day 6 after transduction, and remained depleted at ≥90% for at least 25 days (Figure 2A and Supplementary Figure 1; 13–15; additional data not shown). Co-transduction using the same protocol and shRNA lentiviral stocks led to simultaneous depletion of both WRN and BLM by ≥90% (Figure 2A).
Cell proliferation as a function of WRN/BLM protein content
Depletion of WRN or BLM from GM639 and U-2 OS cells suppressed cell proliferation in both population-based and clonal proliferation assays. Higher percent depletions were associated with stronger suppression for both WRN and BLM. Cell proliferation was more strongly suppressed by BLM than by WRN depletion as a function of percent depletion over the observed depletion range, and comparable slopes were observed for regression lines that related % protein depletion to % proliferative suppression in both population-based and colony-forming assays (Figure 3). Of note, the proliferation of WRN/BLM co-depleted cells did not differ from cells depleted of BLM alone (p = 0.34; Figure 3 and Supplementary Figure 2).
FIGURE 3. WRN and BLM depletion suppress cell proliferation.

A. Proliferation of GM639 human fibroblasts depleted of WRN and/or BLM. Open symbols represent proliferation measured at day 9 (see Figure 1B), and filled symbols proliferation at the same time point in co-depleted cells where WRN (◆) or BLM (●) depletion was determined by Western blot in four independent experiments. The proliferation of BLM- and WRN-depleted cells was significantly different (p = 0.0002), in contrast to BLM- versus WRN/BLM co-depleted cells (p = 0.34). The slopes of regression lines for proliferative suppression as a function of % depletion were respectively for WRN and BLM −0.20 and −0.061 log10 units of percent of control per 1 percent protein depleted. B. Colony formation by WRN- or BLM-depleted cells was significantly suppressed as a function of percent depletion (p = 3.9 × 10−7 for WRN, and p = 0.0016 for BLM), with significantly stronger suppression in BLM versus WRN-depleted cells (p = 1.1×10 −8). There was no difference between BLM- versus WRN/BLM co-depleted cells in colony forming ability (p = 0.45). Slopes of the regression lines for suppression of CFE as a function of WRN or BLM depletion were, respectively, −0.18 and −0.035 log10 units of percent of control per 1% of protein depleted.
Genomic damage after WRN/BLM depletion
As a measure of genomic damage we quantified both basal and replication arrest-induced γ-H2AX levels in WRN and/or BLM-depleted cells. γ-H2AX is a minor histone H2 variant that is phosphorylated on serine 139 in response to replication stress and other types of genomic damage including DNA breakage (21;22). The primary data presented in Figure 4 are means and standard deviations for fold γ-H2AX induction from 5 independent experiments. All of these data and time points were used to build a regression model to determine the rate of change and whether there were differences in mean fold γ-H2AX induction as a function of time and depletion type. These analyses revealed significantly higher γ-H2AX inductions in all depleted cell types (WRN, BLM, or WRN+BLM-co-depleted) as compared with controls, and no significant difference in γ-H2AX induction between cells depleted of BLM alone as opposed to WRN and BLM (Supplementary Table 1). The rate of increase in γ-H2AX staining as a function of time in HU was linear, and did not differ as a function of depleted protein(s). Differences in γ-H2AX staining among depleted cells were not explained by differences in cell cycle phase distribution, as assessed by BrdU labeling (16; additional results not shown). These results indicate that the depletion of WRN or BLM may lead to genomic damage that can be detected by higher levels of γ-H2AX staining. Comparable results were observed in U-2 OS cells, though these were not formally analyzed as we had fewer data than for GM639 cells (additional results not shown).
FIGURE 4. Elevated genomic damage with γ-H2AX staining in HU-arrested, WRN and/or BLM-depleted, cells.
