

Summary
Mutations in superoxide dismutase (SOD1) cause amyotrophic lateral sclerosis (ALS), a neurodegenerative disease characterized by loss of motor neurons. With conformation specific antibodies, we now demonstrate that misfolded mutant SOD1 binds directly to the voltage-dependent anion channel (VDAC1), an integral membrane protein imbedded in the outer mitochondrial membrane. This interaction is found on isolated spinal cord mitochondria and can be reconstituted with purified components in vitro. ADP passage through the outer membrane is diminished in spinal mitochondria from mutant SOD1-expressing ALS rats. Direct binding of mutant SOD1 to VDAC1 inhibits conductance of individual channels when reconstituted in a lipid bilayer. Reduction of VDAC1 activity with targeted gene disruption is shown to diminish survival by accelerating onset of fatal paralysis in mice expressing the ALS-causing mutation SOD1G37R. Taken together, our results establish a direct link between misfolded mutant SOD1 and mitochondrial dysfunction in this form of inherited ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive adult-onset neurodegenerative disorder characterized by the selective loss of upper and lower motor neurons in the brain and spinal cord (Cleveland and Rothstein, 2001). The typical age of onset is between 50 to 60 years, followed by paralysis and ultimately death within 2-5 years after onset (Mulder et al., 1986). Most instances of ALS are sporadic lacking any apparent genetic linkage, but 10% are inherited in a dominant manner. Twenty percent of these familial cases have been attributed to mutations in the gene encoding cytoplasmic Cu/Zn superoxide dismutase (SOD1) (Rosen et al., 1993). Although multiple hypotheses have been proposed to explain mutant SOD1-mediated toxicity (Ilieva et al., 2009), the exact mechanism(s) responsible for motor neuron degeneration remains unsettled.
Mitochondrial dysfunction has been proposed to contribute to disease pathogenesis. Histopathological observations of disturbed mitochondrial structure have been reported in muscle of both sporadic and familial ALS patients (Hirano et al., 1984a; Hirano et al., 1984b; Sasaki and Iwata, 1996, 2007) and in mutant SOD1 mouse models expressing dismutase active (Dal Canto and Gurney, 1994; Higgins et al., 2003; Kong and Xu, 1998; Wong et al., 1995), but not inactive mutants (Bruijn et al., 1997). Moreover, functionality of mitochondria has been reported to be affected in spinal cord and skeletal muscles of human sporadic ALS or familial ALS patients (Dupuis et al., 2003; Echaniz-Laguna et al., 2002; Vielhaber et al., 1999; Wiedemann et al., 2002), as well as in some ALS mouse models (Damiano et al., 2006; Mattiazzi et al., 2002; Nguyen et al., 2009).
A proportion of the predominantly cytosolic SOD1 has been reported to localize to mitochondria in certain contexts. In both rodent models and patient samples, mutant SOD1 is present in fractions enriched for mitochondria derived from affected, but not unaffected, tissues (Bergemalm et al., 2006; Deng et al., 2006; Liu et al., 2004; Mattiazzi et al., 2002; Vande Velde et al., 2008; Vijayvergiya et al., 2005) and a clear temporal correlation between mitochondrial association and disease progression was shown for multiple mutant SOD1s (Liu et al., 2004). Purification of mitochondria, including floatation steps that eliminate protein only aggregates, coupled with protease accessibility has demonstrated mutant SOD1 deposition on the cytoplasmic-facing surface of spinal cord mitochondria (Liu et al., 2004; Vande Velde et al., 2008). Sensitivity to proteolysis and immunoprecipitation with an antibody specific for misfolded SOD1 further indicated that misfolded forms of dismutase active and inactive SOD1 are deposited onto the cytoplasmic face of the outer membrane of spinal cord mitochondria (Vande Velde et al., 2008). This is accompanied by altered accumulated levels of a few mitochondrial proteins, reduced import of multiple mitochondrial proteins, and reduced complex I activity (Miller, Vande Velde and Cleveland, unpublished), albeit none of these appear to be direct effects of mutant SOD1.
Oxidative phosphorylation requires the transport of metabolites, including ADP, ATP and inorganic phosphate across both mitochondrial membranes. Located in the outer mitochondrial membrane, the voltage-dependent anion channel (VDAC), known as mitochondrial porin, assumes a crucial position in the cell, controlling metabolic crosstalk between the mitochondrion and the rest of the cell, thus regulating the metabolic and energetic functions of mitochondria (Shoshan-Barmatz et al., 2006; Shoshan-Barmatz et al., 2008). Of the three VDAC isoforms (VDAC1-3), VDAC1 is the most abundant in most cells. VDAC1 is a primary contributor to ATP/ADP flux across the outer mitochondrial membrane (Colombini, 2004; Lemasters and Holmuhamedov, 2006). Initially named somewhat misleadingly as a channel for anions, it is also responsible for import/export of Ca2+ (Gincel et al., 2001) and other cations (Benz, 1994; Colombini, 2004), adenine nucleotides (Rostovtseva and Colombini, 1997; Rostovtseva and Bezrukov, 1998) and other metabolites (Hodge and Colombini, 1997). Indeed, it has been demonstrated that silencing VDAC1 expression in a cultured cell line using shRNA resulted in reduced ATP production and a decrease in cell growth (Abu-Hamad et al., 2006).
VDAC1 is also a key player in mitochondria-mediated apoptosis. VDAC1 has been implicated in apoptotic-relevant events, due to serving as the target for members of the pro- and anti-apoptotic Bcl2-family of proteins (Arbel and Shoshan-Barmatz, 2009; Shimizu et al., 1999) and due to its function in the release of apoptotic proteins from the intermitochondrial membrane space (Abu-Hamad et al., 2009; Shoshan-Barmatz et al., 2006; Shoshan-Barmatz et al., 2008; Tajeddine et al., 2008). VDAC1 has also been implicated in Parkinson's disease as a direct target for Parkin-mediated polyubiquitylation and mitophagy (Geisler et al., 2010).
Starting from recognition that a proportion of misfolded, mutant SOD1 is bound to the cytoplasmic face of the outer membrane of mitochondria in affected tissues (Liu et al., 2004; Rakhit et al., 2007; Vande Velde et al., 2008), we now identify damage to spinal cord mitochondria to arise through direct binding of misfolded SOD1 onto the cytoplasmic-facing domain of VDAC1, thereby inhibiting its conductance.
Results
Mutant SOD1 and VDAC1 interact in vivo in spinal cord of transgenic SOD1 rats
To investigate potential interactions between mutant SOD1 and VDAC1, mitochondria from rats expressing wild type human SOD1 (hSOD1wt) or either of two different ALS-linked SOD1 mutants, a dismutase active hSOD1G93A and a dismutase inactive hSOD1H46R, were highly purified by repeated centrifugation steps (summarized in Fig. 1A) including a final density gradient flotation step to eliminate any contaminating protein only aggregates (proteins sediment downward in these conditions because of their higher density), as previously described (Vande Velde et al., 2008). Immunoblotting of immunoprecipitates generated after addition of an SOD1 antibody to solubilized mitochondrial lysates revealed that a proportion of VDAC1 was co-precipitated with dismutase active and inactive mutant SOD1, but not wild type SOD1 (Fig. 1B). Parallel immunoprecipitations with a VDAC1 antibody confirmed co-precipitation of both hSOD1G93A and hSOD1H46R with VDAC1 (Fig. 1D). Binding to VDAC1 was a property only of spinal cord mitochondria, as no association of mutant SOD1 was seen with purified brain mitochondria from the same animals using immunoprecipitation with SOD1 (Fig. 1C) or VDAC1 (Fig. 1E) antibodies. This latter finding is consistent with prior efforts that had demonstrated that mutant SOD1 associates with the cytoplasmic face of the outer membrane of mitochondria in spinal cord, but not other tissue types (Liu et al., 2004; Vande Velde et al., 2008). Moreover, mutant SOD1 binding to VDAC1 is inversely correlated with the level of hexokinase-I, a known partner that binds to VDAC1 exposed on the cytoplasmic mitochondrial surface (Abu-Hamad et al., 2008; Azoulay-Zohar et al., 2004; Zaid et al., 2005), with hexokinase accumulating to much higher level in brain than spinal cord mitochondria (Fig. 1F).
