

Abstract
Apoptosis is the best-characterized form of programmed cell death (PCD) and is of fundamental importance in tissue homeostasis. In mammalian systems, there are two major pathways that are involved in the initiation of apoptosis: the “extrinsic” death receptor pathway and the “intrinsic” mitochondrial pathway. Although these pathways act independently to initiate the death machinery in some cellular systems, in many cell types, including numerous tumor cells, there is delicate coordination and cross talk between the extrinsic and intrinsic pathways, which leads to the activation of the executioner caspase cascade. Additionally, there appears to be a fine balance between the caspase-mediated arm of death receptor signaling that engages mitochondria and the caspase-independent arm that promotes vacuole proliferation in many cells. Here, we review our current knowledge about the layers of complexity that are posed by the interactions between death receptor-induced pathways and how they influence mitochondria to regulate cellular life and death decisions.
Keywords: death receptors, mitochondria, Bid, membranes, phospholipases, cardiolipin
INTRODUCTION
Apoptosis, or programmed cell death, is an evolutionarily conserved mechanism for the selective removal of aging, damaged or otherwise unwanted cells.1–7 It is an essential component of many normal physiological processes such as embryogenesis, normal tissue development and the immune response.8 Thus, regulation of apoptosis is critical for tissue homeostasis and its deregulation can lead to a variety of pathological conditions. Inhibition of apoptosis or resistance to apoptosis contributes to carcinogenesis and chemoresistance.2,9–14 On the other hand, enhanced apoptosis is involved in diverse diseases such as myocardial ischemia, neurodegenerative diseases, stroke, septic shock and AIDS.11,12
Apoptosis is primarily mediated through the activation of specific proteases called caspases (cysteinyl, aspastate-specific proteases).2,3,15–17 Caspases are effectors of cell suicide and cleave multiple substrates leading to biochemical and morphological changes that are characteristic to apoptotic cells.5,7 These alterations include cell membrane re-modeling and blebbing, exposure of phosphatidylserine at the external surface of the cell (PS), cell shrinkage with cytoskeletal rearrangements, nuclear condensation and DNA fragmentation.1,3,11,18–20 These morphological changes culminate in the formation of apoptotic bodies that are normally eliminated by phagocytosis.21,22 In mammalian systems, the “extrinsic” death receptor pathway and the “intrinsic” mitochondrial pathway are the two major signaling systems that result in the activation of the executioner caspases, and the consequent induction of cell death.2,3,5,7,18 In the past few years, increasing evidence indicates that the death receptor and mitochondrial pathways are not isolated systems. Instead significant cross-talk and ‘biofeedback’ regulates the apoptotic machinery.7,9,23,24 In this review, we discuss some recent insights into the interconnections between these apoptotic pathways.
THE DEATH RECEPTOR PATHWAY OF APOPTOSIS
The extrinsic apoptotic pathway is activated upon the binding of cytokine ligands (i.e., FasL, TNF and TRAIL) to members of the TNFα receptor super-family, which are usually called the death receptors (i.e., Fas, also called CD95/Apo-1; TNF receptors; TRAIL receptors).2,3,7,18–20 Death receptors contain an intracellular globular interaction domain known as a death domain (DD). Death receptors aggregate at the cell surface following ligand binding to their extracellular domains—possibly to form trimers. This results in the recruitment of adaptor molecules to the aggregated intracellular domains of the receptors. One of the major adaptors to be recruited is Fas Associated Death Domain, FADD, which possesses a DD that interacts either directly with the DD of death receptors, or indirectly through another adaptor molecule, TRADD (TNF Receptor Associated Death Domain). FADD also contains a second protein interaction domain, known as the Death Effector Domain (DED). The DED domain of FADD interacts with the DED of the weakly active zymogen, pro-caspase-8, to form an intracellular multi-protein complex known as the Death Inducing Signaling Complex (DISC).26–30 Once formed, the DISC promotes the proximity-induced activation of caspase-8, which then proceeds to be further processed via an auto-proteolysis mechanism.31,32 Whether still bound to the DISC or released in other intracellular compartments, active caspase-8 activates executioner/effector caspases, such as caspase-3, leading to cell execution via degradation of the nucleus and other intracellular structures.18,29,33,34 This direct activation of caspase-dependent cell execution, which does not require mitochondria, is believed to occur in select cell types, including thymoctyes, that are classified as Type I cells.27,33,35 These cells are able to efficiently activate caspase-8, so that its major target is downstream cleavage and consequent activation of executioner caspases such as caspase-3. This simplified pathway of Type I cells plays an important role in the immune response that is involved in the deletion of transformed cells9,36 and resembles the linear pathway of developmental cell death established in genetic studies of C. elegans.37,38 Nonetheless, programmed cell death (PCD) in C. elegans is distinct in that Bcl-2/Ced-9 is unable to block caspase activation following death receptor stimulation in Type I cells.18,39 Consequently, the simplified extrinsic pathways of mammalian Type I cells is likely to result from a reductionist pattern of evolution.