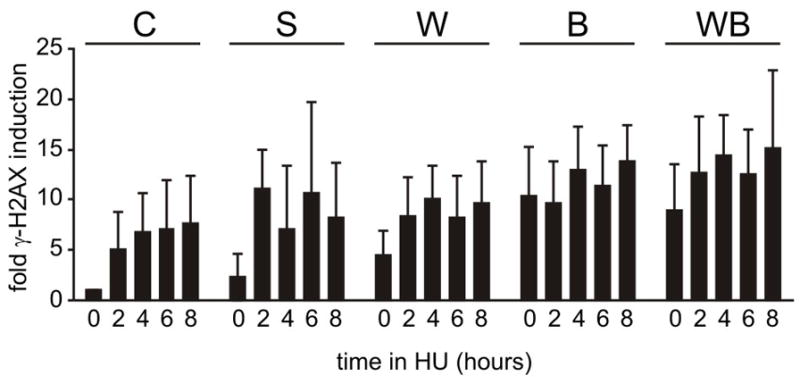
The bar graphs are primary data from γ-H2AX induction experiments using WRN and/or BLM-depleted GM639 fibroblasts and replication arrest in 2 mM HU where controls were pLKO.1 vector-only (C) or scrambled shRNA-expressing cells (S). Regression modeling of these data revealed significantly higher staining in WRN and/or BLM-depleted cells vs either type of control (C,S; p<0.01; Supplementary Table 1), and a linear rate of change in fold γ-H2AX induction as a function of time. BLM- and WRN/BLM co-depleted cells had significantly higher fold inductions in staining than did WRN-depleted cells (p = 0.0016 and p = 2.8 × 10−6, respectively), but did not differ from one another (p = 0.18; Supplementary Table 1). There was no difference in slopes/rate of change in the induction of H2AX staining over time as a function of depletion type (W, B or WB). Error bars represent standard deviations for five independent experiments except for shScr where duplicate values from a single experiment are shown. key: C, pLKO.1 control vector; S, scrambled shRNA control; W, shWRN579; B, shBLM2554; WB, co-depletion with shWRN579 + shBLM2554.
Cell survival after WRN/BLM depletion and DNA damage
We quantified cell survival of depleted, isogenic cell lines after treatment with four different cancer chemotherapeutic agents: the topoisomerase I inhibitor camptothecin (CPT); the DNA crosslinking drug cis-Pt; the ribonucleotide reductase inhibitor hydroxyurea (HU); and the antimetabolite and thymidylate synthase inhibitor 5-fluorouracil (5-FU). Primary data from these analyses, performed as colony-forming efficiency assays using GM639 or U-2 OS cells, are shown in Figure 5. These data were again analyzed by regression modeling in order to use experimental data across different doses and to correct for potential experimental confounders and for multiple testing.
FIGURE 5. Chemotherapeutic drugs selectively kill WRN and/or BLM-depleted cells.

Panels show colony-forming efficiencies (CFEs) of depleted and control GM639 and U-2 OS cell lines after 24 hrs treatment with camptothecin (CPT), hydroxyurea (HU), cisplatin (cis-Pt) or 5-fluorouracil (5-FU). Error bars represent standard deviations for two to five independent experiments. Data were analyzed by regression modeling (Table 1) to account for cell type, depletion type (W, B or WB), agent, dose and between-experiment variation. WRN- or BLM-depleted drug-treated cells had significantly lower survival than controls after treatment with CPT, cis-Pt and 5-FU. WRN-depleted GM639 cells had significantly higher CFEs than did BLM-depleted cells after CPT or HU treatment. BLM- and WRN/BLM co-depleted cells had statistically indistinguishable survivals with the exception of BLM-depleted U-2 OS vs WB co-depleted cells treated with CPT (Table 1). key: C, pLKO.1 control vector; S, scrambled short-hairpin control vector; W, shWRN579; B, shBLM2554; WB, shWRN579 + shBLM2554.
Depletion of WRN or BLM significantly sensitized both GM639 and U-2 OS cells to dose-dependent killing by all four drugs (Figure 5), with the exception of WRN-depleted U-2 OS cells where HU-treated survival was indistinguishable from control cells (p = 0.31; Table 1; see also 16). Depletion of WRN or BLM significantly sensitized GM639 and U-2 OS cells to dose-dependent killing by CPT, cis-Pt and 5-FU, and BLM-depleted cells to HU. Of note, co-depletion of WRN and BLM did not additively or synergistically sensitize depleted cells to killing by any of the 4 drugs tested (Figure 5, Table 1). The p value cutoff for significance in these analyses, corrected for multiple testing to retain a type I error rate of 0.05, was p = 0.00096 (Table 1).
