

Summary
Background
Figitumumab is a fully human IgG2 monoclonal antibody targeting the insulin-like growth-factor-1 receptor (IGF-1R). Preclinical data suggest a dependence on insulin-like growth-factor signalling for sarcoma subtypes, including Ewing’s sarcoma, and early reports show antitumour activity of IGF-1R-targeting drugs in these diseases.
Methods
Between January, 2006, and August, 2008, patients with refractory, advanced sarcomas received figitumumab (20 mg/kg) in two single-stage expansion cohorts within a solid-tumour phase 1 trial. The first cohort (n=15) included patients with multiple sarcoma subtypes, age 18 years or older, and the second cohort (n=14) consisted of patients with refractory Ewing’s sarcoma, age 9 years or older. The primary endpoint was to assess the safety and tolerability of figitumumab. Secondary endpoints included pharmacokinetic profiling and preliminary antitumour activity (best response by Response Evaluation Criteria in Solid Tumours [RECIST]) in evaluable patients who received at least one dose of medication. This study is registered with ClinicalTrials.gov, number NCT00474760.
Findings
29 patients, 16 of whom had Ewing’s sarcoma, were enrolled and received a total of 177 cycles of treatment (median 2, mean 6·1, range 1–24). Grade 3 deep venous thrombosis, grade 3 back pain, and grade 3 vomiting were each noted once in individual patients; one patient had grade 3 increases in aspartate aminotransferase and gammaglutamyltransferase concentrations. This patient also had grade 4 increases in alanine aminotransferase concentrations. The only other grade 4 adverse event was raised concentrations of uric acid, noted in one patient. Pharmacokinetics were comparable between patients with sarcoma and those with other solid tumours. 28 patients were assessed for response; two patients, both with Ewing’s sarcoma, had objective responses (one complete response and one partial response) and eight patients had disease stabilisation (six with Ewing’s sarcoma, one with synovial sarcoma, and one with fibrosarcoma) lasting 4 months or longer.
Interpretation
Figitumumab is well tolerated and has antitumour activity in Ewing’s sarcoma, warranting further investigation in this disease.
Funding
Pfizer Global Research and Development.
Introduction
Systemic treatment options for refractory advanced sarcoma are limited and the prognosis for these patients is poor.1 A substantial proportion of sarcomas seem to be driven by chromosomal translocations or specific pathways, with characteristic molecular events2 that could be new targets for therapy. The insulin-like growth factor (IGF) signalling pathway is involved in sarcomagenesis and regulates cellular growth, proliferation, survival, and transformation.3 The insulin-like growth-factor-1 receptor (IGF-1R) is the key positive regulator for this IGF system.4 IGF-1R is a plasma-membrane-bound heterotetrameric receptor composed of two α-subunits and two β-subunits, linked by disulphide bonds. Binding of IGF-I and IGF-II to the α-subunits activates tyrosine kinases within the β-subunits, leading to autophosphorylation and activation of downstream processes.5 IGF-1R has a crucial role in tumour-cell growth, by mediating mitogenesis and maintaining a transformed phenotype, protecting tumour cells from apoptosis, and reducing growth-factor requirements.3 Impairment of IGF-1R function in vitro results in large-scale apoptosis of tumour cells, and abrogation of tumour growth and metastasis.6 The expression of IGF-1R and its ligands are disregulated in many tumour types.7–9 Inhibiting IGF-1R in different xenograft cancer models results in tumour growth inhibition and increased sensitivity to different cancer therapies.10
These data support the assessment of IGF-1R-targeting drugs in human malignant disease and have led to the development of molecularly targeted anticancer drugs comprising both monoclonal antibodies and small molecules. Studies support the investigation of IGF-1R blockade in several sarcoma subtypes, where this receptor seems to have a central role in disease initiation and maintenance.11 Specifically, IGF-1R has been implicated in growth, metastasis, and angiogenesis in Ewing’s sarcoma12 and rhabdomyosarcoma.13 These data, com bined with case reports describing antitumour activity of IGF-1R-targeting agents in Ewing’s sarcoma, led us to pursue single-stage expansions of a phase 1 study of figitumumab (Pfizer Inc, New London, CT, USA) for the treatment of patients with Ewing’s sarcoma and other sarcomas.
