

Abstract
Compounds that directly disrupt the androgen receptor/steroid receptor coactivator interaction could function as novel inhibitors of androgen signaling that would remain effective in the treatment of prostate cancer that is resistant to conventional endocrine therapies. A structure-based peptidomimetic approach was used to design and synthesize such compounds, based on a pyrimidine-core system. Using fluorescence resonance energy transfer and reporter gene assays, we identified members of this library that disrupt the androgen receptor/steroid receptor coactivator interaction selectively, without affecting the estrogen receptor/steroid receptor coactivator interaction. Unlike the activity of traditional androgen receptor antagonists, such as flutamide and bicalutamide, inhibition by these coactivator binding inhibitors is insurmountable by increased concentrations of androgen agonists and maintains effectiveness even on a mutant androgen receptor that is resistant to traditional antagonists. These findings support the feasibility of targeting the coactivator binding groove of the androgen receptor as an alternative approach to treatment-resistant prostate cancer therapy.
INTRODUCTION
The androgen receptor (AR) is a member of the nuclear hormone receptor superfamily and plays an integral role in primary and secondary male sexual development. While abnormalities resulting in an attenuation of the AR response to endogenous hormones (testosterone and its reduced form, 5α-dihydrotestosterone or DHT) produce male infertility and feminization, excessive stimulation of the AR can also result in pathologies. The most commonly presented diseases of this type are prostate cancer and the related, but benign, prostatic hyperplasia (1). Both of these diseases are responsive to endocrine-based treatments that attempt to suppress tumor/prostate growth either by direct administration of an AR antagonist or by ‘chemical castration’ techniques that result in decreased gonadal production of the endogenous agonist, testosterone.
Traditional AR antagonists, such as flutamide or bicalutamide, act by binding to the ligand binding pocket of the receptor, resulting in a conformational change of the ligand binding domain (LBD) such that helix 12 occludes the binding of coactivators that are required to activate transcription. Consequently, this type of inhibition can be considered a type of indirect, or allosteric modulation of AR activity, because inhibitor binding in the ligand-binding pocket is disabling a protein-protein interaction at a separate site. While treatment with traditional AR antagonists is initially met with suppression of prostate tumor growth, with time (a few months to years), cellular modifications including AR mutations, up-regulation of AR and coactivators, changes in the post-translational modification of AR and accessory proteins, as well as increased androgen production by the suprarenal glands and in the tumors themselves, result in a endocrine-treatment refractory state in which cancer progression occurs despite the presence of an antagonist (2). As a result, new chemical approaches need to be developed to successfully treat this advanced-stage disease (3).
Our laboratory (4–8) and others (9, 10) have recently described the evaluation of small molecules that act as direct protein/protein disruptors of the interaction between the estrogen receptor (ER) LBD and steroid receptor coactivators (SRCs). We have termed these compounds coactivator binding inhibitors or CBIs, and it is hoped that the direct nature of the inhibition caused by this class of compounds—the direct blockade of coactivator binding to AR—will allow for retained inhibitory effectiveness even in instances where traditional allosteric antagonists fail (see Figure 1 for pictorial comparison of traditional antagonists and CBIs). Due to the general homology of the external binding groove of the LBDs of both ER and AR, as shown in crystallographic studies (see Figure 2), and the sharing of coactivators containing the LXXLL consensus sequence (11), we hypothesized that compounds containing structural characteristics similar to those that proved effective as ER CBIs would also antagonize the AR/SRC interaction. Additionally, the ability of the AR LBD to bind preferentially to coregulator proteins and peptides containing bulkier aromatic residues (e.g., 23FQNLF27 and 433WHTLF437 motifs of the AR N-terminal domain with the AR LBD (11, 12)) suggested that AR-selective CBIs could be formed by simple incorporation of larger side chains on already discovered CBI cores. To test this hypothesis, we designed a compound library based on a 2,4,6-trisubstituted pyrimidine core that had proven effective in earlier ER-CBI work and was designed to mimic the i, i+3, and i+4 arrangement of the three interacting residues of both the ER and AR coactivators (see Supplementary Figure 1 for a rationale of this structure-based approach) (8). In addition to the smaller propyl/butyl and isobutyl/isopentyl groups previously studied, we included larger benzyl/phenethyl and naphthalenemethyl/naphthethyl moieties in our design to mimic the phenylalanine and tryptophan residues present in the endogenous AR transcriptional system (see Supplementary Figure 2 for library layout). Synthetic details, compound characterization, and evaluation of the ER/SRC disruptor activity of this library has been presented in a recent article (5).
Figure 1.