Fig. 1. A complex containing mutant SOD1 and VDAC1 from spinal cord mitochondria.
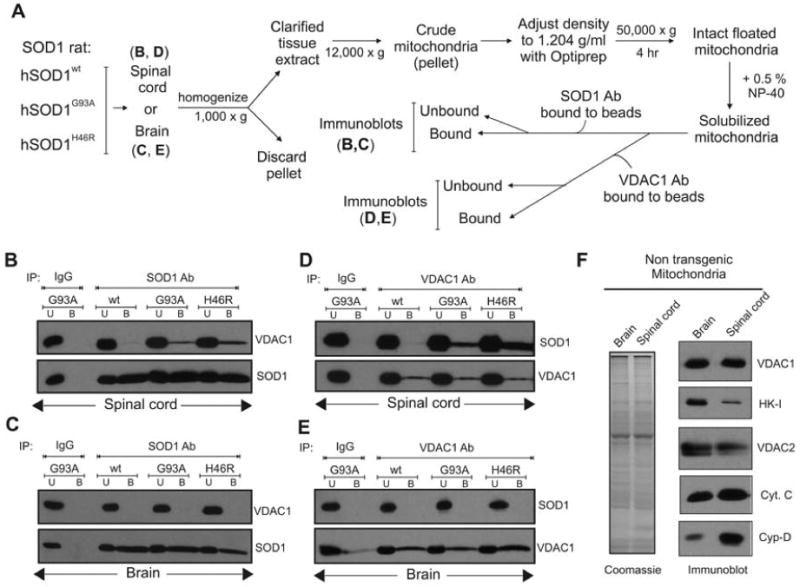
(A) Schematic outlining the different purification steps used. Floated isolated mitochondria from (B, D) hSOD1wt, hSOD1G93A and hSOD1H46R rat spinal cords or (C, E) brain were immunoprecipitated with (B, C) an SOD1 antibody or (D, E) VDAC1 antibody. (B) Immunoblot of the SOD1 immunoprecipitates using VDAC1 antibody indicates that mutant SOD1 proteins hSODG93A and hSOD1H46R coprecipitate VDAC1 (top). SOD1 immunoprecipitation was confirmed by reprobing the membrane with anti-SOD1 antibody (bottom). (C) Immunoblots of SOD1 immunoprecipitates as in (B) except with brain mitochondria. (D) Immunoprecipitation using VDAC1 antibody immunoblotted with SOD1 antibody (top). The membrane was then reprobed for VDAC1 (bottom). (E) Immunoblots of VDAC1 immunoprecipiates as in (D), except with brain mitochondria. Abbreviations: U, unbound fraction (20 %); B, bound fraction. (F) Reduced hexokinase-I levels in spinal cord mitochondria. Polyacrylamide gel analysis of extracts of floated brain and spinal cord mitochondria. (Left) Coomassie stain; (right) immunoblot for VDAC1, hexokinase I (HK-I), VDAC2, cytochrome c (Cyt. C), and cyclophilin D (Cyp-D).
Misfolded mutant SOD1 specifically interacts with VDAC1 in vivo in spinal cord of transgenic SOD1 rats
To test the nature of the interaction between mutant SOD1 and VDAC1, immunoprecipitation was performed with a SOD1 antibody that recognizes a “disease-specific epitope” (DSE) that is unavailable on correctly folded SOD1 (Cashman and Caughey, 2004; Paramithiotis et al., 2003; Urushitani et al., 2007), but is present on misfolded mutant SOD1s in inherited ALS (Rakhit et al., 2007). Using one such antibody (DSE2), age-dependent deposition of mutant SOD1 onto the cytoplasmic face of spinal cord mitochondria has been shown to reflect association of misfolded SOD1 (Vande Velde et al., 2008). We exploited this antibody to examine if the SOD1 associated with VDAC1 is bound through misfolded SOD1. Liver, brain and spinal cord cytosolic and mitochondrial fractions purified from symptomatic rats expressing mutant hSOD1G93A were immunoprecipitated (see schematic in Fig. 2A) with the DSE2 antibody, which recognizes an epitope in the electrostatic loop of hSOD1 (between residues 125-142) that is buried in normally folded SOD1. Misfolded mutant SOD1G93A was not detectable in the soluble fraction of any tissue, but was immunoprecipitated from the spinal cord, but not liver or brain, mitochondrial fractions (Fig. 2B).
Fig. 2. The misfolded mutant SOD1 specifically co-precipitates with VDAC1 in spinal cord mitochondria.
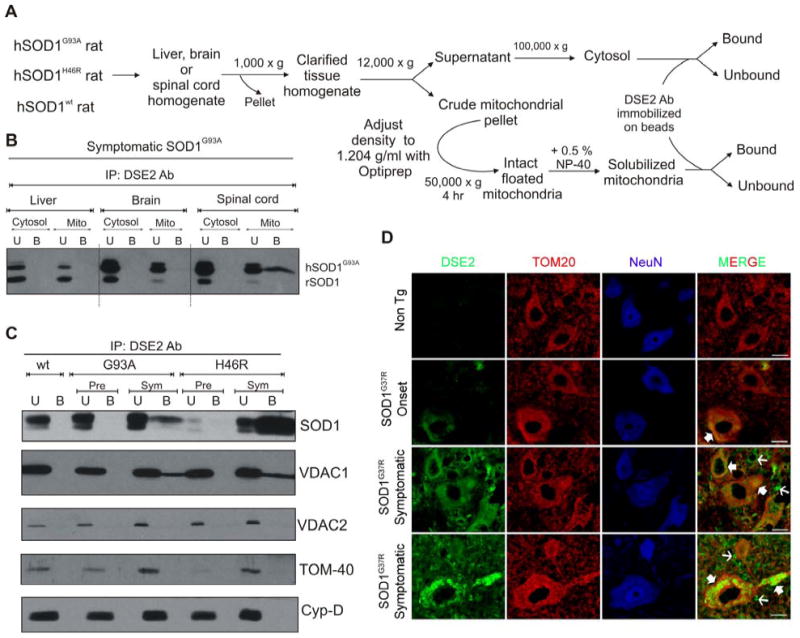
(A) Schematic showing the isolation of cytosolic and mitochondrial fractions. (B) Liver, brain and spinal cord cytosolic and mitochondrial fractions were purified from symptomatic rats expressing hSOD1G93A and the fractions were subjected to immunoprecipitation using DSE2 (3H1), a monoclonal antibody only recognizing misfolded SOD1 (Vande Velde et al., 2008). The immunoprecipitates were immunoblotted using an SOD1 antibody. (C) Isolated floated mitochondria from hSOD1wt, hSOD1G93A and hSOD1H46R rat spinal cords (from pre-symptomatic and symptomatic animals) were immunoprecipitated with DSE2 (3H1), and the immunoprecipitates were immunoblotted using VDAC1, VDAC2, TOM-40 and cyclophilin-D antibodies. SOD1 immunoprecipitation was confirmed by reprobing the membrane with an SOD1 antibody (top). (D) Immunohistochemical detection of misfolded SOD1 using DSE2 antibody shows that misfolded SOD1 (green) colocalizes with TOM20 (red), a mitochondrial outer membrane protein in a subset of spinal cord neurons assessed using NeuN (blue), a neuronal marker as highlighted by filled arrows. DSE2 positive staining can be detected in some neurons at onset and significantly increases with the appearance of disease symptoms.
Of note DSE2 staining is not restricted to neuronal mitochondria but is also detected in non-neuronal cells and the extracellular space as shown with thin arrows. No DSE2 staining was detected in neurons of 1 year old non transgenic control mice (Non Tg). Scale bar: 10 μm. Abbreviation: U, unbound fraction (20 %); B, bound fraction; Pre, pre-symptomatic; Sym, symptomatic.
