

I. SO-CALLED ALZHEIMER DISEASE
In 1907, Alois Alzheimer described the case of a 51 year old woman who presented with a relatively rapidly deteriorating memory along with psychiatric disturbances. She died four years later1. While a variety of progressive and fatal neurological conditions were known at that time, including senile dementia, the early age at onset, and a new pathological finding, the neurofibrillary tangle (NFT), made this condition unique. The justification for Alzheimer disease (AD) as a new nosologic entity, and the motivations of the prominent psychiatrist, Emil Kraepelin, for promoting an evidently new condition, continue to be debated. Nevertheless, AD is today, as it was then, a relentless neurologic deterioration accompanied by hallmark pathology.
Over time, AD was split into two clinical conditions depending upon the age of onset. Alzheimer disease, because of its initial description in a relatively young woman, was a term reserved for a type of “presenile” dementia affecting individuals younger than 65 years of age, whereas a similar dementia in the elderly, i.e., in individuals over 65 years of age, was referred to as senile dementia of the Alzheimer-type after the pioneering studies of Tomlinson, Roth and Blessed2–4. Of historical note, Alzheimer himself was ambivalent about the possibility that this entity was distinct from “dementia senilis,”5 and although these age-related classifications are still sometimes used, AD has never been shown to have a bi-modal age of onset. AD is now generally recognized as a single entity with a prevalence that increases sharply after age 65.
AD must be differentiated from other causes of dementia: Vascular dementia, dementia with Lewy bodies, Parkinson’s disease with dementia, frontotemporal dementia and reversible dementias.
As yet, there is no reliable peripheral biochemical marker for the AD and a definitive diagnosis can only be made on histological examination of the brain at autopsy. positron emission tomography (PET) scanning technology, using the C11-labelled Pittsburgh Compound B (PiB), a derivative of thioflavine T, that binds selectively to amyloid-β (Aβ), has produced conflicting reports6–10. It would appear that PiB binding to Aβ may not always differentiate symptomatic AD from asymptomatic controls with amyloid plaques. Also, the binding of PiB to Aβ depends on the secondary and tertiary structure of the peptide, accounting for the reported false negative result11. While PiB has opened the door to visualizing brain function in a manner relevant to amyloid plaque buildup, more longitudinal studies are needed to determine its relative usefulness.
Refinement of the diagnostic criteria has improved the clinical diagnostic accuracy somewhat, although we may be nearing an endpoint, in light of the mild cognitive impairment (MCI) issue which attempts to diagnosis early neurodegenerative disease, but does so in only a subset of patients12.
II. CLINICAL PRESENTATION
The diagnosis of AD requires clinical evidence of memory loss and impairment of at least one other cognitive domain, with evidence of disturbance in social or occupational function (Table 1). AD must be differentiated from other causes of dementia: Vascular dementia, dementia with Lewy bodies, Parkinson’s disease with dementia, frontotemporal dementia and reversible dementias. Considerable variability in initial clinical presentation may be expected based on the brain regions affected. Likewise, dysfunctions in such domains as speech, personality and judgment, vision, association sensory-motor function, in addition to memory are well known. Nevertheless, the concept of a more or less predictable progression of pathology, and therefore clinical signs, has been in the literature for some time, popularized by Braak and Braak in 199113. Noteworthy is that “transentorhinal” disease followed by “limbic” disease comprise the first two overall pathological stages, followed by an “isocortical” stage with advanced disease. So it is not surprising that dysfunction of memory and personality are affected relatively early. An attempt to refine the early diagnostic features of AD is reflected in the new nosologic entity, MCI, in which patients ostensibly have cognitive dysfunction that can be identified objectively, and are not yet demented12. MCI subtypes refines this concept further, with amnestic/single domain, amnestic/multiple domain, non-amnestic single domain, and nonamnestic/multiple domain categories.
Table 1.
NINCDS/ADRDA clinical criteria for AD.
|
While the attempt to identify early AD is strategic, if not predictable, in terms of targeting the disease for intervention, it is important to point out that MCI is not a distinct entity. Rather, MCI lacks a pathological substrate, genetic predispositions, and discernible treatment benefits, whereas a given patient with this diagnosis may progress to neurodegenerative disease, remain clinically stable for many years, and even improve over time. A given MCI is therefore a case of early AD or not a case of early AD, such that a pool of subjects will, with mathematical certainty, be comprised of a mixture of subjects with genuine AD, subjects with neurodegenerative conditions, and subjects who would not develop neurodegeneration for many years, if ever. Not surprisingly, therefore, the average (of many) cognitive status, and the average pathological correlate, lie somewhere between normal and genuine AD, leaving one with the impression, on casual review, that MCI represents a transition state between normal aging and AD. In actuality, such a view is an artifact of statistics.
Genuine AD, unfortunately, is relentlessly progressive, in spite of all available therapy. Initial insidious memory impairment converts, over months and years, to disorientation, personality and judgment dysfunction, speech abnormalities, and apraxias, among other signs. The ability to care for one’s self is lost over time. With aggressive management, AD patients often live out their final months and sometimes years in a vegetative state, bed bound, with sacral decubiti. The proximate cause of death in most patients is pneumonia, which comes as a merciful ending to a prolonged and tragic illness that has long since robbed the affected patient of their individuality and dignity.
III. EPIDEMIOLOGY
AD constitutes approximately 70% of all dementia cases14, 15. Incidence of AD increases with age, doubling every five to ten years. For persons between ages 65–69, 70–74, 75–79, 80–84, and 85 and older the incidence of AD has been estimated at 0.6%, 1.0%, 2.0%, 3.3%, and 8.4%16. Prevalence also increases exponentially with age, rising from 3% among those 65–74, to almost 50% among those 85 or older17. AD affects 25 million people worldwide. In the US, prevalence was estimated at 5 million in 2007 and, by 2050, is projected to increase to 13 million in the US alone17. Aside from age, other risk factors include family history of dementia, head trauma, genetic factors (e.g., apolipoprotein E [APOE] ε4 allele), being female, low education level, vascular disease, and environmental factors. The above data are in keeping with the projected demographic changes resulting from the “baby boomer” generation reaching old age, and the continued increase in life expectancy. The combination of a declining birth rate and an increased average life span is expected to increase the proportion of the population aged 65 years and older in the US from 12.4% in 2000 to 19.6% in 2030. The number of people aged 65 years and older is expected to increase from 35 million in 2000 to 71 million in 2030. The number of people aged 80 years and older is also expected to double, from 9.3 million in 2000 to 19.5 million in 2030. Globally, the number of older adults (aged 65 years and older) is projected to increase even more dramatically—more than doubling from 420 million in 2000 to 973 million in 2030. Taking into consideration the fact that advanced age is the most significant risk factor for AD, one cannot underestimate the problem this disease poses in terms of its financial and human cost. Indeed, the estimated lifetime cost of persons with AD has been estimated to be $174,000. In 2005, Medicare spent $91 billion for healthcare for persons with AD, with $21 billion associated with long-term care services. The excess cost burden to managed care organizations of persons with AD has been estimated to be more than $4,000 per person per year.
IV. PATHOLOGY
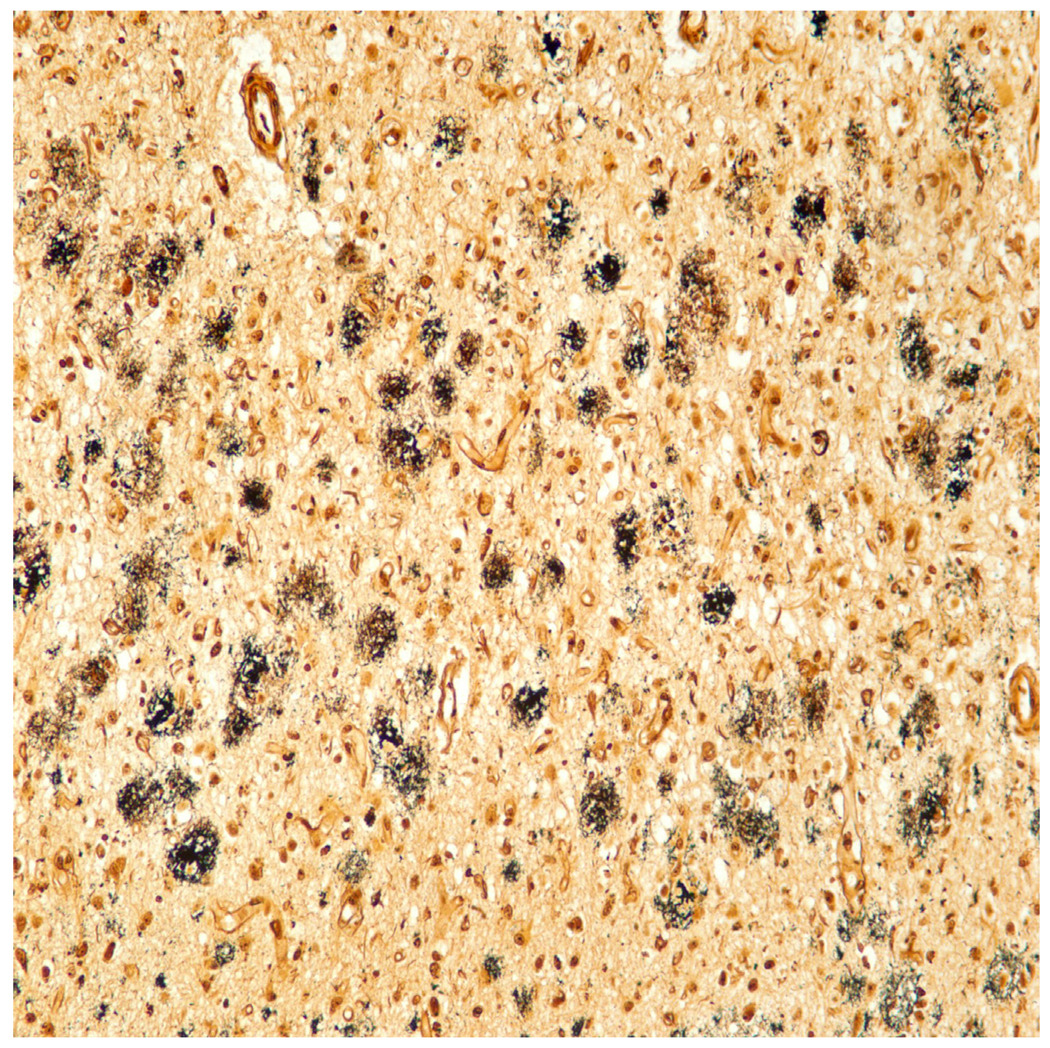
The most common and distinctive “hallmark” lesions present within the diseased brain are the senile plaques and NFTs (Figures 1 and 2). The nosology of senile plaques has evolved over the years to include multiple plaque subtypes, including diffuse, primitive, neuritic, compact, cored, and cotton-wool. The neuritic plaque (Figure 3) in particular has been touted to be the most pathogenically relevant, and often highlighted as the plaque type of significance in the major consensus criteria for the diagnosis of AD at autopsy, although it is interesting that according to CERAD criteria (the most commonly employed plaque-based criteria)18, standard brain regions are assessed via the Bielschowsky silver technique, which is a nonspecific impregnation that reacts to all plaque types. The other major hallmark, the NFT, is occasionally given descriptors such as “globose,” and “pretangles,” although NFT are not semiquantitated in the standard criteria, but rather assigned significance based on location within the brain (see below)19. Because of more detailed characterization of the AD brain using phospho-tau antibodies, other closely related lesions include neuropil threads (thread like accumulations of phospho-tau within neuropil of gray matter and also white matter), and dystrophic neuritis (terminal neuritic swellings) that occur within (and properly define) neuritic plaques.
Figure 1.
Cored “miliary focus” is demonstrated in this Bielshowsky silver impregnation, the technique that Alzheimer used in his original description of Auguste D1. Such foci were known to occur in association with “dementia senilis” prior to Alzheimer’s description5.
Figure 2.

Neurofibrillary tangles, as seen on the Bielshowsky silver stain, were initially characterized by Alzheimer.
Figure 3.

Immunohistochemical stain for phosphorylated tau (monoclonal antibody AT8 which recognizes phosphorylation at residues 202 and 205) shows numerous distended neurites in a classic neuritic plaque. Tau immunohistochemistry is the best indicator of a neuritic plaque, although CERAD recommended Bielschowsky silver impregnation (which detects multiple plaque subtypes) to assess neuritic plaque frequency.
Neuronal and dendritic loss, neuropil threads, dystrophic neurites, granulovacuolar degeneration, Hirano bodies, and cerebrovascular amyloid, are also typical of the AD brain. Synapse loss was described years ago, but has been cited in the literature more frequently in recent years20, 21. Indeed, the common refrain, “synapse loss is the most specific pathological feature of Alzheimer’s disease,” is now accepted dogma, which is due at least in part to the recent attempts to associate synapse loss with low-n soluble Aβ species. It should be pointed out, however, that assessment of the synapse, either by immunohistochemistry or electron microscopy, is strictly investigational and has no role whatsoever in the diagnosis of AD at autopsy, regardless of its putative relationship with various Aβ or, for that matter, tau species22.
The distinction between the pathology of AD and the pathology of aging is problematic, particularly in the elderly23. The spectrum of pathological changes, up to and including Braak stage VI disease, “definite” AD by CERAD criteria, and “high likelihood” AD by NIA-Reagan criteria, have been observed in the cognitively intact. In a recent review23, the interesting observation was made that within the Braak stage VI category, the NFT count was inversely correlated with cognitive status, indicating that diagnosis is most certain, and some would argue only certain, when the pathology is end-stage. This is in marked contrast to virtually all other non-neoplastic conditions in other organ systems, in which specificity decreases the more advanced the pathology22. To illustrate the difficulty in correlating clinical diagnosis and pathological findings, in a study in which neuropathologists were blinded to clinical data, 76% of brains of cognitively intact elderly patients were interpreted as AD brains24.
Interestingly, AD pathology is found prematurely in Down syndrome, a well known phenomenon that constitutes a major point, arguing in favor of the amyloid cascade hypothesis, given that the amyloid-β protein precursor (AβPP) can be found on chromosome 21 and that Down syndrome subjects have an extra copy of chromosome 21. A commonly cited case report further demonstrated a genetic variant of Down syndrome in which the AβPP coding region was not triplicated. Predictably the neuropathology in this case resembled a normal aged control brain25. Familial early onset AD cases may be regarded as similar to sporadic AD although generally with more extensive lesions, including a tendency for white matter lesions and “cotton wool” plaques in some early onset cases associated with exon 9 presenilin-1 mutations26. Interestingly, Lewy body pathology, in addition to classic AD pathology, has also been described in familial early onset cases27. A similar pathological presentation is found in cases of Down syndrome and, of note, most of these pathological features may also be present to a lesser extent in a large proportion of aged non-demented controls, lending support to the possibility that such individuals are in an extremely early pre-clinical stage of AD28.
V. STANDARDIZED PATHOLOGIC CRITERIA FOR AD
The hallmark lesions in AD brains have remained unchanged since Alzheimer’s 1907 description. The plaque component, moreover, was known to be associated with senile dementia prior to Alzheimer’s description22, and was described in the greatest detail by Fischer301. Nevertheless, the two lesions—the senile plaque and the NFT—are the chosen lesions of interest when coming to a diagnosis. A variety of other changes is known to occur but these changes are less specific (e.g., granulovacuolar degeneration, Hirano bodies) or not relevant to pathological interpretation (e.g., synapse loss). Over the years, the spectrum of diseases and age groups associated with the hallmark lesions became more and more apparent, and in 1985, the distinction became strictly quantitative with the Khachaturian criteria for AD29 (Table 2). In this set of criteria, the extent of plaque pathology was quantitated and cross referenced for age, such that older patients required more lesions in order to be given the diagnosis of AD. The capricious relationship with clinical data was known even then, however, as indicated by the suggestion that, in those cases with positive clinical history, the number of lesions might be cut by 50%. Several years later, the consortium to establish a registry for AD, whose purpose was to facilitate research, establish databases and simplify the diagnostic procedure so that virtually anyone with a microscope could undertake the activity18, devised a set of guidelines with the overall theme of the Khachaturian criteria – plaques being the most significant, and the more plaques were required the older the patient – albeit with a more simplified semiquantitative approach (Table 3). The pathological data were then cross referenced with clinical data (presence or absence of dementia), and an interpretation of “no, possible, probably, or definite” AD was obtained. In essence, then, these first two sets of criteria come with the naked admission that either i) the older brain is more tolerant of pathological lesions; or ii) the lesions have nothing to do with etiology. That both cannot be true simultaneously is a mathematical certainty.
Table 2.
Khachaturian Criteria for AD.
|
Table 3.
CERAD Criteria for AD.
| Plaque frequency | ||||
|---|---|---|---|---|
| Age | None | Sparse | Moderate | Frequent |
| <50 | 0 | C | C | C |
| 50–75 | 0 | B | C | C |
| >75 | 0 | A | B | C |
0 = no evidence of AD; A = uncertain; B = suggestive of AD; C = indicative of AD
“Definite AD” = C plus clinical dementia
“Probable AD” = B plus clinical dementia
“Possible AD” = A plus clinical dementia or, B or C and no dementia
Around the same time that the CERAD criteria were devised, Braak and Braak devised a different paradigm (Table 4)19. First, the so-called Braak criteria utilize the NFT rather than the senile plaque. This fact in itself is interesting in that the two competing diagnostic criteria focus on fundamentally different lesions with different regional distributions, and comprised of different proteins from different molecular cascades. The empiric existence of the two sets of criteria, therefore, serves to emphasize the confusion that exists regarding the relationship between diagnosis, lesions, proteins, and etiology. Second, the Braak and Braak approach encompassed much more sophisticated neuroanatomy, compared to the CERAD approach which was designed to be useful even to those with little or no knowledge of neuroanatomy. Third, the cases used in the Braak and Braak study included both demented individuals and aged controls, while the staging system derived from the data was obtained irrespective of the presence or extent of clinical disease. With this in mind, Braak and Braak found that the neurofibrillary degeneration proceeded in stepwise fashion, from the so-called transentorhinal area (neuronal population in the medial temporal lobe bridge, the entorhinal cortex, and temporal neocortex) (stage I–II), to limbic regions (stage III–IV), to isocortical regions (stage V–VI). When compared with clinical data, the majority of stage I–II cases were free of cognitive impairment, the majority of stage V–VI cases demonstrated cognitive impairment, and the stage III–IV patients were more or less evenly divided between those with dementia and those without. The correlation with clinical data raises the obvious question of whether it is legitimate to designate Braak stage I–II subjects as having “Alzheimer’s disease”; nevertheless, the involvement of neurofibrillary pathology, as well as the known superior correlation between neurofibrillary pathology and clinical disease, indicate general improvements over the CERAD, and is why most neuropathologists favor the Braak approach over CERAD criteria.
Table 4.