Cartoon representation of traditional vs. CBI antagonism of a nuclear receptor. (a) Conformation of agonist-bound nuclear receptor ligand binding domain (NR-LBD) with helix 12 (α12) forming part of the steroid receptor coactivator (SRC) binding site. (b) Conformation of antagonist-bound NR in which helix 12 occupies the SRC binding site, disrupting the NR/SRC interaction indirectly. (c) Conformation of agonist-bound NR in which a CBI occupies the SRC binding site, disrupting the NR/SRC interaction directly.
Figure 2.

Comparison of the crystal structures of ERα and AR LBDs bound to LXXLL-containing coactivator peptides. (a) Rendering of agonist bound ERα cocrystallized with a SRC2 NR box II peptide (3erd). (b) Rendering of agonist-bound AR cocrystallized with a SRC2 NR box III peptide (1t63). (c) Overlay of 3erd and 1t63 showing the individual residues that interact with the coactivator peptides (magenta = ER, cyan = AR).
RESULTS AND DISCUSSION
Our initial efforts in screening the synthesized pyrimidine-core library for AR-CBI activity proved frustrating. Furthermore, although we were eventually successful in developing a time-resolved fluorescence resonance energy transfer (TR-FRET)-based assay that closely resembled the TR-FRET assay utilized for the ER system (involving glutathione S-transferase (GST)-tagged AR-LBD, a terbium-bound anti-GST antibody, and fluorescein-labeled SRC3), many of the CBIs with larger aromatic substituents proved insoluble in the buffer required for proper AR-LBD folding. Nonetheless, the activity of smaller alkyl-substituted CBIs (i.e., compound 3 in Table 1) in the AR TR-FRET assay support the feasibility of this approach for AR inhibition and confirm that the pyrimidines bind to the AR-LBD (For AR TR-FRET binding curves and constants, see Supplementary Figure 3).
Table 1.
Pyrimidine-core coactivator binding inhibitors for ERα and AR
| IC50 (μM) | ||||
|---|---|---|---|---|
| Cmpd # | Structure | ERα | AR (wt) | AR (T877A) |
| 1 |

|
7.9 | NB | 7.4 |
| 2 |

|
4.1 | 2.6 | 3.5 |
| 3 |

|
3.6 | 5.6 | 7.1 |
| 4 |

|
3.5 | 3.0 | 3.7 |
| 5 |

|
1.5 | 1.7 | 7.5 |
| 6 |

|
3.5 | 4.9 | 3.6 |
| 7 |

|
>30 | 1.6 | 9.4 |
| 8 |

|
>30 | 3.3 | 11.9 |
| 9 |

|
NBa | 1.9 | 3.5 |
| 10 |
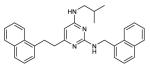
|
NBa | 1.5 | NB |
| 11 |

|
NB | 6.6 | >30 |
| 12 |
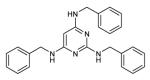
|
NBa | 3.5 | 18.9 |
| 13 |
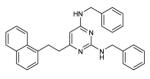
|
NBa | 5.6 | 16.0 |
| 14 |
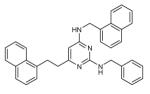
|
NBa | 4.1 | >30 |
All values obtained by luciferase reporter gene assay except where noted: Ki measured by TR-FRET.
Values are averages of duplicate assays generated from 2 or more independent replicates. NB= no binding.
As a consequence of our difficulties in developing a satisfactory in vitro assay for the hydrophobic AR CBIs, we turned to a luciferase reporter gene assay as our primary screen. A similar assay, developed for our work with ER, provided dose-dependent response curves that correspond well with those produced by our ER TR-FRET assay (5). In both cases, human endometrial cancer (HEC-1) cells, which do not endogenously express either nuclear receptor, were used as the eukaryotic hosts and were co-transfected with expression plasmids that code for full-length androgen receptor, an androgen response element/luciferase fusion (MMTV-luc), and pCMV β-galactosidase, used as an internal control. Although we (5) and others (10) have seen general cellular toxicity with compounds that act to disrupt ER/SRC binding at concentrations approximately 10-fold higher than their inhibition constants, the CBIs bearing larger hydrophobic residues seem to be better-tolerated in the cellular environment, and toxicity was not seen at even the highest concentrations assayed. Representative dose-dependent curves for this assay are shown in Figure 3a, and the ER and AR binding constants of select compounds are given in Table 1 (see Supplementary Table 1 for complete listing of assayed pyrimidines and their activities). While the ER and AR luciferase reporter gene assays generally produce values that are repeatable with a standard error of less then 0.5 (duplicate runs on two different days), based on the moderate sensitivity of these assays it is appropriate to view the compounds as either highly active (1–30 μM), moderately active (>30 μM as indicated by decrease in luciferase values at the highest concentration of CBI assayed (20 μM), but for which a mathematical inhibition curve cannot be generated) or inactive at the concentrations assayed (listed as NB in Table 1).
Figure 3.