Figitumumab (previously CP-751,871) is a highly specific, fully human IgG2 monoclonal antibody that inhibits IGF-1R autophosphorylation induced by IGF-I and IGF-II, resulting in receptor internalisation and degradation.10 Phase 1 trials found that figitumumab has a favourable pharmacodynamic profile and is well tolerated as a single agent in patients with solid tumours,14 and in patients with myeloma.15 No dose-limiting toxicities were reported, and dose escalation was terminated at 20 mg/kg because it was not feasible to give higher doses. Figitumumab is also tolerable in combination with paclitaxel and carboplatin16 or docetaxel17 in different solid tumours, at the maximum feasible dose (MFD) of 20 mg/kg once every 3 weeks. However, to our knowledge there have been no specific reports of safety, tolerability, pharmacokinetics, and preclinical activity of IGF-IR monoclonal antibodies in a large series of patients with sarcoma, including young and paediatric patients. Here, we report data from two consecutive expansion cohorts from a phase 1 dose-escalation study of figitumumab in solid tumours.14
Methods
Patients
In the previously reported dose-escalation portion of this phase 1 study, 24 patients received figitumumab doses ranging from 3 to 20 mg/kg, the latter being the MFD and recommended phase 2 dose.14 The current investigation in two expansion cohorts was based on preclinical and clinical evidence for targeting IGF-1R in sarcoma, and the favourable toxicity, pharmacodynamic, and pharmacokinetic profile of figitumumab. Study objectives were to assess safety, tolerability, and pharmacokinetics, and to explore the antitumour activity of figitumumab in a specific sarcoma population, including young adult and paediatric patients. The first cohort included only adult patients with no restriction for sarcoma subtype, and the second cohort was open only to adult and paediatric patients with Ewing’s sarcoma. This study was done at the Royal Marsden NHS Foundation Trust (Sutton, UK), the Mayo Clinic (Rochester, MN, USA), and the University of Michigan Comprehensive Cancer Center (Ann Arbor, MI, USA). The study protocol was approved by the institutional review boards of the participating centres and was conducted in accordance with the Declaration of Helsinki, its amendments, and Good Clinical Practice guidelines.
For the first cohort, eligible patients were 18 years or older with histological or cytological evidence of metastatic or advanced sarcoma of any subtype. During the MFD expansion phase of the study, the protocol was amended to include patients 9 years or older with histological confirmation of Ewing’s sarcoma (the second cohort). All patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and to have progressed on previous standard therapies or to have no optimum standard treatment options. Additional eligibility criteria included: adequate bone-marrow, renal, and hepatic function (absolute neutrophil count ≥1000/μL, haemoglobin ≥80 g/L, platelets >75 000/μL, creatinine clearance >30 mL/min, total bilirubin <1·5×the institution upper limit of normal [ULN], aspartate aminotransferase [AST]/alanine aminotransferase [ALT] <2·5×ULN); full recovery from prior anticancer treatments; and use of adequate contraception in patients with reproductive potential. Written informed consent was obtained from each patient or from a legally acceptable representative before study enrolment. Exclusion criteria included: anticancer therapy or surgery within 4 weeks of first dose of figitumumab on study (or within 8 weeks for mitomycin C or nitrosoureas); palliative irradiation within 10 days; symptomatic or untreated brain metastases; pregnancy or lactation; significant active cardiac disease; concomitant high-dose corticosteroids (≥100 mg prednisone/day or equivalent); and serious active infection or other uncontrolled significant medical or psychiatric illness.
Procedures
Figitumumab was administered intravenously over 5 h, at the MFD of 20 mg/kg, in 500 mL of sodium chloride 0·9%. Patients in the first expansion cohort (adults with all sarcoma subtypes) received figitumumab once every 3 weeks. Patients in the second cohort (adults and children with Ewing’s sarcoma) received figitumumab once every 4 weeks, as approved by local ethics committees to allow the inclusion of paediatric patients 9 years or older. Patients continued on treatment until disease progression or unacceptable toxicity.
Safety assessments, including clinical and laboratory evaluations, were done during each treatment cycle. Treatment-related adverse events were graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 3.0, and qualified as possibly, probably, or definitely related to figitumumab administration. Patients were assessed for subacute and late toxicities up to 150 days after the last study dose. All patients receiving at least one dose of figitumumab were assessed for safety. Tumour response was assessed every two cycles (6–8 weeks) by CT, MRI, or both using Response Evaluation Criteria in Solid Tumours (RECIST; version 1). Patients who had at least one post-treatment scan were eligible for response assessment.
Blood samples were collected in sodium heparin-containing tubes 30 min before antibody infusion and 1 h, 24 h, 3, 7, and 14 days after the end of infusion, during cycles one and four. Additional samples were collected 30 min before and 1 h after infusion during cycles two and three. At cycles five and beyond, samples were collected 30 min before infusion and at the end of the study. Plasma concentrations of figitumumab were analysed by ELISA, as previously described.10 The lower limit of quantitation was 120 ng/mL.