THE MITOCHONDRIAL PATHWAY OF APOPTOSIS
Mitochondria are now thought to be the central intracellular organelles involved in mediating the majority of apoptotic pathways in mammalian cells.25,40–44 In general, mitochondria are engaged via the intrinsic pathway of cell death, which can be initiated by a variety of stress stimuli including UV radiation, γ-irradiation, heat, DNA damage, the actions of some oncoproteins and tumor suppressor genes, viral virulence factors, and most chemotherapeutic agents.40 These diverse forms of stress are sensed or decoded by multiple cytosolic or intra-organellar molecules, which then transduce the signals to mitochondria, resulting in alterations of the outer mitochondrial membrane (OM).25,42,43,45,46 This initial ‘scarring’ of the OM leads to increased permeability to proteins that are normally trapped between the OM and the inner mitochondrial membrane (IM), thus enabling these proteins to escape the mitochondria and diffuse into the cytosol. The IM is a highly convoluted, protein-rich membrane with unusual lipid composition.25,44–47 Oxidative phosphorylation (oxphos) takes place within the IM.25,48,49 Because of the crucial importance of oxphos in producing cellular ATP, which is also essential for apoptosis signaling (apoptosome formation, see below), genuine apoptotic stimuli normally do not affect the properties of the IM. However, a number of drugs and agents are able to activate a multiprotein complex that promotes the formation of a large channel in the IM, the so-called permeability transition pore (PTP).25,42,48–50 Changes in mitochondrial membrane potential—ΔΨm—the energy source of oxphos—are often observed in apoptotic cells and are interpreted to derive from the PTP opening. However, most frequently these changes occur after the initial loss of OM permeability, thereby reflecting a mixture of caspase-mediated and caspase-independent damages, including the opening of the PTP.43
The first, and likely the most important, event in the intrinsic pathway of apoptosis is the loss of OM integrity and consequent release of mitochondrial proteins. Among the various proteins that leak out of mitochondria,51,52 a few, such as cytochrome c, play a prominent role in promoting the caspase cascade of cell execution, and are cumulatively called ‘apoptogenic factors.’ There appears to be a hierarchical release of apoptogenic factors during cell death signaling, with cytochrome c, Omi/Htr2A and Smac/Diablo being released first with seemingly comparable kinetics.51 The subsequent release of AIF (apoptosis inducing factor) and endoG53,54 is linked to more severe damage of mitochondrial membranes, including IM alteration. It is important to note that a direct apoptogenic role has only been demonstrated for cytochrome c. Specifically, cytochrome c is indispensable for the activation of Apoptosis Protease Activating Factor-1 (Apaf-1) and subsequent formation of the apoptosome—the ultimate cellular ‘death machine’.54
The apoptosome works like a large platform for recruiting and facilitating the self-activation of pro-caspase-9, the apical caspase of the intrinsic pathway of apoptosis.55–61 There is a clear similarity in the activation of caspase-8 by the DISC and the activation of caspase-9 by the apoptosome, in that both systems rely on large multiprotein complexes to promote local accumulation of zymogens that initiate an auto-catalytic process of caspase activation.55–61 The apoptosome, however, requires additional regulatory factors to fully activate the caspase cascade. Included amongst these factors is Smac/Diablo, a protein that is able to interact with several Inhibitor of Apoptosis Proteins (IAP’s) and dislodge them from their inhibitory interaction with pro-caspase-9 and other caspases.58–61 As aforementioned, Smac/Diablo is also present in mitochondria (directly attached to the OM) and is efficiently released as soon as OM integrity is perturbed following intrinsic cell death stimuli.51,59 There is an important evolutionary parallel between the action of Smac/Diablo and death genes in Drosophila.62 For instance reaper acts by sequestering IAP’s from their constitutive inhibition of pro-caspases.63–66 However, it is not understood why reaper and similarly acting genes are not located in insect mitochondria, while Smac/Diablo is normally sequestered within these organelles in mammalian (and presumably other vertebrate) cells. This separation might reflect the recruitment of mitochondria in the fundamental system of cell suicide of vertebrate cells.
There is some debate regarding the apoptogenic role of some proteins that escape mitochondria during cell death, in particular AIF and Omi. Genetic defects in these proteins induce a phenotype comparable to that of mitochondrial diseases,65,67–69 rather than the reduction in cell death that would be expected following ablation of a genuine pro-apoptotic action, cf. Apaf-1 knockout.70 It is thus possible that both AIF and Omi are normally required for mitochondrial homeostasis and that once OM permeability is increased, they ‘accidentally’ leave mitochondria and redistribute to other compartments. They may then contribute to some fine-tuning aspects of downstream execution. Hence, the relevance of these and other (e.g., Endo G) mitochondrial proteins to death receptor-mediated apoptosis is likely to be minimal.
In the great majority of cells (Type II cells), extracellular death signals engage mitochondria in a way that is fundamentally equivalent to the intrinsic pathway.4,7,16,35 In these cells, signals emanating from the activated DISC bifurcate into two arms, one of which directly engages mitochondria via a sequence of events that are mediated by the apical caspases (8 and 10). Whereas the major target of active caspase-8 in Type I cells is caspase-3 and other executioner caspases, caspase-8 activation is slower or less efficient in type II cells and promotes the cleavage of non-caspase substrates such as Bid, as discussed further below. While caspase-8 is being slowly activated and begins to cleave these intracellular ‘messengers’ of mitochondrial engagement, a second signaling pathway is initiated by the DISC.7,71 This pathway involves the action of RIP, a serine kinase, and perhaps other ill-defined factors that regulate a caspase-independent sequence of events that lead to vacuole-mediated cell death.71–73 This is especially true when caspase-8 activation is blocked.74–78 Caspase-independent death signaling is particularly evident in the presence of peptide inhibitors of caspases, such as z-VAD, and produces a cellular morphology that is comparable to autophagic cell death.74,75 A similar morphology is often described as ‘necrotic’ in TNF or Fas-mediated death of certain cell types, wherein caspase-8 is either inactivated or not engaged.77,79 This ‘necrotic’ cell death pathway is becoming an increasingly hot topic in apoptosis research, and warrants additional discussion.
VACUOLE-MEDIATED CELL DEATH (NECROSIS): THE OLDEST WAY TO CELL SUICIDE?
In many Type II cells, there appears to be a fine balance between the caspase-mediated arm of death signaling that engages mitochondria and the caspase-independent arm that promotes vacuole proliferation, with at least two points of mutual regulation. Specifically, cleavage of RIP by caspase-8 results in the inactivation of the downstream cascade that promotes vacuole proliferation, thereby shutting down the caspase-independent pathway.72 A second subtler point is that the engagement of mitochondria appears to occur prior to overt activation of caspase-8 and affects membrane lipids rather than specific protein targets.25,43–45,47 In essence, caspase-independent reactions mediated by the DISC promote an alteration in the homeostasis of phosphatidylcholine (PC), the major membrane lipid in cells. This leads to a cascade of metabolic effects on other lipids, including the mitochondrial lipid cardiolipin (CL), which is crucial for mediating the pro-apoptotic action of Bid and its cleaved form, tBid. Hence, the caspase-independent arm meets the caspase-dependent arm of death receptor signaling at the level of the mitochondrial membrane lipids (Fig. 1), and proceeds to converge into effective permeabilization of the OM and perturbation of mitochondrial structure and function. In this respect, death receptor-mediated apoptosis has a double-hit way of attacking mitochondria to facilitate the release of their apoptogenic factors and thus amplify the cascade of cell execution. Therefore, OM perturbation is effectively the central cross-road at which various pathways for mammalian cell suicide intersect.