Summary of Braak Staging.
| Anatomical Stage | Braak Stage |
Medial Temporal Tau Pathology |
Isocortical Tau Pathology |
|---|---|---|---|
| Transentorhinal* | I–II | Limited to transentorhinal and entorhinal involvement |
Minimal |
| Limbic | III–IV | Transentorhinal, entorhinal, Ammon’s horn involvement |
Moderate |
| Isocortical | V–VI | Extensive medial temporal involvement |
Advanced |
Note that in the original Braak study, none of the patients in the transentorhinal stage were clinically demented during life. About half of the subjects in the limbic stage were demented, and all in the isocortical stage were demented.
The slightly more recent NIA-Reagan criteria, devised evidently as a compromise among the schools of thought (since CERAD represents de facto support for the amyloid cascade hypothesis, and the Braak method similarly supports the primacy of phosphorylated tau), encompasses both CERAD and Braak criteria, and depending on the extent of pathology, divides individual cases into “low likelihood,” “intermediate likelihood,” and “high likelihood” of AD (Table 5)30.
Table 5.
NIA-Reagan Criteria
| “High Likelihood” (that “dementia is due to Alzheimer disease lesions”) |
| “frequent” neuritic plaque score (CERAD), and Braak stage V–VI |
| “Intermediate Likelihood” |
| “Moderate” neuritic plaques (CERAD), Braak Stage III–IV. |
| “Low Likelihood” |
| “limited” pathology, i.e., CERAD infrequent, Braak Stage I/II |
VI. GROSS PATHOLOGICAL FEATURES
Classically, the brains of individuals with AD are atrophic with widened sulci and narrowed gyri (Figures 4 and 5). However, while atrophy is often prominent in younger patients where there is greater disparity with control cases, in more elderly individuals, especially in the seventh decade and beyond, there is considerable overlap in the degree of atrophy between diseased and age-matched controls31. Therefore, atrophy, while of cursory diagnostic value in younger individuals, is not used in the routine pathological diagnosis of the disease in the majority of cases. Nonetheless, the brains of individuals with AD, where atrophy is most pronounced in the association cortices, do tend to be about 8–15% smaller than age-matched controls.
Figure 4.

Gross brain of a 73 year old patient with Braak stage IV AD who was ambulatory but demented at the time of death. Note the generalized atrophy, ventricular dilatation (“hydrocephalus ex vacuo”), and medial temporal atrophy as well.
Figure 5.

Close up view of the hippocampal formation of the same patient as in Figure 4, showing dilatation of the temporal horn and hippocampal atrophy. An atherosclerotic plaque involving the posterior cerebral artery is also present.
A. Microscopic lesions
1. Regional susceptibility
The pathological hallmarks of AD, and identical lesions in cases of Down syndrome, are most frequently found within the association regions of the cerebral cortex, and within the medial temporal lobes19. The earliest pathological lesions tend to occur in the medial temporal, particularly the amygdale, subiculum, hippocampal CA-1 region, entorhinal cortex, and transentorhinal regions. Early changes in the olfactory bulbs are also described32, although these are not routinely sampled in AD cases, and are not part of standardized neuropathological work up or staging. On metabolic imaging, early abnormalities have been described in the precuneous and the posterior cingulate region33, although the pathological changes in these regions are generally less severe than the amygdala34.
The amygdala regions appear to be especially susceptible to the pathological changes that characterize AD such that this region tends to contain abundant plaque as well as neurofibrillary pathology35. The hippocampus and subiculum, on the other hand, typically contain abundant neurofibrillary degeneration compared to the neocortex, whereas the plaque pathology is generally less compared to the neocortex19. According to Braak, neurofibrillary degeneration occurs first in the transentorhinal region, followed by the limbic region, and then the isocortex. As such, brains from the nondemented elderly typically have neurofibrillary pathology in the medial temporal lobe, limited neurofibrillary pathology in the neocortex, and variable (but often extensive) plaque pathology and cerebral amyloid angiopathy. The general finding of the earliest and most severe neurofibrillary pathology involving brain regions responsible for declarative memory highlights the long known concept that neurofibrillary degeneration is a better correlate of clinical disease than plaque pathology. Moreover, a high NFT count in the neocortex is virtually synonymous with clinical dementia23, whereas abundant plaque pathology is often seen in elderly individuals who are cognitively intact36.
Of note, experimental bilateral hippocampal damage results in learning and memory impairments, similar to those experienced by early sufferers of AD37, 38. Supporting the early involvement of the hippocampal/amygdaloidal region in disease pathogenesis, aged normal primates39, 40, showing only limited pathology, always show lesions within the hippocampal/amygdaloidal regions. Furthermore, the hippocampus, the subiculum, and CA1 show greater quantitative pathology, i.e., NFT, senile plaques, and granulovacuolar degeneration, than other regions within the hippocampal formation41–43. Neuronal populations containing specific neurotransmitters or their receptors may underlie this selective vulnerability which may be mediated by local cytoarchitecture and circuitry44, 45. Indeed, degeneration of selective hippocampal regions would result in functional isolation of the hippocampus and, as such, accounts for the marked short-term memory impairment evident in the early stages of the disease44. Further damage to subcortical regions is believed to be due to the specific cytoarchitecture of the cortex and its reciprocal projections. There is comparative evidence which supports this postulate, the most convincing being that neurons from the ventral brainstem tegmentum projecting to the amygdala and cortex are lost in AD46. Moreover, neurons within the tegmentum that project to different areas show a similar selective degeneration, i.e., dorsal regions of the locus ceruleus connected to cortical regions are affected, whereas ventral regions projecting to the cerebellum, basal ganglia, and spinal cord are largely spared47. The distribution of pathological changes is closely correlated with cortical and subcortical connections suggesting transynaptic degeneration48.
Early work suggested that the degree of cognitive impairment in AD correlated with the density of characteristic lesions, i.e., senile plaques43, 49 and NFT43. However, more recent studies indicate that, despite increasing dementia, the number of senile plaques and NFT can remain stable for many years50, 51. In a recent analysis of the Baltimore Longitudinal study on aging, the presence of pathology did not predict an increased likelihood of cognitive decline, indicating again the stable nature of pathological lesions and the limited knowledge of the kinetics of such lesions over time36. Therefore, although senile plaques and NFT are the classic pathological hallmarks of AD, their numbers are not a good indicator of the severity of the disease process, except for the most advanced numbers of NFT. Instead, loss of pyramidal neurons50or indicators of neuronal connectivity, such as synapse loss52or neuritic alterations53, appear to correlate best with the decline in cognitive function. This may not be surprising since compromised neuronal function may precede, and be more widespread than, lesion formation and neuronal death.
Several hypotheses have been proposed to account for neuritic/synapse abnormalities in AD, including but not limited to, Aβ toxicity21, axonal transport deficiencies54–56, and oxidative stress57–59. These pathological factors have in common a profound effect on membrane properties including fluidity, replenishment, integrity and calcium flux, aspects of which are all altered in AD55, 60–62.
While synapse loss is now suggested as the most proximate cause of cognitive dysfunction, this appears to be a conclusion in search of data (Figure 6)63. It is important to note that, of all disorders of the central nervous system (CNS), the majority lack a substrate of structural pathology. Indeed, it may be said that all non-neoplastic dysfunctions of the CNS affect function of the synapse. The evidence for the synapse loss postulate is based on older electron microscopic studies64, involving counting of synapses (a labor intensive and nakedly error prone exercise), in addition to immunohistochemical data looking at various markers from presynaptic to postsynaptic functions65. Here again there is the issue of a conclusion in search of data. The data itself is voluminous, and yet may not be better than assessing gross atrophy, or metabolic dysfunction, on functional brain imaging. Perhaps for these reasons, it is more often being posited that synaptic dysfunction is the true substrate, in which case the normal brain would be the pathological substrate of AD.
Figure 6.

Synapse by electron microscopy. Note the presynaptic vesicles and the dense junctional complex. Assessment of the synapse in neurodegenerative disease is strictly investigational, and has no role in the neuropathological diagnosis.
Encouraging this newer focus on the synapse in AD appears to be the historically poor correlation between amyloid load and dementia22, 23, and the postulated good correlation between synapse loss and AD. These data evidently provided the impetus for a review, and ultimately an amendment, of the original amyloid cascade model, as the synapse and toxic (soluble) Aβ are now inextricably intertwined with each other21, 63. More data, however, is still necessary, since so-called oligomer species cannot be directly visualized or otherwise measured in vivo, either histopathologically or on functional brain imaging. Moreover, they can be identified only after homogenization and high speed centrifugation. The evidence for their role, therefore, is by definition indirect, which in turn increases the importance of experimental and in vitro studies, in that such studies comprise the whole of the data in this regard. In the end, we are left with the following: low-n soluble oligomeric Aβ species, (obtained from extracts of AβPP overexpressing Chinese hamster ovary cells), when injected into the ventricles of experimental mice, cause poor performance on cognitive-behavioral testing; and when brains from such animals are excised, the hippocampus shows electrophysiological abnormalities suggestive of synaptic damage, and abnormalities of dendritic arborization are seen using such techniques as the Golgi silver impregnation (of limited value at best in human brain examination)66. It is also somewhat ironic that the pathological lesions (amyloid plaque cores and vascular amyloid) that were initially purified and sequenced leading directly the amyloid cascade hypothesis, are now regarded as epiphenomena, and are in fact discarded. The same can also be said for NFT, which facilitated the identification of tau and yet are now discarded in favor of toxic tau intermediates in the more recent pathogenic studies.
In older and now less relevant studies, the importance of neuritic pathology was suggested by the better correlation of so-called neuritic plaques with disease, in both AD and Down syndrome67, 68, and by the association between so-called diffuse senile plaques in normal aging, although some suggest that such individuals, had they lived longer, would have developed AD69, 70. Since the kinetics of progression of pathology in AD is almost entirely unknown, such statements have almost no meaning. Nevertheless, the presence of AβPP in dystrophic neuritis suggests that they may be the source of the Aβ found in the senile plaques71. A mechanism establishing dystrophic neurites as the source of Aβ has not been firmly accepted and the role of dystrophic neurites in Aβ deposits remains controversial.
While the distribution of senile plaques varies from individual to individual and also within discrete regions of the brain, NFT, as well as neuropil threads, exhibit a characteristic distribution pattern. This pattern can be divided into six discrete neuropathological stages that follow a predicted pattern between involved brain regions and from individual to individual19.
2. Senile plaques
Described in Alzheimer’s original report as “miliary foci” (Figure 1), senile plaques are spherical extracellular lesions 10–200 µm in diameter that were originally identified and classified by the use of the Bielschowsky silver technique, which is still used today1, 22. It is also noteworthy that such miliary foci had been described prior to Alzheimer’s report, by Blocq and Marinesco in an elderly epileptic, as well as Beljahow, and Redlich and Neri, in association with senile dementia22. So the association between dementia in the elderly and senile plaques was not a novel observation in Alzheimer’s original report. Rather, the presence of NFT, and more importantly the early age of onset, justified the new nosological entity.
The presence of generic amyloid was confirmed by Divry in 1927302 with Congo red staining, but it was not until the advent of highly specific antisera72, 73 and monoclonal antibodies74 that the specific protein species comprising the amyloid was assigned to Aβ, a metabolic product of AβPP (Figure 7). In addition, the application of antisera revealed an extent and severity of plaque pathology that was greater than had previously been appreciated. Senile plaques can be classified into multiple types as noted above, although for practical purposes only two are routinely used in neuropathological analyses, namely diffuse and neuritic.
Figure 7.

Congo red stain for generic amyloid highlights senile plaques in a case of AD, although in general Congo red staining is far less sensitive and Bielschowsky silver staining or Aβ immunohistochemistry.
Diffuse senile plaques represent the earliest cerebral lesions in AD50 and it has been proposed, but not proved, that these diffuse deposits progress to classic and compact plaques with increasing neuritic involvement (Figure 8). Whether they are a substrate for normal aging, or MCI, is conjectural. However, even in severe, long standing cases of AD or Down syndrome, diffuse senile plaques account for the majority of Aβ protein-immunoreactive material75, 76. Furthermore, while the chronology from diffuse to classical senile plaques is supported indirectly by neuropathological studies, this does not necessarily represent an evolution from one type to another and could equally well represent different mechanisms of senile plaque formation that follows different morphogenic kinetics.
Figure 8.

Diffuse plaques in the cerebral cortex with Aβ (4G8) immunohistochemistry.
The typical neuritic, classical, or compact plaques are spherical extracellular lesions 10–50 µm in diameter, with an average diameter of approximately 30 µm. Diffuse senile plaques tend to be more heterogeneous in size with a frequency that decreases with increasing size such that the majority of diffuse deposits are less than 20 µm in diameter77. Senile plaques of all types are distributed predominantly within the cerebral cortex and tend to be distributed evenly within the various neocortical areas. Interestingly, plaque pathology tends to decrease in the medial temporal allocortical areas19. Some studies have suggested that different regions within the brain often contain only one or two particular types of senile plaque even in severe cases of AD. This observation could represent a chronological order of development and/or the alternative morphologies of senile plaques resulting from their interaction with different neuronal populations78, 79.
A cross-β secondary structure has been observed from X-ray diffraction analysis of Aβ isolated from senile plaques, which accounts for the congophilic staining properties, i.e., birefringence under plane-polarized light after staining with Congo red79 Neuritic senile plaques contain a central core made of 6–10 nm Aβ protein filaments, arranged as bundles radiating from the center80. The core is surrounded by an argyrophilic rim of dystrophic synapses and neuritis (mainly axons) often containing paired helical filaments and altered membranes. Aβ protein and its precursor, AβPP, as well as tau and neurofilament proteins are found in this peripheral region of the senile plaque71, 81, 82.
Sometimes overlooked in neuritic plaques is the invariable presence of activated microglia (Figure 9). Their presence, while probably a reactive phenomena, comprises the basis for the hypothesis that AD is a disease of excessive inflammation within the brain83. Indeed, substantial data now exists that indicate an integral role of inflammatory cytokines such as TNF-α, interleukins, and complement in the disease process. Similarly, substantial data now exist in support of a role for the astrocyte as an inflammatory mediator83.
Figure 9.

H&E stained section of a cored, neuritic plaque. Note the presence of activated microglia to left and below the amyloid core.
There are several hypotheses concerning the pathological mechanism(s) responsible for the genesis of senile plaques. For example, a primary neuronal event has been postulated by many authors, wherein abnormal neurites develop first, with Aβ protein deposition being secondary84. However, the finding of Aβ fibrils in all senile plaques, together with the observation of Aβ deposition in white matter areas85 makes this postulate unlikely if there is a chronological evolution of senile plaques. Nevertheless, it is still quite possible that amorphous and compact senile plaques have separate etiologies86.
Alternatively, Aβ deposition may be the primary event in senile plaque formation87. It has been suggested that a breakdown of the blood-brain barrier may underlie this process and that abnormal congophilic capillaries are found in senile plaque cores88. However, these findings have been refuted by observations that congophilic vessels and plaques are often distributed separately and by observations on young Down syndrome patients where deposits of Aβ are often found in the neural parenchyma in the absence of vascular deposits40. Moreover, morphometric assessments have revealed no obligatory relationships between senile plaques and blood vessels77, 89 over the range of AD cases, although it is interesting to note a promiment perivascular plaque pattern in isolated cases, referred to in the older literature as “dyshoric angiopathy.” Whether this pattern speaks to sporadic AD overall is doubtful, but its existence raises interesting questions regarding the potential vascular source for Aβ.
Another suggested source of Aβ, microglia85, is consistent with claims that abnormal microglial cells are at the center of early senile plaque-like lesions90 and the close proximity of astrocytic and pericytic basement membranes to Aβ deposits88. Nonetheless, these findings are controversial and others have not found such a close association between microglia and senile plaque cores69, 91.
Aβ deposits in the form of senile plaques are not limited to AD and are also found in several other conditions including Down syndrome, dementia pugilistica, diffuse Lewy body disease, acute traumatic brain injury, as well as in normal aging of humans and other higher mammals such as the beagle92. Abundant Aβ deposits may also be seen in so-called dementia with Lewy bodies, while the extent of neurofibrillary pathology varies from scant to abundant. Given that cortical Lewy body pathology is indistinct with routine histopathology and silver impregnation techniques, and with the advent of anti-ubiquitin and more recently anti-α-synuclein immunohistochemistry, many cases of so called plaque only AD are probably dementia with Lewy bodies93. However, unlike NFT that are associated with a number of degenerative conditions, Aβ deposition is generally limited to AD and advanced age94.
3. Neurofibrillary Tangles
NFT are a major microscopic lesion of AD and are located primarily in large pyramidal neurons of Ammon’s horn and the cerebral neocortex, although neurofibrillary pathology is also encountered in deep structures, including midbrain and pontine tegmentum, basal nucleus of Meynert, and hypothalamus (Figure 10)19. Like Aβ filaments, a cross-β structure is observed by X-ray diffraction analysis of isolated NFT filaments which accounts for their characteristic congophilic and argyrophilic histochemical staining95. Morphologically, NFT are classically described as consisting of numerous paired helical filaments96which are composed of two axially opposed helical filaments with a diameter of 10 nm and a half-period of 80 nm80, 97, 98. Each individual filament is thought to consist of ribbon-like arrays of 'globular' domains99, 100. Based on negatively stained purified paired helical filaments and ultrathin sections, each filament is suggested to consist of either two or four protofilaments; however, image analysis studies suggest that such observations are a result of globular domains lining up and such ambiguity has led several investigators to reject the idea of protofilaments as components of the paired helical filament fibrils. While the term paired helical filaments is widely accepted, some studies suggest that a twisted ribbon-like structure composed of two aligned parallel components, rather than separate filaments, more accurately describes the ultrastructure of the filaments101, 102. In addition to paired helical filaments, there are a host of antigenically related fibrillar structures, including straight filaments103, that comprise the NFT. This includes the paired helical filaments found in association with dystrophic neurites55, 104.
Figure 10.

Immunohistochemical stain for tau (AT8) showing a neurofibrillary tangle. Note the neuropil threads in the background.
Despite intense efforts to understand the molecular composition of paired helical filaments, biochemical studies have been severely hampered by their extreme insolubility and by difficulties in obtaining a homogeneous sample105, 106. Therefore, nearly all of the published studies to date on the biochemical composition of paired helical filaments are qualitative and provide little or no quantitative data on the proportion of material assayed. Nonetheless, it is widely accepted that the microtubule associated protein tau is the major proteinaceous element of NFT. However, biochemical, immunochemical, amino acid composition and protein sequencing techniques have demonstrated the presence of the following, as reviewed in Smith107:
Cytoskeletal elements including tau, neurofilaments, high molecular weight microtubule-associated protein MAP2, vimentin, and tropomyosin.
Protease-related elements including ubiquitin, α1-antichymotrypsin, α1-antitrypsin, cathepsins B and D, trypsin and elastase.
Proteoglycans including heparan, chondroitin, and keratin sulfate proteoglycans.
Inflammatory molecules including acute phase proteins, cytokines and complement molecules (reviewed in108).
Amyloidogenic-related molecules including AβPP, presenilin, and apolipoprotein E.
Serum-related molecules including P-component.
Adducts of oxidative stress including advanced glycation and lipid peroxidation109, 110.