Statistical analysis
Patients were followed up for analysis until June 17, 2009. Plasma concentration–time data for figitumumab were analysed by non-compartmental methods.18 For treatment cycles one and four, the area under the plasma concentration–time curve (AUC) from time 0 to the last sampling timepoint with quantifiable concentration within a cycle (AUClast), and from time 0 to the end of a cycle (AUC0–tau), was found with the linear/log trapezoidal approximation. The accumulation ratio was calculated as the ratio of cycle four AUC0–tau to cycle one AUC0–tau. Pharmacokinetic parameters in patients with sarcoma were compared with those in non-sarcoma patients in the (previously reported) solid-tumour cohort of this trial.14,19 Analysis of pharmacokinetic parameters was done with WinNonlin software, version 5.2.
This study is registered with ClinicalTrials.gov, number NCT00474760.
Role of the funding source
This study was proposed and written by the investigators with the support of the sponsor. The investigators and the sponsor together were responsible for data collection and analysis of the raw data. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
29 patients with sarcoma (21 males and eight females) were enrolled between January, 2006, and August, 2008. Patient characteristics and demographics for each expansion cohort are summarised in table 1. Median age was 30 years (range 12–63) and six patients were 18 years or younger. The most common histological subtype was Ewing’s sarcoma (16 patients). All but one patient received at least one previous chemotherapy regimen and the median number of chemotherapy regimens received was three. Two patients had previous peripheral-blood stem-cell transplant (PBSCT); 21 had prior radiotherapy, and 20 had received excisional surgery for primary or metastatic tumours. A total of 177 cycles of figitumumab had been given at the time of this analysis (mean 6·1, median 2, range 1–24).
Table 1.
Baseline characteristics (N=29)
| N | |
|---|---|
| Age (years) * | |
| ≤18 years old | 6 |
| Sex | |
| Male | 21 |
| Female | 8 |
| ECOG performance status | |
| 0 | 15 |
| 1 | 14 |
| Sarcoma histological subtype | |
| Ewing’s sarcoma | 16 |
| Synovial sarcoma | 5 |
| DSRCT | 3 |
| Alveolar rhabdomyosarcoma | 2 |
| Fibrosarcomas | 2 |
| Myxoid chondrosarcoma | 1 |
| Previous treatments | |
| Excisional surgery (resected/partially resected) | 20 |
| Previous chemotherapy | 28 |
| 1 regimen | 3 |
| 2 regimens | 6 |
| ≥3 regimens | 19 |
| PBSCT | 2 |
| Allogenic | 1 |
| Autologous | 1 |
| Previous radiotherapy | 21 |
| Neoadjuvant | 9 |
| Adjuvant | 9 |
| Metastatic disease | 9 |
N=number of patients. ECOG=Eastern Cooperative Oncology Group. DSRCT=desmoplastic small round cell tumour. PBSCT=peripheral-blood stem-cell transplant.
Median age=30 years (range 12-63).
All patients were eligible for safety and tolerability assessment. Adverse events were mostly mild-to-moderate in severity and reversible. Severe (grade 3) adverse events included deep venous thrombosis (n=1), vomiting (n=1), and back pain (n=1). Clinically significant laboratory abnormalities included grade 4 raised uric acid concentration in one patient, and grade 3–4 raised concentrations of AST, ALT, and gamma glutamyltransferase (GGT) in a patient receiving concomitant fluconazole, paracetamol, and lorazepam. Table 2 lists all reported adverse events of grade 2 or higher. Grade 1 hyperglycaemia (6·5–8·9 mmol/L) occured in four patients and another patient developed grade 2 hyperglycaemia (9·0–13·9 mmol/L) requiring metformin and glibencamide. Insulin levels were assessed in 14 of the 29 patients. Repeated echocardiographic assessments did not show substantial changes in cardiac valve function or left ventricular ejection fraction (LVEF) in any patient on the study, including 21 patients who were pretreated with anthracyclines. A pre pubertal male participant, who is still on treatment and has so far received 24 cycles of figitumumab, did not experience initial growth delay but did have delayed onset of puberty. An investigation showed hypothyroidism and primary testicular failure, possibly related to prior chemotherapy exposure. The participant’s growth rate accelerated after introduction of thyroxine. Sex-hormone replacement is being postponed in this patient to prevent a suboptimum growth spurt while he remains on a planned further year of IGF-1R inhibition.
Table 2.