Reporter gene assays of wild type AR and T877A AR mutant activity. Representative compounds show dose-dependent inhibition of full-length androgen receptor (a) or T877A full-length androgen receptor mutant (c) action. Compounds were also assayed against wild type AR (b) or T877A AR (d) activated with both 1 nM and 100 nM DHT to show the independence of CBI action from ligand concentration.
To establish that the pyrimidine CBIs cause inhibition by binding to the surface of the receptor and not by displacing DHT from the ligand binding pocket, repeat reporter gene assays were performed on select compounds in the presence of both 1 nM and 100 nM DHT. If competitive inhibition occurred at the ligand binding pocket, a right shift in the inhibition curve and increase in IC50 would result from the increased DHT concentration, while no shift would be expected if the compound competed directly with coactivator, rather than DHT. As seen in Figure 3b, active CBI 12 shows the same IC50 at both 1 nM and 100 nM DHT, while the traditional non-steroidal AR antagonist 2-hydroxyflutamide (OH-Flu) shows a complete loss of inhibitory activity at the higher DHT concentration. To provide further evidence that the pyrimidine-core CBIs do not act by binding to the internal binding site of the AR-LBD, a competitive radiometric binding assay was performed on a subset of active compounds using tritium-labeled methyltrienolone (R1881), a potent AR agonist, as a tracer and standard. The results from this assay also confirm that the pyrimidine-core compounds cannot be acting as traditional antagonists, because their affinity for the ligand binding pocket is not sufficient to explain their cell-based inhibitory potency (see Supplementary Table 1 for specific relative binding affinities).
While it is difficult to establish detailed structure-activity relationships from results of the cell-based assay, the data do exhibit a number of important trends. We had previously shown that the 2,4-diamino-6-alkylpyrimidines synthesized in this library bind almost exclusively to ERα and not ERβ (5), and this work further demonstrates the ability of members of the relatively small pyrimidine-core compounds to selectively bind different nuclear receptors, depending on the nature of the appending groups. In general, pyrimidine CBIs containing less bulky substituents—up to a total of two aromatic rings (either one naphthalene or two benzene groups)—bind to ERα and AR with comparable affinity (i.e., compounds 2–6 in Table 1). There are a few notable exceptions to this, namely compounds 1 and S44, which fail to inhibit the wild type AR/SRC interaction even at the highest assayed concentrations. (Interestingly, activity is restored when the AR T877A mutant is employed, as detailed below. This suggests that at higher concentrations, 1 and S44 would also prove to be active CBIs for wild type AR.)
While these results are gratifying, more impressive is the wide array of AR-specific CBIs that were found in this series (i.e., compounds 7–14 in Table 1). In agreement with our initial hypothesis, pyrimidines containing multiple bulky substituents (more than two aromatic rings) bound with complete selectivity to AR, and activity was seen in compounds as large as those containing two naphthalene and one benzene moieties (compound 14 in Table 1). These results reflect those previously found with peptide libraries, which showed that AR can bind to peptides ranging from those containing the general LXXLL binding motif of the SRCs to those having multiple phenylalanine or tryptophan residues, even ones encompassing motifs as large as WXXVW, which were found in phage display peptide libraries (13–16). These earlier reports also indicate that peptides containing these larger residues do not show any measurable binding to the ERα coactivator binding groove. Together, these results are a striking example of small-molecule peptidic mimicry, in which the exchange of side-arms on the heterocycle core results in selectivity corresponding to the analogous exchange of side-chains on peptides or proteins.