TABLE 1. Regression modeling identifies significance differences in cell survival of WRN and/or BLM-depleted cells after DNA damage.
Colony-forming efficiency data in figure 5 were used for a statistical analysis of colony-forming efficiency that employed regression modeling to account for cell type, depletion type (W, B or WB), agent, dose and between-experiment variation. Control and depleted cells were treated for 24 hrs with camptothecin (CPT), hydroxyurea (HU), cisplatin (cis-Pt) or 5-fluorouracil (5-FU).
| agent† | comparison samples‡ | cell line | ||
|---|---|---|---|---|
| GM639 | U-2 OS | |||
| S1 | S2 | p value§ | p value | |
| CPT |
C | W | <1.0×10−16* | 2.7×10−6* |
| C | B | <1.0×10−16* | 2.2×10−6* | |
| C | WB | <1.0×10−16* | 4.9×10−7* | |
| W | B | 6.0×10−6* | 0.064 | |
| W | WB | 0.0025 | 0.55 | |
| B | WB | 0.81 | 9.5×10−5* | |
| HU |
C | W | 0.0012‖ | 0.31 |
| C | B | 3.0×10−9* | 5.7×10−10* | |
| C | WB | 7.0×10−11* | 4.0×10−16* | |
| W | B | 5.0×10−4* | 0.14 | |
| W | WB | 4.0×10−5* | 0.10 | |
| B | WB | 0.04 | 0.37 | |
|
cis-Pt |
C | W | <1.0×10−16* | <1.0×10−16* |
| C | B | <1.0×10−16* | <1.0×10−16* | |
| C | WB | <1.0×10−16* | <1.0×10−16* | |
| W | B | 0.0025 | <1.0×10−16* | |
| W | WB | 0.0006* | <1.0×10−16* | |
| B | WB | 0.22 | <1.0×10−16* | |
| 5-FU |
C | W | <1.0×10−16* | <1.0×10−16* |
| C | B | <1.0×10−16* | <1.0×10−16* | |
| C | WB | <1.0×10−16* | <1.0×10−16* | |
| W | B | 0.013 | 0.52 | |
| W | WB | 0.21 | 0.13 | |
| B | WB | 0.11 | 0.03 | |
key:
P values < 0.00096 are significant after Bonferroni correction for multiple testing.
sample pairs were tested for significance: C = pLKO.1 vector-transduced; W=WRN-depleted; B=BLM-depleted; WB = WRN/BLM-depleted.
the p value for significance after Bonferroni correction for multiple testing was 0.00096.
Statistically significant differences are marked with an asterisk (*).
This P value was calculated from 0.5 mmol/L HU data only in order to avoid introducing artefact.
Discussion
In order to determine how WRN and BLM influence the response to chemotherapeutic drugs, we quantified cell proliferation, genomic damage as assessed by γ-H2AX induction and cell survival after treating WRN- and/or BLM-depleted cells with four different DNA-damaging chemotherapeutic drugs. Isogenic, WRN/BLM-depleted or co-depleted TP53(+) and TP53(−) cell lines were used together with regression modeling to control for important variables including cell line, depleted protein and % depletion, drug and dose. Co-depletion analyses also allowed us to analyze for the first time the functional redundancy of WRN and BLM in isogenic human cell line pairs.
WRN or BLM depletion alone suppressed cell proliferation in both TP53(+) and TP53(−) cell lines (Figure 3), and increased genomic damage as assessed by γ-H2AX induction both prior to and after HU-mediated replication arrest (Figure 4). WRN or BLM-depleted cells were sensitized to dose-dependent cell killing by CPT, cis-PT and 5-FU (see Figure 5 data and Table 1 statistical analysis). WRN-depleted GM639 cells had significantly higher survivals after CPT or HU treatment than did isogenic BLM-depleted cells. Of note, depletion of BLM from WRN-depleted cells sensitized them to HU-mediated cell killing. Conversely, BLM-depleted U-2 OS cells were refractory to CPT killing, but could be sensitized to CPT-mediated cell killing by the depletion of WRN (Figure 5 data and Table 1 statistical analysis).