Number of adverse events of grade 2 or higher attributed to figitumumab (N=29)
| Grade 2 | Grade 3 | Grade 4 | Total (%) | |
|---|---|---|---|---|
| Deep venous thrombosis | .. | 1 | .. | 3·5 |
| Anorexia/decreased appetite | 2 | .. | .. | 6·9 |
| Arthralgia | 2 | .. | .. | 6·9 |
| Diarrhoea | 2 | .. | .. | 6·9 |
| Back pain | 1 | 1 | .. | 6·9 |
| Fatigue | 3 | .. | .. | 10·4 |
| Headache | 3 | .. | .. | 10·4 |
| Limb cramps | 2 | .. | .. | 6·9 |
| Skin reactions (rash/urticaria/ infection/eczema) |
4 | .. | .. | 13·7 |
| Candidiasis (oral or vaginal) | 2 | .. | .. | 6·9 |
| Vomiting | 1 | 1 | .. | 6·9 |
| Weight loss | 2 | .. | .. | 6·9 |
| Laboratory abnormalities | ||||
| Raised uric acid concentration | .. | .. | 1 | 3·5 |
| Raised ALT concentration | .. | .. | 1* | 3·5 |
| Raised AST concentration | .. | 1* | .. | 3·5 |
| Anaemia/low haemoglobin | 2 | .. | .. | 6·9 |
| Raised GGT concentration | 2 | 1* | .. | 10·4 |
| Lymphocyte count decrease | 2 | .. | .. | 6·9 |
ALT=alanine aminotransferase. AST=aspartate aminotransferase. GGT=gammaglutamyltransferase.
Grade 3 raised GGT and AST concentrations, and grade 4 ALT, were reported in the same patient; these adverse events were initially attributed to figitumumab, but resolved upon discontinuation of concomitant paracetamol, fluconazole, and lorazepam.
Figitumumab plasma concentrations decreased in a similar and multi-exponential pattern for all patients in this trial (figure 1). Plasma concentrations at the end of infusion (C1h) and the AUC0–tau of figitumumab during treatment cycles one and four are shown in table 3. A moderate accumulation in plasma exposure was seen in most patients after figitumumab dosing of once every 3 or 4 weeks (figure 1). The apparent elimination half-life (t1/2) in these patients was not found, because sampling within 21-day or 28-day cycles did not sufficiently capture the terminal disposition phase in most patients. Although there were only six paediatric patients treated in this study, no apparent differences in pharmacokinetics were noted in this small cohort (data not shown).
Figure 1.
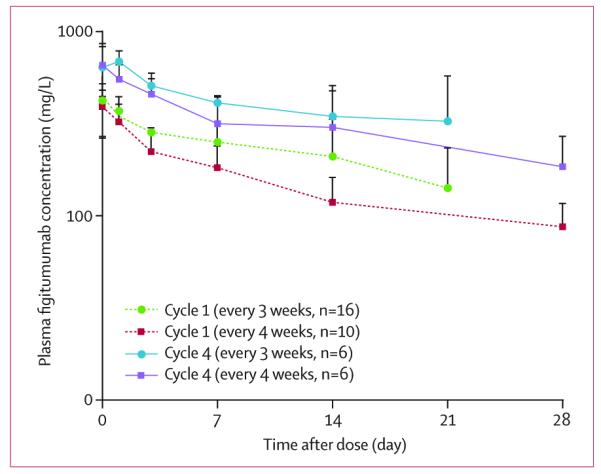
Cycle 1 and cycle 4 mean (SD) plasma concentration–time profiles of figitumumab in patients with sarcoma after dosing every 3 or 4 weeks
Table 3.
Plasma exposure for figitumumab (mean [SD]) given at 20 mg/kg, according to dosing regimen
| Cycle 1 |
Cycle 4 |
Accumulation ratio | |||||
|---|---|---|---|---|---|---|---|
| N | Clhr (mg/L) | AUC0-tau (mgxh/L) | N | C1hr (mg/L) | AUC0-tau (mgxh/L) | ||
| Every 3 weeks | 16 | 419 (99) | 118 000 (48 700)* | 6 | 633 (216) | 218 000 (41 100)† | 1·6 (0·6)† |
| Every 4 weeks | 10 | 385 (91) | 100 000 (27 700) | 6 | 650 (171) | 207 000 (77 200) | 2·0 (04) |
N=number of patients. C1hr=plasma concentrations at the end of infusion. AUC0-tau=area under the plasma concentration-time curve from time 0 to the end of the cycle.
11 patients.
3 patients.
28 of 29 patients had a pre-treatment and at least one post-treatment scan, and were assessable for tumour response by RECIST; the remaining patient (with Ewing’s sarcoma) was discontinued from the study after one treatment cycle due to rapid clinical deterioration attributed to disease progression. 22 of these 28 evaluable patients had measurable disease by RECIST (figure 2). The patients with non-measurable disease were assessed as having stable disease (n=1) or progressive disease (n=5). Of the 28 assessable patients, two confirmed responses were seen (both in patients with Ewing’s sarcoma): one pathological complete response, which was confirmed at surgical resection of the residual lesion (figure 3A), and one partial response with decrease or clearance of the metastatic lung deposits (a decrease in a non-target lesion in the mediastinum was also seen in this patient, after 45 Gy of radiation therapy in 15 fractions, in addition to figitumumab treatment; figure 3B). Six additional patients with Ewing’s sarcoma had disease stabilisation lasting from 4 to more than 16·1 months (table 4). One patient with Ewing’s sarcoma had a partial response in target lesions, but developed new symptomatic bone metastasis on bone scan that led to a classification of progressive disease. Among the 13 re maining patients, one with fibrosarcoma and one with synovial sarcoma had prolonged stable disease lasting for 9·8 and 8·9 months, respectively (table 4).