Solubilized spinal cord mitochondria purified from presymptomatic and symptomatic rats expressing either of two different SOD1 mutants, dismutase active hSOD1G93A and dismutase inactive hSOD1H46R, as well as hSOD1wt were immunoprecipitated with the DSE2 antibody and co-immunoprecipitated components identified by immunoblotting. An age-dependent increase in misfolded SOD1 was seen for both mutants, with a significantly higher proportion of the dismutase inactive SOD1H46R in a misfolded conformation. In samples from symptomatic animals, VDAC1 coprecipitated together with the misfolded mutant SOD1, as revealed by immunoblotting of immunoprecipitates (Fig. 2C). This association was selective for VDAC1, as misfolded mutant SOD1 did not co-immunoprecipitate with any of three other mitochondrial proteins examined (Fig. 2C), including two additional outer mitochondrial membrane proteins with domains facing the cytoplasm: TOM40, the 40 kDa component of Transport across the Outer Membrane (TOM) complex mediating all protein import from the cytoplasm to the mitochondria, and VDAC2, a second voltage-dependent anion channel isoform that has been estimated to represent 7 % (kidney) to 25 % (brain) of accumulated VDAC (Yamamoto et al., 2006). It also did not co-precipitate cyclophilin-D, an important component of the permeability transition pore.
Furthermore, in order to determine which cells accumulate the misfolded form of SOD1, we performed immunostaining using the DSE2 antibody. Spinal cords from loxSOD1G37R mice at different stages of the disease were subjected to immunostaining with DSE2 antibody (Fig. 2D). The accumulation of misfolded SOD1 dramatically increased with disease progression. Although little accumulation of misfolded SOD1 is found by disease onset, it was preferentially found within motor neurons. During disease progression, a dramatic increase of misfolded SOD1 was apparently accumulated in other cells as well and probably also extracellularly. Throughout disease a proportion of the misfolded SOD1 was co-localized with mitochondria of motor neurons and other cells, starting at onset and increasing with disease progression (Fig. 2D).
Binding of mutant SOD1 directly inhibits VDAC1 channel conductance
To test if binding of mutant SOD1 affects VDAC1 function, VDAC1 was purified from spinal cords of non-transgenic rats (Fig. 3A) and reconstituted into a planar lipid bilayer (Fig. 3A) using conditions previously demonstrated to yield polarized VDAC1 membrane insertion such that the VDAC1 surface exposed on the cis side is the surface exposed to the cytosol when inserted into the mitochondrial outer membrane (Gincel et al., 2001; Israelson et al., 2005). Activity of individual channels was measured as a function of time by the ions passing across the bilayer in response to an applied voltage gradient. This revealed that in the absence of SOD1, VDAC1 was stably in a fully open state (4 nS at 1 M KCl (Shoshan-Barmatz et al., 2006)) and remained so for extended periods.
Fig. 3. Mutant, but not wild type, SOD1 interacts with bilayer-reconstituted VDAC1 to reduce its channel conductance.
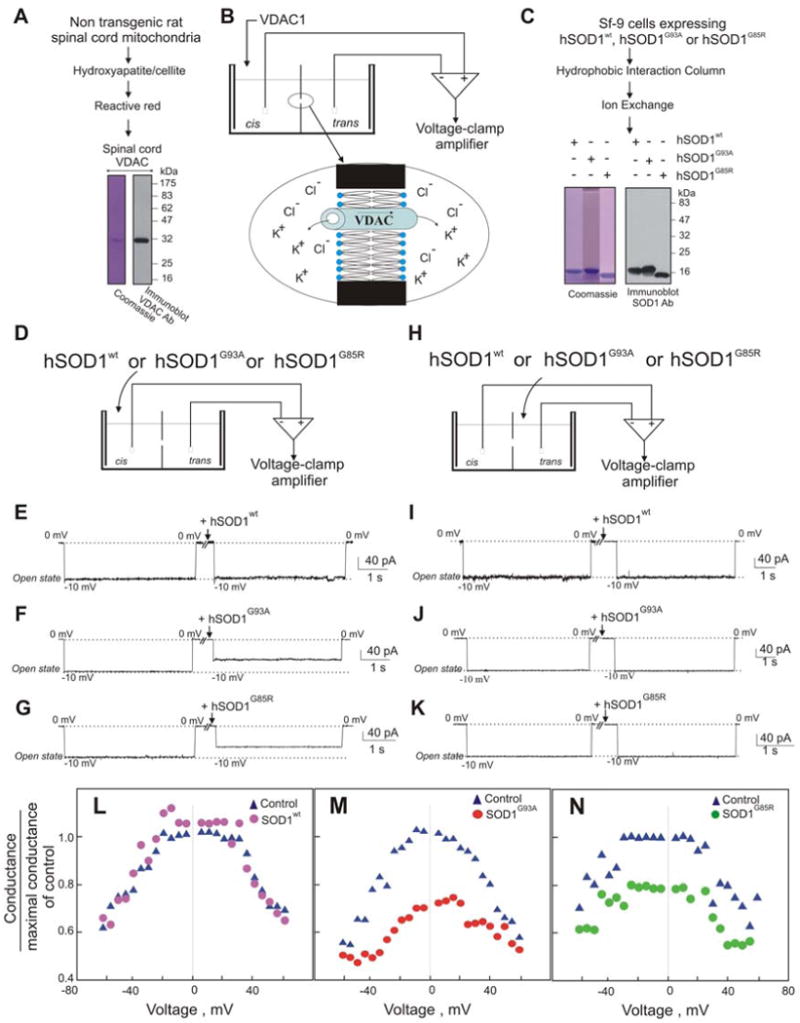
(A) Coomassie Blue staining and immunoblot of purified VDAC1 purified from rat spinal cord. (B) Schematic presentation showing the planar lipid bilayer reconstitution and channel conductance assay system. Purified spinal cord VDAC1 was reconstituted into a planar lipid bilayer, and channel currents through VDAC1 were recorded. (C) Coomassie Blue staining and immunoblot of purified recombinant hSOD1wt, hSOD1G93A and hSOD1G85R expressed in insect cells using baculovirus. (D-K) Currents through VDAC1 in response to a voltage step from 0 to −10 mV were recorded before and 2 min after the addition (to 2 μg/ml final) of purified recombinant (E) hSOD1wt, (F) hSOD1G93A or (G) hSOD1G85R to the cis side of the bilayer. (G-K) Currents through VDAC1 as in (D-K), except after SOD1 addition to the trans side of the bilayer. The dotted lines indicate current levels in the maximal and zero conductance states. These examples are representative of the results from 3-4 independent reconstitution experiments. (L-N) Mutant SOD1 effect on VDAC1 channel activity at different voltages. Average steady-state conductance of VDAC1 before and after addition of (L) hSOD1wt, (M) hSOD1G93A, or (N) hSOD1G85R, determined as a function of voltage with a multi-channel recording.
Mutant SOD1 proteins hSOD1G93A, hSOD1G85R, as well as hSOD1wt, were expressed using baculovirus and purified (Fig. 3C) (Hayward et al., 2002). Wild type SOD1, even at the highest added concentration (8 μg/ml), had no effect on VDAC1 conductance when added on either cis or trans sides of the membrane (Fig. 3E,I). However, addition of purified recombinant hSOD1G93A or hSOD1G85R (Fig. 3C) substantially reduced VDAC1 channel conductance (Fig. 3F,G). Both mutant SOD1s modified VDAC1 conductance only when added to the cis side (Fig. 3F,G), but not the trans side (Fig. 3J,K) of the bilayer, indicating that mutant SOD1 interacts with what would correspond to the cytosolic face of VDAC1 inserted into the outer mitochondrial membrane. Use of multichannel recordings revealed that not only did mutant SOD1 significantly lower the maximum voltage gated conductance of individual channels, it also provoked a stable, reduced level of VDAC1 conductance at all applied voltages (Fig. 3L-N). In order to determine if this interaction is specific for mutant SOD1, the effect of another aggregating protein (α-synuclein) was tested on bilayers containing reconstituted VDAC1. Even when added to levels 25 times greater than an amount of mutant SOD1 that markedly affected VDAC1 conductance (Fig. 3F,G), neither wild type nor mutant α-synuclein affected VDAC1 channel activity at any voltage (Fig. 1S).