These results substantially extend and clarify our understanding of the proliferation and drug sensitivity phenotypes of WRN or BLM-deficient human cells. Previous work by us and others had documented reduced proliferative potential and DNA damage sensitivity of WRN-deficient, patient-derived fibroblasts, peripheral blood lymphocytes or B-lymphoblastoid cell lines to cis-Pt and CPT. Other reports had documented the selective killing of WRN-deficient cells by 4-nitroquinoline 1-oxide, mitomycin-C and 8-methoxy-psoralen + UV light using chromosomal breakage, colony formation or flow cytometric assays (12;16;23–30). The are inconsistent reports of the HU sensitivity of WRN-deficient cells, together with 1 or more reports of no selective sensitivity of WRN-deficient, often patient-derived, cells after UV damage or adriamycin, daunomycin, etoposide, trans-Pt, beneril or mitoxantron treatment (27;31). Fewer reports have documented the drug sensitivity of BLM-deficient cells. BLM-deficient human lymphoblasts and fibroblast cell lines were reported to be selectively killed by UV light or HU (32) and, less consistently, by CPT (25;33;34). Blm-mutant mouse ES cells appear to be hypersensitive to the intercalating agent ICRF-193, but in contrast to BLM-deficient human cells are mildly resistant to CPT and strongly resistant to HU (35).
One unexpected new finding with both mechanistic and clinical implications in our analyses was the marked 5-FU sensitivity of both WRN and BLM-deficient, TP53(+) and TP53(−) cells. There were few prior suggestions that this widely used chemotherapeutic anti-metabolite might selectively kill RecQ helicase-deficient human cells. Although the mode of action of FU is still poorly understood (36;37), loss of WRN or BLM could promote 5-FU cell killing by interfering with DNA replication or by inducing error-prone, homology-dependent recombination. Both DNA replication and recombination have different requirements for RecQ helicase function as discussed above. It should be possible to provide additional mechanistic insight into the 5-FU-mediated killing of WRN or BLM-deficient cells. DNA replication could be analyzed at the single molecule level with methods we developed and used to demonstrate replication defects in WRN-deficient human cells (15). Similar approaches have also been used to demonstrate replication defects in BLM-deficient human cells (9;38). Recombination defects in 5-FU-treated WRN or BLM-deficient cells could be quantified and analyzed at the molecular level using recombination reporter substrates of the type originally employed to identify the recombination resolution defect in WRN-deficient cells (see, e.g., 12).
Our results provide a first analysis of functional redundancy between WRN and BLM in human cells. Prior analyses of functional redundancy had used Wrn/Blm double-mutant mice, or avian DT-40 cells. In contrast, no patient has been identified who lacks more than one of the human RecQ helicases. Wrn/Blm-mutant mice do develop strong cellular and organismal phenotypes that resemble WS, but only after ≥3 generations in a telomerase-deficient background (39;40). These experiments thus support the idea that WRN and BLM may act on short or disrupted telomeres to suppress DNA damage responses, genetic instability and cellular senescence (41;42). Wrn/Blm-mutant avian DT-40 cells display a proliferation defect and are hypersensitive to CPT (43). Wrn-deficient DT-40 cells, in contrast, are only mildly sensitive to CPT, cis-Pt, 4-NQO, and MMS (44;45), whereas Blm-deficient DT-40 cells have a proliferative defect and are selectively sensitive to etoposide, bleomycin, 4NQO, UV (UVC) irradition, X-irradiation and HU (46;47).
We found that co-depletion of WRN and BLM from human cells suppressed cell proliferation, led to higher γ-H2AX staining both prior to and after HU arrest (Figures 3,4) and led to dose-dependent killing of both TP53(+) or TP53(−) cells by all four chemotherapeutic agents we tested (Figure 5). An important new finding in these analyses was lack of additive or synergistic defects in WRN/BLM co-depleted cells. This finding indicates that WRN and BLM may act in common pathway to suppress genomic damage and ensure cell survival after chemotherapeutic DNA damage. The stronger organismal and cellular phenotype of observed after loss of BLM suggests further that BLM may have a disproportionate role in this common functional pathway, or may have additional functions that were not readily revealed in our assays (3;4). One prediction from this model is that somatic cells, stem cells and tissue from Bloom syndrome patients will display higher levels of cell turnover, mutagenesis and telomere erosion than comparable cells or tissues from WS patients (48).