To test the ability of CBIs to circumvent clinically relevant hormone-refractory conditions, a reporter gene assay similar to that described above was developed involving a full-length AR containing the LNCaP mutation. This mutation confers agonist activity to many weak AR ligands, including the non-steroidal antagonist hydroxyflutamide (OH-Flu), due to a point mutation in the ligand binding pocket (T877A), and it is present in ~30% of patients with metastatic disease who have been treated with this drug (17). As anticipated, in this model both OH-Flu and DHT act as agonists, while many of the pyrimidine CBIs retain antagonistic activity comparable to that observed with the wild type receptor (See Figure 3c for representative traces and Table 1 and Supplementary Table 1 for binding constants). A sampling of these compounds were also assayed on the LNCaP mutant activated with both 1 nM and 100 nM DHT; the insignificant shift in the inhibition curve again provides evidence that these compounds do not effect inhibition by interaction at the ligand binding site (Figure 3d). Impressively, all but three of the AR-active compounds (10, 11, and 14 in Table 1) remain efficacious in the LNCaP model. All of these mutant-inactive compounds contain two large naphthyl substituents, and this data, coupled with the demonstrated ability of the LNCaP AR-LBD to accommodate the small ER-selective compounds, 1 and S44, as noted above, suggests that the T877A mutation introduces subtle, yet significant, differences to the coactivator binding, producing an overall more size-restrictive binding site. It should be noted that previous work with peptides showing differential selectivity between WT and T877A coactivator grooves has also been reported. (16) Supplementary Figure 4 shows a crystallographic comparison of wild type and T877A mutant AR coactivator binding grooves, demonstrating their significant structural homology.
In summary, we have utilized a structure-based peptidomimetic approach to design and synthesize a pyrimidine-core CBI library, the larger members of which selectively disrupt the AR/SRC interaction. The feasibility of this approach to effectively treat even a form of endocrine-insensitive prostate disease has also been demonstrated in an LNCaP model of prostate cancer. Finally, efforts are currently underway to not only increase the affinity of these compounds for AR, but also to improve their solubility through incorporation of various heterocycles (e.g., pyridine) and the addition of polar substituents (i.e., −OH, −NH2, etc) to the peripheral aromatic rings of the CBIs, which will facilitate evaluation of these compounds in animal models.
METHODS
TR-FRET CBI assay for wild-type and T877A mutant androgen receptors
A wild-type androgen receptor rat protein (GST-tagged) was purchased from Invitrogen and included both the hinge domain and the ligand binding domain with an amino acid sequence identical to that of the human sequence. The T877A mutant androgen receptor human protein (GST-tagged) was also purchased from Invitrogen and included the ligand binding domain (amino acids 606–902, with exception of T877A). These proteins, bound to a terbium-labeled anti-GST antibody, acted as the donor in the FRET assay. The fluorescein-labeled SRC3 NRD protein fragment was prepared according to previously published protocols (5).
The protocol below describes the TR-FRET assay using the wild-type receptor; the T877A mutant AR TR-FRET assay is conducted in the same manner with only substitution of the mutant receptor for wild type. Specifically, 5 μL of a stock solution of AR-GST (40 nM), dihydrotestosterone (4 μM), and terbium-labeled anti-GST antibody (Invitrogen) (40 nM) in TR-FRET coregulator buffer (Invitrogen; proprietary formula) were placed in separate wells of a black 96-well Molecular Devices HE high efficiency microplate (Molecular Devices, Inc.). In a second 96-well Nunc polypropylene plate (Nalge Nunc International), a 0.02 M solution of each coactivator binding inhibitor was serially diluted in a 1:10 fashion into DMF. Each concentration of coactivator binding inhibitor or vehicle was then diluted 1:10 into TR-FRET coregulator buffer, and 10 μL of this solution was added to the stock androgen receptor solution in the 96-well plate. After a five-minute incubation, 5 μL of 200 nM fluorescein-SRC3-NRD was added to each well. This mixture was allowed to incubate for 20 minutes at room temperature in the dark. TR-FRET was measured using an excitation filter at 340/10 nm, and emission filters for terbium and fluorescein at 495/20 and 520/25 nm, respectively. The final concentrations of the reagents were as follows: AR (10 nM), terbium-labeled ant-GST antibody (10 nM), dihydrotestosterone (1 μM), coactivator binding inhibitor (0–1 mM), SRC3-NRD (50 nM).