ADP transport across the outer mitochondrial membrane is reduced in spinal cords of mutant SOD1 rats
Since both dismutase active and inactive SOD1 mutant proteins reduced VDAC1 channel conductance for K+ and Cl- (Fig. 3), we next tested whether mitochondrial conductance across the outer mitochondrial membrane was affected in animals chronically expressing mutant SOD1. To do this, we examined the uptake into mitochondria of adenine nucleotides (Fig. 4A) which are known to be transported by VDAC1 (Lemasters and Holmuhamedov, 2006; Rostovtseva and Colombini, 1997). Freshly isolated spinal cord and liver mitochondria from SOD1G93A rats were incubated (for 1 min.) with radiolabeled [3H]ADP and the amount of imported ADP was measured by scintillation counting after rapid filtration to remove the unincorporated ADP. Co-incubation with 1 mM of the VDAC1 inhibitor DIDS (4, 4′-diisothiocyanostilbene-2, 2′-disulfonic acid) demonstrated that ∼2/3rd of the ADP uptake was through VDAC1 (Fig. 4B,C). Compared to mitochondria from non-transgenic animals, uptake of ADP by spinal cord mitochondria from SOD1 mutant expressing animals was selectively and progressively inhibited, yielding ∼40% inhibition of VDAC1-dependent uptake (∼25% overall inhibition of ADP uptake) by a symptomatic stage (Fig. 4B). Inhibition of ADP uptake was selective to spinal mitochondria as liver mitochondria from the same hSOD1G93A animals retained normal ADP import at all ages examined (Fig. 4C).
Fig. 4. ADP transport across the outer mitochondrial membrane is reduced in mitochondria from spinal cord of SOD1G93A ALS rats.
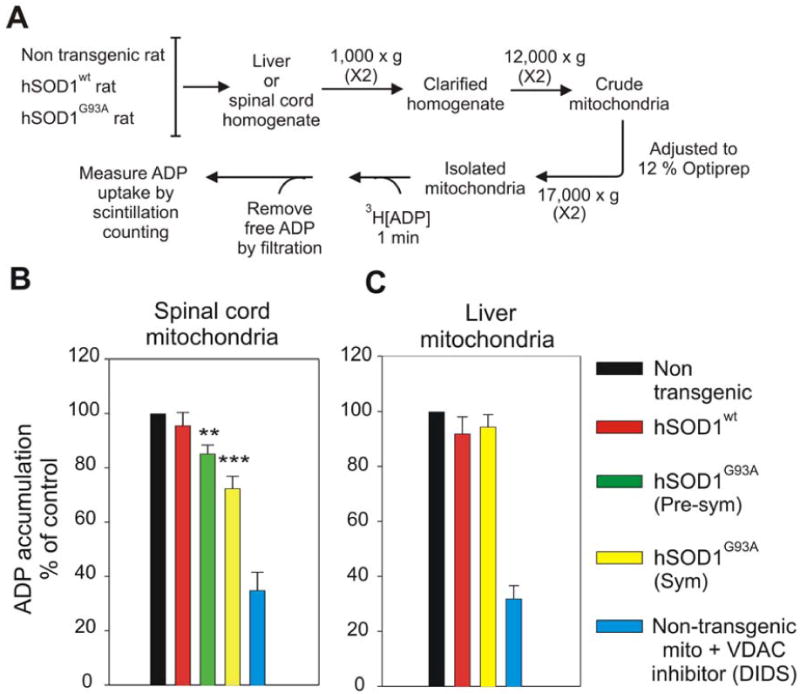
(A) Schematic presentation of method for measuring ADP accumulation into isolated mitochondria as measured using radiolabeled [3H]ADP. (B,C) Mitochondria were isolated from (B) spinal cord and (C) liver of non-transgenic, hSOD1wt, hSOD1G93A pre-symptomatic and hSOD1G93A symptomatic rats. Student's t test was used and p < 0.001 (marked by three asterisks) and p < 0.01 (marked by two asterisks) were considered statistically significant. Values represent the means ± SEM of three to four independent experiments.
Mutant SOD1 binding to mitochondria in vitro diminishes ADP but not Ca2+ uptake
To test if inhibition of ADP import seen in spinal cord mitochondria from mutant SOD1 animals could be generated solely from mutant SOD1 binding to the cytoplasmic face of those mitochondria, purified recombinant SOD1 proteins (hSOD1wt, hSOD1G93A and hSOD1G85R) (Fig. 3C) were added to mitochondria purified from spinal cords or livers of non transgenic rats (Fig. 5A). Although a proportion of each of the recombinant SOD1s associated with both spinal cord and liver mitochondria (Fig. 5D), accumulation of radiolabeled Ca2+ (presumably through the action of the mitochondrial calcium uniporter) into spinal cord or liver mitochondria was not affected by the addition of wild type or mutant SOD1 (Fig. 5C). On the other hand, VDAC1-mediated ADP accumulation into the same spinal cord or liver mitochondria was inhibited by both hSOD1G93A and hSOD1G85R mutants, but not hSOD1wt (Fig. 5B). This inhibition corresponded to a proportion of misfolded SOD1 associated with those mitochondria after incubation with either mutant, but not wild type SOD1, as demonstrated by immunoprecipitation of intact mitochondria with the DSE2 antibody to misfolded SOD1 (Fig. 5E). In contrast, wild type SOD1 associated with the same mitochondria was not recognized by this misfolded SOD1 antibody (Fig. 5E), consistent with its retention of normal folding and/or import into those mitochondria (Fig. 5E).
Fig. 5. Mutant SOD1 proteins affect ADP but not Ca2+ accumulation into mitochondria.
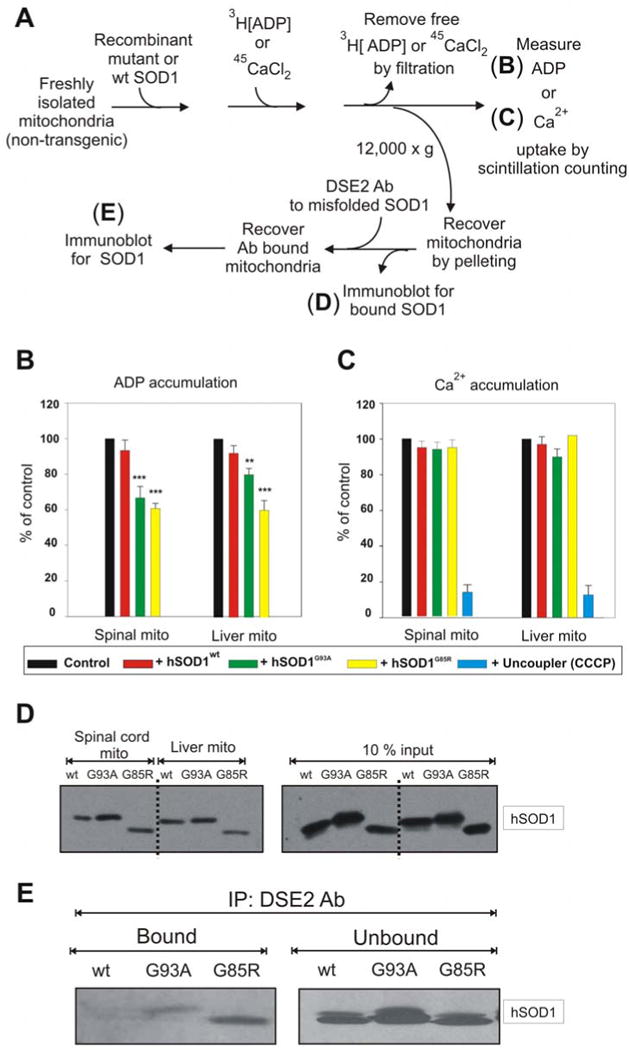
(A) ADP or Ca2+ accumulation into isolated mitochondria was measured using a filter trap assay with radiolabeled 45CaCl2 or [3H]ADP. Mitochondria were isolated from fresh spinal cords and livers of non-transgenic rats. (B) ADP and (C) Ca2+ accumulation were measured before and after the addition of 3 μM (50 μg/ml) hSOD1wt, hSOD1G93A or hSOD1G85R purified proteins. Student's t test was used and p < 0.001 (marked by three asterisks) and p < 0.01 (marked by two asterisks) were considered statistically significant. Values represent the means ± SEM of three independent experiments. (D) Purified hSOD1wt, hSOD1G93A or hSOD1G85R were incubated with liver or spinal cord mitochondrial fractions purified from a non-transgenic rat for 20 min at 37°C. The samples were then washed 3 times and the mitochondrial pellet was subjected to immunoblot using an SOD1 antibody. (E) Purified hSOD1wt, hSOD1G93A or hSOD1G85R was incubated for 20 min at 37°C with spinal cord mitochondria purified from non-transgenic rats. The samples were then washed 3 times and the mitochondrial pellet was subjected to immunoprecipitation using DSE2 (3H1) antibody, a monoclonal antibody only recognizing misfolded SOD1. The immunoprecipitates were immunoblotted using an SOD1 antibody.