NFT are not restricted to AD and are also found in association with a variety of other conditions including, but not limited to, postencephalic Parkinson’s disease, Parkinsonian dementia complex of Guam, Down syndrome, progressive supranuclear palsy, corticobasal degeneration, dementia pugilistica as well as normal aging. However, while accumulations of neurofilaments are found in neuronal perikarya in experimental aluminum encephalopathy and dialysis encephalopathy, these structures are morphologically and chemically distinct from NFT111, 112.
VII. CEREBRAL AMYLOID ANGIOPATHY
Cerebral amyloid angiopathy (CAA), as reviewed in Castellani et al.113, is a heterogeneous clinicopathological entity, sharing in common the deposition of protein within the walls of cerebral vessels (Figure 11). By definition, the protein accumulating in CAA, regardless of the clinical disease state with which it is associated, is aberrantly folded, with excessive β-sheet content and a tendency for the formation of fibrils that are highly insoluble. While systemic amyloidosis was recognized as far back as the mid-19th century and, interestingly, derives its name erroneously from CNS corpora amylacea114, CAA was described in 1938, more than a decade after the description by Divry of amyloid deposition in senile plaques. So far, no significant sequence homology has been detected between the various amyloid proteins that would suggest a common underlying primary structure.
Figure 11.

Immunohistochemical stain for Aβ in an individual who died of complications of lobar intracerebral hemorrhage. The patient was living independent with no evidence of cognitive dysfunction prior to death.
Of the twenty or so proteins that are now known to be associated with amyloid deposits (systemic and central nervous system combined), seven have been shown to cause CAA. These include AβPP (sporadic CAA, CAA related to AD and Down syndrome, hereditary cerebral hemorrhage with amyloidosis—Dutch type), cystatin C (hereditary cerebral hemorrhage with amyloidosis—Icelandic type), transthyretin (meningovascular amyloidosis), Gelsolin (familial amyloidosis, Finnish type), prion protein (Gerstmann–Straussler–Scheinker syndrome with Y145 stop mutation), ABri precursor protein (familial British dementia), and ADan precursor protein (familial Danish dementia). All but Aβ are associated only rarely with CAA, while CAA due to Aβ deposits is rather common in elderly individuals, being the most common “cause” of non-hypertensive intracerebral hemorrhage in this age group. In light of the fact that vascular deposition of Aβ, with or without AD, comprises the overwhelming majority of CAA cases, the issue of the relationship between CAA and AD naturally arises. Some arguments favoring the view that CAA plays a major role in the cognitive decline in AD are the following: i) the frequency and severity of CAA is increased in AD; ii) amyloid angiopathy produces ischemia and hemorrhage that could then cause cognitive dysfunction; iii) even when mild, CAA alters vascular physiology such that end-organ consequences are not precluded; iv) extensive CAA alone can cause dementia in some subjects with hereditary CAA (e.g., hereditary cerebral hemorrhage with angiopathy—Dutch type (HCHWA-D), D694N Iowa pedigree, familial British dementia); v) sporadic CAA with dementia and severe plaque-like Aβ angiopathy has been reported resembling the CAA Iowa pedigree; and vi) senile dementia has been reported in association with Aβ protein angiopathy and tau perivascular pathology, but not neuritic plaques, in patients homozygous for the APOE ε4 allele.
On the other hand, since about 90% of AD subjects have CAA, some AD subjects must then be entirely absent of CAA. The progression of AD pathophysiology, therefore, can and does occur without CAA, and there is no evidence in the form of prospective analysis suggesting that the progression of cognitive decline in those subjects without CAA differs from subjects with CAA. In one retrospective study, subjects with AD plus CAA had a lower score on cognitive testing than AD subjects without CAA115, but the possibility that this finding reflected more advanced AD stage was not excluded. Moreover, in non-demented subjects, the presence of CAA had no effect on cognitive ability. Some subjects with hereditary cerebral hemorrhage with amyloidosis—Dutch type suffer cognitive decline; however, the critical factor in these subjects appears to be vascular stenosis116 rather than the presence of amyloid per se. Indeed, subjects in these kindreds often have a prodigious vascular amyloid burden, but do not suffer cognitive decline, or, for that matter, pathology more closely associated with cognitive decline (e.g., neurofibrillary pathology)116. The appropriateness of equating subjects with exceedingly rare point mutations and constitutive production of aberrant gene products, with sporadic, late onset AD might also be questioned. The empirical fact that elderly patients presenting with lobar intracerebral hemorrhage often live independently and are not significantly demented is also worth noting. Indeed, consensus criteria for the clinical diagnosis of CAA-related hemorrhage do not include presence or absence of dementia117.
In one study, CAA and cerebrovascular end-organ pathology occurred at a similar frequency between AD and controls118. Within the setting of AD, hemorrhage and ischemic lesions were more likely in subjects with severe CAA in another study119, however this relationship was not independent of atherosclerotic disease and was also not correlated with AD stage. While the statistics with regard to AD stage, CAA, and complications of CAA remain to be worked out, it is entirely possible that CAA complicated by lobar intracerebral hemorrhage has no correlation with AD, or is even negatively correlated with AD, compared to the cognitively intact, elderly population. The fundamental differences in pathogenesis of CAA and AD are also reflected in the Aβ chemistry and ApoE interactions. While the APOE ε4 allele has been identified as a risk factor for sporadic and AD-related CAA, the APOE ε2 allele, normally “protective” for development of AD, is associated with an increased risk of CAA-related hemorrhage120. Moreover, the dominant Aβ species in sporadic CAA appears to be Aβ1–40, whereas the Aβ1–42 species, the “pathogenic Aβ,” dominates the Aβ content in senile plaques121. Conveniently, the high Aβ1–40content in vessels with CAA has been explained by the notion that Aβ1–42provides a nidus or “seed” for attachment of soluble Aβ1–40, although no direct evidence for this seeding hypothesis has been presented. Generally neglected in clinicopathological studies relating CAA to AD, or CAA to cognition, is a correlation between CAA and neurofibrillary pathology (e.g., Braak stage). This is particularly noteworthy in light of the close correlation between neuroanatomical distribution of neurofibrillary pathology and the corresponding signs and symptoms of AD19, 122, 123. The same correlation does not exist for amyloid deposits. There are no studies to suggest, for example, that CAA colocalizes with the limbic structures, such as the pre-αpyramidal neurons of the entorhinal cortex, that undergo neurofibrillary degeneration early in disease and correlate closely with memory impairment19.
Pfeifer et al.124 published an intriguing account of the effects of passive Aβ peptide immunization. In this study, ten APP23 mice were passively immunized by weekly intraperitoneal injections of 0.5 mg of mouse monoclonal immunoglobulin G1 antibody that recognizes amino acids 3–6 of human Aβ. Five months of treatment yielded a significant reduction in amyloid load. This reduction was attributed to a decrease in diffuse parenchymal amyloid (down 33% compared to controls by stereological analysis). Interestingly, the frequency and severity of CAA per se were not affected by immunization, yet the immunized mice suffered a twofold increase in the frequency of intracerebral hemorrhage, and the hemorrhage was generally present in regions also affected by CAA. They suggested in effect that CAA is a contraindication to Aβ immunotherapy and that results could be improved by screening patients for CAA. Among potential mechanisms suggested were local inflammation and increased vascular permeability, although no direct evidence for either of these mechanisms was provided. Avoided was the suggestion that interference with vascular amyloid itself may have been responsible for vascular rupture, since the amyloid burden appeared the same for immunized and non-immunized mice. On the other hand, the colocalization of CAA and hemorrhage, with the absence of other pathology, tends to suggest that vascular changes of a mechanical nature contributed to the hemorrhage. Extrapolation of such studies to sporadic CAA and AD are complicated by the obviously artificial environment. If anything, such models bare closest resemblance to the rare kindreds with AβPP mutations. However, assuming a close parallel between APP23 mice and sporadic CAA in aged humans, the results appear more to suggest that Aβ deposition is a homeostatic mechanism, such that disruption of this process is deleterious125.
Recent studies have provided evidence that Aβ production is an adaptive phenomenon in response to oxidative stress and actually blunts free radical injury126. Thus Aβ deposition in blood vessel walls, as well as Aβ production in the form of senile plaques, is more likely a response to injury, whereas the neurodegenerative process is one of complex neurochemistry at the neuronal level that includes oxidative stress and aberrant cell cycle activation126. The finding that vascular atrophy in AD is independent of Aβ deposits127 argues further against a primary toxic effect of Aβ. Rather, logic would dictate that chronic deposition of Aβ, which occurs over a period of many years in the setting of human disease, requires exquisite balance, such that disruption of this process compromises vascular integrity. The Pfeifer study tends to support this concept, as does the fact that the majority of AD subjects with varying amounts of CAA, fail to suffer vascular rupture and intracerebral hemorrhage, and when they do, a risk independent of atherosclerotic and arteriosclerotic disease is difficult to establish. Thus, in the vast majority of AD subjects, CAA is more of a parenchymal “decoration” than a clinicopathological entity. This being the case, the failure of Aβ immunization and indeed adverse consequences as encountered in human and experimental studies, is not surprising.
The concept that vasculature is important to the functioning of the end-organs, including brain, has been known since William Harvey’s assertions in the 17thcentury. So it is reasonable to speculate that if the vasculature is overwhelmed by proteinaceous material, end-organ damage may occur (e.g., HCHWA-D). The question with respect to AD is whether CAA has a primary, or anything other than nominal, role in the parenchymal degeneration that characterizes AD. The strongest evidence of such a process rests in rare familial conditions characterized by exorbitant amounts of aberrantly synthesized precursor protein; the premise of this evidence, however, that these conditions reflect sporadic disease, is dubious, whereas the clinicopathological association between sporadic CAA and cognitive decline is weak and often contradictory. If one views AD pathogenesis outside the narrow prism of amyloid toxicity, it becomes apparent that amyloid fibril formation, initiated in response to cellular stress (e.g., oxidative stress), is a manifestation of parenchymal adaptation. If amyloid is excessive (e.g., in HCHWA-D), blood flow might be compromised. But within the spectrum of sporadic and late onset AD subjects, CAA is little more than a curiosity in comparison with the parenchymal pathophysiology.
VIII. OTHER INCLUSIONS
A. Hirano Bodies
Hirano bodies are intra-neuronal eosinophilic rod-like inclusions, 15–30 µm in length, which are located primarily in the hippocampus and whose frequency increases with age and in AD128 (Figure 12). The precise chemical composition of Hirano bodies is unknown, however the presence of actin, α-actinin, vinculin, and tropomyosin have been demonstrated to be diffusely distributed throughout the structure suggesting that a breakdown of the microfilament system is involved in their formation129. While initially identified within hippocampal tissue of patients with the amyotrophic lateral sclerosis-Parkinsonism-Dementia complex of Guam, Hirano bodies are a common feature of AD, normal aging, alcoholism, Pick disease, and Kuru. Thus, like many pathologic inclusions, an initial enthusiasm for lesion specificity has been tempered by subsequent studies indicating, in essence, their nonspecific nature. Their role in AD pathogenesis is doubtful, although their accumulation as an age associated phenomenon is without dispute.
Figure 12.

H&E stained section of the hippocampus showing a Hirano body in the perikaryon of a pyramidal neuron.
B. Granulovacuolar Degeneration
Granulovacuolar degeneration, electron dense argyrophilic cores or granules within a membrane-limited vacuole approximately 3–5 µm in diameter, are frequently located in the cytoplasm of pyramidal neurons of the hippocampal formation, but also found in smaller numbers in the neocortex of AD brains (Figure 13). Immunocytochemical studies suggest that granulovacuolar degeneration is a proteolytic disorder as manifested by ubiquitin immunolabeling130, 131, or cytoskeletal abnormality of elements such as tubulin132. Granulovacuolar degeneration is not specific for AD and also occurs in the brains of aged individuals, albeit at a lower frequency, as well as in Pick disease, adult Down syndrome and other neurological conditions.
Figure 13.

H&E stained section of hippocampus showing granulovacuolar degeneration affecting a pyramidal neuron. A Hirano body is also present.
In a recent study, we found that the granulovacuolar degeneration is also comprised of phosphorylated 40S ribosomal subunit, phospho-ribosomal S6133. S6 is also present in so-called stress granules in a variety of stress models, evidently precipitated by phosphorylation of the eukaryotic initiation factor eIF2a134. Stress granules are thought to serve as a protective and decision mechanism for RNAs. Thus, granulovacuolar degeneration may be the human homologue to stress granules in other systems. The crystallization of this change as a signature either of neurodegeneration or neuroprotection requires further study.
IX. PATHOLOGICAL PROTEINS
A. Amyloid-β Protein Precursor
The major protein component of senile plaque cores and vascular amyloid is a small polypeptide of approximately 4.2 kDa termed Aβ72, 135 which molecular genetic studies have shown to be part of a much larger amyloid-β protein precursor (AβPP) encoded on chromosome 21. The first mRNA sequence obtained encoded AβPP695, a 695 amino acid polypeptide136. The AβPP gene contains 18 exons with the Aβ sequence being interrupted by an intron137. Two exons are involved in alternate splicing variants, one encodes a sequence of 56 amino acids which shows a considerable degree of conservation throughout mammalian evolution138and is highly homologous in sequence and function to the Kunitz family of serine protease inhibitors139. The other exon encodes 19 amino acids which has homology with the MRC OX-2 antigen found on the surface of neurons and thymocytes140. Alternative splicing occurs whereby several larger versions of the AβPP are generated that contain the serine protease inhibitor domain139, 141, 142. The four possible mRNAs encoding the Aβ peptide have been detected in rat and human brain, i.e., AβPP695, AβPP714, AβPP751, and AβPP770143, 144.
B. Amyloid-β
As previously discussed, extracellular Aβ fibrils are a major component of congophilic angiopathy135, senile plaque cores72 and may also form a constituent of the chemically complex intracellular NFT145. Aβ is characteristically congophilic, argyrophilic, thioflavin S positive and highly insoluble in most protein solvents—a feature that is used in purification strategies72. While formic acid is known to solubilize the Aβ cores of senile plaques and markedly enhance the immunocytochemical detection of Aβ in tissue sections146, such pretreatment abolishes the ability of Aβ to be detected by modified Bielschowsky, periodic acid-methenamine silver and Congo red staining indicating that formic acid results in structural and/or biochemical changes147.
The first amino acid composition data of the protein from isolated senile plaque cores indicated that the protein was probably unique, having an unusually high level of valine, and containing very little threonine148. Sequencing of the 28 amino-terminal residues showed that the protein is indeed unique and has no significant homology to other known proteins. The full length polypeptide was later shown to be composed of 42–43 amino acids derived from the larger precursor AβPP by comparison with the cDNA sequence136. Raman spectroscopy revealed a nearly identical composition of highly purified plaque cores and amyloid peptide149.
Amino acid composition data for plaque cores, vessel amyloid and paired helical filament preparations led to the suggestion that these pathological structures all contained the same protein, i.e., Aβ protein150. However, while sequence data from paired helical filaments showed sequence identity to Aβ protein, both a lack of quantitation and likely contamination by senile plaque or vessel amyloid made this particular report far from conclusive. Nonetheless, immunocytochemical analyses have revealed the presence of Aβ in NFT145, 151.
Biochemical and sequence analyses reveal senile plaque Aβ protein species ending in residue 40 or 42 and, based on several independent lines of evidence, it is suspected that the longer Aβ42 form is crucial in the pathogenesis of AD21. For example, Aβ42 is the initial and major component of senile plaque deposits and aggregates at a faster rate than Aβ40. Moreover, several mutations of AβPP and, as will be discussed later, presenilin 1 and 2, associated with familial AD cases result in an increased secretion of Aβ, particularly Aβ42.
Using NMR data, the three-dimensional structure of the Aβ1–39 peptide has been obtained. Overall, the structure is defined by an extended structure (residues 1–9), α-helix (residues 10–26), reverse turn (residues 27–30), and α-helix (residues 31–39). The presence of two, rather than one, α-helical regions supports the proposed mechanism, in which the helix between residues 1–25 breaks-down first at pH 4–7, due to unfavorable electrostatic interactions of the Glu22 side-chain with the α-helix macrodipole, resulting in production of an amyloid-like β-sheet structure152. The presence of a reverse turn within the Asn27-Lys28-Gly29-Ala30 is nearly identical to that suggested for the β-sheet structure for an Aβ10–43 peptide153.
The Aβ protein present in AD is identical to that found in similar lesions in Down syndrome72, 135. There may be a certain degree of N-terminal heterogeneity found in senile plaque core Aβ which is slightly different in the two diseases, although this may be the result of the use of proteolytic procedures used in the isolation of senile plaques72. Alternately, some degree of amino-terminal heterogeneity might reflect alternate processing of the precursor protein to generate truncated Aβ forms154. Aβ derived from vessels does not seem to show the same degree of N-terminal heterogeneity and starts predominantly at either residue 1 or 2155.
Vascular Aβ is readily amenable to sequencing and is soluble in guanidine hydrochloride135 while Aβ isolated from senile plaque cores is insoluble in guanidine hydrochloride72 and cannot be directly sequenced due to a blocked amino terminus156. The blocked amino terminus is presumably removed if the isolation procedure includes proteolytic procedures prior to sequence analysis but casts doubt on the biological validity of heterogeneous amino termini. These differences between senile plaque and vessel Aβ might indicate a chronological mechanism with Aβ first appearing in the parenchyma156 or, instead, alternate mechanisms of Aβ metabolism and/or catabolism in the vessels and brain parenchyma157, 158.
C. Pittsburgh Compound B (PiB)
Until recently, a definitive diagnosis of AD could be made only by brain biopsy or at autopsy because only by these methods was it possible to demonstrate the presence of amyloid plaques and NFTs, to determine whether pathology may have contributed to the clinical phenotype in a patient with dementia. The now available PET scanning with the 11C-labelled Pittsburgh compound B (PiB), a benzothiazole derivative of thioflavin T, is a non-invasive method to assess cerebral amyloid. This compound readily crosses the blood brain barrier and binds selectively to brain Aβ deposits which may be in the form of neuritic (but not diffuse, non-fibrillar) plaque and cerebral amyloid angiopathy159. It is inevitable that its use will soon change the way physicians diagnose and treat disorders associated with aggregation of Aβ in the brain.
Results of early studies using this scanning method, however, have been somewhat conflicting. It would appear that PiB binding to amyloid may not always differentiate symptomatic AD from asymptomatic controls with amyloid plaque10, 160, 161. PiB labeling confirms that Aβ deposition in familial AD is different from that in sporadic AD162. It is suggested that amyloid deposits as detected by PiB binding in the asymptomatic patient is a means of detecting preclinical AD associated with progression to AD163. The lack of binding to non-fibrillar amyloid plaque, however, can give false negative results9. A study using PiB7 gives support to the theory of ‘cognitive reserve’ and the relationship of education to amyloid burden in the clinical presentation of AD.
While PiB is a new tool in the study of AD that has opened the door to visualizing in vivo brain function in a manner relevant to amyloid plaque build up, more longitudinal studies are needed to determine its usefulness. It has the potential to improve our understanding of the natural history of amyloid deposits, give us further insights into AD pathophysiology and help evaluate the amyloid hypothesis of AD development. Its greatest value may lie in that it allows one to directly measure the efficacy of anti-amyloid therapy.