Luciferase Reporter Gene Assay
Human endometrial cancer (HEC-1) cells were maintained in culture and transfected in 24 well plates as previously described (18). HBSS (50 μL/well), Holo-transferrin (Sigma T1408) (20 μL/well), and lipofectin (Invitrogen #18292-011) (5 μL/well) were incubated together at room temperature for 5 minutes. A DNA mixture containing 200 ng of pCMVβ-galactosidase as an internal control, 500 ng of the androgen-responsive reporter gene plasmid MMTV-Luc, and 100 ng of full-length androgen receptor expression vector with 75 μL HBSS per well was added to the first mixture and allowed to incubate for 20 minutes at room temperature. After changing the cell media to Opti-MEM (350 μL/well), 150 μL of the transfection mixture was added to each well. The cells were incubated at 37 °C in a 5% CO2 containing incubator for 6 h before the medium was replaced with fresh medium containing 5% charcoal-dextran-treated calf serum and the desired concentrations of ligands. Luciferase reporter gene activity was assayed 24 h after ligand addition as described, (18) and values from duplicate wells at each concentration were plotted to generate binding curves. Compounds were described as having inhibition constants of “>30” μM if the most concentrated data point (20 μM) showed any decrease from maximal values, but enough information was not present to generate a binding curve.
In the initial screen, compounds were assayed in a dose-response format at concentrations ranging from 0.6 μM to20 μM; their inhibitory potential was determined by performing the assay in the presence of 10−7 M dihydrotestosterone (DHT). Upon validation of antagonistic activity, mechanism of action was examined by repeating the compound titration in the presence of both 10−7 and 10−9 M DHT with an expectation that changing the concentration of DHT 100-fold does not change the IC50 of true coactivator binding inhibitors.
Androgen receptor binding assays
Relative binding affinities were determined by competitive radiometric binding assays with 10 nM [3H]R1881 as tracer ([17α-methyl-3H]methyltrienolone, 17β-hydroxy-17α-methyl-estra-4,9,11-trien-3-one 70–87 Ci/mmol, PerkinElmer), as a modification of methods previously described (19-21). The source of AR was purified, recombinant rat, ligand binding domain purchased from Invitrogen. Incubations were done at 0 °C for 18–24 h, and hydroxyapatite (Bio-Rad) was used to absorb the purified receptor-ligand complexes. The binding affinities are expressed as relative binding affinity (RBA) values, where the RBA of R1881 is 100%; under these conditions, the Kd of R1881 for AR is ca. 0.6 nM. The determination of these RBA values is reproducible in separate experiments with a CV of 0.3.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge support of this research from the National Institutes of Health (PHS 5R37DK15556). J. Gunther received additional support from a David Robertson Fellowship, an NIH Fellowship NRSA 1 F30 ES016484-01 and Training Grant NRSA 5 T32 GM070421. A.Parent received support from an Illinois Distinguished Fellowship and a Robert D. Doolen Fellowship. The wild-type AR and MMTV-luc AR-response element plasmids were generously provided by D. McDonnell and the T877A mutant AR plasmid by E. Wilson. Radiometric binding assays were performed by K. Carlson.
Footnotes
Supporting Information Available: Supplemental material including figures and a complete table of analyzed compounds and biological results are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Chatterjee B. The role of the androgen receptor in the development of prostatic hyperplasia and prostate cancer. Mol. Cell. Biochem. 2003;253:89–101. doi: 10.1023/a:1026057402945. [DOI] [PubMed] [Google Scholar]