Figure 2. Waterfall plot of best responses, as percentage decrease in tumour size by RECIST criteria, of the target lesions in 22 evaluable patients.

Green bars are patients with disease progression (increase in target-lesion size); blue bars are patients with a decrease in size of the target lesions. The numbers above/below the bars represent the number of treatment cycles patients received before withdrawal due to disease progression. RECIST=Response Evaluation Criteria in Solid Tumours. DSRC=desmoplastic small round cell tumour. *Remains on study.†Partial response in soft-tissue disease but progressive disease with a new bone lesion. ‡Patient with a partial response after treatment and complete response after surgery. §Reduction in target lesions after two cycles of treatment and radiotherapy to the mediastinum.
Figure 3. CT scan of a 12-year-old patient with Ewing’s sarcoma, at baseline and after six cycles of treatment (A), and a 24-year-old patient with Ewing’s sarcoma with ongoing partial response (B).

In A, target lesion decreased by about 60% after six cycles, with a remaining central calcification. The residual mass was resected and confirmed as a pathological complete response. Non-target lesions also disappeared. In B, several target lung nodules disappeared after four cycles of treatment, qualifying for partial response. The non-target lesion in the mediastinum decreased by 60% (this also followed 45 Gy of radiation therapy in 15 fractions).
Table 4.
Duration and type of response according to histology subtype
| Time responding (months) | |
|---|---|
| Complete response (N=1) | |
| Ewing’s sarcoma | 21·5* |
| Partial response (N=1) | |
| Ewing’s sarcoma | 11·4* |
| SD ≥9 months (N=4) | |
| Ewing’s sarcoma | 16·1* |
| Ewing’s sarcoma | 12·5* |
| Ewing’s sarcoma | 10·2 |
| Fibrosarcoma | 9·8 |
| SD ≥6 months (N=2) | |
| Ewing’s sarcoma | 7·1 |
| Synovial sarcoma | 8·9 |
| SD ≥3 months (N=2) | |
| Ewing’s sarcoma | 5·6 |
| Ewing’s sarcoma | 4·0 |
Ongoing figitumumab treatment. N=number of patients. SD=stable disease.
11 of the 22 patients with measurable disease by RECIST had some degree of tumour reduction (figure 2). Apart from the reported complete response and partial response, five other patients with Ewing’s sarcoma patients with measurable disease had shrinkage of the target tumour lesions. Small decreases in tumour size were also seen in patients with chondrosarcoma, fibrosarcoma, and synovial sarcoma. The non-progression rate at 3 months was 34% (95% CI 10–58) and at 6 months was 28% (5–51), for all patients in this study. In the Ewing’s sarcoma subgroup, eight of 15 patients evaluable for response were free of disease progression at 3 months, and six of 15 patients at 6 months.
Discussion
To our knowledge, this is the first study assessing the anti-IGF-1R monoclonal antibody, figitumumab, in a specific cohort of sarcoma patients. This study confirmed that figitumumab at 20 mg/kg every 3–4 weeks is safe for patients with sarcoma and is associated with a favourable pharmacokinetic profile. Additionally, objective antitumour activity was seen in patients with Ewing’s sarcoma.
Targeted agents have improved the treatment of some sarcoma subtypes, with inhibitors of stem-cell factor receptor (KIT) having activity in gastrointestinal stromal tumours (GIST)20 and platelet-derived growth-factor receptor in dermatofibrosarcoma protruberans.21 However, clinical benefit with targeted agents in other sarcoma subtypes has been elusive.22 Targeted therapies can produce dramatic tumour shrinkage when cancer-cell survival is dependent on the target’s expression or activation, but the expression of a target does not necessarily correlate with response to its inhibition.23 IGF-1R is widely expressed and implicated as having a key role in several sarcoma subtypes, including Ewing’s sarcoma and rhabdomyosarcoma.11 This role is supported by clinical case reports of antitumour activity with IGF-1R targeting antibodies in patients with Ewing’s sarcoma.24,25 Our study was not powered to formally detect antitumour activity as an endpoint. However, we report such activity of figitumumab in Ewing’s sarcoma as evidenced by objective responses and non-progression. Non-progression is widely considered to be an indicator of activity in sarcoma,26 and these results compare favourably with those of other targeted agents. Preliminary data for the mTOR inhibitor, deferolimus, have shown a non-progression rate of 30% at 16 weeks in bone sarcomas.27 The mTOR inhibitor was deemed active and a phase 3 trial comparing deferolimus with placebo, as maintenance therapy, has begun. Furthermore, a recent trial in various sarcoma subtypes showed that imatinib has no activity in patients with Ewing’s sarcoma, with no objective responses and no patient remaining on the drug longer than 4 months.28 Minor disease activity was also seen with figitumumab in other sarcoma subtypes, which supports the rationale of targeting the IGF1-R pathway in sarcoma and the investigation of figitumumab in larger trials.