D. Tau Protein
The identification of phosphorylated tau as the major protein component of NFT led to a host of scientific investigations related to tau metabolism, mechanisms of phosphorylation and dephosphorylation, role of tau processing in disease, and in general the concept that phosphorylated tau was inherently toxic, such that the amelioration of phosphorylated tau from the aged brain may successfully treat AD. Lost in the process, however, were clinicopathological findings indicating that phosphorylated tau typically occupies viable cells and accumulates with age, often in significant amounts. More recent experimental and human data further call into question the notion of primary toxicity, while the presence of sequestered, redox-active heavy metals within neurofibrillary pathology suggests that tau lesions may be a manifestation of protection, or cellular adaptation with age.
The structure of the NFT as noted above was elucidated by Kidd in 1963 with the initial description of paired helical filaments96. Some ultrastructural details of NFTs were subsequently added to the literature164, but it was not until 1986 that NFTs were purified and the microtubule associated protein tau determined as the major protein component165. This finding initiated a substantial focus on tau pathophysiology; however, since the identification of tau in NFTs occurred about two years after the identification of Aβ in senile plaques, and since Down syndrome and familial AD had a genetic link to Aβ, it is not surprising that Aβ assumed primacy over tau as the important pathogenic, if not etiologic, factor in AD.
Tau is relatively abundant in neurons but is present in all nucleated cells and functions physiologically to bind microtubules and stabilize microtubule assembly for polymerization. The tau gene, comprised of over 100 kb and containing 16 exons22, contains consensus binding sites for transcription factors such as AP2 and SP1. In the adult brain, alternative splicing of tau nuclear RNA transcribed on exons 2, 3, and 10, results in six tau isoforms, having either three or four peptide repeats of 31 or 32 residues in the C terminal region encoded on exon 10, comprising the microtubule binding domain or differing in the expression of zero, one, or two inserts encoded on exons two and three. These tau isoforms, as well as their phosphorylation status, change during development such that 3 repeat tau with no inserts is expressed in the fetus and early postnatal infant, while heterogeneous isoforms are expressed in the adult brain. This switch in RNA splicing also corresponds to a reduction in tau phosphorylation. During neurodegeneration, tau is abnormally phosphorylated at proline directed serine/threonine phosphorylation sites, which can be detected using specific antisera, including Ser-202/Thr-205 (AT8 site), Ser-214 and/or Ser-212 (AT100 site), Thr-231 and/or Ser-235 (TG3 site), and Ser-396/Ser-404 (PHF-1 site). As noted above, the profile of alternative tau splicing differs among pathological phenotypes, such that tau accumulation in AD is a mixture of 3R and 4R tau, Pick disease tends to be 3R tau, corticobasal degeneration and progressive supranuclear palsy tends to be 4R tau, and so-called argyrophilic grain disease accumulates small inclusions comprised of 3R tau. The general term “tauopathy” encompasses the broad classification of neurodegenerative diseases that accumulate phosphorylated tau.
The correlation between regional distribution of phosphorylated tau and clinical signs suggests a close relationship between tau and AD pathogenesis22, 23. The increased tau phosphorylation that accompanies AD may result in separation of tau from the microtubule, possibly aided by other factors (Aβ, oxidative stress, inflammatory mediators), and sequestration in NFTs and neuropil threads. The loss of normal tau function (stabilization and maintenance of microtubules), combined with a toxic gain of function, could compromise axonal transport and contribute to the synaptic degeneration that is now a central theme. Interestingly, the concept of NFT toxicity, like senile plaque toxicity, is increasingly being challenged. In one transgenic model, mice expressing a repressible human tau developed NFTs, neuronal loss, and behavioral impairments. After tau suppression, the behavioral deficits stabilized, yet NFTs continued to accumulate, suggesting that they are not sufficient to cause cognitive decline or neuronal death166. In another AD-like model, axonal pathology with accumulation of tau preceded plaque deposition167, while studies of a P301S tauopathy model demonstrated microglial activation and synapse loss prior to NFT formation168. Thus, the proponents of tau phosphorylation appear to be headed toward an analogy of the updated Aβ cascade model, by suggesting that toxicity of hyperphosphorylated tau relates to the presence of toxic tau intermediates. This was predictable but does not completely address the deficiencies in the original hypothesis. Indeed, it is important to note that NFTs (and presumable “intermediates”) exist within the cytoplasm of viable neurons169. Only in advanced disease are large numbers of extracellular NFTs identified, and when they are encountered, the largest numbers appear in the CA-1 region of the hippocampus—an area that is selectively vulnerable to ischemic injury.
In another study, NFTs (and presumably tau oligomers) survive within the cytoplasm of neurons for decades170, arguing strongly against any primary toxicity of phosphorylated tau. In another study, neurons with NFTs showed intact microtubules171, calling into question the concept that accumulation of phosphorylated tau and destabilization of microtubule assembly go hand in hand.
X. ETIOLOGY
A. Age
An often ignored aspect is that age represents, by far, the single greatest risk factor in the etiology of AD. Indeed, even in genetically-predisposed individuals, the disease essentially does not occur in middle age. Therefore, regardless of whether one is genetically predisposed or not, aging is essential factor in AD, strongly suggesting that an age-related process is involved in the development of the disease. This age-related penetrance is not restricted to AD and is also a risk factor in a number of other chronic diseases including other neurodegenerative diseases, cancer, atherosclerosis, arthritis and emphysema indicating the possibility that there may be common etiologies with diverse consequences.
Nevertheless, it remains an open question whether AD is purely a manifestation of advanced age, as has been suggested, and whether there remains some merit is separating “senile dementia” as was known at the time of Alzheimer, from “Alzheimer disease.” The literature has long hinted at the possibility that AD pathology is more severe in younger affected patients, although additional data have always managed to refute this claim5. Nevertheless, a progressive neurological deterioration, beginning in the 6th decade with no family history, does occur. It differs dramatically from the slowly progressive memory loss in a patient in the 9th or 10th decade with concomitant atherosclerotic disease and the confounding effect of a multitude of comorbid illnesses, their complications, and a long medication list.
The difficulties in diagnosing AD in the very old (i.e., the spectrum of neuropathology in the cognitively intact elderly ranges from minimal changes to the advanced stage of pathologically “proven” AD) has also been known since the original AD descriptions. Indeed, in one study, neuropathologists blinded to clinical history diagnosed AD in 76% of elderly subjects who were otherwise not clinically demented during life24. These data seem to emphasize again a discernible difference, clinical and pathological, between AD and senile dementia, the former being precipitated by a specific cause as yet unknown, and the latter being a manifestation of advanced age per se.
B. Genetics
In a substantial proportion of individuals with AD there is evidence for a significant genetic component (Table 6). First degree relatives of AD patients have a higher lifetime incidence of AD than the general population, and 15–35% of patients with AD have affected first-degree relatives172. Using the traditional, now unfavored, arbitrary distinction between early- (<65 years) and late- (>65 years) onset, it has been shown that the early familial form of the disease tends to be more aggressive86, with particular familial forms having a characteristic age of onset. Unfortunately, the actual incidence of genetically-linked AD is likely confounded by a number of factors, viz.: (a) incorrect diagnosis as sporadic AD if other family members were short lived; (b) long lived families developing ‘familial Alzheimer disease’; (c) local environmental factors causing clusters of ‘familial Alzheimer disease’ (e.g., Parkinsonism-dementia on Guam173; (d) multiple gene interactions with the environment; (e) incorrect diagnosis of AD due to inadequate postmortem data.
Table 6.
Genetic Factors Predisposing to AD.
| Chromosome 21 | AβPP mutation | ↑ Aβ42 peptide |
| Chromosome 14 | Presenilin-1 mutation | ↑ Aβ42 peptide* |
| Chromosome 1 | Presenilin-2 mutation | ↑ Aβ42 peptide |
| Down syndrome | AβPP overexpression | ↑ Aβ42 peptide |
| Chromosome 19 | APOE polymorphism | ↑ Aβ40 plaques, CAA |
| Chromosome 8 | CLU polymorphism | Aβ toxicity |
| Chromosome 11 | PICALM polymorphism | Synaptic trafficking??? |
Recent studies suggest that in familial cases, only the Aβ42/40 ratio is increased, while the absolute levels decrease
Nonetheless, a number of genetic loci have been implicated in familial cases of AD.
1. Chromosome 21
The location of the AβPP gene on chromosome 21 may provide a link between the pathology observed in individuals with Down syndrome (trisomy 21), and that seen in AD174. A relatively recent case report of an older subject with Down syndrome, whose third chromosome 21 component lacked the AβPP coding region and whose brain lacked advanced AD pathology, is further evidence of the concept of AβPP overexpression resulting in gain of function toxicity. Moreover, some transgenic mice overexpressing mutated AβPP form senile plaques (e.g., Tg2576), show age related deficiencies on cognitive-behavioral testing and show synaptic deficits, supporting a pathogenic role for mutated AβPP. Neuronal loss, on the other hand, has not been convincingly demonstrated, tau phosphorylation does not occur unless there are additional pathogenic mutations incorporated into the model, and, in some studies, “clearing” of the Aβ failed to reverse the cognitive deficits.
A small number of early-onset familial AD cases are linked to mutations in AβPP. To date, 89 families affected by germline AβPP mutations have been described worldwide (http://www.molgen.ua.ac.be/ADMutations/default.cfm). Interestingly, such cases comprise only about 9% of familial AD families, while the most common germline mutations linked to familial early onset disease are linked to presenilin 1 (~40%). The mechanism(s) by which a mutated version of AβPP precipitates the onset of AD with very near 100% penetrance is unknown. Mutations in AβPP have traditionally been thought to increase the amount of Aβ produced and/or the proportion of Aβ1–42175, although more recent data cast some doubt on this concept. In one study, an increase in the Aβ1–42: Aβ1–40 ratio was not due to an absolute increase in Aβ1–42 but rather a greater decrease in Aβ1–40176 compared to Aβ1–42 (also decreased, but less so). Thus, the possibility cannot be ruled out that germline mutation results in reduced Aβ1–42 synthesis with an altered steady state resulting in an accumulation in brain nevertheless.
2. Chromosome 19
One of the major risk factors for the development of AD is carrying the ε4 allele of APOE either in heterozygous or homozygous state177. The mechanism by which ApoE influences the pathogenesis of AD is unknown; however, ApoE, a serum cholesterol transport protein, is a component of both senile plaques and NFT178, 179. One intriguing suggestion is that the differential risk posed by the apolipoprotein genotypes is mediated, in part, through the interaction of the apolipoproteins with other proteins that are involved in the pathogenesis of AD including tau and Aβ180. Indeed, ApoE, presumably by sequestration through protein binding, eliminates the neurotoxicity of Aβ in hippocampal cultures181. Interestingly, oxidative stress, a major etiological consideration discussed below, would also be important in the action of ApoE, since the formation of ApoE/Aβ complexes is increased in oxygenated buffer and completely abolished under reducing condition180. Moreover, oxidized ApoE ε4 forms complexes with Aβ at a significantly higher rate than in similar experiments with ApoE ε3. Therefore, the oxidation of ApoE, alone or bound with Aβ protein, might affect receptor affinity and/or other catabolic interactions by a mechanism analogous to the oxidation of low density lipoprotein in diabetic renal disease182.
3. Chromosome 14/Chromosome 1
The majority (~70%) of early-onset familial AD cases are associated with mutations in two genes, presenilin 1 and presenilin 2, located on chromosomes 14 and 1, respectively177. To date, over thirty different pathogenic mutations in these genes have been described from over sixty unrelated kindred183. It is now generally accepted that the presenilins comprise part of the γ-secretase complex, necessary for the synthesis of Aβ peptides. Mutations at these sites evidently alter Aβ processing in favor of increased synthesis and deposition, although as noted above this may be an oversimplification. Nevertheless, familial early onset AD cases associated with presenilin mutations are noteworthy in the extensive deposits of Aβ throughout the brain, including white matter and extensively within blood vessels22.
There is considerable homology between the gene products of presenilin 1 and presenilin-2 which are transmembrane proteins of 463 and 448 amino acids, respectively, with between six and nine hydrophobic membrane spanning domains177.
It may be noted that in light of the prodigious accumulations of Aβ in presenilin-linked familial AD, and the requirement for the existence of γ-secretase, the relationship between presenilins and AβPP, and indeed the assignment of γ-secretase function to presenilins, was for practical purposes pre-ordained, and the task of scientists given to the amyloid cascade hypothesis became the discovery of data that supported the enzyme-substrate paradigm, rather than determining whether or not a relationship existed in the first place. In this vein, it is perhaps not surprising that supporting evidence has been found in abundance, and that it is now second nature to view the presenilins and γ-secretase as the same. This is in spite of the evidence being based on in vivo data from worms, flies, and transgenic mice, and that little is known about the structure of γ-secretase, the mechanism it utilizes for proteolysis, or the regulation of cleavage22.
4. Other Genetic Factors
A recent genome-wide association study of AD involving over 16,000 individuals replicated the known association with APOE and also identified two loci that were not previously diseaseassociated: CLU (which encodes clusterin) and PICALM (phosphatidylinositol-binding clathrin assembly protein, also known as CALM, clathrin assembly lymphoid-myeloid leukemia gene)184.
As pointed out by Harold et al.184, clusterin is a multifunctional molecule. It interacts with the soluble form of Aβ in animal models of disease and binds soluble Aβ in a specific and reversible manner, forming complexes that have been shown to cross the blood-brain barrier. Notably, ApoE also appears to act as a molecular chaperone for Aβ and influences when Aβ aggregates and deposits; ApoE also influences Aβ conformation and toxicity. In a similar manner similar to ApoE, clusterin may regulate both the toxicity of Aβ and its conversion into insoluble forms.
Furthermore, ApoE and clusterin have been shown to cooperate in suppressing Aβ deposition, and they may critically modify Aβ clearance at the blood-brain barrier, which could suggest a role for clusterin in the amyloidogenic pathway. Levels of ApoE protein appear to be inversely proportional to APOE ε4 allele dose levels, with protein levels reduced in ε4 homozygotes compared with heterozygotes. Conversely, clusterin levels are increased in proportion to APOE ε4 allele dose levels, suggesting an induction of clusterin in individuals with low ApoE levels185.
Also as noted by Harold et al.184, PICALM is ubiquitously expressed in all tissue types with prominent expression in neurons, where it is nonselectively distributed at the pre- and postsynaptic structures. It has been shown that like BIN1, the PICALM protein is involved in clathrin-mediated endocytosis, an essential step in the intracellular trafficking of proteins and lipids such as nutrients, growth factors and neurotransmitters. Of relevance to AD, PICALM appears to be involved in directing the trafficking of VAMP2. VAMP2 is a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein that has a prominent role in the fusion of synaptic vesicles to the presynaptic membrane in neurotransmitter release, a process that is crucial to neuronal function. Genetically directed changes in PICALM function may lead to synaptic dysfunction, possibly through synaptic vesicle cycling, thereby increasing risk for AD. Alternatively, PICALM could influence AD risk through AβPP processing via endocytic pathways, resulting in changes in Aβ levels.
C. Other Factors
1. Education
No other area of study in aging and dementia is accompanied by more hand waving than is the discussion of education and risk for dementia. The idea of a protective effect from mental activity dates as far back as Cicero in the 2nd century B.C. - “it is our duty to resist old age; to compensate for its defects by a watchful care; to fight against it as we would fight against disease…Old men retain their mental faculties, provided their interest and application continue.”186. Santiago Ramon y Cajal took this concept one step further when he suggest that axons could sprout collaterals by rigorous mental activity, something he termed “gymnastique cerebrale.” The “use it or lose it” concept has always had an appeal, and perhaps provides some peace of mind for the overeducated individuals in the medical and scientific communities. Thus, renewed efforts in recent years have taken an epidemiological approach, and it has been found in different studies that the risk for dementia increases with decreased educational level187 or may not increase the risk for dementia188, 189. In the former study, the education is said to enhance the “cognitive reserve” such that a given level of pathology will take longer to have a clinical effect in those who spent more time educating themselves. Generally lacking from such studies, however, is the ability to control for comorbid traits that accompany the lesser educated and the lower socioeconomic strata, such as diet, alcoholism, cigarette smoking, co-existing atherosclerotic disease, etc. The overall concepts of cognitive reserve and the “mentally active” state are moreover ill defined at the neurochemical level. Education may indicate more reading per se, and the committing of more items to memory, but not necessarily more mental activity at the neurochemical level. Indeed, while cognitive function seems to decline most steeply in less educated individuals190, a more subtle indicator of complex brain thought, such as linguistic ability in early life might be a better indicator of risk than that of educational attainment alone191.
2. Anti-Inflammatory Drugs
The lower than expected prevalence rate of AD in patients with rheumatoid arthritis192 indicates that long term treatment with anti-inflammatory agents including non-steroidal anti-inflammatory drugs (NSAIDs) might be protective against the development of AD. Indeed, clinical studies have shown that individuals taking anti-inflammatory drugs reduce their risk of developing AD and that patients with AD who take anti-inflammatory drugs perform better in neuropsychological tests including the Mini-Mental Status Examination193. Therefore, anti-inflammatory drugs appear to lower the incidence as well as slow the progression of AD suggesting the possibility that inflammation may play a role in the etiology of the disease.
3. Head Trauma
The risk of AD that is attributable to head trauma might be as high as 15%194. Since the 1920s, it has been also known that the repetitive, concussive and subconcussive brain trauma associated with boxing may produce a progressive neurological deterioration. The generally accepted term for this process is dementia pugilistica, although more recent literature is tending toward the nosological phrase “chronic traumatic encephalopathy”195. Dementia pugilistica is associated clinically with memory disturbances, behavioral and personality changes, parkinsonism, and speech and gait abnormalities, and in some cases is indistinguishable clinically from AD. Co-existing vascular disease and nutritional disturbances such as Wernicke-Korsakoff syndrome may complicate the overall picture. Dementia pugilistica is characterized pathologically by atrophy of the cerebral hemispheres, particularly the medial temporal lobes, although the thalamus, mammillary bodies, and brainstem are also described, as is ventricular dilatation, fenestrated cavum septum pellucidum, and cavum septum pellucidum.
The microscopic pathology lends itself to the controversy of the primacy of tau versus Aβ in the disease process, as extensive neurofibrillary pathology and, inconveniently, limited amyloid pathology is the rule in dementia pugilistica195. Microscopically, there are extensive NFT, astrocytic tangles, and spindle-shaped and threadlike neurites throughout the brain. In addition, the neurofibrillary degeneration in dementia pugilistica is somewhat unique in its preferential involvement of the superficial cortical layers, irregular patchy distribution in the frontal and temporal cortices, propensity for sulcal depths, prominent perivascular, periventricular, and subpial distribution, and marked accumulation of tau-immunoreactive astrocytes.
Deposition of Aβ, when it does occur (fewer than half of the cases), is of the diffuse type. The obvious conclusion therefore is that dementia pugilistica represents primarily an abnormality of tau metabolism, and that Aβ deposits, when they do occur, are incidental, age related phenomena. Such a conclusion, however, is blunted by the amyloid cascade theorists who still favor an upstream Aβ abnormality and a downstream tau phosphorylation196. Part of the logic lies in the fact that acute elevations are Aβ, in addition to AβPP, are described in head trauma, and the accompanying temptation to relate acute head injury to chronic sequelae. Regardless, the mental gymnastics required to ascribe dementia pugilistica as an amyloid cascade disease emphasizes the difficulty, if not the impossibility, with which such embedded hypotheses are set aside.