Reduced VDAC1 activity diminishes survival of mutant SOD1G37R mice by accelerating disease onset
Since we have established that 1) mutant SOD1 interacts directly with VDAC1 thereby inhibiting VDAC1 conductance (Fig. 3), 2) spinal cord mitochondria from SOD1 mutant animals have progressive loss of ADP uptake, and 3) misfolded mutant SOD1 binds to normal mitochondria in vitro accompanied by selective loss of ADP conductance (Fig. 5), we examined how reduced level and activity of VDAC1 affect disease course in SOD1G37R mutant mice. To do this, we exploited mice heterozygous for disruption of the VDAC1 gene (producing what is effectively a null allele – (Weeber et al., 2002)). These mice accumulate about half the normal level of VDAC1 protein (Fig. 2S), while overall ADP conductance of spinal mitochondrial isolated from VDAC1+/- mice is reduced by ∼25% (Fig. 3S) relative to wild type mice. After mating with SOD1G37R mice, sex matched cohorts of mice and their littermates carrying the SOD1G37R transgene and either one or two active VDAC1 alleles were obtained and followed for disease onset, progression and survival. Measurement of ADP conductance of spinal mitochondria from SOD1G37R/VDAC1+/- mice revealed a reduction to a level comparable to that corresponding to complete deletion of VDAC1 (Fig. 3S).
A simple and objective measure of disease onset and early disease progression was applied by initiation of weight loss, reflecting denervation-induced muscle atrophy. While timing of progression from onset through either early (Fig. 6E) or late (Fig. 6F) disease phases was only modestly affected by reduction of VDAC1 levels, disease onset (Fig. 6A, D) and progression to an early disease point (Fig. 6B) were accelerated by 41 and 45 days, respectively, in SOD1G37R/VDAC1+/- mice (183 ± 22 and 230 ± 28 d) compared with their SOD1G37R littermates (224 ± 19 and 275 ± 25 d). Moreover, age at which end stage disease was reached was also reduced by an average of 59 days (Fig. 6C; SOD1G37R/VDAC1+/- mice (310 ± 42 d) compared with their SOD1G37R littermates (369 ± 32 d)). A similar reduction in age of onset and life span was also observed for SOD1G37R/VDAC1-/- mice (Fig. 4S), demonstrating that reduction in VDAC1 activity does affect SOD1 mutant-dependent pathogenesis, primarily by accelerating an early step in disease onset or spread.
Fig. 6. Reduction of VDAC1 levels accelerates disease onset and diminishes survival in the hSOD1G37R mouse model of ALS.
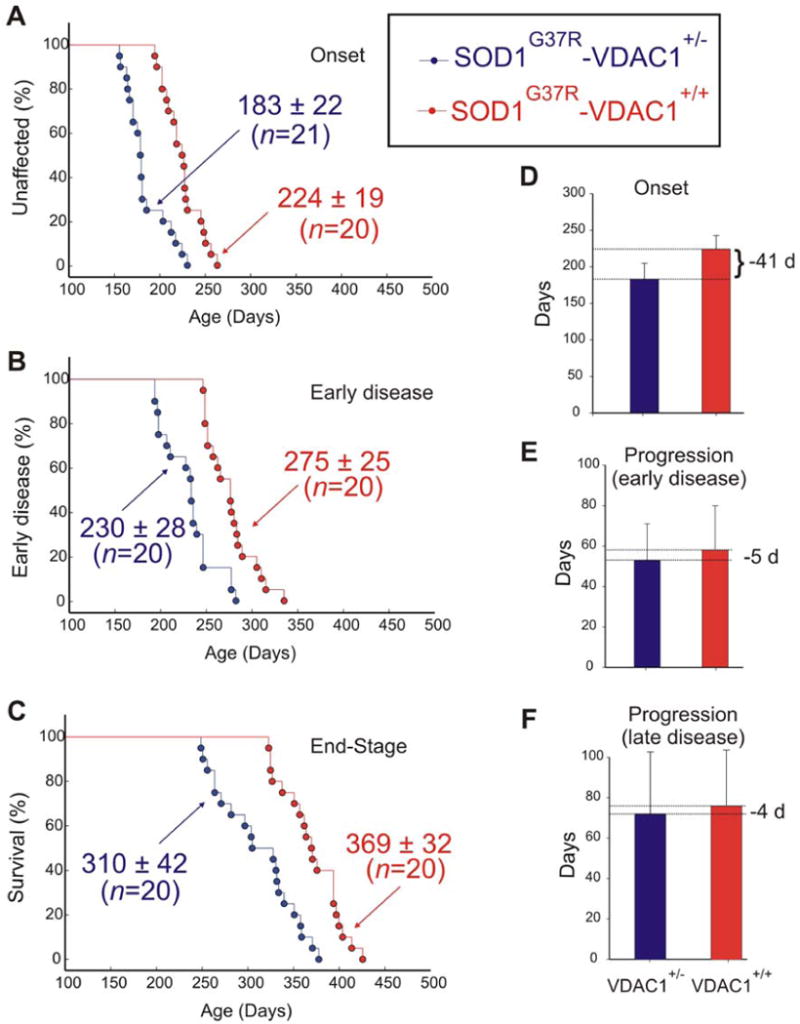
Ages of (A) disease onset (determined as the time when mice reached peak body weight), (B) early disease (determined as the time when mice lost 10% of maximal weight) and (C) disease end stage (determined as the time when the animal could not right itself within 20 seconds when placed on its side) of SOD1G37R-VDAC1+/- (blue) and SOD1G37R-VDAC1+/+ littermates (red). Mean ages ± s.d. is provided. (D, E, F) Mean onset (D), mean duration of early disease (from onset to 10% weight loss; E) and mean duration of late disease (from 10% weight loss to end-stage; F). Error bars denote s.d. See also Fig. S1.
Discussion
We have demonstrated here in floated spinal cord mitochondria from mutant SOD1 expressing animals that both misfolded dismutase active or inactive SOD1 mutants bind directly and selectively to the cytoplasmically exposed face of VDAC1. Both dismutase active and dismutase inactive, but not wild type, SOD1 binding to VDAC1 reduces channel conductance, as demonstrated for K+ and Cl- ions by electrophysiological recording and for ADP by inhibition of normal ADP accumulation into mitochondria. Channel conductance was not affected in liver mitochondria (where misfolded SOD1 does not accumulate). Mutant association and conductance inhibition is replicated in spinal cord mitochondria purified from mutant expressing animals beginning pre-symptomatically and increasing in severity during disease progression contemporaneous with increased accumulation of misfolded mutant SOD1. The clear implication from this is that only the misfolded portion of SOD1 is able to affect the channel, thereby partially blocking metabolite flux across the outer mitochondrial membrane. Reduced conductance by VDAC1 will decrease ATP synthesis, increase the ADP/ATP ratio in the cytosol, and reduce membrane potential (as outlined in Fig. 7). Chronic mitochondrial dysfunction can in turn drive generation of damaging reactive oxygen species that could drive further SOD1 misfolding through chemical damage to it, as has been previously documented selectively in spinal cords from mutant SOD1 animals (Liu et al., 2004; Vande Velde et al., 2008). Thus, our evidence demonstrates that reduced VDAC1 conductance, and correspondingly reduced respiration rate (Lemasters and Holmuhamedov, 2006), are direct components of intracellular damage from mutant SOD1.
Fig. 7. Effects of misfolded SOD1 binding to VDAC1.