Figure 1.
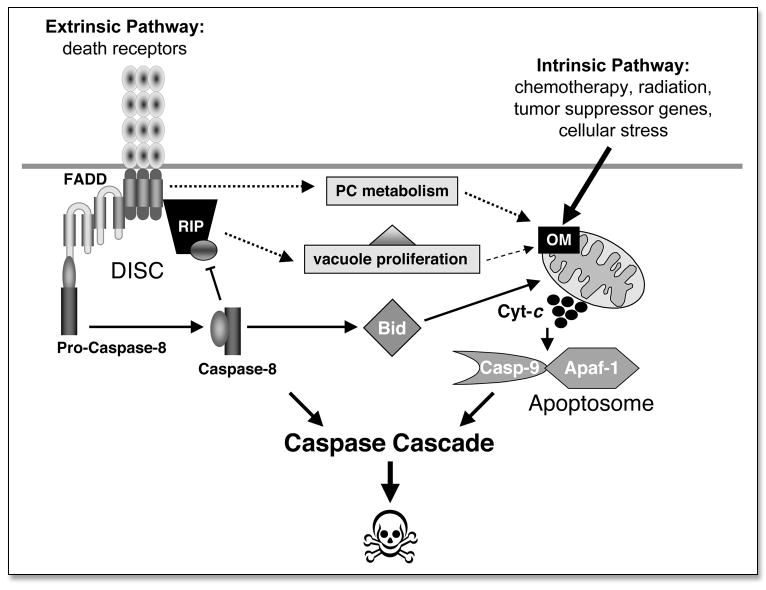
An overview of the extrinsic and intrinsic pathways leading to programmed cell death.
The fact that mitochondria appear to not be involved in the developmental cell death of Drosophila and C. elegans cannot be used as an argument to disqualify the pivotal role of mitochondrial membranes in vertebrate pathways of cell death.8 It may well be that the simplified systems observed in invertebrates represent either deviations or over-simplifications of older pathways that have evolved since the symbiotic events that led to the emergence of eukaryotic cells.80,81 The ancestral method of programmed cell death that is observed in unicellular organisms, such as the slime molds, involves vacuole proliferation and massive autophagic degradation80,81 with morphological features that are similar to caspase-independent, death-receptor induced cell death of mammalian cells.74,77,82–85 It appears the main reason that this vacuole-mediated form of PCD has been generally qualified as ‘necrotic’ 76,77,82,85 is that these cells become positive for propodium iodide staining, which is the conventional assay of plasma-membrane integrity. Although the same staining is obtained from injured cells that undergo uncontrolled ‘classical’ necrosis (characterized by a large swelling of mitochondria and other organelles), cells that die by vacuole-mediated PCD mechanisms do not succumb to the same necrotic morphology. In particular, their mitochondria are progressively degraded by vacuoles or by autophagic organelles and they maintain oxphos function for prolonged times.76,77,82,85 Therefore, the uptake of membrane impermeable dyes like propidium iodide by mammalian cells that are dying via caspase-independent mechanisms reflects enhanced endocytosis and membrane traffic linked to vacuole proliferation. Subsequent lysis of the internalized, dye-filled endocytotic vacuoles will produce intracellular staining, even if the overall integrity of the cell membrane surface has not been compromised. In other words, the dynamics of membrane traffic will be dramatically altered in autophagic cells in comparison to cells dying from caspase-mediated mechanisms. In support of this possibility, increasing evidence shows that death receptor signaling induces enhanced endocytosis in a variety of cell types, and that caspase-8 is able to regulate some key steps in membrane traffic and receptor internalization.86–89 Future studies will clarify the importance and the relevance of endocytic membrane traffic in death receptor mediated apoptosis, especially with respect to the engagement of mitochondria and their membranes.80
BID: A LINK BETWEEN THE INTRINSIC AND EXTRINSIC PATHWAYS
OM permeability and the mitochondrial pathway are crucially regulated by diverse pro- and anti-apoptotic members of the Bcl-2 family.90 The anti-apoptotic members include Bcl-2, Bcl-XL and Mcl-1, whereas the pro-apoptotic members encompass Bax and Bak, as well as the BH3-only proteins, Bid and Bim.91 In particular, mitochondria are targeted by key pro-apoptotic proteins such as Bax and Bid.25,92–95 Bid is a potent pro-apoptotic protein that is normally located in the cytosol, but also shuttles through the surfaces of intra-cellular membranes due to its intrinsic lipid-interacting capacity. Upon cleavage by caspase-8, cleaved Bid (and in particular its C-terminal fragment, tBid) acquires a strong propensity to bind to mitochondria, where it promotes effective OM permeabilization and the release of apoptogenic factors.92,93,96 The mitochondrial ‘receptor’ for caspase-cleaved Bid is considered to be cardiolipin (CL), a mitochondrial lipid, or metabolites of CL.25,44–46
Although it is now generally accepted that tBid constitutes the fundamental link between the DISC and mitochondria,92 some observations suggest that parallel signals could be delivered to mitochondria during death receptor-mediated apoptosis. Analysis of mitochondrial membrane permeability after ex vivo activation of Fas in murine primary tissues has indicated that OM damage leading to the initial release of cytochrome c occurs prior to overt activation of caspase-8.97 In addition, it is now clear that induction of apoptosis by caspase-8 is amplified through mitochondrial release of cytochrome c.98 In a recent paper,99 Bax was demonstrated to be absolutely required for TRAIL-induced apoptosis, whereas a second report showed that Fas-induced apoptosis is independent of Bax.100 Moreover, additional evidence indicates that the DISC produces early responses that appear to be caspase-independent.101,102 Excluding the mentioned insights regarding phospholipid metabolism, the mechanism by which alternative pathways emanating from death receptors reach mitochondria remains essentially unknown.