From a more fundamental perspective, further studies of traumatic brain injury and its relationship to chronic disease are clearly warranted, from the simple standpoint that dementia pugilistica, or chronic traumatic encephalopathy, represents a distinct, slowly progressive tauopathy with a clear environmental etiology195.
XI. TREATMENT STRATEGIES
A. Cholinergics
The nucleus basalis of Meynert, a distinct population of basal forebrain neurons, is a major source of cholinergic innervation to the cerebral cortex. In 1982, Whitehouse and colleagues found that these neurons are selectively (>70%) degenerated in patients with AD197, 198. Furthermore, the loss of cholinergic function has been found to be closely related to cognitive dysfunction199. Over the last two decades, a number of therapeutics targeting cholinesterase inhibition, choline precursors, postsynaptic cholinergic stimulation with muscarinic agonists, and presynaptic cholinergic stimulation with nicotinic agonists has been investigated200. These drugs stabilize cognitive decline for up to 3–6 months201–203, although no modification of disease duration or general disease progression has been accomplished.
Acetylcholinesterase inhibitors (ACIs) are the best developed therapy and are used for mild to moderate disease. The mechanism by which ACIs slow progression of disease is thought to be by decreasing levels of AβPP and production of Aβ and amyloidogenic compounds204, 205. Tacrine was the first widely used ACI206. A 30-week randomized control clinical trial showed a significant dose related improvement in cognitive function207, 208. However, subsequent studies were less impressive and a short half-life, hepatotoxicity, and cholinergic side effects have limited the use of this drug. Second generation cholinergics, including donepezil (trade name Aricept®, Eisai Company and Pfizer Inc.), galantamine (Hoechst Marion Roussel Inc., Shire Pharmaceutical Group, and Janssen Pharmaceutical, trade names Reminyl® and Nivalin, U.S. trade name Razadyne®) and rivastigmine (trade name Exelon®, Novartis Pharmaceuticals) have since been developed. These drugs have fewer side effects, longer half-lives, and greater efficacy. In a meta-analysis of 29 randomized placebo-controlled trials, patients on cholinesterase inhibitors improved 0.1 standard deviations (SDs) on activity of daily living (ADL) scales and 0.09 SDs on instrumental ADL scales compared with placebo. This effect is similar to preventing 2 months per year decline in a patient with AD209. Typically, ACIs are started at low doses to minimize side effects such as facial flushing, dyspepsia, nausea, vomiting, and diarrhea. The dose is then titrated up to the maximum tolerated dose210.
Transdermal patch studies are currently under investigation and may provide more optimal drug delivery. The Investigation of Transdermal Exelon® in Alzheimer’s Disease (IDEAL) Study, a 24-week, double-blind, double-dummy, placebo- and active-controlled study in patients with Mini-Mental Status Exam (MMSE) scores of 10–20, recently showed similar efficacy of the rivastigmine patch compared to high dose capsules211. Precursor (lecithin and choline) and nicotine supplementation have not been found to improve overall cognition199. Direct activation of postsynaptic cholinergic receptors by muscarinic agonists may decrease secretion of Aβ and increase secretion of nerve growth factor210. In addition, substances capable of blocking the nonsynaptic (nonspecific) cholinesterases; called butyrylcholinesterases, are currently being explored212.
B. Neurotropins
A number of studies investigating growth factor effects on the adult CNS began yielding positive results in the 1980s. Beginning in 1986, the infusion of nerve growth factor (NGF) into the adult rat brain was found to completely prevent the death of basal forebrain cholinergic neurons both spontaneously and after injury213–216. Over the next decade, studies in rodent models went further to show improvement in cognitive deficits213, 217–220. Primate studies in the 1990s confirmed the ability of NGF to prevent degeneration of basal forebrain cholinergic neurons in monkeys221–226. However, the inability of the NGF protein to cross the blood brain barrier, its short half-life, and its broad ranging effect of biological signals has made efficacious delivery of these neurotropins difficult to attain in the clinical setting. Administration of mouse NGF in AD patients and clinical trials of growth factors in patients with either amyotrophic lateral sclerosis or Parkinson’s disease failed secondary to adverse side effects and lack of efficacy227–230. In response to this dilemma, multiple CNS gene delivery studies were initiated. Ex vivo preclinical studies demonstrated targeted, efficacious, sustained responses to NGF with no associated toxicity221, 223, 226, 231–234. In 2001, human clinical trials were initiated. A Phase I clinical trial with eight early stage AD patients was performed wherein autologous NGF secreting cells were injected into the nucleus basalis of three dose cohorts. In the patients who received injection without complication, there were no adverse events observed over 2 years and no signs of non-targeted NGF spread235. Serial PET scans in four subjects demonstrated reversal of the pattern of cortical metabolic decline normally seen in AD236. Cognitive decline was reduced on both the MMSE and Alzheimer’s Disease Assessment Scale-Cognitive (ADASCog) scales, though interpretation of these results was limited by small sample size, lack of placebo, and lack of blinding. In vivo gene delivery utilizing the adeno-associated virus (AAV) and other vectors has been found to be successful in animal studies237, 238. Results from a Phase I clinical trial of AAVNGF gene delivery in early to mid stage AD are pending and plans for a Phase II multicenter, sham surgery-controlled trial of AAV-NGF gene transfer are underway239.
C. Antioxidants
Multiple lines of evidence indicate that oxidative stress is an important pathogenic process associated with aging and AD, while markers of oxidative stress have been shown to precede pathological lesions in AD, including senile plaques and NFT109, 240–242. Antioxidants may thus blunt the cognitive decline in AD or slow disease progression243–245. The Alzheimer’s Disease Cooperative Study compared selegiline, a-tocopherol, or both with placebo246 Given the delay in progression to adverse outcome in the treatment groups, an American Academy of Neurology practice parameter states that vitamin E likely delays time to clinical worsening. Vitamin E in combination with vitamin C is also associated with a decrease in the prevalence and incidence of AD247
D. Statins
Cerebral Aβ levels have been shown to be decreased in vivo with simvastatin248 and results in the specific neuropathologic change in statin use of decreased NFT burden at autopsy249 The Adult Changes in Thought (ACT) Study also showed a significant protective effect of statins against dementia; however, a later analysis of a larger sample from ACT suggested a protective effect in subjects who began statin use before age 80250, 251. In line with these results, numerous early epidemiologic studies indicated that the use of statins significantly reduces the risk of AD252–255. However, other large prospective cohort studies and two large randomized placebo-controlled trials of statins for coronary heart disease prevention failed to provide evidence of a protective effect against cognitive decline256–259. The pattern of results obtained thus far suggests that statins may slow progression of the neurodegenerative process, but may not be able to reverse neuronal degeneration once it has occurred.
F. Non-steroidal anti-inflammatory drugs (NSAIDs)
Aβ deposition and plaque formation are associated with an innate immune response that includes activation of complement260, secretion of pro-inflammatory cytokines, expression of chemokines, and excretion of nitric oxide which mediates apoptosis261, 262. NSAIDs downregulate pro-inflammatory signals, microglia, and astrocytes and may reduce risk of AD by lowering Aβ1–42 production263. The Baltimore Longitudinal Study of Aging showed reduced risk for AD with NSAID use proportional to duration of use264. Case control studies of individuals taking NSAIDs for arthritis, a small clinical trial of indomethacin, and a number of familial studies also indicated protection from development of AD or progression of disease265, 266. Recently, a large population-based, longitudinal study, the Cache County Study on Memory Health and Aging, suggested that NSAIDs may help prevent cognitive decline if started in midlife. However, randomized controlled trials of various NSAIDs have failed to show a significant effect on the rate of cognitive decline in patients with dementia or the rate of conversion to AD in patients with MCI267–269. Moreover, a randomized controlled primary prevention trial of NSAIDs in AD, the AD Anti-inflammatory Prevention Trial (ADAPT), was terminated in 2004 secondary to concerns regarding cardiovascular risk270. Given the associated risks, cyclooxygenase-II inhibitors and NSAIDs are currently not recommended for the treatment or prevention of AD.
F. Hormone replacement therapy
Estrogen enhances cerebral blood flow, prevents atrophy of cholinergic neurons, reduces oxidative stress, and modulates the effects of nerve growth factors271. It may also reduce neuronal injury by decreasing formation of Aβ. Three prospective, population-based epidemiologic studies suggested that postmenopausal estrogen replacement therapy (HRT) may delay the onset of AD. One randomized clinical trial and a meta-analysis showed some improvement in cognitive function272–276. However, other randomized trials actually showed an increase in the development of dementia in healthy individuals and no improvement in cognitive or functional outcome277–279. As such, further work is clearly needed to explore the use of HRT in AD and whether any effects are direct or indirect280, 281.
G. Blocking of excitotoxicity
Glutamate is the principle excitatory neurotransmitter in cortical and hippocampal neurons. Glutamine synthetase is oxidized in the brains of individuals with AD, leading to excess glutamate. Excessive activation of NMDA receptors by glutamate increases the vulnerability of CNS neurons leading to neuronal degeneration. Memantine (trade name Namanda, Forest) blocks glutamate gated NMDA channels, thereby blocking pathological activation and preserving physiological activation282–285. In contrast with ACIs, which are approved for only the early and intermediate stages of disease, memantine is approved for treating the advanced stages of AD.
Data regarding memantine use are equivocal. A randomized clinical trial of 252 patients with MMSE scores of 3–14 found that memantine significantly reduces deterioration on multiple accounts286. Adverse event rates were similar to placebo. However, this was not confirmed by a doubleblind, placebo-controlled trial involving 350 patients. This trial showed no statistically significant treatment benefit at study end point on any primary or secondary outcome measure after 24 weeks287. A Cochrane systematic review reported a small beneficial effect at 6 months in moderate to severe AD on cognition, ADLs, and behavior supported by clinical impression of change. In patients with mild to moderate dementia, pooled data from three unpublished studies indicated marginal benefit at 6 months on intention to treat cognition, which were barely detectable clinically. There was no effect on behavior, ADLs, or observed case analysis of cognition288. There is evidence that combination therapy with donepezil may provide better outcomes with regards to cognition, ADLs, global outcome, and behavior as illustrated in a 24-week trial in 322 patients with MMSE scores from 5 to 14289. Memantine may also be helpful in other neurodegenerative conditions, such as Huntington disease, AIDS-related dementia, and vascular dementia. The most common side effect of memantine is dizziness, with confusion and hallucinations occurring at lower frequency.
H. Diet
The Mediterranean diet was recently demonstrated to be associated with lower AD risk. A subsequent study has now shown that the Mediterranean diet is also associated with lower mortality in AD with a possible dose response effect. This diet is characterized by high intake of vegetables, fruits, and cereals; a low-to-moderate intake of saturated fatty acids, moderately high intake of fish, low-to-moderate intake of dairy products, low intake of meat and poultry, and a moderate amount of ethanol290.
I. The Aβ vaccine trials
Removal of excess Aβ from the brain occurs by a reactive T cell response. In 1999, active immunization of transgenic mice producing human Aβ was shown to produce a significant reduction of Aβ plaques291. The PDAPP transgenic mouse was the model used and immunization with Aβ was administered prior to the onset of AD-type neuropathologies or once these changes were already established. Immunization of the young animals prevented the development of Aβ plaque formation, neuritic dystrophy and astrogliosis. Treatment of the older animals reduced the extent and progression of these neuropathologies291. There is evidence for a number of mechanisms of plaque removal: 1) solubilization by binding of antibody to Aβ; 2) phagocytosis of opsonized Aβ by microglial cells; and 3) the “sink” hypothesis in which Aβ antibodies remain in the plasma and extract Aβ from the brain by altering equilibrium across the blood brain barrier292 to regain functional effects following immunization273, 274. Following these animal trials, clinical trials of the immunogen AN-1792 [Aβ1–42 with adjuvant (QS-21)] were performed by Elan Pharmaceuticals/Wyeth. Phase IIa clinical trials were suspended when 6% of patients in the active treatment group developed meningoencephalitis characterized by subacute neurologic deterioration, lymphocytic pleocytosis, and white matter abnormalities on imaging293. Five patients receiving the trial drug died, with one death (secondary to cerebral infarction) considered to be related to study treatment294.
J. Immunotherapy
In the attempt to avoid adverse T cell mediated immune response, many vaccination modalities under current investigation are directed towards the humoral response. Nasal immunization of an AD mouse model with AdPEDI-(Aβ1–6) demonstrated a predominantly IgG1 response and reduced Aβ load in the brain295. Transcutaneous immunization has also been studied in mice with aggregated Aβ1–42 plus the adjuvant cholera toxin. This animal study showed significant decreases in cerebral Aβ1–40,42 levels in the setting of increased circulating levels of Aβ1–40,42 without the side effects of brain T cell infiltration or microhemorrhage296. Aβ derivatives in alum adjuvant also promote humoral immunity and decrease the Aβ burden297. Preliminary clinical results indicate that IVIg has the potential to deliver a controlled immune attack on the Aβ peptide298. Clinical trials testing the safety and tolerability of the monoclonal antibody Bapineuzumab (AAB-001), an Aβ peptide antibody, are being conducted by Wyeth and Elan in 200 patients with mild to moderate AD. A small imaging study in Europe is ongoing to determine whether existing plaques are reduced while on this therapy (AAB-001). In addition to passive immunotherapy, Elan and Wyeth have initiated a clinical trial for an active immunization (ACC-001). This immunoconjugate is hoped to be effective while avoiding potential side effects by inducing a highly specific antibody response to Aβ (NCT00479557).
K. Secretase effectors
Memapsin 2 (β-secretase, BACE1) is the protease that initiates cleavage of AβPP leading to the production of Aβ. Immunization of transgenic AD mice (Tg2576) with BACE has also resulted in Aβ reduction and cognitive improvement in the absence of an inflammatory response299. A nonselective γ-secretase inhibitor, LY450139 (Eli Lilly) was evaluated in PDAPP transgenic mice and found to result in a 60% reduction of Aβ40 in plasma, CSF, and the hippocampus. There was also a reduction in the median value for Aβ plaque burden in the cortex and hippocampus. Two clinical trials showed dose dependent reduction of plasma Aβ, however, following the inhibitory phase, plasma Aβ levels exceeded baseline levels. GI side effects and eosinophilia were also properties of concern (LY450139 Dihydrate). Tarenflurbil (MPC-7869; Myriad Pharmaceuticals) is a γ-secretase modulator in the class of drugs referred to as selective amyloid lowering agents (SALAs). As these drugs do not interfere with γ-secretase substrates, they are well tolerated. A Phase II study in 207 patients with mild to moderate AD showed improvement on ADLs, global function, and cognition in patients with mild AD, with a significant plasma concentration to response relationship. There was no benefit observed in patients with moderate AD. A Phase III study of Tarenflurbil in patients with mild AD is ongoing300.
XII. FUTURE DEVELOPMENTS/RESEARCH AVENUES
As the number of studies and research proposals espousing “therapeutic implications” expands at an exponential rate, what is perhaps most striking is the absence of progress in altering the course of AD. The only marginal beneficial treatment currently achievable involves the enhancement of cholinergic neurotransmission, while the neurodegenerative process continues unabated. This is in spite of thousands of research articles and copious knowledge of molecular cascades that are presumed pathogenic. The situation is not unlike that in Parkinson’s disease, in which treatment with L-Dopa is beneficial in providing symptomatic relief by replacing defective neurotransmission, but does not slow the loss of neurons.
In the end, it seems likely that the next major advance will occur either as a multidisciplinary multimodality approach, attacking every conceivable facet of neurodegeneration, or else will occur by accident. The scope of the problem, however, is too pervasive and too costly to ignore, and is a clarion call for cooperation among clinicians, neuropathologists, biochemists, cell biologists and molecular biologists, so we might prevent our “second childhood.”