- 2.Taplin M-E. Drug Insight: role of the androgen receptor in the development and progression of prostate cancer. Nat. Clin. Pract. Oncol. 2007;4:236–244. doi: 10.1038/ncponc0765. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr. Opin. Pharmacol. 2008;8:440–448. doi: 10.1016/j.coph.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LaFrate AL, Gunther JR, Carlson KE, Katzenellenbogen JA. Synthesis and biological evaluation of guanylhydrazone coactivator binding inhibitors for the estrogen receptor. Bioorg. Med. Chem. 2008;16:10075–10084. doi: 10.1016/j.bmc.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parent AA, Gunther JR, Katzenellenbogen JA. Blocking Estrogen Signaling After the Hormone: Pyrimidine-Core Inhibitors of Estrogen Receptor-Coactivator Binding. J. Med. Chem. 2008;51:6512–6530. doi: 10.1021/jm800698b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gunther JR, Moore TW, Collins ML, Katzenellenbogen JA. Amphipathic Benzenes Are Designed Inhibitors of the Estrogen Receptor a/Steroid Receptor Coactivator Interaction. ACS Chem. Biol. 2008;3:282–286. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou H-B, Collins ML, Gunther JR, Comninos JS, Katzenellenbogen JA. Bicyclo[2.2.2]octanes: Close structural mimics of the nuclear receptor-binding motif of steroid receptor coactivators. Bioorg. Med. Chem. Lett. 2007;17:4118–4122. doi: 10.1016/j.bmcl.2007.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA. Design, Synthesis, and in Vitro Biological Evaluation of Small Molecule Inhibitors of Estrogen Receptor a Coactivator Binding. J. Med. Chem. 2004;47:600–611. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 9.Becerril J, Hamilton AD. Helix mimetics as inhibitors of the interaction of the estrogen receptor with coactivator peptides. Angew. Chem., Int. Ed. 2007;46:4471–4473. doi: 10.1002/anie.200700657. [DOI] [PubMed] [Google Scholar]
- 10.Shao D, Berrodin TJ, Manas E, Hauze D, Powers R, Bapat A, Gonder D, Winneker RC, Frail DE. Identification of novel estrogen receptor a antagonists. J. Steroid Biochem. Mol. Biol. 2004;88:351–360. doi: 10.1016/j.jsbmb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Chmelar R, Buchanan G, Need EF, Tilley W, Greenberg NM. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int. J. Cancer. 2007;120:719–733. doi: 10.1002/ijc.22365. [DOI] [PubMed] [Google Scholar]
- 12.He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 2000;275:22986–22994. doi: 10.1074/jbc.M002807200. [DOI] [PubMed] [Google Scholar]
- 13.Chang C.-y., Abdo J, Hartney T, McDonnell DP. Development of peptide antagonists for the androgen receptor using combinatorial peptide phage display. Mol. Endocrinol. 2005;19:2478–2490. doi: 10.1210/me.2005-0072. [DOI] [PubMed] [Google Scholar]
- 14.Estebanez-Perpina E, Moore JMR, Mar E, Delgado-Rodrigues E, Nguyen P, Baxter JD, Buehrer BM, Webb P, Fletterick RJ, Guy RK. The Molecular Mechanisms of Coactivator Utilization in Ligand-dependent Transactivation by the Androgen Receptor. J. Biol. Chem. 2005;280:8060–8068. doi: 10.1074/jbc.M407046200. [DOI] [PubMed] [Google Scholar]
- 15.Hun E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. Recognition and accommodation at the androgen receptor coactivator binding interface. PLoS Biol. 2004;2:1303–1312. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozers M. Szatkowski, Marks BD, Gowda K, Kupcho KR, Ervin KM, De Rosier T, Qadir N, Eliason HC, Riddle SM, Shekhani MS. The androgen receptor T877A mutant recruits LXXLL and FXXLF peptides differently than wild-type androgen receptor in a time-resolved fluorescence resonance energy transfer assay. Biochemistry. 2007;46:683–695. doi: 10.1021/bi061321b. [DOI] [PubMed] [Google Scholar]
- 17.Taplin M-E, Bubley GJ, Ko Y-J, Small EJ, Upton M, Rajeshkumar B, Balk SP. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–2515. [PubMed] [Google Scholar]
- 18.Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS. Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor-alpha or estrogen receptor-beta. Endocrinology. 1999;140:800–804. doi: 10.1210/endo.140.2.6480. [DOI] [PubMed] [Google Scholar]
- 19.Katzenellenbogen JA, Johnson HJ, Jr., Myers HN. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 20.Brandes SJ, Katzenellenbogen JA. Fluorinated androgens and progestins: molecular probes for androgen and progesterone receptors with potential use in positron emission tomography. Mol. Pharmacol. 1987;32:391–403. [PubMed] [Google Scholar]
- 21.Liu A, Carlson KE, Katzenellenbogen JA. Synthesis of high-affinity fluorine-substituted ligands for the androgen receptor. Potential agents for imaging prostatic cancer by positron emission tomography. J. Med. Chem. 1992;35:2113–2129. doi: 10.1021/jm00089a024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.