ADDITIONAL SUBSTRATES OF APICAL CASPASES THAT TRANSMIT DEATH SIGNALS TO MITOCHONDRIA
Besides Bid and possibly other BH3-only members of the Bcl-2 family, a few other non-caspase substrates of apical caspases have been reported to promote the engagement of mitochondria in the process of death receptor-mediated apoptosis. One recent example is BAP31, a membrane protein that is involved in the export of glycosylated proteins103 and is cleaved by caspase-8.104 BAP31 is usually considered to be an ER resident protein, but is also clearly present in the Golgi of primary tissues,105 presumably reflecting constitutive trafficking of ER-Golgi Intermediate Compartment (ERGIC) vesicles.103 Although some Golgi proteins have been recently found to be substrates of executioner caspases,106 only BAP31 appears to be a specific target of apical caspases that are activated by death receptors.103 Cleavage of the cytosolic coiled-coil domain of BAP31 induces pro-apoptotic activity that is transmitted to mitochondria and seems to involve increased membrane fission.107 This has generated the ‘two-hit model’ for caspase-8-dependent alteration of mitochondria, wherein one hit is mediated by BAP31 and the other by Bid.107 The model implies that other ERGIC-associated proteins could transmit damage to mitochondria. This is likely to be dependent on the cleaved coiled-coil domain of BAP31 recruiting similar coiled-coil proteins with membrane-tethering or fusion properties.80 Our unpublished results, however, do not sustain this possibility. These studies indicate that cleavage of BAP31 may be a late event in Fas-mediated apoptosis. Future studies will establish the precise role, if any, of BAP31 and other non-mitochondrial membrane proteins in the modulation of death receptor-mediated apoptosis.108
LIPID DEGRADATION IN APOPTOSIS SIGNALING
As was summarized in a recent review by MacEwan,109 phospholipase activation has long been considered to be involved in death receptor-mediated apoptosis. The protective effects of PLA2 inhibitors in TNF-mediated cell death in vivo are similar to the initial findings regarding Ca-dependent phospholipase A2 (cPLA2).110 Phosphorylation by stress kinases can activate cPLA2, contributing to caspase-independent lipid degradation reactions. However, cPLA2 is also known to be inactivated by caspase cleavage,109,111 thereby providing another example of cross-talk between the caspase-driven and the caspase-independent bifurcation of death receptor signaling. Several other lipid degrading enzymes have been reported to be engaged in death receptor-mediated apoptosis, including Ca-independent PLA2 (iPLA2),111,112 PC-specific PLC,113,114 phosholipase D115 and sphingomyelinases.116–119 To date, none of these enzymes have been found to be indispensable for apoptosis induction, especially in vivo. Moreover, mammalian PC-PLC has yet to be identified at the molecular level. However, it is likely that multiple lipid-degrading reactions follow the initial alteration in lipid metabolism, particularly of PC (Fig. 1).80 Furthermore, various lipid degradation reactions, including that typical of sphingomyelinases, are facilitated by the enhanced traffic of endocytic vesicles and lysosomal vacuoles that is promoted by death receptor activation.88 The most upstream reactions emanating from death receptors, which then trigger the cascade of metabolic changes in membrane lipids remains to be identified.
MITOCHONDRIA AND CANCER TREATMENT
Defective apoptosis is one of the hallmarks of tumorigenicity and is implicated in multiple stages of cancer development and progression.2,14,120 Additionally, the ability of tumor cells to evade apoptosis plays a significant role in promoting resistance to conventional chemotherapy and radiation therapy.7,9,10,12,121,122 Many oncogenes that deregulate the cell cycle also trigger apoptosis to eliminate cells that are proliferating inappropriately. As mitochondria play important roles in cellular energy metabolism, free radical formation and programmed cell death, defects in mitochondrial function are suspected to contribute to the development and progression of cancer and to resistance to therapy.110,123–129
The role of mitochondria in cellular energy metabolism was reported by Warburg over half a century ago and was termed the “Warburg effect.” The Warburg effect indicated that a key event in carcinogenesis is the development of an “injury” to the respiratory machinery.130 This results in compensatory increases in glycolytic ATP production to satisfy the energy needs of malignant cells. Preferential reliance on glycolysis over the more energetically efficient process of oxidative metabolism has been correlated with tumor progression in several cancer types.131 Since the initial report of the Warburg effect, a number of cancer-related mitochondrial defects have been identified.110,128,129,132 These defects include altered expression and activity of respiratory chain subunits and glycolytic enzymes, changes in oxidation of NADH-linked substrates and mutations in mitochondrial DNA. Thus, the differences in energy metabolism between normal cells and cancer cells constitute a biochemical basis for the development of therapeutic strategies that might selectively kill cancer cells in their inherently compromised respiratory state.
As mitochondria are potent integrators and coordinators of apoptotic signaling pathways, induction of apoptosis in many cell types lead to the induction of mitochondrial membrane permeabilization (MMP).40,128 MMP is the event that defines the point-of-no-return in most programmed cell death models and is subject to complex regulation by pre-mitochondrial signal transduction pathways. These pathways involve DISC-dependent and DISC-independent mechanisms, members of the Bcl-2 family of proteins and changes in the composition of mitochondrial membranes.4,25,40–44,50,98,125,127,128 In response to MMP, pro-apoptotic factors are released into the cytosol to trigger the execution of cell death. Under pathologic conditions, tumor cells escape from apoptosis and/or become resistant to treatment by affecting various components of the apoptotic machinery and through the inhibition of MMP.124,125,127,129 Therefore, targeting and overcoming abnormalities in tumor cells that suppress MMP could generate a potent pro-apoptotic stimulus. Moreover, since MMP is a relatively early event in the apoptotic program, methods to detect this process can be useful in assessing the response to chemotherapy.