Repeated administration of figitumumab was well tolerated in this study; only four cases of mild hyperglycaemia were reported and one case of moderate hyperglycaemia, which required oral antidiabetic agents. The mechanism for the observed hyperglycaemia is unclear, although IGF-1R may be involved in glucose metabolism via cross-talk and heterodimerisation with the insulin receptor.29–32 This observation, and the increased plasma insulin levels reported after figitumumab treatment,14,15 suggest compensatory insulin secretion and associated insulin resistance, the latter possibly secondary to increased IGF-1 and growth-hormone levels.3,33 No cardiac toxicity was reported in this study; this is noteworthy since three-quarters of the patients were pretreated with anthracyclines, and despite the potential cardiotoxicity suggested by expression of IGF-1R on cardiac myocytes.
Theoretically, figitumumab would be expected to have an inhibitory effect on IGF and growth-hormone-mediated growth, which is supported by the identification of patients with genetic defects in the IGF-1 axis such as IGF-1 deficiency.34 No specific assessments of bone age or hormone levels were undertaken in this study; however, detailed investigations of growth and hormone levels are being done in an ongoing phase 2 study of figitumumab in paediatric patients with Ewing’s sarcoma. In our study, a heavily pretreated, prepubertal male patient (12 years old) has remained on treatment for almost 2 years at the time of this analysis. His growth rate during the first year on study was at the lower limit of normal and declined during his second year on treatment, with delayed puberty. Investigations suggested a complex aetiology, including acquired hypothyroidism and primary testicular failure. Although his growth rate improved on thyroxine replacement and his sex-hormone deficiency has not yet been treated, a possible contribution of figitumumab cannot be ruled out. A large group of actively growing children would need to be treated and followed for several months to accurately address the effect of IGF-1R targeted therapy on growth during childhood and puberty. However, this particular patient shows that growth can occur during prolonged treatment with figitumumab.
Figitumumab is a fully human anti-IGF-1R IgG2 antibody with a longer half-life than anti-IGF-1R IgG1 antibodies.10 Pharmacokinetic data from previous dose-escalation studies support the administration of figitumumab every 3 weeks.14,15 The present study shows that a similar accumulation in plasma figitumumab was achieved at a dose of 20 mg/kg every 4 weeks, supporting the feasibility of this dosing regimen. Pharmacokinetic parameters in patients with sarcoma were comparable with those previously reported in patients with other solid tumour types.14,19 These findings differentiate figitumumab from IgG1 therapeutic antibodies to IGF-1R, which have shorter half-lives and require more frequent administration.24,25,35 Additionally, figitumumab does not seem to support antibody-dependent cellular cytotoxicity or complement activity, which has been described for IgG1 antibodies. Therefore, the activity of figitumumab should be purely attributed to its ability to inhibit IGF-1R.
Whether translocation type has any value for predicting response in this group of patients is unclear. Defining predictive biomarkers for IGF-1R therapy should be a priority for the further development of these agents and needs to be addressed prospectively in larger studies. Although IGF-1R protein expression was not assessed in this trial, expression alone is probably not sufficiently predictive of clinical benefit from IGF-1R targeting drugs, and a suite of biomarkers will likely be required for patient selection.
In conclusion, our results show that figitumumab can be safe for both adult and paediatric sarcoma patients, and has single-agent antitumour activity in different sarcoma subtypes, including Ewing’s sarcoma. Phase 2 studies of figitumumab and other anti-IGF-R agents in Ewing’s sarcoma and other sarcoma subtypes are now completing accrual and rational combinations with other treatments are also being pursued.
Acknowledgments
The ASCO Foundation awarded DO with a 2008 ASCO Annual Meeting Merit Award for the presentation of this work. Editorial assistance was provided by Siân Marshall, ACUMED (Tytherington, UK) and was funded by Pfizer Inc. This study was funded by Pfizer Inc. The Drug Development Unit of the Royal Marsden NHS Foundation Trust and The Institute of Cancer Research (ICR) receive support from Cancer Research UK and the National Institute for Health Research Biomedical Research Centre. The ICR also receives support from the Experimental Cancer Medicine Centre.