ACKNOWLEDGMENTS
Work in the author’s laboratory has been supported by grant support from the National Institutes of Health (AG026151, AG028679, AG031364), American Health Assistance Foundation, American Federation for Aging Research, and Alzheimer’s Association.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Alzheimer A. Uber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeits Psychiat PsychischYGerichtlich Med. 1907;64:146–148. [Google Scholar]
- 2.Roth M, Tomlinson BE, Blessed G. Correlation between scores for dementia and counts of 'senile plaques' in cerebral grey matter of elderly subjects. Nature. 1966;209:109–110. doi: 10.1038/209109a0. [DOI] [PubMed] [Google Scholar]
- 3.Roth M, Tomlinson BE, Blessed G. The relationship between quantitative measures of dementia and of degenerative changes in the cerebral grey matter of elderly subjects. Proc R Soc Med. 1967;60:254–260. [PMC free article] [PubMed] [Google Scholar]
- 4.Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci. 1970;11:205–242. doi: 10.1016/0022-510x(70)90063-8. [DOI] [PubMed] [Google Scholar]
- 5.Berrios G. Alzheimer's disease: A conceptual history. International Journal of Geriatric Psychiatry. 1990;5:355–365. [Google Scholar]
- 6.Wiley CA, Lopresti BJ, Venneti S, Price J, Klunk WE, DeKosky ST, et al. Carbon 11-labeled Pittsburgh Compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol. 2009;66:60–67. doi: 10.1001/archneurol.2008.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roe CM, Mintun MA, D'Angelo G, Xiong C, Grant EA, Morris JC. Alzheimer disease and cognitive reserve: variation of education effect with carbon 11-labeled Pittsburgh Compound B uptake. Arch Neurol. 2008;65:1467–1471. doi: 10.1001/archneur.65.11.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klunk WE. Biopsy support for the validity of Pittsburgh compound B positron emission tomography with a twist. Arch Neurol. 2008;65:1281–1283. doi: 10.1001/archneur.65.10.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah AR, et al. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol. 2009;66:1557–1562. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leinonen V, Alafuzoff I, Aalto S, Suotunen T, Savolainen S, Nagren K, et al. Assessment of beta-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh Compound B. Arch Neurol. 2008;65:1304–1309. doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- 12.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 13.Braak H, Rub U, Schultz C, Del Tredici K. Vulnerability of cortical neurons to Alzheimer's and Parkinson's diseases. J Alzheimers Dis. 2006;9:35–44. doi: 10.3233/jad-2006-9s305. [DOI] [PubMed] [Google Scholar]
- 14.Fratiglioni L, De Ronchi D, Aguero-Torres H. Worldwide prevalence and incidence of dementia. Drugs Aging. 1999;15:365–375. doi: 10.2165/00002512-199915050-00004. [DOI] [PubMed] [Google Scholar]
- 15.Small GW, Rabins PV, Barry PP, Buckholtz NS, DeKosky ST, Ferris SH, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer's Association, and the American Geriatrics Society. JAMA. 1997;278:1363–1371. [PubMed] [Google Scholar]
- 16.Hebert LE, Scherr PA, Beckett LA, Albert MS, Pilgrim DM, Chown MJ, et al. Age-specific incidence of Alzheimer's disease in a community population. JAMA. 1995;273:1354–1359. [PubMed] [Google Scholar]
- 17.Zhu CW, Sano M. Economic considerations in the management of Alzheimer's disease. Clin Interv Aging. 2006;1:143–154. doi: 10.2147/ciia.2006.1.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 19.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 20.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 22.Castellani RJ, Lee HG, Zhu X, Perry G, Smith MA. Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol. 2008;67:523–531. doi: 10.1097/NEN.0b013e318177eaf4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brayne C, Richardson K, Matthews FE, Fleming J, Hunter S, Xuereb JH, et al. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J Alzheimers Dis. 2009;18:645–658. doi: 10.3233/JAD-2009-1182. [DOI] [PubMed] [Google Scholar]
- 25.Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC, et al. Molecular mapping of Alzheimer-type dementia in Down's syndrome. Ann Neurol. 1998;43:380–383. doi: 10.1002/ana.410430316. [DOI] [PubMed] [Google Scholar]
- 26.Brooks WS, Kwok JB, Kril JJ, Broe GA, Blumbergs PC, Tannenberg AE, et al. Alzheimer's disease with spastic paraparesis and 'cotton wool' plaques: two pedigrees with PS-1 exon 9 deletions. Brain. 2003;126:783–791. doi: 10.1093/brain/awg084. [DOI] [PubMed] [Google Scholar]
- 27.Janssen JC, Lantos PL, Fox NC, Harvey RJ, Beck J, Dickinson A, et al. Autopsy-confirmed familial early-onset Alzheimer disease caused by the l153V presenilin 1 mutation. Arch Neurol. 2001;58:953–958. doi: 10.1001/archneur.58.6.953. [DOI] [PubMed] [Google Scholar]
- 28.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 29.Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 30.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- 31.Terry RD, Peck A, DeTeresa R, Schechter R, Horoupian DS. Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann Neurol. 1981;10:184–192. doi: 10.1002/ana.410100209. [DOI] [PubMed] [Google Scholar]
- 32.Perry G, Castellani RJ, Smith MA, Harris PL, Kubat Z, Ghanbari K, et al. Oxidative damage in the olfactory system in Alzheimer's disease. Acta Neuropathol. 2003;106:552–556. doi: 10.1007/s00401-003-0761-7. [DOI] [PubMed] [Google Scholar]
- 33.Whitwell JL, Shiung MM, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, et al. MRI patterns of atrophy associated with progression to AD in amnestic mild cognitive impairment. Neurology. 2008;70:512–520. doi: 10.1212/01.wnl.0000280575.77437.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frisoni GB, Lorenzi M, Caroli A, Kemppainen N, Nagren K, Rinne JO. In vivo mapping of amyloid toxicity in Alzheimer disease. Neurology. 2009;72:1504–1511. doi: 10.1212/WNL.0b013e3181a2e896. [DOI] [PubMed] [Google Scholar]
- 35.Kromer Vogt LJ, Hyman BT, Van Hoesen GW, Damasio AR. Pathological alterations in the amygdala in Alzheimer's disease. Neuroscience. 1990;37:377–385. doi: 10.1016/0306-4522(90)90408-v. [DOI] [PubMed] [Google Scholar]
- 36.Driscoll I, Resnick SM, Troncoso JC, An Y, O'Brien R, Zonderman AB. Impact of Alzheimer's pathology on cognitive trajectories in nondemented elderly. Ann Neurol. 2006;60:688–695. doi: 10.1002/ana.21031. [DOI] [PubMed] [Google Scholar]
- 37.Mahut H, Zola-Morgan S, Moss M. Hippocampal resections impair associative learning and recognition memory in the monkey. J Neurosci. 1982;2:1214–1220. doi: 10.1523/JNEUROSCI.02-09-01214.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zola-Morgan S, Squire LR, Amaral DG. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci. 1986;6:2950–2967. doi: 10.1523/JNEUROSCI.06-10-02950.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Struble RG, Price DL, Jr, Cork LC, Price DL. Senile plaques in cortex of aged normal monkeys. Brain Res. 1985;361:267–275. doi: 10.1016/0006-8993(85)91298-3. [DOI] [PubMed] [Google Scholar]
- 40.Mann DM, Esiri MM. The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down's syndrome. J Neurol Sci. 1989;89:169–179. doi: 10.1016/0022-510x(89)90019-1. [DOI] [PubMed] [Google Scholar]
- 41.Hirano A, Zimmerman HM. Silver impregnation of nerve cells and fibers in celloidin sections. A simple impregnation technique. Arch Neurol. 1962;6:114–122. doi: 10.1001/archneur.1962.00450200028003. [DOI] [PubMed] [Google Scholar]
- 42.Ball MJ. Topographic distribution of neurofibrillary tangles and granulovacuolar degeneration in hippocampal cortex of aging and demented patients. A quantitative study. Acta Neuropathol. 1978;42:73–80. doi: 10.1007/BF00690970. [DOI] [PubMed] [Google Scholar]
- 43.Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56:343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- 44.Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer's disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 45.Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR. Perforant pathway changes and the memory impairment of Alzheimer's disease. Ann Neurol. 1986;20:472–481. doi: 10.1002/ana.410200406. [DOI] [PubMed] [Google Scholar]
- 46.Mann DM, Yates PO, Marcyniuk B. Dopaminergic neurotransmitter systems in Alzheimer's disease and in Down's syndrome at middle age. J Neurol Neurosurg Psychiatry. 1987;50:341–344. doi: 10.1136/jnnp.50.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcyniuk B, Mann DM, Yates PO. The topography of cell loss from locus caeruleus in Alzheimer's disease. J Neurol Sci. 1986;76:335–345. doi: 10.1016/0022-510x(86)90179-6. [DOI] [PubMed] [Google Scholar]
- 48.Pearson RC, Esiri MM, Hiorns RW, Wilcock GK, Powell TP. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci U S A. 1985;82:4531–4534. doi: 10.1073/pnas.82.13.4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968;114:797–811. doi: 10.1192/bjp.114.512.797. [DOI] [PubMed] [Google Scholar]
- 50.Mann DM, Marcyniuk B, Yates PO, Neary D, Snowden JS. The progression of the pathological changes of Alzheimer's disease in frontal and temporal neocortex examined both at biopsy and at autopsy. Neuropathol Appl Neurobiol. 1988;14:177–195. doi: 10.1111/j.1365-2990.1988.tb00880.x. [DOI] [PubMed] [Google Scholar]
- 51.Bennett DA, Cochran EJ, Saper CB, Leverenz JB, Gilley DW, Wilson RS. Pathological changes in frontal cortex from biopsy to autopsy in Alzheimer's disease. Neurobiol Aging. 1993;14:589–596. doi: 10.1016/0197-4580(93)90043-b. [DOI] [PubMed] [Google Scholar]
- 52.Masliah E, Terry RD, DeTeresa RM, Hansen LA. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett. 1989;103:234–239. doi: 10.1016/0304-3940(89)90582-x. [DOI] [PubMed] [Google Scholar]
- 53.Masliah E, Terry RD, Alford M, DeTeresa R. Quantitative immunohistochemistry of synaptophysin in human neocortex: an alternative method to estimate density of presynaptic terminals in paraffin sections. J Histochem Cytochem. 1990;38:837–844. doi: 10.1177/38.6.2110586. [DOI] [PubMed] [Google Scholar]
- 54.Suzuki K, Terry RD. Fine structural localization of acid phosphatase in senile plaques in Alzheimer's presenile dementia. Acta Neuropathol. 1967;8:276–284. doi: 10.1007/BF00688828. [DOI] [PubMed] [Google Scholar]
- 55.Praprotnik D, Smith MA, Richey PL, Vinters HV, Perry G. Filament heterogeneity within the dystrophic neurites of senile plaques suggests blockage of fast axonal transport in Alzheimer's disease. Acta Neuropathol. 1996;91:226–235. doi: 10.1007/s004010050420. [DOI] [PubMed] [Google Scholar]
- 56.Smith MA, Perry G. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1997;56:217. [PubMed] [Google Scholar]
- 57.Perry G, Nunomura A, Hirai K, Zhu X, Perez M, Avila J, et al. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer's and other neurodegenerative diseases? Free Radic Biol Med. 2002;33:1475–1479. doi: 10.1016/s0891-5849(02)01113-9. [DOI] [PubMed] [Google Scholar]
- 58.Castellani RJ, Honda K, Zhu X, Cash AD, Nunomura A, Perry G, et al. Contribution of redox-active iron and copper to oxidative damage in Alzheimer disease. Ageing Res Rev. 2004;3:319–326. doi: 10.1016/j.arr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 59.Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- 60.Bosman GJ, Bartholomeus IG, de Grip WJ. Alzheimer's disease and cellular aging: membrane-related events as clues to primary mechanisms. Gerontology. 1991;37:95–112. doi: 10.1159/000213253. [DOI] [PubMed] [Google Scholar]
- 61.Nitsch RM, Blusztajn JK, Pittas AG, Slack BE, Growdon JH, Wurtman RJ. Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci U S A. 1992;89:1671–1675. doi: 10.1073/pnas.89.5.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roth GS, Joseph JA, Mason RP. Membrane alterations as causes of impaired signal transduction in Alzheimer's disease and aging. Trends Neurosci. 1995;18:203–206. doi: 10.1016/0166-2236(95)93902-a. [DOI] [PubMed] [Google Scholar]
- 63.Tanzi RE. The synaptic Abeta hypothesis of Alzheimer disease. Nat Neurosci. 2005;8:977–979. doi: 10.1038/nn0805-977. [DOI] [PubMed] [Google Scholar]
- 64.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 65.Arendt T. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:167–179. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- 66.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy GM, Jr, Eng LF, Ellis WG, Perry G, Meissner LC, Tinklenberg JR. Antigenic profile of plaques and neurofibrillary tangles in the amygdala in Down's syndrome: a comparison with Alzheimer's disease. Brain Res. 1990;537:102–108. doi: 10.1016/0006-8993(90)90345-c. [DOI] [PubMed] [Google Scholar]
- 68.Rifenburg RP, Perry G. Dystrophic neurites define diffuse as well as core-containing senile plaques in Alzheimer's disease. Neurodegeneration. 1995;4:235–237. [PubMed] [Google Scholar]
- 69.Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci U S A. 1991;88:7552–7556. doi: 10.1073/pnas.88.17.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morris JC, Storandt M, McKeel DW, Jr, Rubin EH, Price JL, Grant EA, et al. Cerebral amyloid deposition and diffuse plaques in "normal" aging: Evidence for presymptomatic and very mild Alzheimer's disease. Neurology. 1996;46:707–719. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- 71.Perry G, Lipphardt S, Mulvihill P, Kancherla M, Mijares M, Gambetti P, et al. Amyloid precursor protein in senile plaques of Alzheimer disease. Lancet. 1988;2:746. doi: 10.1016/s0140-6736(88)90219-x. [DOI] [PubMed] [Google Scholar]
- 72.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong CW, Quaranta V, Glenner GG. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc Natl Acad Sci U S A. 1985;82:8729–8732. doi: 10.1073/pnas.82.24.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allsop D, Landon M, Kidd M, Lowe JS, Reynolds GP, Gardner A. Monoclonal antibodies raised against a subsequence of senile plaque core protein react with plaque cores, plaque periphery and cerebrovascular amyloid in Alzheimer's disease. Neurosci Lett. 1986;68:252–256. doi: 10.1016/0304-3940(86)90152-7. [DOI] [PubMed] [Google Scholar]
- 75.Ikeda S, Allsop D, Glenner GG. Morphology and distribution of plaque and related deposits in the brains of Alzheimer's disease and control cases. An immunohistochemical study using amyloid beta-protein antibody. Lab Invest. 1989;60:113–122. [PubMed] [Google Scholar]
- 76.Ikeda S, Yanagisawa N, Allsop D, Glenner GG. Evidence of amyloid beta-protein immunoreactive early plaque lesions in Down's syndrome brains. Lab Invest. 1989;61:133–137. [PubMed] [Google Scholar]
- 77.Kawai M, Cras P, Perry G. Serial reconstruction of beta-protein amyloid plaques: relationship to microvessels and size distribution. Brain Res. 1992;592:278–282. doi: 10.1016/0006-8993(92)91686-9. [DOI] [PubMed] [Google Scholar]
- 78.Pro JD, Smith CH, Sumi SM. Presenile Alzheimer disease: amyloid plaques in the cerebellum. Neurology. 1980;30:820–825. doi: 10.1212/wnl.30.8.820. [DOI] [PubMed] [Google Scholar]
- 79.Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J. Spectrum of morphological appearance of amyloid deposits in Alzheimer's disease. Acta Neuropathol. 1989;78:337–347. doi: 10.1007/BF00688170. [DOI] [PubMed] [Google Scholar]
- 80.Kidd M. Alzheimer's Disease--an Electron Microscopical Study. Brain. 1964;87:307–320. doi: 10.1093/brain/87.2.307. [DOI] [PubMed] [Google Scholar]
- 81.Allsop D, Ikeda S, Bruce M, Glenner GG. Cerebrovascular amyloid in scrapie-affected sheep reacts with antibodies to prion protein. Neurosci Lett. 1988;92:234–239. doi: 10.1016/0304-3940(88)90067-5. [DOI] [PubMed] [Google Scholar]
- 82.Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, et al. Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci U S A. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McGeer PL, Rogers J, McGeer EG. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271–276. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- 84.Hardy JA, Mann DM, Wester P, Winblad B. An integrative hypothesis concerning the pathogenesis and progression of Alzheimer's disease. Neurobiol Aging. 1986;7:489–502. doi: 10.1016/0197-4580(86)90086-2. [DOI] [PubMed] [Google Scholar]
- 85.Rudelli RD, Ambler MW, Wisniewski HM. Morphology and distribution of Alzheimer neuritic (senile) and amyloid plaques in striatum and diencephalon. Acta Neuropathol. 1984;64:273–281. doi: 10.1007/BF00690393. [DOI] [PubMed] [Google Scholar]
- 86.Bondareff W, Mountjoy CQ, Roth M, Rossor MN, Iversen LL, Reynolds GP. Age and histopathologic heterogeneity in Alzheimer's disease. Evidence for subtypes. Arch Gen Psychiatry. 1987;44:412–417. doi: 10.1001/archpsyc.1987.01800170026005. [DOI] [PubMed] [Google Scholar]
- 87.Glenner GG. Congophilic microangiopathy in the pathogenesis of Alzheimer's syndrome (presenile dementia) Med Hypotheses. 1979;5:1231–1236. doi: 10.1016/0306-9877(79)90005-7. [DOI] [PubMed] [Google Scholar]
- 88.Miyakawa T, Shimoji A, Kuramoto R, Higuchi Y. The relationship between senile plaques and cerebral blood vessels in Alzheimer's disease and senile dementia. Morphological mechanism of senile plaque production. Virchows Arch B Cell Pathol Incl Mol Pathol. 1982;40:121–129. doi: 10.1007/BF02932857. [DOI] [PubMed] [Google Scholar]
- 89.Kawai M, Kalaria RN, Harik SI, Perry G. The relationship of amyloid plaques to cerebral capillaries in Alzheimer's disease. Am J Pathol. 1990;137:1435–1446. [PMC free article] [PubMed] [Google Scholar]
- 90.Probst A, Brunnschweiler H, Lautenschlager C, Ulrich J. A special type of senile plaque, possibly an initial stage. Acta Neuropathol. 1987;74:133–141. doi: 10.1007/BF00692843. [DOI] [PubMed] [Google Scholar]
- 91.DiPatre PL, Gelman BB. Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol. 1997;56:143–149. doi: 10.1097/00005072-199702000-00004. [DOI] [PubMed] [Google Scholar]
- 92.Selkoe DJ, Bell DS, Podlisny MB, Price DL, Cork LC. Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer's disease. Science. 1987;235:873–877. doi: 10.1126/science.3544219. [DOI] [PubMed] [Google Scholar]
- 93.Hansen LA, Masliah E, Galasko D, Terry RD. Plaque-only Alzheimer disease is usually the lewy body variant, and vice versa. J Neuropathol Exp Neurol. 1993;52:648–654. doi: 10.1097/00005072-199311000-00012. [DOI] [PubMed] [Google Scholar]
- 94.Mann DM, Jones D. Deposition of amyloid (A4) protein within the brains of persons with dementing disorders other than Alzheimer's disease and Down's syndrome. Neurosci Lett. 1990;109:68–75. doi: 10.1016/0304-3940(90)90539-l. [DOI] [PubMed] [Google Scholar]
- 95.Kirschner DA, Abraham C, Selkoe DJ. X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-beta conformation. Proc Natl Acad Sci U S A. 1986;83:503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kidd M. Paired helical filaments in electron microscopy of Alzheimer's disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 97.Wisniewski HM, Narang HK, Terry RD. Neurofibrillary tangles of paired helical filaments. J Neurol Sci. 1976;27:173–181. doi: 10.1016/0022-510x(76)90059-9. [DOI] [PubMed] [Google Scholar]