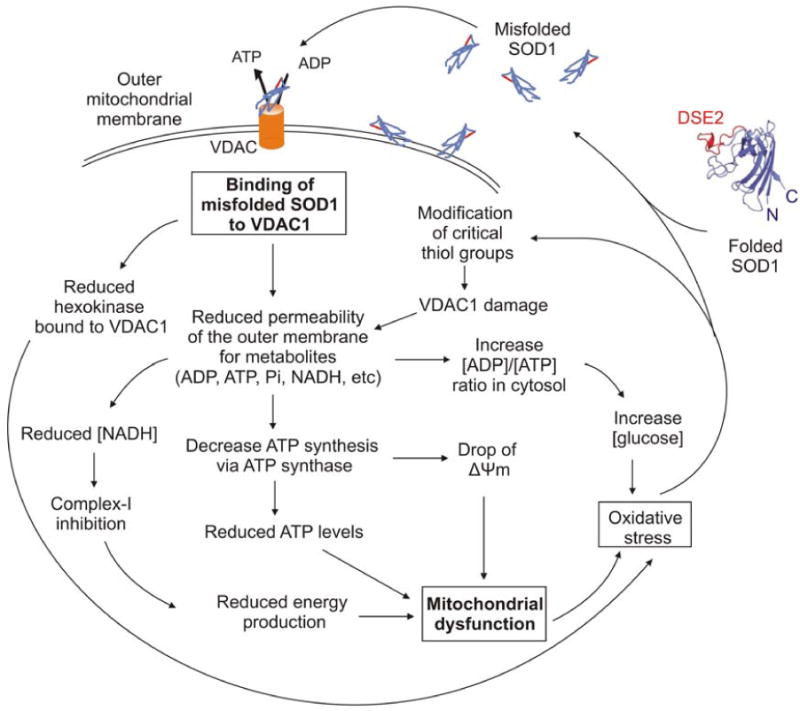
Schematic model showing the effects of misfolded SOD1 binding to VDAC1. Misfolded SOD1 is proposed to inhibit VDAC1 conductance and suppress both uptake and release of mitochondrial metabolites. This reduction in metabolites flux would result in reduced energy production and oxidative stress leading to mitochondrial dysfunction.
Moreover, not only does mutant SOD1 lower VDAC1-dependent ADP conductance by half as much as does complete VDAC1 deletion (Fig. 3S), further reduction in conductance (by VDAC1 gene inactivation) significantly accelerates disease onset (but not progression), reducing survival by more than two months for both VDAC1 heterozygous and homozygous mice. Intracellular targets for SOD1 damage beyond VDAC1 have been proposed (Ilieva et al., 2009), including aberrant glutamate handling from delayed synaptic glutamate recovery by astrocytes (Rothstein et al., 1995), mutant damage in the extracellular space following aberrant co-secretion with chromogranin (Urushitani et al., 2006), endoplasmic reticulum stress from inhibition of the ERAD pathway by mutant SOD1 binding to the integral membrane protein derlin (Nishitoh et al., 2008), and excessive production by microglia of extracellular superoxide following mutant SOD1 binding to the small G protein Rac1 and its subsequent stimulation of NAPDH oxidase (Harraz et al., 2008). Moreover, it was recently proposed that misfolded SOD1 damage to mitochondria can induce morphological changes and cytochrome c release in the presence of Bcl-2 (Pedrini et al., 2010). To those hypotheses, we proposed that the partial blockage of the VDAC1 channel by direct association with misfolded SOD1 would make motor neurons more vulnerable to any of these additional stresses derived either from mutant SOD1 acting within motor neurons, astrocytes, microglia and possibly additional neighboring non-neuronal cells. Indeed, in the presence of reduced VDAC1 conductance such pathways can play roles in pathogenesis, as we have shown that mutant SOD1-mediated disease still ensues in VDAC1 null mice.
Surprisingly, in the absence of VDAC1, we have found a 60% residual ADP conductance which seems most likely to be contributed by compensatory VDACs or VDAC-like activity(ies). Although no other VDAC isoform is known to be overexpressed in VDAC1 null mice, VDAC2 has been shown to exist in two forms that differ in conductance and selectivity (Xu et al., 1999). It is plausible that in the absence of VDAC1, VDAC2 exists predominantly in a high conductance state, as a compensatory mechanism. This mechanism should now be tested by purifying VDAC2 from VDAC1 knockout mouse, and testing its channel properties in lipid bilayers.
The compromise in mutant SOD1-mediated VDAC1 conductance that we have found offers a mechanistic explanation for alteration in mitochondrial electron transfer chain complexes and the capacity to consume oxygen and synthesize ATP previously reported in one mutant SOD1 expressing mouse line (Jung et al., 2002; Kirkinezos et al., 2005; Mattiazzi et al., 2002). The recent report that association of hSOD1G93A and hSOD1G85R with motor neuron mitochondria reduces capacity of the electron transfer chain to limit Ca2+-induced Ψm depolarization (Nguyen et al., 2009) is also fully compatible with altered adenine nucleotide transport across the outer mitochondrial membrane as the initiating deficit. So too is the report of reduced ability of mitochondria from SOD1G93A and SOD1G85R mice to survive repetitive Ca2+ addition (Damiano et al., 2006).
VDAC1 has been proposed to be the mediator for ROS release from the intermitochondrial spaces to the cytosol (Han et al., 2003; Madesh and Hajnoczky, 2001). Moreover, hexokinase (known to interact with VDAC1) has been shown in cell culture to decrease ROS release when over-expressed, thereby reducing intracellular levels of ROS (Ahmad et al., 2002; da-Silva et al., 2004). The relatively low level of hexokinase in spinal cord as compared to that in brain (Fig. 1F) may therefore be a component of selective vulnerability. This is also consistent with the selective association of misfolded mutant SOD1 with VDAC1 on the cytoplasmic face of mitochondria from spinal cord, but not liver or brain. Although both tissues accumulate high levels of mutant SOD1 (Liu et al., 2004; Vande Velde et al., 2008), prior findings show that misfolded mutant SOD1 is bound to the cytoplasmic face of spinal cord mitochondria, while apparently imported into the intermembrane space of mitochondria from cortex of the same animals and not associated with liver mitochondria at all (Vande Velde et al., 2008). Another factor likely underlying the differences in mutant SOD1 association with mitochondria, and therefore potentially factors underlying selective vulnerability, is that mitochondria from different tissues (and which retain different functional properties) have different protein compositions (Bailey et al., 2007; Mootha et al., 2003), including hexokinase levels. This is accompanied by intrinsic differences in O2−· production, lipid peroxidation, DNA oxidation and Ca2+ accumulation capacity (Sullivan et al., 2004).
Our finding that VDAC1 is one of the targets for misfolded SOD1 within the nervous system raises substantial implications for the mechanism underlying premature degeneration and death of motor neurons. A variety of apoptotic stimuli are known to trigger cell death by modulation of VDAC1 (Abu-Hamad et al., 2008; Shoshan-Barmatz et al., 2006; Tsujimoto and Shimizu, 2002; Yagoda et al., 2007; Zaid et al., 2005; Zamzami and Kroemer, 2003; Zheng et al., 2004), implicating VDAC1 as a component of the apoptotic machinery. Although VDAC1 proteins have been reported to be dispensable for Ca2+ and oxidative stress-induced permeability transition pore (PTP) opening (Baines et al., 2007), siRNA-mediated reduction in VDAC1 has supported VDAC1 as an indispensable protein for endostatin-, cisplatin- and selenite-induced oxidative stress induced PTP opening and apoptosis (Tajeddine et al., 2008; Tomasello et al., 2009; Yuan et al., 2008). Moreover, VDAC1 was recently shown to be involved in staurosporine- and ceramide-induced cell death downstream of BAD and BCL-XL (Roy et al., 2009) and curcumin induced apoptosis by cooperating with Bax in the release of AIF from mitochondria (Scharstuhl et al., 2009). Since VDAC1 is one of several targets for a cholesterol-like small molecule (TRO19622) that can protect motor neurons from SOD1 mutant-mediated death in culture and modestly delay disease onset in SOD1 mutant mice (Bordet et al., 2007), it now seems likely that its efficacy may be through direct effect on VDAC1.
Finally, it is well established that although motor neurons are the final targets in ALS, mutant damage within astrocytes and microglia contributes to driving rapid disease progression (Beers et al., 2006; Boillee et al., 2006a; Boillee et al., 2006b; Clement et al., 2003; Yamanaka et al., 2008a; Yamanaka et al., 2008b). In this context, we show here that little accumulation of misfolded SOD1 is found by disease onset, preferentially within motor neurons. However, during disease progression a dramatic increase of misfolded SOD1 is observed accumulated in other cells as well and probably extracellularly. Interestingly, mitochondrial dysfunction(s) within mutant astrocytes has been reported to cause acute motor neuron death in astrocyte-motor neuron co-cultures (Cassina et al., 2008) and astrocytes expressing mutant SOD1 have been reported to induce mitochondrial dysfunction within motor neurons (Bilsland et al., 2008). Coupling these findings with the appearance of aberrant mitochondria within motor neurons in multiple animal models of SOD1 mutant mediated ALS (Bendotti et al., 2001; Jaarsma et al., 2001; Kong and Xu, 1998; Wong et al., 1995) and the association of mutant SOD1 with mitochondria within affected tissues, we propose that misfolded SOD1 association directly with VDAC1 represents a primary event of damage within motor neurons.