Our results provide a strong rationale for developing the human RecQ helicases as novel cancer therapeutic biomarkers and targets. RecQ helicase mutations are uncommon in human tumors, but epigenetic loss of expression appears to be frequent in common adult epithelial malignancies such as colorectal cancer (6;7). Thus RecQ expression profiling could identify tumors that could be selectively killed by widely used chemotherapeutic agents such as 5-FU, cis-Pt or CPT that selectively kill RecQ-deficient cells. Of note, as show above, these drugs selectively kill WRN and/or BLM-deficient cells regardless of TP53 status. Targeting human RecQ helicases may provide a second way to improve cancer chemotherapy. Direct inhibition of WRN or BLM should confer a drug sensitivity profile similar to that observed in RecQ-deficient, transfotrmed human cells. However, this strategy in its simplest form would not confer tumor-specific cell killing in vivo. An alternative approach would be to identify drugs or small molecules that inhibit survival pathways required for cell viability in the absence of WRN or BLM (49;50). Agents that selectively killed RecQ-deficient tumor cells might be useful as monotherapies, or in conjunction with lower doses of conventional chemotherapy. Our results and these additional approaches thus might allow highly effective therapies to be designed for cancer patients with RecQ helicase-deficient tumors.
Supplementary Material
Acknowledgments
We thank the Rabinovitch Lab (Department of Pathology, University of WA, Seattle, WA) for help with flow cytometric analyses, Alden F.M. Hackmann for data assembly and graphics support and Carla Grandori and Kiran Dhillon for help in establishing shRNA technology.
Support: This work was supported by an NCI PO1 Award to R.J.M., Jr. (CA77852), and a Mary Gates Undergraduate Research Fund Award to F.J.M.
Footnotes
Conflicts of interest: none to declare.
References
- 1.Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–20. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009 Sep;9(9):644–54. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 3.Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner’s syndrome: A review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine. 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- 4.German J. Bloom syndrome: a Mendelian prototype of somatic mutational disease. Medicine. 1993;72:393–406. [PubMed] [Google Scholar]
- 5.Wang LL, Levy ML, Lewis RA, Chintagumpala MM, Lev D, Rogers M, et al. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Hum Genet. 2001;102:11–7. doi: 10.1002/1096-8628(20010722)102:1<11::aid-ajmg1413>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 6.Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proceedings of the National Academy of Sciences. 2006 Jun 6;103(23):8822–7. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, Liu Z, Yamamoto H, et al. WRN promoter methylation possibly connects mucinous differentiation, microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod Pathol. 2007 Dec 14;21:150–8. doi: 10.1038/modpathol.3800996. [DOI] [PubMed] [Google Scholar]
- 8.Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374:577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao VA, Conti C, Guirouilh-Barbat J, Nakamura A, Miao ZH, Davies SL, et al. Endogenous γ-H2AX-ATM-Chk2 checkpoint activation in Bloom’s Syndrome helicase-deficient cells is related to DNA replication arrested forks. Mol Cancer Res. 2007 Jul 1;5(7):713–24. doi: 10.1158/1541-7786.MCR-07-0028. [DOI] [PubMed] [Google Scholar]
- 10.Xu X, Rochette PJ, Feyissa EA, Su TV, Liu Y. MCM10 mediates RECQ4 association with MCM2–7 helicase complex during DNA replication. EMBO J. 2009 Oct 7;28(19):3005–14. doi: 10.1038/emboj.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thangavel S, Mendoza-Maldonado R, Tissino E, Sidorova JM, Yin J, Wang W, et al. The human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol. 2009;30(6):1382–96. doi: 10.1128/MCB.01290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Homologous recombination resolution defect in Werner syndrome. Mol Cell Biol. 2002;22(20):6971–8. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pontén J, Saksela E. Two established in vitro cell lines from human mesenchymal tumours. Int J Cancer. 1967;2:434–47. doi: 10.1002/ijc.2910020505. [DOI] [PubMed] [Google Scholar]