- 98.Terry RD, Gonatas NK, Weiss M. Ultrastructural Studies in Alzheimer's Presenile Dementia. Am J Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- 99.Wischik CM, Crowther RA, Stewart M, Roth M. Subunit structure of paired helical filaments in Alzheimer's disease. J Cell Biol. 1985;100:1905–1912. doi: 10.1083/jcb.100.6.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Crowther RA, Wischik CM. Image reconstruction of the Alzheimer paired helical filament. EMBO J. 1985;4:3661–3665. doi: 10.1002/j.1460-2075.1985.tb04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pollanen MS, Markiewicz P, Bergeron C, Goh MC. Twisted ribbon structure of paired helical filaments revealed by atomic force microscopy. Am J Pathol. 1994;144:869–873. [PMC free article] [PubMed] [Google Scholar]
- 102.Pollanen MS, Markiewicz P, Goh MC. Paired helical filaments are twisted ribbons composed of two parallel and aligned components: image reconstruction and modeling of filament structure using atomic force microscopy. J Neuropathol Exp Neurol. 1997;56:79–85. doi: 10.1097/00005072-199701000-00008. [DOI] [PubMed] [Google Scholar]
- 103.Perry G, Mulvihill P, Manetto V, Autilio-Gambetti L, Gambetti P. Immunocytochemical properties of Alzheimer straight filaments. J Neurosci. 1987;7:3736–3738. doi: 10.1523/JNEUROSCI.07-11-03736.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer's disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351–355. doi: 10.1016/0304-3940(86)90288-0. [DOI] [PubMed] [Google Scholar]
- 105.Selkoe DJ, Ihara Y, Salazar FJ. Alzheimer's disease: insolubility of partially purified paired helical filaments in sodium dodecyl sulfate and urea. Science. 1982;215:1243–1245. doi: 10.1126/science.6120571. [DOI] [PubMed] [Google Scholar]
- 106.Smith MA, Siedlak SL, Richey PL, Nagaraj RH, Elhammer A, Perry G. Quantitative solubilization and analysis of insoluble paired helical filaments from Alzheimer disease. Brain Res. 1996;717:99–108. doi: 10.1016/0006-8993(95)01473-x. [DOI] [PubMed] [Google Scholar]
- 107.Smith MA. Alzheimer disease. Int Rev Neurobiol. 1998;42:1–54. doi: 10.1016/s0074-7742(08)60607-8. [DOI] [PubMed] [Google Scholar]
- 108.Kalaria RN. The immunopathology of Alzheimer's disease and some related disorders. Brain Pathol. 1993;3:333–347. doi: 10.1111/j.1750-3639.1993.tb00761.x. [DOI] [PubMed] [Google Scholar]
- 109.Castellani RJ, Harris PL, Sayre LM, Fujii J, Taniguchi N, Vitek MP, et al. Active glycation in neurofibrillary pathology of Alzheimer disease: N(epsilon)-(carboxymethyl) lysine and hexitol-lysine. Free Radic Biol Med. 2001;31:175–180. doi: 10.1016/s0891-5849(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 110.Castellani RJ, Perry G, Siedlak SL, Nunomura A, Shimohama S, Zhang J, et al. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett. 2002;319:25–28. doi: 10.1016/s0304-3940(01)02514-9. [DOI] [PubMed] [Google Scholar]
- 111.Klatzo I, Wisniewski H, Streicher E. Experimental Production of Neurofibrillary Degeneration. I. Light Microscopic Observations. J Neuropathol Exp Neurol. 1965;24:187–199. doi: 10.1097/00005072-196504000-00002. [DOI] [PubMed] [Google Scholar]
- 112.Burks JS, Alfrey AC, Huddlestone J, Norenberg MD, Lewin E. A fatal encephalopathy in chronic haemodialysis patients. Lancet. 1976;1:764–768. doi: 10.1016/s0140-6736(76)91608-1. [DOI] [PubMed] [Google Scholar]
- 113.Castellani RJ, Smith MA, Perry G, Friedland RP. Cerebral amyloid angiopathy: major contributor or decorative response to Alzheimer's disease pathogenesis. Neurobiol Aging. 2004;25:599–602. doi: 10.1016/j.neurobiolaging.2003.12.019. discussion 3–4. [DOI] [PubMed] [Google Scholar]
- 114.Castellani RJ, Siedlak SL, Fortino AE, Perry G, Ghetti B, Smith MA. Chitin-like polysaccharides in Alzheimer's disease brains. Curr Alzheimer Res. 2005;2:419–423. doi: 10.2174/156720505774330555. [DOI] [PubMed] [Google Scholar]
- 115.Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology. 2002;58:1629–1634. doi: 10.1212/wnl.58.11.1629. [DOI] [PubMed] [Google Scholar]
- 116.Natte R, Maat-Schieman ML, Haan J, Bornebroek M, Roos RA, van Duinen SG. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol. 2001;50:765–772. doi: 10.1002/ana.10040. [DOI] [PubMed] [Google Scholar]
- 117.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56:537–539. doi: 10.1212/wnl.56.4.537. [DOI] [PubMed] [Google Scholar]
- 118.Jellinger KA, Attems J. Incidence of cerebrovascular lesions in Alzheimer's disease: a postmortem study. Acta Neuropathol. 2003;105:14–17. doi: 10.1007/s00401-002-0634-5. [DOI] [PubMed] [Google Scholar]
- 119.Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- 120.Nicoll JA, Burnett C, Love S, Graham DI, Dewar D, Ironside JW, et al. High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol. 1997;41:716–721. doi: 10.1002/ana.410410607. [DOI] [PubMed] [Google Scholar]
- 121.Wilson CA, Doms RW, Lee VM. Intracellular APP processing and A beta production in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:787–794. doi: 10.1097/00005072-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 122.Hyman BT. The neuropathological diagnosis of Alzheimer's disease: clinical-pathological studies. Neurobiol Aging. 1997;18:S27–S32. doi: 10.1016/s0197-4580(97)00066-3. [DOI] [PubMed] [Google Scholar]
- 123.Giannakopoulos P, Gold G, Duc M, Michel JP, Hof PR, Bouras C. Impaired processing of famous faces in Alzheimer's disease is related to neurofibrillary tangle densities in the prefrontal and anterior cingulate cortex. Dement Geriatr Cogn Disord. 2000;11:336–341. doi: 10.1159/000017263. [DOI] [PubMed] [Google Scholar]
- 124.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, et al. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- 125.Atwood CS, Bishop GM, Perry G, Smith MA. Amyloid-beta: a vascular sealant that protects against hemorrhage? J Neurosci Res. 2002;70:356. doi: 10.1002/jnr.10388. [DOI] [PubMed] [Google Scholar]
- 126.Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol. 2006;111:503–509. doi: 10.1007/s00401-006-0071-y. [DOI] [PubMed] [Google Scholar]
- 127.Perry G, Castellani RJ, Hirai K, Smith MA. Reactive oxygen species mediate cellular damage in Alzheimer disease. J Alzheimers Dis. 1998;1:45–55. doi: 10.3233/jad-1998-1103. [DOI] [PubMed] [Google Scholar]
- 128.Hirano A, Dembitzer HM, Kurland LT, Zimmerman HM. The fine structure of some intraganglionic alterations. Neurofibrillary tangles, granulovacuolar bodies and "rod-like" structures as seen in Guam amyotrophic lateral sclerosis and parkinsonism-dementia complex. J Neuropathol Exp Neurol. 1968;27:167–182. [PubMed] [Google Scholar]
- 129.Galloway PG, Perry G, Gambetti P. Hirano body filaments contain actin and actinassociated proteins. J Neuropathol Exp Neurol. 1987;46:185–199. doi: 10.1097/00005072-198703000-00006. [DOI] [PubMed] [Google Scholar]
- 130.Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, et al. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and Mallory bodies in alcoholic liver disease. J Pathol. 1988;155:9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- 131.Ii K, Ito H, Tanaka K, Hirano A. Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J Neuropathol Exp Neurol. 1997;56:125–131. doi: 10.1097/00005072-199702000-00002. [DOI] [PubMed] [Google Scholar]
- 132.Price DL, Altschuler RJ, Struble RG, Casanova MF, Cork LC, Murphy DB. Sequestration of tubulin in neurons in Alzheimer's disease. Brain Res. 1986;385:305–310. doi: 10.1016/0006-8993(86)91077-2. [DOI] [PubMed] [Google Scholar]
- 133.Castellani RJ, Gupta Y, Siedlak SL, Harris PLR, Coller JM, Perry G, Zhu X, Tabaton M, Smith MA. Granulovacuolar degeneration in aging and Alzheimer’s disease are the human homologue of stress granules precipitated by oxidative injury. Submitted. [Google Scholar]
- 134.Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10:430–436. doi: 10.1038/nrm2694. [DOI] [PubMed] [Google Scholar]
- 135.Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 136.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 137.Lemaire HG, Salbaum JM, Multhaup G, Kang J, Bayney RM, Unterbeck A, et al. The PreA4(695) precursor protein of Alzheimer's disease A4 amyloid is encoded by 16 exons. Nucleic Acids Res. 1989;17:517–522. doi: 10.1093/nar/17.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yamada T, Sasaki H, Dohura K, Goto I, Sakaki Y. Structure and expression of the alternatively-spliced forms of mRNA for the mouse homolog of Alzheimer's disease amyloid beta protein precursor. Biochem Biophys Res Commun. 1989;158:906–912. doi: 10.1016/0006-291x(89)92808-8. [DOI] [PubMed] [Google Scholar]
- 139.Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer's disease. Nature. 1988;331:528–530. doi: 10.1038/331528a0. [DOI] [PubMed] [Google Scholar]
- 140.Weidemann A, Konig G, Bunke D, Fischer P, Salbaum JM, Masters CL, et al. Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- 141.Kitaguchi N, Takahashi Y, Tokushima Y, Shiojiri S, Ito H. Novel precursor of Alzheimer's disease amyloid protein shows protease inhibitory activity. Nature. 1988;331:530–532. doi: 10.1038/331530a0. [DOI] [PubMed] [Google Scholar]
- 142.Ponte P, Gonzalez-DeWhitt P, Schilling J, Miller J, Hsu D, Greenberg B, et al. A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature. 1988;331:525–527. doi: 10.1038/331525a0. [DOI] [PubMed] [Google Scholar]
- 143.Kang J, Muller-Hill B. Differential splicing of Alzheimer's disease amyloid A4 precursor RNA in rat tissues: PreA4(695) mRNA is predominantly produced in rat and human brain. Biochem Biophys Res Commun. 1990;166:1192–1200. doi: 10.1016/0006-291x(90)90992-v. [DOI] [PubMed] [Google Scholar]
- 144.Golde TE, Estus S, Usiak M, Younkin LH, Younkin SG. Expression of beta amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer's disease using PCR. Neuron. 1990;4:253–267. doi: 10.1016/0896-6273(90)90100-t. [DOI] [PubMed] [Google Scholar]
- 145.Hyman BT, Van Hoesen GW, Beyreuther K, Masters CL. A4 amyloid protein immunoreactivity is present in Alzheimer's disease neurofibrillary tangles. Neurosci Lett. 1989;101:352–355. doi: 10.1016/0304-3940(89)90559-4. [DOI] [PubMed] [Google Scholar]
- 146.Kitamoto T, Ogomori K, Tateishi J, Prusiner SB. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 1987;57:230–236. [PubMed] [Google Scholar]
- 147.Yamaguchi H, Nakazato Y, Hirai S, Shoji M, Harigaya Y. Electron micrograph of diffuse plaques. Initial stage of senile plaque formation in the Alzheimer brain. Am J Pathol. 1989;135:593–597. [PMC free article] [PubMed] [Google Scholar]
- 148.Allsop D, Landon M, Kidd M. The isolation and amino acid composition of senile plaque core protein. Brain Res. 1983;259:348–352. doi: 10.1016/0006-8993(83)91273-8. [DOI] [PubMed] [Google Scholar]
- 149.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, et al. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry (Mosc) 2003;42:2768–2773. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 150.Kidd M, Allsop D, Landon M. Senile plaque amyloid, paired helical filaments, and cerebrovascular amyloid in Alzheimer's disease are all deposits of the same protein. Lancet. 1985;1:278. doi: 10.1016/s0140-6736(85)91054-2. [DOI] [PubMed] [Google Scholar]
- 151.Perry G, Richey PL, Siedlak SL, Smith MA, Mulvihill P, DeWitt DA, et al. Immunocytochemical evidence that the beta-protein precursor is an integral component of neurofibrillary tangles of Alzheimer's disease. Am J Pathol. 1993;143:1586–1593. [PMC free article] [PubMed] [Google Scholar]
- 152.Zagorski MG, Barrow CJ. NMR studies of amyloid beta-peptides: proton assignments, secondary structure, and mechanism of an alpha-helix----beta-sheet conversion for a homologous, 28-residue, N-terminal fragment. Biochemistry. 1992;31:5621–5631. doi: 10.1021/bi00139a028. [DOI] [PubMed] [Google Scholar]
- 153.Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K. Aggregation and secondary structure of synthetic amyloid beta A4 peptides of Alzheimer's disease. J Mol Biol. 1991;218:149–163. doi: 10.1016/0022-2836(91)90881-6. [DOI] [PubMed] [Google Scholar]
- 154.Higgins LS, Murphy GM, Jr, Forno LS, Catalano R, Cordell B. P3 beta-amyloid peptide has a unique and potentially pathogenic immunohistochemical profile in Alzheimer's disease brain. Am J Pathol. 1996;149:585–596. [PMC free article] [PubMed] [Google Scholar]
- 155.Pardridge WM, Vinters HV, Yang J, Eisenberg J, Choi TB, Tourtellotte WW, et al. Amyloid angiopathy of Alzheimer's disease: amino acid composition and partial sequence of a 4,200-dalton peptide isolated from cortical microvessels. J Neurochem. 1987;49:1394–1401. doi: 10.1111/j.1471-4159.1987.tb01005.x. [DOI] [PubMed] [Google Scholar]
- 156.Selkoe DJ, Abraham CR, Podlisny MB, Duffy LK. Isolation of low-molecular-weight proteins from amyloid plaque fibers in Alzheimer's disease. J Neurochem. 1986;46:1820–1834. doi: 10.1111/j.1471-4159.1986.tb08501.x. [DOI] [PubMed] [Google Scholar]
- 157.Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 158.Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, et al. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- 159.Bacskai BJ, Frosch MP, Freeman SH, Raymond SB, Augustinack JC, Johnson KA, et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431–434. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- 160.Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 161.Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Villemagne VL, Ataka S, Mizuno T, Brooks WS, Wada Y, Kondo M, et al. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol. 2009;66:1537–1544. doi: 10.1001/archneurol.2009.285. [DOI] [PubMed] [Google Scholar]
- 163.Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66:1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tellez-Nagel I, Wisniewski HM. Ultrastructure of neurofibrillary tangles in Steele-Richardson-Olszewski syndrome. Arch Neurol. 1973;29:324–327. doi: 10.1001/archneur.1973.00490290064007. [DOI] [PubMed] [Google Scholar]
- 165.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 168.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 169.Lee HG, Perry G, Moreira PI, Garrett MR, Liu Q, Zhu X, et al. Tau phosphorylation in Alzheimer's disease: pathogen or protector? Trends Mol Med. 2005;11:164–169. doi: 10.1016/j.molmed.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 170.Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- 171.Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, et al. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am J Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Breitner JC, Folstein MF. Familial Alzheimer Dementia: a prevalent disorder with specific clinical features. Psychol Med. 1984;14:63–80. doi: 10.1017/s0033291700003081. [DOI] [PubMed] [Google Scholar]
- 173.Rodgers-Johnson P, Garruto RM, Yanagihara R, Chen KM, Gajdusek DC, Gibbs CJ., Jr Amyotrophic lateral sclerosis and parkinsonism-dementia on Guam: a 30-year evaluation of clinical and neuropathologic trends. Neurology. 1986;36:7–13. doi: 10.1212/wnl.36.1.7. [DOI] [PubMed] [Google Scholar]
- 174.Schweber M. A possible unitary genetic hypothesis for Alzheimer's disease and Down syndrome. Ann N Y Acad Sci. 1985;450:223–238. doi: 10.1111/j.1749-6632.1985.tb21495.x. [DOI] [PubMed] [Google Scholar]
- 175.Selkoe DJ. Alzheimer's disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 176.Hellstrom-Lindahl E, Viitanen M, Marutle A. Comparison of Abeta levels in the brain of familial and sporadic Alzheimer's disease. Neurochem Int. 2009;55:243–252. doi: 10.1016/j.neuint.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 178.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 179.Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 180.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Whitson JS, Mims MP, Strittmatter WJ, Yamaki T, Morrisett JD, Appel SH. Attenuation of the neurotoxic effect of A beta amyloid peptide by apolipoprotein E. Biochem Biophys Res Commun. 1994;199:163–170. doi: 10.1006/bbrc.1994.1209. [DOI] [PubMed] [Google Scholar]
- 182.Gupta S, Rifici V, Crowley S, Brownlee M, Shan Z, Schlondorff D. Interactions of LDL and modified LDL with mesangial cells and matrix. Kidney Int. 1992;41:1161–1169. doi: 10.1038/ki.1992.177. [DOI] [PubMed] [Google Scholar]
- 183.Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer's disease. Neurobiol Dis. 1996;3:159–168. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- 184.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Holtzman DM. In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology. J Mol Neurosci. 2004;23:247–254. doi: 10.1385/JMN:23:3:247. [DOI] [PubMed] [Google Scholar]
- 186.Berchtold NC, Cotman CW. Evolution in the conceptualization of dementia and Alzheimer's disease: Greco-Roman period to the 1960s. Neurobiol Aging. 1998;19:173–189. doi: 10.1016/s0197-4580(98)00052-9. [DOI] [PubMed] [Google Scholar]
- 187.Katzman R. Education and the prevalence of dementia and Alzheimer's disease. Neurology. 1993;43:13–20. doi: 10.1212/wnl.43.1_part_1.13. [DOI] [PubMed] [Google Scholar]
- 188.Beard CM, Kokmen E, Offord KP, Kurland LT. Lack of association between Alzheimer's disease and education, occupation, marital status, or living arrangement. Neurology. 1992;42:2063–2068. doi: 10.1212/wnl.42.11.2063. [DOI] [PubMed] [Google Scholar]
- 189.Bonaiuto S, Rocca WA, Lippi A, Giannandrea E, Mele M, Cavarzeran F, et al. Education and occupation as risk factors for dementia: a population-based case-control study. Neuroepidemiology. 1995;14:101–109. doi: 10.1159/000109785. [DOI] [PubMed] [Google Scholar]
- 190.Butler SM, Ashford JW, Snowdon DA. Age, education, and changes in the Mini-Mental State Exam scores of older women: findings from the Nun Study. J Am Geriatr Soc. 1996;44:675–681. doi: 10.1111/j.1532-5415.1996.tb01831.x. [DOI] [PubMed] [Google Scholar]
- 191.Snowdon DA, Kemper SJ, Mortimer JA, Greiner LH, Wekstein DR, Markesbery WR. Linguistic ability in early life and cognitive function and Alzheimer's disease in late life Findings from the Nun Study. JAMA. 1996;275:528–532. [PubMed] [Google Scholar]
- 192.Jenkinson ML, Bliss MR, Brain AT, Scott DL. Rheumatoid arthritis and senile dementia of the Alzheimer's type. Br J Rheumatol. 1989;28:86–88. doi: 10.1093/rheumatology/28.1.86-b. [DOI] [PubMed] [Google Scholar]
- 193.Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology. 1995;45:51–55. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- 194.Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, et al. The association between head trauma and Alzheimer's disease. Am J Epidemiol. 1990;131:491–501. doi: 10.1093/oxfordjournals.aje.a115523. [DOI] [PubMed] [Google Scholar]
- 195.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Franz G, Beer R, Kampfl A, Engelhardt K, Schmutzhard E, Ulmer H, et al. Amyloid beta 1–42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology. 2003;60:1457–1461. doi: 10.1212/01.wnl.0000063313.57292.00. [DOI] [PubMed] [Google Scholar]
- 197.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 198.Whitehouse PJ. Quality of life: the bridge from the cholinergic basal forebrain to cognitive science and bioethics. J Alzheimers Dis. 2006;9:447–453. doi: 10.3233/jad-2006-9s351. [DOI] [PubMed] [Google Scholar]
- 199.Farlow MR, Evans RM. Pharmacologic treatment of cognition in Alzheimer's dementia. Neurology. 1998;51:S36–S44. doi: 10.1212/wnl.51.1_suppl_1.s36. discussion S65–S67. [DOI] [PubMed] [Google Scholar]