Experimental Procedures
Transgenic Rats and Mice
Transgenic rats expressing hSOD1wt (Chan et al., 1998), hSOD1G93A (Howland et al., 2002) and hSOD1H46R (Nagai et al., 2001) were as originally described. All animal procedures were consistent with the requirements of the Animal Care and Use Committee of the University of California.
Mice heterozygous for the mutant human SOD1G37R transgene (LoxSOD1G37R) (Boillee et al., 2006b) were crossed with mice heterozygous for a VDAC1 gene disruption (Weeber et al., 2002). Mice were genotyped by PCR for the presence of the mutant SOD1 transgene (Williamson and Cleveland, 1999) and using a four-primer multiplex PCR for the presence of VDAC1 (Weeber et al., 2002), as previously described.
For survival experiments, SOD1G37R, VDAC1+/- mice were always compared with their contemporaneously produced SOD1G37R, VDAC1+/+ littermates. Time of disease onset was retrospectively determined as the time when mice reached peak body weight, early disease was defined at the time when denervation-induced muscle atrophy had produced a 10% loss of maximal weight, and end-stage was determined by paralysis so severe that the animal could not right itself within 20 seconds when placed on its side, an endpoint frequently used for SOD1 mutant mice and one that was consistent with the requirements of the Animal Care and Use Committee of the University of California.
Subcellular Fractionation
Mitochondria were purified as previously described (Vande Velde et al., 2008). Tissues were homogenized on ice in 5 volumes of ice-cold homogenization buffer (HB) composed of 210 mM mannitol, 70 mM sucrose, 1 mM EDTA-(Tris) and 10 mM Tris-HCl (pH 7.2). Homogenates were centrifuged at 1000 × g for 10 min. Supernatants were recovered, and pellets were washed with ½ volume HB and centrifuged at 1000 × g. Supernatants were pooled and centrifuged at 12,000 × g for 15 min to yield a crude mitochondrial pellet. The supernatant was used to make cytosolic fractions by further centrifugation at 100,000 × g for 1 hour. The mitochondria were gently resuspended in HB and then adjusted to 1.204 g/ml Optiprep (iodixanol) and loaded on the bottom of a polycarbonate tube. Mitochondria were overlaid with an equal volume of 1.175 g/ml and 1.079 g/ml Optiprep and centrifuged at 50,000 × g for 4 h (SW-55; Beckman). Mitochondria were collected at the 1.079/1.175 g/ml interface and washed once to remove the Optiprep. Optiprep stock solution was diluted in 250 mM sucrose, 120 mM Tris-HCl (pH 7.4), 6 mM EDTA plus protease inhibitors.
For activity assays, spinal cords were homogenized in 5 volumes of ice-cold homogenization buffer (HB) on ice. Homogenates were centrifuged at 1000 × g for 5 min. Supernatants were recovered, and centrifuged again at 1000 × g for 5 min. Supernatants were centrifuged at 12,000 × g for 10 min to yield crude mitochondrial pellets. These mitochondria were gently resuspended in HB and then adjusted to 12 % Optiprep (iodixanol) and centrifuged at 17,000 × g for 10 min (SW-55; Beckman). The majority of the myelin (at the top of the sample) was removed and the mitochondria were washed once with HB (without EDTA) to remove the Optiprep.
Liver was homogenized in 5 volumes of ice-cold homogenization buffer (HB) on ice. Homogenates were centrifuged at 1000 × g for 5 min. Supernatants were recovered, and centrifuged again at 1000 × g for 5 min. Supernatant was centrifuged at 12,000 × g for 10 min to yield a crude mitochondrial pellet. These mitochondria were resuspended in HB (without EDTA) and centrifuged again at 12,000 × g for 10 min. The pellet was resuspended in a small volume of HB without EDTA.
VDAC Channel Recording and Analysis
Reconstitution of VDAC into a planar lipid bilayer (PLB), single channel current recording, and data analysis were carried out as previously described (Gincel et al., 2001). Briefly, PLB were prepared from soybean asolectin dissolved in n-decane (50 mg/ml). Only PLB with a resistance greater than 100 GΩ, were used. Purified protein (about 1 ng) was added to the cis chamber. After one or a few channels were inserted into the PLB, the excess protein was removed by perfusion of the cis chamber with 20 volumes of a solution to prevent further incorporation. Currents were recorded under voltage-clamp using a Bilayer Clamp BC-525B amplifier (Warner Instrument Corp.). The currents were measured with respect to the trans side of the membrane (ground). The currents were low-pass, filtered at 1 kHz and digitized on-line using a Digidata 1200 interface board and pCLAMP 6 software (Axon Instruments, Inc.). Sigma Plot 6.0 scientific software (Jandel Scientific) was used for curve fitting. All experiments were performed at room temperature.
Supplementary Material
Acknowledgments
We would like to thank Neil Cashman (University of British Columbia) for generously providing us with DSE2 antibodies, William Craigen (Baylor College of Medicine) for VDAC1 knockout mice and Larry Hayward (UMass Medical School) for wild type and mutant SOD1 baculovirus stock. This work has been supported by grants from the NIH (R37 NS27036). A.I. has been supported by EMBO Long-Term Fellowship and by a postdoctoral fellowship from IsrALS. D.W.C. receives salary support from the Ludwig Institute for Cancer Research.
Footnotes
Please see Supplemental Information for the following experimental procedures: Protein Purification, Immunoprecipitation, DSE2 antibodies, Immunostaining, Ca2+ and ADP accumulation by mitochondria and Immunoblotting.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, Ben-Romano R, Friedman O, Shoshan-Barmatz V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. Journal of cell science. 2009;122:1906–1916. doi: 10.1242/jcs.040188. [DOI] [PubMed] [Google Scholar]
- Abu-Hamad S, Sivan S, Shoshan-Barmatz V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc Natl Acad Sci U S A. 2006;103:5787–5792. doi: 10.1073/pnas.0600103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem. 2008;283:13482–13490. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Ahmad S, Schneider BK, Allen CB, Chang LY, White CW. Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. American journal of physiology. 2002;283:L573–584. doi: 10.1152/ajplung.00410.2001. [DOI] [PubMed] [Google Scholar]
- Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel-1-based peptides interact with Bcl2 to prevent anti-apoptotic activity. J Biol Chem. 2010;285:6053–6062. doi: 10.1074/jbc.M109.082990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377:347–355. doi: 10.1042/BJ20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AO, Miller TM, Dong MQ, Vande Velde C, Cleveland DW, Yates JR. RCADiA: simple automation platform for comparative multidimensional protein identification technology. Anal Chem. 2007;79:6410–6418. doi: 10.1021/ac070585g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, Silani V, De Biasi S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci. 2001;191:25–33. doi: 10.1016/s0022-510x(01)00627-x. [DOI] [PubMed] [Google Scholar]
- Benz R. Permeation of hydrophilic solutes through mitochondrial outer membranes: review on mitochondrial porins. Biochim Biophys Acta. 1994;1197:167–196. doi: 10.1016/0304-4157(94)90004-3. [DOI] [PubMed] [Google Scholar]
- Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Rehnmark A, Marklund SL. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J Neurosci. 2006;26:4147–4154. doi: 10.1523/JNEUROSCI.5461-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR. Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J Neurochem. 2008;107:1271–1283. doi: 10.1111/j.1471-4159.2008.05699.x. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006b;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bordet T, Buisson B, Michaud M, Drouot C, Galea P, Delaage P, Akentieva NP, Evers AS, Covey DF, Ostuni MA, et al. Identification and characterization of cholest-4-en-3-one, oxime ( TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. The Journal of pharmacology and experimental therapeutics. 2007;322:709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Cashman NR, Caughey B. Prion diseases--close to effective therapy? Nat Rev Drug Discov. 2004;3:874–884. doi: 10.1038/nrd1525. [DOI] [PubMed] [Google Scholar]
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de Leon A, Robinson KM, Mason RP, Beckman JS, Barbeito L, Radi R. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28:4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Colombini M. VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem. 2004;256-257:107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- da-Silva WS, Gomez-Puyou A, de Gomez-Puyou MT, Moreno-Sanchez R, De Felice FG, de Meis L, Oliveira MF, Galina A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J Biol Chem. 2004;279:39846–39855. doi: 10.1074/jbc.M403835200. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96:1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF, Meininger V, Loeffler JP. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. Faseb J. 2003;17:2091–2093. doi: 10.1096/fj.02-1182fje. [DOI] [PubMed] [Google Scholar]