CONCLUSIONS AND FUTURE PROSPECTS
Death receptor signaling to mitochondria appears to be significantly more complex than was originally suggested by simple cas-pase-8 activation. Death receptors activate multiple caspase-8-dependent and caspase-8-independent pathways, some of which lead to alterations in the mitochondria. For example, death receptor induced signals modulate the activities of signal transduction molecules and regulators of mitochondria such as Bcl-2 family proteins, lead to changes in mitochondrial membrane lipid metabolism, induce intracellular membrane remodeling, and instigate MMP. Together, these events promote the release of apoptogenic factors from mitochondria. The release of these factors can also lead to diverse morphological end points. Overt activation of caspase-8, on the other hand, leads specifically to rapid changes in mitochondria that promote morphological alterations that are characteristics of apoptosis. Necrosis and autophagy can also be activated in response to death receptor signaling and provide alternative mechanisms for achieving cell death. Indeed, it is suggested that various forms of PCD are triggered in response to cellular transformation and this provides a challenge for tumor cell progression and invasion. A current hurdle in the field is the identification of death receptor signaling pathways that selectively induce the apoptosis of tumor cells. Perhaps, death receptor signaling pathways that regulate changes in mitochondrial membrane lipids and the release of apoptogenic factors can be targets for the development of therapeutic agents. Additionally, the alternative programmed cell death pathway, i.e., death-receptor induced autophagy, is not well characterized and can provide a basis for tumor-selective therapeutic targets.
Acknowledgments
The suggestions of R. Shalini are most appreciated. Work in Khosravi-Far laboratory is supported by Public Health Service grant, 1 HL080192 from the National Institute of Health, DAMD, 17-02-10298 and DAMD, 17-02-10299 from the Department of Defense and RSG 03-012-01-CCG from American Cancer Society.
References
- 1.Lawen A. Apoptosis-an introduction. Bioessays. 2003;25:888–96. doi: 10.1002/bies.10329. [DOI] [PubMed] [Google Scholar]
- 2.Ozoren N, El-Deiry WS. Cell surface Death Receptor signaling in normal and cancer cells. Semin Cancer Biol. 2003;13:135–47. doi: 10.1016/s1044-579x(02)00131-1. [DOI] [PubMed] [Google Scholar]
- 3.Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–44. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 1998;10:545–51. doi: 10.1016/s0952-7915(98)80222-7. [DOI] [PubMed] [Google Scholar]
- 5.Strasser A, O’Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–45. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 6.Degli Esposti M. To die or not to die—The quest of the TRAIL receptors. J Leukoc Biol. 1999;65:535–42. doi: 10.1002/jlb.65.5.535. [DOI] [PubMed] [Google Scholar]
- 7.Abe K, Kurakin A, Mohseni-Maybodi M, Kay B, Khosravi-Far R. The complexity of TNF-related apoptosis-inducing ligand. Ann N Y Acad Sci. 2000;926:52–63. doi: 10.1111/j.1749-6632.2000.tb05598.x. [DOI] [PubMed] [Google Scholar]
- 8.Vaux DL, Korsmeyer SJ. Cell death in development. Cell. 1999;96:245–54. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 9.Zornig M, Hueber A, Baum W, Evan G. Apoptosis regulators and their role in tumorigenesis. Biochim Biophys Acta. 2001;1551:F1–37. doi: 10.1016/s0304-419x(01)00031-2. [DOI] [PubMed] [Google Scholar]
- 10.Daniel PT, Wieder T, Sturm I, Schulze-Osthoff K. The kiss of death: promises and failures of death receptors and ligands in cancer therapy. Leukemia. 2001;15:1022–32. doi: 10.1038/sj.leu.2402169. [DOI] [PubMed] [Google Scholar]
- 11.Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1:19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 12.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 13.Sheikh MS, Huang Y. Death receptors as targets of cancer therapeutics. Curr Cancer Drug Targets. 2004;4:97–104. doi: 10.2174/1568009043481597. [DOI] [PubMed] [Google Scholar]
- 14.Burns TF, el-Deiry WS. Cell death signaling in maligancy. Cancer Treat Res. 2003;115:319–43. doi: 10.1007/0-306-48158-8_13. [DOI] [PubMed] [Google Scholar]
- 15.Stegh AH, Peter ME. Apoptosis and caspases. Cardiol Clin. 2001;19:13–29. doi: 10.1016/s0733-8651(05)70192-2. [DOI] [PubMed] [Google Scholar]
- 16.Algeciras-Schimnich A, Shen L, Barnhart BC, Murmann AE, Burkhardt JK, Peter ME. Molecular ordering of the initial signaling events of CD95. Mol Cell Biol. 2002;22:207–20. doi: 10.1128/MCB.22.1.207-220.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–6. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 18.Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- 19.Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254:439–59. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- 20.Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol. 1999;11:255–60. doi: 10.1016/s0955-0674(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 21.Geske FJ, Gerschenson LE. The biology of apoptosis. Hum Pathol. 2001;32:1029–38. doi: 10.1053/hupa.2001.28250. [DOI] [PubMed] [Google Scholar]
- 22.Wallach D. Apoptosis. Placing death under control. Nature. 1997;388:123, 125–6. doi: 10.1038/40516. [DOI] [PubMed] [Google Scholar]
- 23.Reed JC. Mechanisms of apoptosis. Am J Pathol. 2000;157:1415–30. doi: 10.1016/S0002-9440(10)64779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Yuan J. Deciphering the pathways of life and death. Curr Opin Cell Biol. 1999;11:261–6. doi: 10.1016/s0955-0674(99)80035-0. [DOI] [PubMed] [Google Scholar]
- 25.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 26.Abe K, Kurakin A, Mohseni-Maybodi M, Kay B, Khosravi-Far R. The complexity of TNF-related apoptosis-inducing ligand. Ann NY Acad Sci. 2000;926:52–63. doi: 10.1111/j.1749-6632.2000.tb05598.x. Review. [DOI] [PubMed] [Google Scholar]