- 200.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 201.Giacobini E. Cholinesterase inhibitors stabilize Alzheimer's disease. Ann N Y Acad Sci. 2000;920:321–327. doi: 10.1111/j.1749-6632.2000.tb06942.x. [DOI] [PubMed] [Google Scholar]
- 202.Giacobini E. Do cholinesterase inhibitors have disease-modifying effects in Alzheimer's disease? CNS Drugs. 2001;15:85–91. doi: 10.2165/00023210-200115020-00001. [DOI] [PubMed] [Google Scholar]
- 203.Giacobini E. Long-term stabilizing effect of cholinesterase inhibitors in the therapy of Alzheimer' disease. J Neural Transm Suppl. 2002:181–187. doi: 10.1007/978-3-7091-6139-5_17. [DOI] [PubMed] [Google Scholar]
- 204.Giacobini E, Mori F, Lai CC. The effect of cholinesterase inhibitors on the secretion of APPS from rat brain cortex. Ann N Y Acad Sci. 1996;777:393–398. doi: 10.1111/j.1749-6632.1996.tb34451.x. [DOI] [PubMed] [Google Scholar]
- 205.Mori F, Lai CC, Fusi F, Giacobini E. Cholinesterase inhibitors increase secretion of APPs in rat brain cortex. Neuroreport. 1995;6:633–636. doi: 10.1097/00001756-199503000-00012. [DOI] [PubMed] [Google Scholar]
- 206.Summers WK. Tacrine, and Alzheimer's treatments. J Alzheimers Dis. 2006;9:439–445. doi: 10.3233/jad-2006-9s350. [DOI] [PubMed] [Google Scholar]
- 207.Knapp MJ, Knopman DS, Solomon PR, Pendlebury WW, Davis CS, Gracon SI. A 30-week randomized controlled trial of high-dose tacrine in patients with Alzheimer's disease. The Tacrine Study Group. JAMA. 1994;271:985–991. [PubMed] [Google Scholar]
- 208.Whitehouse PJ. Cholinergic therapy in dementia. Acta Neurol Scand Suppl. 1993;149:42–45. doi: 10.1111/j.1600-0404.1993.tb04254.x. [DOI] [PubMed] [Google Scholar]
- 209.Trinh NH, Hoblyn J, Mohanty S, Yaffe K. Efficacy of cholinesterase inhibitors in the treatment of neuropsychiatric symptoms and functional impairment in Alzheimer disease: a meta-analysis. JAMA. 2003;289:210–216. doi: 10.1001/jama.289.2.210. [DOI] [PubMed] [Google Scholar]
- 210.Mulugeta E, Karlsson E, Islam A, Kalaria R, Mangat H, Winblad B, et al. Loss of muscarinic M4 receptors in hippocampus of Alzheimer patients. Brain Res. 2003;960:259–262. doi: 10.1016/s0006-8993(02)03542-4. [DOI] [PubMed] [Google Scholar]
- 211.Winblad B, Grossberg G, Frolich L, Farlow M, Zechner S, Nagel J, et al. IDEAL: a 6-month, double-blind, placebo-controlled study of the first skin patch for Alzheimer disease. Neurology. 2007;69:S14–S22. doi: 10.1212/01.wnl.0000281847.17519.e0. [DOI] [PubMed] [Google Scholar]
- 212.Kamal MA, Klein P, Yu QS, Tweedie D, Li Y, Holloway HW, et al. Kinetics of human serum butyrylcholinesterase and its inhibition by a novel experimental Alzheimer therapeutic, bisnorcymserine. J Alzheimers Dis. 2006;10:43–51. doi: 10.3233/jad-2006-10108. [DOI] [PubMed] [Google Scholar]
- 213.Fischer W, Wictorin K, Bjorklund A, Williams LR, Varon S, Gage FH. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329:65–68. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- 214.Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kromer LF. Nerve growth factor treatment after brain injury prevents neuronal death. Science. 1987;235:214–216. doi: 10.1126/science.3798108. [DOI] [PubMed] [Google Scholar]
- 216.Williams LR, Varon S, Peterson GM, Wictorin K, Fischer W, Bjorklund A, et al. Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc Natl Acad Sci U S A. 1986;83:9231–9235. doi: 10.1073/pnas.83.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cooper JD, Salehi A, Delcroix JD, Howe CL, Belichenko PV, Chua-Couzens J, et al. Failed retrograde transport of NGF in a mouse model of Down's syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci U S A. 2001;98:10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Holtzman DM, Li Y, Chen K, Gage FH, Epstein CJ, Mobley WC. Nerve growth factor reverses neuronal atrophy in a Down syndrome model of age-related neurodegeneration. Neurology. 1993;43:2668–2673. doi: 10.1212/wnl.43.12.2668. [DOI] [PubMed] [Google Scholar]
- 219.Markowska AL, Koliatsos VE, Breckler SJ, Price DL, Olton DS. Human nerve growth factor improves spatial memory in aged but not in young rats. J Neurosci. 1994;14:4815–4824. doi: 10.1523/JNEUROSCI.14-08-04815.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tuszynski MH, Gage FH. Bridging grafts and transient nerve growth factor infusions promote long-term central nervous system neuronal rescue and partial functional recovery. Proc Natl Acad Sci U S A. 1995;92:4621–4625. doi: 10.1073/pnas.92.10.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Emerich DF, Winn SR, Harper J, Hammang JP, Baetge EE, Kordower JH. Implants of polymer-encapsulated human NGF-secreting cells in the nonhuman primate: rescue and sprouting of degenerating cholinergic basal forebrain neurons. J Comp Neurol. 1994;349:148–164. doi: 10.1002/cne.903490110. [DOI] [PubMed] [Google Scholar]
- 222.Koliatsos VE, Clatterbuck RE, Nauta HJ, Knusel B, Burton LE, Hefti FF, et al. Human nerve growth factor prevents degeneration of basal forebrain cholinergic neurons in primates. Ann Neurol. 1991;30:831–840. doi: 10.1002/ana.410300613. [DOI] [PubMed] [Google Scholar]
- 223.Kordower JH, Winn SR, Liu YT, Mufson EJ, Sladek JR, Jr, Hammang JP, et al. The aged monkey basal forebrain: rescue and sprouting of axotomized basal forebrain neurons after grafts of encapsulated cells secreting human nerve growth factor. Proc Natl Acad Sci U S A. 1994;91:10898–10902. doi: 10.1073/pnas.91.23.10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Tuszynski MH, U HS, Amaral DG, Gage FH. Nerve growth factor infusion in the primate brain reduces lesion-induced cholinergic neuronal degeneration. J Neurosci. 1990;10:3604–3614. doi: 10.1523/JNEUROSCI.10-11-03604.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tuszynski MH, Sang H, Yoshida K, Gage FH. Recombinant human nerve growth factor infusions prevent cholinergic neuronal degeneration in the adult primate brain. Ann Neurol. 1991;30:625–636. doi: 10.1002/ana.410300502. [DOI] [PubMed] [Google Scholar]
- 226.Tuszynski MH, Roberts J, Senut MC, U HS, Gage FH. Gene therapy in the adult primate brain: intraparenchymal grafts of cells genetically modified to produce nerve growth factor prevent cholinergic neuronal degeneration. Gene Ther. 1996;3:305–314. [PubMed] [Google Scholar]
- 227.A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. ALS CNTF Treatment Study Group. Neurology. 1996;46:1244–1249. doi: 10.1212/wnl.46.5.1244. [DOI] [PubMed] [Google Scholar]
- 228.A controlled trial of recombinant methionyl human BDNF in ALS: The BDNF Study Group (Phase III) Neurology. 1999;52:1427–1433. doi: 10.1212/wnl.52.7.1427. [DOI] [PubMed] [Google Scholar]
- 229.Eriksdotter JonhagenM, Nordberg A, Amberla K, Backman L, Ebendal T, Meyerson B, et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer's disease. Dement Geriatr Cogn Disord. 1998;9:246–257. doi: 10.1159/000017069. [DOI] [PubMed] [Google Scholar]
- 230.Kordower JH, Palfi S, Chen EY, Ma SY, Sendera T, Cochran EJ, et al. Clinicopathological findings following intraventricular glial-derived neurotrophic factor treatment in a patient with Parkinson's disease. Ann Neurol. 1999;46:419–424. doi: 10.1002/1531-8249(199909)46:3<419::aid-ana21>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 231.Chen KS, Gage FH. Somatic gene transfer of NGF to the aged brain: behavioral and morphological amelioration. J Neurosci. 1995;15:2819–2825. doi: 10.1523/JNEUROSCI.15-04-02819.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Conner JM, Darracq MA, Roberts J, Tuszynski MH. Nontropic actions of neurotrophins: subcortical nerve growth factor gene delivery reverses age-related degeneration of primate cortical cholinergic innervation. Proc Natl Acad Sci U S A. 2001;98:1941–1946. doi: 10.1073/pnas.98.4.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Rosenberg MB, Friedmann T, Robertson RC, Tuszynski M, Wolff JA, Breakefield XO, et al. Grafting genetically modified cells to the damaged brain: restorative effects of NGF expression. Science. 1988;242:1575–1578. doi: 10.1126/science.3201248. [DOI] [PubMed] [Google Scholar]
- 234.Smith DE, Roberts J, Gage FH, Tuszynski MH. Age-associated neuronal atrophy occurs in the primate brain and is reversible by growth factor gene therapy. Proc Natl Acad Sci U S A. 1999;96:10893–10898. doi: 10.1073/pnas.96.19.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11:551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- 236.Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer's Disease Treatment Studies. Am J Psychiatry. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- 237.Blesch A, Conner J, Pfeifer A, Gasmi M, Ramirez A, Britton W, et al. Regulated lentiviral NGF gene transfer controls rescue of medial septal cholinergic neurons. Mol Ther. 2005;11:916–925. doi: 10.1016/j.ymthe.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 238.Klein RL, Hirko AC, Meyers CA, Grimes JR, Muzyczka N, Meyer EM. NGF gene transfer to intrinsic basal forebrain neurons increases cholinergic cell size and protects from age-related, spatial memory deficits in middle-aged rats. Brain Res. 2000;875:144–151. doi: 10.1016/s0006-8993(00)02634-2. [DOI] [PubMed] [Google Scholar]
- 239.Tuszynski MH. Nerve growth factor gene therapy in Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21:179–189. doi: 10.1097/WAD.0b013e318068d6d2. [DOI] [PubMed] [Google Scholar]
- 240.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 242.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 243.Jama JW, Launer LJ, Witteman JC, den Breeijen JH, Breteler MM, Grobbee DE, et al. Dietary antioxidants and cognitive function in a population-based sample of older persons. The Rotterdam Study. Am J Epidemiol. 1996;144:275–280. doi: 10.1093/oxfordjournals.aje.a008922. [DOI] [PubMed] [Google Scholar]
- 244.Perrig WJ, Perrig P, Stahelin HB. The relation between antioxidants and memory performance in the old and very old. J Am Geriatr Soc. 1997;45:718–724. doi: 10.1111/j.1532-5415.1997.tb01476.x. [DOI] [PubMed] [Google Scholar]
- 245.Rottkamp CA, Nunomura A, Hirai K, Sayre LM, Perry G, Smith MA. Will antioxidants fulfill their expectations for the treatment of Alzheimer disease? Mech Ageing Dev. 2000;116:169–179. doi: 10.1016/s0047-6374(00)00124-x. [DOI] [PubMed] [Google Scholar]
- 246.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 247.Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, Tschanz JT, et al. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol. 2004;61:82–88. doi: 10.1001/archneur.61.1.82. [DOI] [PubMed] [Google Scholar]
- 248.Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, et al. Simvastatin strongly reduces levels of Alzheimer's disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Li G, Larson EB, Sonnen JA, Shofer JB, Petrie EC, Schantz A, et al. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology. 2007;69:878–885. doi: 10.1212/01.wnl.0000277657.95487.1c. [DOI] [PubMed] [Google Scholar]
- 250.Li G, Higdon R, Kukull WA, Peskind E, Van Valen Moore K, Tsuang D, et al. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology. 2004;63:1624–1628. doi: 10.1212/01.wnl.0000142963.90204.58. [DOI] [PubMed] [Google Scholar]
- 251.Li G, Kukull WA, Peskind E, McCormick W, Bowen JD, Teri L, et al. Differential effect of statins on risk of AD by age, sex and APOE genotype: findings from a community-based prospective cohort study. Alzheimer Dis Assoc Disord. 2006;20:S103–S104. [Google Scholar]
- 252.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 253.Rockwood K, Kirkland S, Hogan DB, MacKnight C, Merry H, Verreault R, et al. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol. 2002;59:223–227. doi: 10.1001/archneur.59.2.223. [DOI] [PubMed] [Google Scholar]
- 254.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 255.Yaffe K, Barrett-Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol. 2002;59:378–384. doi: 10.1001/archneur.59.3.378. [DOI] [PubMed] [Google Scholar]
- 256.MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. doi: 10.1016/S0140-6736(02)09327-3. [DOI] [PubMed] [Google Scholar]
- 257.Rea TD, Breitner JC, Psaty BM, Fitzpatrick AL, Lopez OL, Newman AB, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62:1047–1051. doi: 10.1001/archneur.62.7.1047. [DOI] [PubMed] [Google Scholar]
- 258.Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomized controlled trial. Lancet. 2002;360:1623–1630. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- 259.Zandi PP, Sparks DL, Khachaturian AS, Tschanz J, Norton M, Steinberg M, et al. Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry. 2005;62:217–224. doi: 10.1001/archpsyc.62.2.217. [DOI] [PubMed] [Google Scholar]
- 260.Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, et al. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 262.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Breitner JC, Gau BA, Welsh KA, Plassman BL, McDonald WM, Helms MJ, et al. Inverse association of anti-inflammatory treatments and Alzheimer's disease: initial results of a co-twin control study. Neurology. 1994;44:227–232. doi: 10.1212/wnl.44.2.227. [DOI] [PubMed] [Google Scholar]
- 264.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 265.Andersen K, Launer LJ, Ott A, Hoes AW, Breteler MM, Hofman A. Do nonsteroidal anti-inflammatory drugs decrease the risk for Alzheimer's disease? The Rotterdam Study. Neurology. 1995;45:1441–1445. doi: 10.1212/wnl.45.8.1441. [DOI] [PubMed] [Google Scholar]
- 266.Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 267.Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 268.Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005;30:1204–1215. doi: 10.1038/sj.npp.1300690. [DOI] [PubMed] [Google Scholar]
- 269.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 270.Cardiovascular and cerebrovascular events in the randomized, controlled Alzheimer's Disease Anti-Inflammatory Prevention Trial (ADAPT) PLoS Clin Trials. 2006;1:e33. doi: 10.1371/journal.pctr.0010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Goutte C, Tsunozaki M, Hale VA, Priess JR. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci U S A. 2002;99:775–779. doi: 10.1073/pnas.022523499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 273.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 274.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 275.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 277.Becher B, Prat A, Antel JP. Brain-immune connection: immuno-regulatory properties of CNS-resident cells. Glia. 2000;29:293–304. [PubMed] [Google Scholar]
- 278.Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 279.Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–422. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Casadesus G, Milliken EL, Webber KM, Bowen RL, Lei Z, Rao CV, et al. Increases in luteinizing hormone are associated with declines in cognitive performance. Mol Cell Endocrinol. 2007;269:107–111. doi: 10.1016/j.mce.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 281.Webber KM, Casadesus G, Marlatt MW, Perry G, Hamlin CR, Atwood CS, et al. Estrogen bows to a new master: the role of gonadotropins in Alzheimer pathogenesis. Ann N Y Acad Sci. 2005;1052:201–209. doi: 10.1196/annals.1347.020. [DOI] [PubMed] [Google Scholar]
- 282.Kornhuber J, Bormann J, Retz W, Hubers M, Riederer P. Memantine displaces [3H]MK-801 at therapeutic concentrations in postmortem human frontal cortex. Eur J Pharmacol. 1989;166:589–590. doi: 10.1016/0014-2999(89)90384-1. [DOI] [PubMed] [Google Scholar]
- 283.Misztal M, Frankiewicz T, Parsons CG, Danysz W. Learning deficits induced by chronic intraventricular infusion of quinolinic acid--protection by MK-801 and memantine. Eur J Pharmacol. 1996;296:1–8. doi: 10.1016/0014-2999(95)00682-6. [DOI] [PubMed] [Google Scholar]
- 284.Wenk GL, Danysz W, Roice DD. The effects of mitochondrial failure upon cholinergic toxicity in the nucleus basalis. Neuroreport. 1996;7:1453–1456. doi: 10.1097/00001756-199606170-00001. [DOI] [PubMed] [Google Scholar]
- 285.Zajaczkowski W, Quack G, Danysz W. Infusion of (+) -MK-801 and memantine -- contrasting effects on radial maze learning in rats with entorhinal cortex lesion. Eur J Pharmacol. 1996;296:239–246. doi: 10.1016/0014-2999(95)00716-4. [DOI] [PubMed] [Google Scholar]
- 286.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 287.van Dyck CH, Tariot PN, Meyers B, Malca Resnick E. A 24-week randomized, controlled trial of memantine in patients with moderate-to-severe Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21:136–143. doi: 10.1097/WAD.0b013e318065c495. [DOI] [PubMed] [Google Scholar]
- 288.McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev. 2006 doi: 10.1002/14651858.CD003154.pub5. CD003154. [DOI] [PubMed] [Google Scholar]
- 289.Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–324. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 290.Scarmeas N, Stern Y, Tang MX, Mayeux R, Luchsinger JA. Mediterranean diet and risk for Alzheimer's disease. Ann Neurol. 2006 doi: 10.1002/ana.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 292.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 293.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 294.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 295.Kim HD, Tahara K, Maxwell JA, Lalonde R, Fukuiwa T, Fujihashi K, et al. Nasal inoculation of an adenovirus vector encoding 11 tandem repeats of Abeta1-6 upregulates IL-10 expression and reduces amyloid load in a Mo/Hu APPswe PS1dE9 mouse model of Alzheimer's disease. J Gene Med. 2007;9:88–98. doi: 10.1002/jgm.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Nikolic WV, Bai Y, Obregon D, Hou H, Mori T, Zeng J, et al. Transcutaneous beta-amyloid immunization reduces cerebral beta-amyloid deposits without T cell infiltration and microhemorrhage. Proc Natl Acad Sci U S A. 2007;104:2507–2512. doi: 10.1073/pnas.0609377104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Asuni AA, Boutajangout A, Scholtzova H, Knudsen E, Li YS, Quartermain D, et al. Vaccination of Alzheimer's model mice with Abeta derivative in alum adjuvant reduces Abeta burden without microhemorrhages. Eur J Neurosci. 2006;24:2530–2542. doi: 10.1111/j.1460-9568.2006.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Solomon B. Intravenous immunoglobulin and Alzheimer's disease immunotherapy. Curr Opin Mol Ther. 2007;9:79–85. [PubMed] [Google Scholar]
- 299.Chang WP, Downs D, Huang XP, Da H, Fung KM, Tang J. Amyloid-beta reduction by memapsin 2 (beta-secretase) immunization. FASEB J. 2007;21:3184–3196. doi: 10.1096/fj.06-7993com. [DOI] [PubMed] [Google Scholar]
- 300.Christensen DD. Changing the course of Alzheimer's disease: anti-amyloid diseasemodifying treatments on the horizon. Prim Care Companion J Clin Psychiatry. 2007;9:32–41. doi: 10.4088/pcc.v09n0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Fischer O. Miliäre nekrosen mit drusigen wucherungen der neurofibrillen, eine regelmässige verä nderung der hirnrinde bei seniler demenz. Monatsschr Psychiatr Neurol. 1907;22:361–372. [Google Scholar]
- 302.Divry P. Etude histochimique des plaques séniles. J Belg Neurol Psychiatr. 1927;9:649–657. [Google Scholar]