- 14.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006 Mar 24;124(6):1283–98. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 15.Sidorova JM, Li N, Folch A, Monnat RJ., Jr The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle. 2008;7:796–807. doi: 10.4161/cc.7.6.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhillon KK, Sidorova J, Saintigny Y, Poot M, Gollahon K, Rabinovitch PS, et al. Functional role of the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell. 2007 Feb 3;6(1):53–61. doi: 10.1111/j.1474-9726.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- 17.Swanson C, Saintigny Y, Emond MJ, Monnat RJ., Jr The Werner syndrome protein has separable recombination and viability functions. DNA Repair. 2004;3:475–82. doi: 10.1016/j.dnarep.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Opresko PL, Calvo JP, von Kobbe C. Role for the Werner syndrome protein in the promotion of tumor cell growth. Mech Ageing Dev. 2007;128(7–8):423–36. doi: 10.1016/j.mad.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Venkatesan RN, Treuting PM, Fuller ED, Goldsby RE, Norwood TH, Gooley TA, et al. Mutation at the polymerase active site of mouse DNA polymerase g increases genomic instability and accelerates tumorigenesis. Mol Cell Biol. 2007 Nov 1;27(21):7669–82. doi: 10.1128/MCB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kronmal RA. Spurious correlation and the fallacy of the ratio standard. Journal of the Royal Statistical Society Series A. 1993;156:379–92. [Google Scholar]
- 21.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009 Oct 22;461(7267):1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura AJ, Rao VA, Pommier Y, Bonner WM. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle. 2010;9(2):389–97. doi: 10.4161/cc.9.2.10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gebhart E, Bauer R, Raub U, Schinzel M, Ruprecht KW, Jonas JB. Spontaneous and induced chromosomal instability in Werner syndrome. Hum Genet. 1988;80:135–9. doi: 10.1007/BF00702855. [DOI] [PubMed] [Google Scholar]
- 24.Ogburn CE, Oshima J, Poot M, Chen R, Hunt KE, Gollahon KA, et al. An apoptosis-inducing genotoxin differentiates heterozygotic carriers for Werner helicase mutations from wild-type and homozygous mutants. Hum Genet. 1997;101:121–5. doi: 10.1007/s004390050599. [DOI] [PubMed] [Google Scholar]
- 25.Okada M, Goto M, Furuichi Y, Sugimoto M. Differential effects of cytotoxic drugs on mortal and immortalized B-lymphoblastoid cell lines from normal and Werner’s syndrome patients. Biol Pharm Bull. 1998;21:235–9. doi: 10.1248/bpb.21.235. [DOI] [PubMed] [Google Scholar]
- 26.Poot M, Gollahon KA, Rabinovitch PS. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Human Genetics. 1999;104:10–4. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- 27.Poot M, Yom JS, Whang SH, Kato JT, Gollahon KA, Rabinovitch PS. Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB J. 2001 Mar 5;15:1224–6. doi: 10.1096/fj.00-0611fje. [DOI] [PubMed] [Google Scholar]
- 28.Poot M, Silber JR, Rabinovitch PS. A novel flow cytometric technique for drug cytotoxicity gives results comparable to colony-forming assays. Cytometry. 2002;48:1–5. doi: 10.1002/cyto.10101. [DOI] [PubMed] [Google Scholar]
- 29.Poot M, Gollahon KA, Emond MJ, Silber JR, Rabinovitch PS. Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: implications for the disease phenotype. FASEB J. 2002;16:757–8. doi: 10.1096/fj.01-0906fje. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Lopez AM, Jackson DA, Iborra F, Cox LS. Asymmetry of DNA replication fork progression in Werner’s syndrome. Aging Cell. 2002 Oct 27;1(1):30–9. doi: 10.1046/j.1474-9728.2002.00002.x. [DOI] [PubMed] [Google Scholar]