- 27.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–41. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 28.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 29.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 30.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–67. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 31.Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA. 1999;96:10964–7. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Chang HY, Baltimore D. Autoproteolytic activation of pro-caspases by oligomerization. Mol Cell. 1998;1:319–25. doi: 10.1016/s1097-2765(00)80032-5. [DOI] [PubMed] [Google Scholar]
- 33.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–87. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozoren N, El-Deiry WS. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia. 2002;4:551–7. doi: 10.1038/sj.neo.7900270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hickman JA. Apoptosis and tumourigenesis. Curr Opin Genet Dev. 2002;12:67–72. doi: 10.1016/s0959-437x(01)00266-0. [DOI] [PubMed] [Google Scholar]
- 37.Vaux DL. Apoptosis timeline. Cell Death Differ. 2002;9:349–54. doi: 10.1038/sj.cdd.4400990. [DOI] [PubMed] [Google Scholar]
- 38.Horvitz HR. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999;59:1701s–6s. [PubMed] [Google Scholar]
- 39.Scaffidi C, Kirchhoff S, Krammer PH, Peter ME. Apoptosis signaling in lymphocytes. Curr Opin Immunol. 1999;11:277–85. doi: 10.1016/s0952-7915(99)80045-4. [DOI] [PubMed] [Google Scholar]
- 40.Kroemer G. Mitochondrial control of apoptosis: an introduction. Biochem Biophys Res Commun. 2003;304:433–5. doi: 10.1016/s0006-291x(03)00614-4. [DOI] [PubMed] [Google Scholar]
- 41.Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192:131–7. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- 42.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 43.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 44.Sorice M, Circella A, Cristea IM, Garofalo T, Renzo LD, Alessandri C, Valesini G, Esposti MD. Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ. 2004;11:1133–45. doi: 10.1038/sj.cdd.4401457. [DOI] [PubMed] [Google Scholar]
- 45.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–42. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 46.Esposti MD, Cristea IM, Gaskell SJ, Nakao Y, Dive C. Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ. 2003;10:1300–9. doi: 10.1038/sj.cdd.4401306. [DOI] [PubMed] [Google Scholar]
- 47.Esposti MD. Lipids, cardiolipin and apoptosis: a greasy licence to kill. Cell Death Differ. 2002;9:234–6. doi: 10.1038/sj.cdd.4400997. [DOI] [PubMed] [Google Scholar]
- 48.Degli Esposti M. Measuring mitochondrial reactive oxygen species. Methods. 2002;26:335–40. doi: 10.1016/S1046-2023(02)00039-7. [DOI] [PubMed] [Google Scholar]
- 49.Shrivastava P, Pantano C, Watkin R, McElhinney B, Guala A, Poynter ML, Persinger RL, Budd R, Janssen-Heininger Y. Reactive nitrogen species-induced cell death requires Fas-dependent activation of c-Jun N-terminal kinase. Mol Cell Biol. 2004;24:6763–72. doi: 10.1128/MCB.24.15.6763-6772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waterhouse NJ, Goldstein JC, Kluck RM, Newmeyer DD, Green DR. The (Holey) study of mitochondria in apoptosis. Methods Cell Biol. 2001;66:365–91. doi: 10.1016/s0091-679x(01)66017-5. [DOI] [PubMed] [Google Scholar]
- 51.Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–74. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- 52.Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, Goldberg M, Allen T, Barber MJ, Green DR, et al. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol. 1999;147:809–22. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Penninger JM, Kroemer G. Mitochondria, AIF and caspases--rivaling for cell death execution. Nat Cell Biol. 2003;5:97–9. doi: 10.1038/ncb0203-97. [DOI] [PubMed] [Google Scholar]
- 54.Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003;22:4385–99. doi: 10.1093/emboj/cdg423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill MM, Adrain C, Martin SJ. Portrait of a killer: the mitochondrial apoptosome emerges from the shadows. Mol Interv. 2003;3:19–26. doi: 10.1124/mi.3.1.19. [DOI] [PubMed] [Google Scholar]
- 56.Chinnaiyan AM. The apoptosome: heart and soul of the cell death machine. Neoplasia. 1999;1:5–15. doi: 10.1038/sj.neo.7900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salvesen GS, Renatus M. Apoptosome: the seven-spoked death machine. Dev Cell. 2002;2:256–7. doi: 10.1016/s1534-5807(02)00137-5. [DOI] [PubMed] [Google Scholar]
- 58.Shi Y. Apoptosome: the cellular engine for the activation of caspase-9. Structure (Camb) 2002;10:285–8. doi: 10.1016/s0969-2126(02)00732-3. [DOI] [PubMed] [Google Scholar]
- 59.Cain K, Bratton SB, Cohen GM. The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie. 2002;84:203–14. doi: 10.1016/s0300-9084(02)01376-7. [DOI] [PubMed] [Google Scholar]
- 60.Adams JM, Cory S. Apoptosomes: engines for caspase activation. Curr Opin Cell Biol. 2002;14:715–20. doi: 10.1016/s0955-0674(02)00381-2. [DOI] [PubMed] [Google Scholar]
- 61.Baliga B, Kumar S. Apaf-1/cytochrome c apoptosome: an essential initiator of caspase activation or just a sideshow? Cell Death Differ. 2003;10:16–8. doi: 10.1038/sj.cdd.4401166. [DOI] [PubMed] [Google Scholar]
- 62.Hay BA. Understanding IAP function and regulation: a view from Drosophila. Cell Death Differ. 2000;7:1045–56. doi: 10.1038/sj.cdd.4400765. [DOI] [PubMed] [Google Scholar]
- 63.Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun. 2003;304:499–504. doi: 10.1016/s0006-291x(03)00622-3. [DOI] [PubMed] [Google Scholar]
- 64.Bergmann A, Yang AY, Srivastava M. Regulators of IAP function: coming to grips with the grim reaper. Curr Opin Cell Biol. 2003;15:717–24. doi: 10.1016/j.ceb.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 65.Martin SJ. Destabilizing influences in apoptosis: sowing the seeds of IAP destruction. Cell. 2002;109:793–6. doi: 10.1016/s0092-8674(02)00802-4. [DOI] [PubMed] [Google Scholar]