Footnotes
Contributors DO, MLP, DY, IJ, AG, JSB, and PH were responsible for the conception and design of the study. AG was responsible for financial support. DO, SHO, SMS, GNB, KP-J, IJ, MS, JSB, and PH were responsible for the provision of study materials or patients. DO, SP-V, LRM, MLP, GNB, DY, IJ, JSB, and PH were responsible for the collection and assembly of data. DO, SP-V, LRM, SHO, MLP, DY, IJ, FPW, AG, MS, JSB, and PH were responsible for data analysis and interpretation. DO, SP-V, LRM, MLP, DY, KP-J, IJ, AG, MS, JSB, and PH contributed to writing the manuscript; all authors gave their final approval to the manuscript.
Conflicts of interest JSB and PH have received research funding from, and have served in a consultant or advisory role for, Pfizer Oncology. DY, MLP, and AG are employees of Pfizer Oncology and own stock in the company. IJ has received honoraria from Pfizer Oncology. All other authors declared no conflicts of interest.
References
- 1.Hartmann JT. Systemic treatment options for patients with refractory adult-type sarcoma beyond anthracyclines. Anticancer Drugs. 2007;18:245–54. doi: 10.1097/CAD.0b013e3280124e41. [DOI] [PubMed] [Google Scholar]
- 2.Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer. 2003;3:685–94. doi: 10.1038/nrc1168. [DOI] [PubMed] [Google Scholar]
- 3.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–28. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 4.Samani AA, Yakar S, LeRoith D, et al. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- 5.Massague J, Czech MP. The subunit structures of two distinct receptors for insulin-like growth factors I and II and their relationship to the insulin receptor. J Biol Chem. 1982;257:5038–45. [PubMed] [Google Scholar]
- 6.Reiss K, D’Ambrosio C, Tu X, et al. Inhibition of tumour growth by a dominant negative mutant of the insulin-like growth factor I receptor with a bystander effect. Clin Cancer Res. 1998;4:2647–55. [PubMed] [Google Scholar]
- 7.Hankinson SE, Willett WC, Colditz GA, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–96. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- 8.Cardillo MR, Monti S, Di Silverio F, et al. Insulin-like growth factor(IGF)-I, IGF-II and IGF type I receptor (IGFR-I) expression in prostatic cancer. Anticancer Res. 2003;23:3825–35. [PubMed] [Google Scholar]
- 9.Durai R, Yang W, Gupta S, et al. The role of the insulin-like growth factor system in colorectal cancer: review of current knowledge. Int J Colorectal Dis. 2005;20:203–20. doi: 10.1007/s00384-004-0675-4. [DOI] [PubMed] [Google Scholar]
- 10.Cohen BD, Baker DA, Soderstrom C, et al. Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res. 2005;11:2063–73. doi: 10.1158/1078-0432.CCR-04-1070. [DOI] [PubMed] [Google Scholar]
- 11.Rikhof B, de Jong S, Suurmeijer AJ, et al. The insulin-like growth factor system and sarcomas. J Pathol. 2009;217:469–82. doi: 10.1002/path.2499. [DOI] [PubMed] [Google Scholar]
- 12.Manara MC, Landuzzi L, Nanni P, et al. Preclinical in vivo study of new insulin-like growth factor-I receptor-specific inhibitor in Ewing’s sarcoma. Clin Cancer Res. 2007;13:1322–30. doi: 10.1158/1078-0432.CCR-06-1518. [DOI] [PubMed] [Google Scholar]
- 13.Scotlandi K, Manara MC, Nicoletti G, et al. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res. 2005;65:3868–76. doi: 10.1158/0008-5472.CAN-04-3192. [DOI] [PubMed] [Google Scholar]
- 14.Haluska P, Shaw HM, Batzel GN, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res. 2007;13:5834–40. doi: 10.1158/1078-0432.CCR-07-1118. [DOI] [PubMed] [Google Scholar]
- 15.Lacy MQ, Alsina M, Fonseca R, et al. Phase I, pharmacokinetic and pharmacodynamic study of the anti-insulinlike growth factor type 1 receptor monoclonal antibody CP-751,871 in patients with multiple myeloma. J Clin Oncol. 2008;26:3196–203. doi: 10.1200/JCO.2007.15.9319. [DOI] [PubMed] [Google Scholar]
- 16.Karp DD, Paz-Ares LG, Novello S, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol. 2009;27:2516–22. doi: 10.1200/JCO.2008.19.9331. [DOI] [PubMed] [Google Scholar]
- 17.de Bono JS, Attard G, Adjei A, et al. Potential applications for circulating tumor cells expressing the insulin-like growth factor-I receptor. Clin Cancer Res. 2007;13:3611–16. doi: 10.1158/1078-0432.CCR-07-0268. [DOI] [PubMed] [Google Scholar]