- 31.Karmakar P, Bohr VA. Cellular dynamics and modulation of WRN protein is DNA damage specific. Mech Ageing Dev. 2005 Nov;126(11):1146–58. doi: 10.1016/j.mad.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Davies SL, North PS, Dart A, Lakin ND, Hickson ID. Phosphorylation of the Bloom’s Syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004 Feb 1;24(3):1279–91. doi: 10.1128/MCB.24.3.1279-1291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poot M, Hoehn H. DNA topoisomerases and the DNA lesion in human genetic instability syndromes. Toxicology Letters. 1993 Apr;67(1–3):297–308. doi: 10.1016/0378-4274(93)90063-4. [DOI] [PubMed] [Google Scholar]
- 34.Rao VA, Fan AM, Meng L, Doe CF, North PS, Hickson ID, et al. Phosphorylation of BLM, dissociation from topoisomerase IIIα, and colocalization with γ-H2AX after topoisomerase I-induced replication damage. Mol Cell Biol. 2005 Oct 15;25(20):8925–37. doi: 10.1128/MCB.25.20.8925-8937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marple T, Kim TM, Hasty P. Embryonic stem cells deficient for Brca2 or Blm exhibit divergent genotoxic profiles that support opposing activities during homologous recombination. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2006 Dec 1;602(1–2):110–20. doi: 10.1016/j.mrfmmm.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Parker WB, Cheng YC. Metabolism and mechanism of action of 5-fluorouracil. Pharmacology & Therapeutics. 1990;48(3):381–95. doi: 10.1016/0163-7258(90)90056-8. [DOI] [PubMed] [Google Scholar]
- 37.Wyatt M, Wilson D. Participation of DNA repair in the response to 5-fluorouracil. Cellular and Molecular Life Sciences. 2009 Mar 1;66(5):788–99. doi: 10.1007/s00018-008-8557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies SL, North PS, Hickson ID. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol. 2007 Jul;14(7):677–9. doi: 10.1038/nsmb1267. [DOI] [PubMed] [Google Scholar]
- 39.Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, et al. Telomere shortening exposes functions for the mouse Werner and Bloom Syndrome genes. Mol Cell Biol. 2004 Oct 1;24(19):8437–46. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004 Aug;36(8):877–82. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 41.Multani AS, Chang S. WRN at telomeres: implications for aging and cancer. J Cell Sci. 2007 Mar 1;120(5):713–21. doi: 10.1242/jcs.03397. [DOI] [PubMed] [Google Scholar]
- 42.Opresko PL. Telomere ResQue and preservation--Roles for the Werner syndrome protein and other RecQ helicases. Mech Ageing Dev. 2008;129(1–2):79–90. doi: 10.1016/j.mad.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 43.Imamura O, Fujita K, Itoh C, Takeda S, Furuichi Y, Matsumoto T. Werner and Bloom helicases are involved in DNA repair in a complementary fashion. Oncogene. 2002;21:954–63. doi: 10.1038/sj.onc.1205143. [DOI] [PubMed] [Google Scholar]
- 44.Kawabe Yi, Seki M, Yoshimura A, Nishino K, Hayashi T, Takeuchi T, et al. Analyses of the interaction of WRNIP1 with Werner syndrome protein (WRN) in vitro and in the cell. DNA Repair. 2006 Jul 13;5(7):816–28. doi: 10.1016/j.dnarep.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 45.Otsuki M, Seki M, Kawabe Yi, Inoue E, Dong YP, Abe T, et al. WRN counteracts the NHEJ pathway upon camptothecin exposure. Biochemical and Biophysical Research Communications. 2007 Apr 6;355(2):477–82. doi: 10.1016/j.bbrc.2007.01.175. [DOI] [PubMed] [Google Scholar]
- 46.Imamura O, Fujita K, Shimamoto A, Tanabe H, Takeda S, Furuichi Y, et al. Bloom helicase is involved in DNA surveillance in early S phase in vertebrate cells. Oncogene. 2001;20:1143–51. doi: 10.1038/sj.onc.1204195. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi T, Seki M, Inoue E, Yoshimura A, Kusa Y, Tada S, et al. Vertebrate WRNIP1 and BLM are required for efficient maintenance of genome stability. Genes & Genetic Systems. 2008;83:95–100. doi: 10.1266/ggs.83.95. [DOI] [PubMed] [Google Scholar]
- 48.Kudlow BA, Kennedy BK, Monnat RJ. Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007 May;8(5):394–404. doi: 10.1038/nrm2161. [DOI] [PubMed] [Google Scholar]
- 49.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008 Mar;8(3):193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 50.Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009 Mar 6;136(5):823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.