- 66.Verhagen AM, Vaux DL. Cell death regulation by the mammalian IAP antagonist Diablo/Smac. Apoptosis. 2002;7:163–6. doi: 10.1023/a:1014318615955. [DOI] [PubMed] [Google Scholar]
- 67.Cande C, Vahsen N, Garrido C, Kroemer G. Apoptosis-inducing factor (AIF): caspase-independent after all. Cell Death Differ. 2004;11:591–5. doi: 10.1038/sj.cdd.4401400. [DOI] [PubMed] [Google Scholar]
- 68.Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–74. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 69.Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–44. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 70.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–37. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 71.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 72.Barcia RN, Valle NS, McLeod JD. Caspase involvement in RIP-associated CD95-induced T cell apoptosis. Cell Immunol. 2003;226:78–85. doi: 10.1016/j.cellimm.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 73.Weglarczyk K, Baran J, Zembala M, Pryjma J. Caspase-8 activation precedes alterations of mitochondrial membrane potential during monocyte apoptosis induced by phagocytosis and killing of Staphylococcus aureus. Infect Immun. 2004;72:2590–7. doi: 10.1128/IAI.72.5.2590-2597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu L, Lenardo MJ, Baehrecke EH. Autophagy and caspases: A new cell death program. Cell Cycle. 2004;3:1124–6. [PubMed] [Google Scholar]
- 75.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 76.Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18:7719–30. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- 77.Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151:1247–56. doi: 10.1083/jcb.151.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 79.Vanden Berghe T, Denecker G, Brouckaert G, Vadimovisch Krysko D, D’Herde K, Vandenabeele P. More than one way to die: methods to determine TNF-induced apoptosis and necrosis. Methods Mol Med. 2004;98:101–26. doi: 10.1385/1-59259-771-8:101. [DOI] [PubMed] [Google Scholar]
- 80.Cristea IM, Degli Esposti M. Membrane lipids and cell death: an overview. Chem Phys Lipids. 2004;129:133–60. doi: 10.1016/j.chemphyslip.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 81.Ameisen JC. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ. 2002;9:367–93. doi: 10.1038/sj.cdd.4400950. [DOI] [PubMed] [Google Scholar]
- 82.Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol. 2004;36:2405–19. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 83.Zakeri Z, Lockshin RA. Cell death during development. J Immunol Methods. 2002;265:3–20. doi: 10.1016/s0022-1759(02)00067-4. [DOI] [PubMed] [Google Scholar]
- 84.Lockshin RA, Zakeri Z. Caspase-independent cell death? Oncogene. 2004;23:2766–73. doi: 10.1038/sj.onc.1207514. [DOI] [PubMed] [Google Scholar]
- 85.Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353–60. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schutze S, Machleidt T, Adam D, Schwandner R, Wiegmann K, Kruse ML, Heinrich M, Wickel M, Kronke M. Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J Biol Chem. 1999;274:10203–12. doi: 10.1074/jbc.274.15.10203. [DOI] [PubMed] [Google Scholar]
- 87.Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21:415–28. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 88.Algeciras-Schimnich A, Barnhart BC, Peter ME. Apoptosis-independent functions of killer caspases. Curr Opin Cell Biol. 2002;14:721–6. doi: 10.1016/s0955-0674(02)00384-8. [DOI] [PubMed] [Google Scholar]
- 89.Higuchi M, Singh S, Chan H, Aggarwal BB. Protease inhibitors differentially regulate tumor necrosis factor-induced apoptosis, nuclear factor-kappa B activation, cytotoxicity, and differentiation. Blood. 1995;86:2248–56. [PubMed] [Google Scholar]
- 90.Korsmeyer SJ. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 1999;59:1693s–1700s. [PubMed] [Google Scholar]
- 91.Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- 92.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–33. [PubMed] [Google Scholar]
- 93.Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2:63–7. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 94.Esposti MD. The roles of Bid. Apoptosis. 2002;7:433–40. doi: 10.1023/a:1020035124855. [DOI] [PubMed] [Google Scholar]
- 95.Zamzami N, Kroemer G. Apoptosis: Mitochondrial membrane permeabilization—The (w)hole story? Curr Biol. 2003;13:R71–3. doi: 10.1016/s0960-9822(02)01433-1. [DOI] [PubMed] [Google Scholar]
- 96.van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 2002;9:1031–42. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- 97.Esposti MD, Erler JT, Hickman JA, Dive C. Bid, a widely expressed proapoptotic protein of the Bcl-2 family, displays lipid transfer activity. Mol Cell Biol. 2001;21:7268–76. doi: 10.1128/MCB.21.21.7268-7276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuwana T, Smith JJ, Muzio M, Dixit V, Newmeyer DD, Kornbluth S. Apoptosis induction by caspase-8 is amplified through the mitochondrial release of cytochrome c. J Biol Chem. 1998;273:16589–94. doi: 10.1074/jbc.273.26.16589. [DOI] [PubMed] [Google Scholar]
- 99.LeBlanc H, Lawrence D, Varfolomeev E, Totpal K, Morlan J, Schow P, Fong S, Schwall R, Sinicropi D, Ashkenazi A. Tumor-cell resistance to death receptor--induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat Med. 2002;8:274–81. doi: 10.1038/nm0302-274. [DOI] [PubMed] [Google Scholar]
- 100.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Siegmund D, Mauri D, Peters N, Juo P, Thome M, Reichwein M, Blenis J, Scheurich P, Tschopp J, Wajant H. Fas-associated death domain protein (FADD) and caspase-8 mediate up-regulation of c-Fos by Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) via a FLICE inhibitory protein (FLIP)-regulated pathway. J Biol Chem. 2001;276:32585–90. doi: 10.1074/jbc.M100444200. [DOI] [PubMed] [Google Scholar]