- Echaniz-Laguna A, Zoll J, Ribera F, Tranchant C, Warter JM, Lonsdorfer J, Lampert E. Mitochondrial respiratory chain function in skeletal muscle of ALS patients. Ann Neurol. 2002;52:623–627. doi: 10.1002/ana.10357. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J. 2001;358:147–155. doi: 10.1042/0264-6021:3580147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schoneich C, Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH., Jr Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Xu Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003;4:16. doi: 10.1186/1471-2202-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Donnenfeld H, Sasaki S, Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984a;43:461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- Hirano A, Nakano I, Kurland LT, Mulder DW, Holley PW, Saccomanno G. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984b;43:471–480. doi: 10.1097/00005072-198409000-00002. [DOI] [PubMed] [Google Scholar]
- Hodge T, Colombini M. Regulation of metabolite flux through voltage-gating of VDAC channels. J Membr Biol. 1997;157:271–279. doi: 10.1007/s002329900235. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. The Journal of cell biology. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelson A, Arzoine L, Abu-hamad S, Khodorkovsky V, Shoshan-Barmatz V. A photoactivable probe for calcium binding proteins. Chem Biol. 2005;12:1169–1178. doi: 10.1016/j.chembiol.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem. 2002;83:535–545. doi: 10.1046/j.1471-4159.2002.01112.x. [DOI] [PubMed] [Google Scholar]
- Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, Bradley WG, Moraes CT. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25:164–172. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator--thinking outside the box. Biochim Biophys Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Madesh M, Hajnoczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. The Journal of cell biology. 2001;155:1003–1015. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- Mulder DW, Kurland LT, Offord KP, Beard CM. Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology. 1986;36:511–517. doi: 10.1212/wnl.36.4.511. [DOI] [PubMed] [Google Scholar]
- Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH, Jr, Itoyama Y. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KT, Garcia-Chacon LE, Barrett JN, Barrett EF, David G. The {Psi}m depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc Natl Acad Sci U S A. 2009;106:2007–2011. doi: 10.1073/pnas.0810934106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22:1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramithiotis E, Pinard M, Lawton T, LaBoissiere S, Leathers VL, Zou WQ, Estey LA, Lamontagne J, Lehto MT, Kondejewski LH, et al. A prion protein epitope selective for the pathologically misfolded conformation. Nat Med. 2003;9:893–899. doi: 10.1038/nm883. [DOI] [PubMed] [Google Scholar]
- Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH, Jr, Naniche N, Kia A, Trotti D, Pasinelli P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet. 2010 doi: 10.1093/hmg/ddq202. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhit R, Robertson J, Vande Velde C, Horne P, Ruth DM, Griffin J, Cleveland DW, Cashman NR, Chakrabartty A. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med. 2007;13:754–759. doi: 10.1038/nm1559. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rostovtseva T, Colombini M. VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function. Biophys J. 1997;72:1954–1962. doi: 10.1016/S0006-3495(97)78841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva TK, Bezrukov SM. ATP transport through a single mitochondrial channel, VDAC, studied by current fluctuation analysis. Biophys J. 1998;74:2365–2373. doi: 10.1016/S0006-3495(98)77945-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Roy SS, Madesh M, Davies E, Antonsson B, Danial N, Hajnoczky G. Bad Targets the Permeability Transition Pore Independent of Bax or Bak to Switch between Ca(2+)-Dependent Cell Survival and Death. Mol Cell. 2009;33:377–388. doi: 10.1016/j.molcel.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Dendritic synapses of anterior horn neurons in amyotrophic lateral sclerosis: an ultrastructural study. Acta Neuropathol. 1996;91:278–283. doi: 10.1007/s004010050426. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66:10–16. doi: 10.1097/nen.0b013e31802c396b. [DOI] [PubMed] [Google Scholar]
- Scharstuhl A, Mutsaers HA, Pennings SW, Russel FG, Wagener FA. Involvement of VDAC, Bax and ceramides in the efflux of AIF from mitochondria during curcumin-induced apoptosis. PLoS One. 2009;4:e6688. doi: 10.1371/journal.pone.0006688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. The Voltage-Dependent Anion Channel (VDAC): Function in Intracellular Signalling, Cell Life and Cell Death. Curr Pharm Des. 2006;12:2249–2270. doi: 10.2174/138161206777585111. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Keinan N, Zaid H. Uncovering the role of VDAC in the regulation of cell life and death. Journal of bioenergetics and biomembranes. 2008;40:183–191. doi: 10.1007/s10863-008-9147-9. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Keller JN, Lovell M, Sodhi A, Hart RP, Scheff SW. Intrinsic differences in brain and spinal cord mitochondria: Implication for therapeutic interventions. J Comp Neurol. 2004;474:524–534. doi: 10.1002/cne.20130. [DOI] [PubMed] [Google Scholar]
- Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L, Araujo N, Pinna G, Larochette N, Zamzami N, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene. 2008;27:4221–4232. doi: 10.1038/onc.2008.63. [DOI] [PubMed] [Google Scholar]
- Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina S, Thoraval D, Crouzet M, Ichas F, De Pinto V, De Giorgi F. Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. 2009;19:1363–1376. doi: 10.1038/cr.2009.98. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S. The voltage-dependent anion channel: an essential player in apoptosis. Biochimie. 2002;84:187–193. doi: 10.1016/s0300-9084(02)01370-6. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104:2495–2500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielhaber S, Winkler K, Kirches E, Kunz D, Buchner M, Feistner H, Elger CE, Ludolph AC, Riepe MW, Kunz WS. Visualization of defective mitochondrial function in skeletal muscle fibers of patients with sporadic amyotrophic lateral sclerosis. J Neurol Sci. 1999;169:133–139. doi: 10.1016/s0022-510x(99)00236-1. [DOI] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber EJ, Levy M, Sampson MJ, Anflous K, Armstrong DL, Brown SE, Sweatt JD, Craigen WJ. The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J Biol Chem. 2002;277:18891–18897. doi: 10.1074/jbc.M201649200. [DOI] [PubMed] [Google Scholar]
- Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem. 2002;80:616–625. doi: 10.1046/j.0022-3042.2001.00731.x. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Xu X, Decker W, Sampson MJ, Craigen WJ, Colombini M. Mouse VDAC isoforms expressed in yeast: channel properties and their roles in mitochondrial outer membrane permeability. J Membr Biol. 1999;170:89–102. doi: 10.1007/s002329900540. [DOI] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Yamada A, Watanabe M, Yoshimura Y, Yamazaki N, Yoshimura Y, Yamauchi T, Kataoka M, Nagata T, Terada H, Shinohara Y. VDAC1, having a shorter N-terminus than VDAC2 but showing the same migration in an SDS-polyacrylamide gel, is the predominant form expressed in mitochondria of various tissues. J Proteome Res. 2006;5:3336–3344. doi: 10.1021/pr060291w. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Boillee S, Roberts EA, Garcia ML, McAlonis-Downes M, Mikse OR, Cleveland DW, Goldstein LS. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci U S A. 2008a;105:7594–7599. doi: 10.1073/pnas.0802556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008b;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Fu Y, Wang X, Shi H, Huang Y, Song X, Li L, Song N, Luo Y. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. Faseb J. 2008;22:2809–2820. doi: 10.1096/fj.08-107417. [DOI] [PubMed] [Google Scholar]
- Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell death and differentiation. 2005;12:751–760. doi: 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
- Zamzami N, Kroemer G. Apoptosis: mitochondrial membrane permeabilization--the (w)hole story? Curr Biol. 2003;13:R71–73. doi: 10.1016/s0960-9822(02)01433-1. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, Almasan A, Tang H, Chen Q. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene. 2004;23:1239–1247. doi: 10.1038/sj.onc.1207205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.