- 18.Gibaldi M, Perrier D. Non compartmental analysis based on statistical moment therapy. Marcel Dekker; New York: 1982. [Google Scholar]
- 19.Haluska P, Worden F, Olmos D, et al. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother Pharmacol. 2009 doi: 10.1007/s00280-009-1083-9. published online Aug 2, 2009. DOI: 10.1007/s00280-009-1083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumours. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 21.Rubin BP, Schuetze SM, Eary JF, et al. Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. J Clin Oncol. 2002;20:3586–91. doi: 10.1200/JCO.2002.01.027. [DOI] [PubMed] [Google Scholar]
- 22.Ray-Coquard I, Le Cesne A, Whelan JS, et al. A phase II study of gefitinib for patients with advanced HER-1 expressing synovial sarcoma refractory to doxorubicin-containing regimens. Oncologist. 2008;13:467–73. doi: 10.1634/theoncologist.2008-0065. [DOI] [PubMed] [Google Scholar]
- 23.Carden CP, Molife LR, de Bono JS. Predictive biomarkers for targeting insulin-like growth factor-I (IGF-I) receptor. Mol Cancer Ther. 2009;8:2077–78. doi: 10.1158/1535-7163.MCT-09-0641. [DOI] [PubMed] [Google Scholar]
- 24.Tolcher AW, Rothenberg ML, Rodon J, et al. A phase I pharmacokinetic and pharmacodynamic study of AMG 479, a fully human monoclonal antibody against insulin-like growth factor type 1 receptor (IGF-1R), in advanced solid tumours. Proc Am Soc Clin Oncol. 2007;25 doi: 10.1200/JCO.2009.23.6745. abstr 3002. [DOI] [PubMed] [Google Scholar]
- 25.Benjamin R, Gore L, Dias C, et al. Activity of R1507, a fully humanized monoclonal antibody IGF-1R (insulin-like growth factor receptor)antagonist, in patients with Ewing’s sarcoma noted in a phase I study. 13th Annual Meeting of the Connective Tissue Oncology Society; Seattle, WA, USA. Oct 31–Nov 3, 2007; abstr 932. [Google Scholar]
- 26.Van Glabbeke M, Verweij J, Judson I, et al. Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur J Cancer. 2002;38:543–49. doi: 10.1016/s0959-8049(01)00398-7. [DOI] [PubMed] [Google Scholar]
- 27.Chawla SP, Tolcher AW, Staddon AP, et al. Updated results of a phase II trial of AP23573, a novel mTOR inhibitor, in patients (pts) with advanced soft tissue or bone sarcomas. Proc Am Soc Clin Oncol. 2006;24:9505. [Google Scholar]
- 28.Chugh R, Wathen JK, Maki RG, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J Clin Oncol. 2009;27:3148–53. doi: 10.1200/JCO.2008.20.5054. [DOI] [PubMed] [Google Scholar]
- 29.Guler HP, Zapf J, Froesch ER. Short-term metabolic effects of recombinant human insulin-like growth factor I in healthy adults. N Engl J Med. 1987;317:137–40. doi: 10.1056/NEJM198707163170303. [DOI] [PubMed] [Google Scholar]
- 30.Clemmons DR. Involvement of insulin-like growth factor-I in the control of glucose homeostasis. Curr Opin Pharmacol. 2006;6:620–25. doi: 10.1016/j.coph.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Moses AC, Young SC, Morrow LA, et al. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes. 1996;45:91–100. doi: 10.2337/diab.45.1.91. [DOI] [PubMed] [Google Scholar]
- 32.Pennisi P, Gavrilova O, Setser-Portas J, et al. Recombinant human insulin-like growth factor-I treatment inhibits gluconeogenesis in a transgenic mouse model of type 2 diabetes mellitus. Endocrinology. 2006;147:2619–30. doi: 10.1210/en.2005-1556. [DOI] [PubMed] [Google Scholar]
- 33.del Rincon JP, Iida K, Gaylinn BD, et al. Growth hormone regulation of p85alpha expression and phosphoinositide 3-kinase activity in adipose tissue: mechanism for growth hormone-mediated insulin resistance. Diabetes. 2007;56:1638–46. doi: 10.2337/db06-0299. [DOI] [PubMed] [Google Scholar]
- 34.Laron Z, Anin S, Klipper-Aurbach Y, et al. Effects of insulin-like growth factor on linear growth, head circumference, and body fat in patients with Laron-type dwarfism. Lancet. 1992;339:1258–61. doi: 10.1016/0140-6736(92)91594-x. [DOI] [PubMed] [Google Scholar]
- 35.Hidalgo M, Gomez M Tirado, Lewis N, et al. A phase I study of MK-0646, a humanized monoclonal antibody against the insulin-like growth factor receptor type 1 (IGF1R) in advanced solid tumour patients in a q2 wk schedule. Proc Am Soc Clin Oncol. 2008;26 abstr 3520. [Google Scholar]