- 102.Gougeon ML, Kroemer G. Charming to death: caspase-dependent or -independent? Cell Death Differ. 2003;10:390–2. doi: 10.1038/sj.cdd.4401199. [DOI] [PubMed] [Google Scholar]
- 103.Annaert WG, Becker B, Kistner U, Reth M, Jahn R. Export of cellubrevin from the endoplasmic reticulum is controlled by BAP31. J Cell Biol. 1997;139:1397–410. doi: 10.1083/jcb.139.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–26. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bell AW, Ward MA, Blackstock WP, Freeman HN, Choudhary JS, Lewis AP, Chotai D, Fazel A, Gushue JN, Paiement J, et al. Proteomics characterization of abundant Golgi membrane proteins. J Biol Chem. 2001;276:5152–65. doi: 10.1074/jbc.M006143200. [DOI] [PubMed] [Google Scholar]
- 106.Machamer CE. Golgi disassembly in apoptosis: cause or effect? Trends Cell Biol. 2003;13:279–81. doi: 10.1016/s0962-8924(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 107.Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160:1115–27. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chandra D, Choy G, Deng X, Bhatia B, Daniel P, Tang DG. Association of active caspase 8 with the mitochondrial membrane during apoptosis: potential roles in cleaving BAP31 and caspase 3 and mediating mitochondrion-endoplasmic reticulum cross talk in etoposide-induced cell death. Mol Cell Biol. 2004;24:6592–607. doi: 10.1128/MCB.24.15.6592-6607.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.MacEwan DJ. TNF ligands and receptors--a matter of life and death. Br J Pharmacol. 2002;135:855–75. doi: 10.1038/sj.bjp.0704549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 2004;23:2746–56. doi: 10.1038/sj.onc.1207513. [DOI] [PubMed] [Google Scholar]
- 111.Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem. 1998;273:13870–7. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 112.Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway. Proteolytic fragment of type IVA cytosolic phospholipase A2alpha inhibits stimulus-induced arachi-donate release, whereas that of type VI Ca2+-independent phospholipase A2 augments spontaneous fatty acid release. J Biol Chem. 2000;275:18248–58. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]
- 113.Cifone MG, Roncaioli P, De Maria R, Camarda G, Santoni A, Ruberti G, Testi R. Multiple pathways originate at the Fas/APO-1 (CD95) receptor: sequential involvement of phosphatidylcholine-specific phospholipase C and acidic sphingomyelinase in the propagation of the apoptotic signal. EMBO J. 1995;14:5859–68. doi: 10.1002/j.1460-2075.1995.tb00274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Kronke M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphin-gomyelin breakdown. Cell. 1992;71:765–76. doi: 10.1016/0092-8674(92)90553-o. [DOI] [PubMed] [Google Scholar]
- 115.Han JS, Hyun BC, Kim JH, Shin I. Fas-mediated activation of phospholipase D is coupled to the stimulation of phosphatidylcholine-specific phospholipase C in A20 cells. Arch Biochem Biophys. 1999;367:233–9. doi: 10.1006/abbi.1999.1250. [DOI] [PubMed] [Google Scholar]
- 116.Gulbins E. Regulation of death receptor signaling and apoptosis by ceramide. Pharmacol Res. 2003;47:393–9. doi: 10.1016/s1043-6618(03)00052-5. [DOI] [PubMed] [Google Scholar]
- 117.Gulbins E, Grassme H. Ceramide and cell death receptor clustering. Biochim Biophys Acta. 2002;1585:139–45. doi: 10.1016/s1388-1981(02)00334-7. [DOI] [PubMed] [Google Scholar]
- 118.Testi R. Sphingomyelin breakdown and cell fate. Trends Biochem Sci. 1996;21(12):468–71. doi: 10.1016/s0968-0004(96)10056-6. [DOI] [PubMed] [Google Scholar]
- 119.Kronke M, Adam-Klages S. Role of caspases in TNF-mediated regulation of cPLA(2) FEBS Lett. 2002;531:18–22. doi: 10.1016/s0014-5793(02)03407-5. [DOI] [PubMed] [Google Scholar]
- 120.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 121.Barnhart BC, Legembre P, Pietras E, Bubici C, Franzoso G, Peter ME. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J. 2004;23:3175–85. doi: 10.1038/sj.emboj.7600325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.El-Deiry WS. Role of oncogenes in resistance and killing by cancer therapeutic agents. Curr Opin Oncol. 1997;9:79–87. doi: 10.1097/00001622-199701000-00013. [DOI] [PubMed] [Google Scholar]
- 123.Kasibhatla S, Tseng B. Why target apoptosis in cancer treatment? Mol Cancer Ther. 2003;2:573–80. [PubMed] [Google Scholar]
- 124.Hersey P, Zhang XD. Overcoming resistance of cancer cells to apoptosis. J Cell Physiol. 2003;196:9–18. doi: 10.1002/jcp.10256. [DOI] [PubMed] [Google Scholar]
- 125.Bettaieb A, Dubrez-Daloz L, Launay S, Plenchette S, Rebe C, Cathelin S, Solary E. Bcl-2 proteins: targets and tools for chemosensitisation of tumor cells. Curr Med Chem Anti-Canc Agents. 2003;3:307–18. doi: 10.2174/1568011033482396. [DOI] [PubMed] [Google Scholar]
- 126.Costantini P, Jacotot E, Decaudin D, Kroemer G. Mitochondrion as a novel target of anti-cancer chemotherapy. J Natl Cancer Inst. 2000;92:1042–53. doi: 10.1093/jnci/92.13.1042. [DOI] [PubMed] [Google Scholar]
- 127.Kim R, Emi M, Tanabe K, Toge T. Therapeutic potential of antisense Bcl-2 as a chemosensitizer for cancer therapy. Cancer. 2004;101:2491–502. doi: 10.1002/cncr.20696. [DOI] [PubMed] [Google Scholar]
- 128.Brenner C, Le Bras M, Kroemer G. Insights into the mitochondrial signaling pathway: what lessons for chemotherapy? J Clin Immunol. 2003;23:73–80. doi: 10.1023/a:1022541009662. [DOI] [PubMed] [Google Scholar]
- 129.Debatin KM, Poncet D, Kroemer G. Chemotherapy: targeting the mitochondrial cell death pathway. Oncogene. 2002;21(57):8786–803. doi: 10.1038/sj.onc.1206039. [DOI] [PubMed] [Google Scholar]
- 130.Warburg E. Therapeutic imperative and evaluation of therapeutics. Ugeskr Laeger. 1951;113:86–8. [PubMed] [Google Scholar]
- 131.Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P, et al. ‘The metabolism of tumours’: 70 years later. Novartis Found Symp. 2001;240:251–60. discussion 260-4. [PubMed] [Google Scholar]
- 132.Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]