

Abstract
Thousands of biochemical interactions are available for download from curated databases such as Reactome, Pathway Interaction Database and other sources in the Biological Pathways Exchange (BioPAX) format. However, the BioPAX ontology does not encode the necessary information for kinetic modeling and simulation. The current standard for kinetic modeling is the System Biology Markup Language (SBML), but only a small number of models are available in SBML format in public repositories. Additionally, reusing and merging SBML models presents a significant challenge, because often each element has a value only in the context of the given model, and information encoding biological meaning is absent. We describe a software system that enables a variety of operations facilitating the use of BioPAX data to create kinetic models that can be visualized, edited, and simulated using the Virtual Cell (VCell), including improved conversion to SBML (for use with other simulation tools that support this format).
1. Introduction
1.1. Motivation
Currently, a great deal of information about signaling pathways (ranging from complete pathways, to just molecules participating in such pathways, or to just individual interactions) can be obtained in standardized formats from multiple online resources. The Biological Pathways Exchange standard (BioPAX, [1], [2], http://biopax.org) allows extracting qualitative information from Reactome database ([3], http://www.reactome.org/), Pathway Interaction Database (http://pid.nci.nih.gov/), BioCyc collection of Pathway/Genome databases ([4], http://biocyc.org) and more (for current listing see http://biopax.org). A growing number of tools for analysis and visualization of interaction networks support the BioPAX standard – e.g. Cytoscape (http://cytoscape.org, [5]), cPath database http://cbio.mskcc.org/software/cpath, [6]), PathCase (http://nashua.case.edu/PathwaysWeb), VisANT (http://visant.bu.edu, [7]). However, the current standard for kinetic modeling is Systems Biology Markup Language, SBML ([8], http://sbml.org). Both BioPAX and SBML are used to encode key information about the participants in biochemical pathways, their modifications, locations and interactions, but only SBML can be used directly for kinetic modeling, because elements are included in SBML specifically for the context of a quantitative theory. In contrast, concepts in BioPAX are more abstract. SBML-encoded models typically contain all data necessary for simulations, such as molecular species and their concentrations, reactions among these species, and kinetic laws for these reactions. This data is uniquely identified within a given SBML model, but often it has no value if considered outside of it: there is no way to compare the Species element with name S1 of model 1 with the Species element with name S1 of model 2 in many SBML files. The recent introduction in SBML of the sboTerm attribute to support the Systems Biology Ontology (SBO), and the standardization of the Annotation elements, solves this problem only partially – since these are optional, and relatively new. SBML does not require the use of sboTerm in order to encode relationships, or the use of Annotation to uniquely identify model elements outside of the model itself (by the use of references to controlled vocabularies). Moreover, when the Annotation element is being used, SBML does not enforce any constraints on its content, and therefore, for example, two Species elements that are uniquely identified within the model by different id attributes, may have the same identification information included in Annotation elements (for example, phosphorylated and unphosphorylated forms may be linked to the same external database reference). It is the liberty and the burden of the SBML producer to properly curate the models in a comprehensive and consistent way. Currently, there are few resources that provide publicly accessible SBML models that consistently include such information. Meanwhile, most pathways available from public repositories in BioPAX format, while not having the necessary kinetic information required for simulation, do typically include unique identification of all elements through external references, as well as additional information regarding relationships between the elements of the pathway which allow for automated reasoning. Providing a modeling framework that uses data in BioPAX format and facilitates conversion to SBML would solve two big problems: (i) use of abundant sources of well-curated quantitative data, and (ii) creating easily reusable quantitative models.
1.2. BioPAX, SBML, and SBO
Systems Biology Markup Language (SBML) is designed mainly to enable the exchange of quantitative models of biochemical networks between different simulation software packages with little or no human intervention. One feature of simulation-centric XML standards, such as SBML and CellML [9], is that no hierarchy of different types of molecular species or different types of interactions is necessary to be encoded. A simulation software simply needs a list of things of the same kind, called Species, and a list of things of the same kind, called Reactions, uniquely identified within the model, and mathematical information such as kinetic laws, initial conditions, etc., in order to reproduce a certain simulation result. Additional information that can help a human understand the meaning of the model elements and their relationships, as well as unique identification of elements across different models, can simply be ignored by the simulator in the context of the specific model to be simulated. In practice, it became apparent, that while one can reliably port SBML models between different software tools, true reusability is limited.
As long as the data is small enough to be tweaked by hand, flat and simple formats are most welcome. The user knows what each symbol means and therefore the software does not need to. But this is changing: as projects grow, the need is growing to combine data from different sources and to process them by software sophisticated enough to know that there is some sort of difference between a complex of proteins and a small molecule. SBML has evolved to provide the means for this. As of Level 2, Revision 3, it includes direct support for SBO, which is a new and comprehensive ontology that covers both general biological relationships as well as model-specific ones. Additionally, support for the use of external controlled vocabularies and other namespaces has been standardized. Unfortunately, most models in SBML currently use few or none of these features. For example, the largest public resource of curated SBML models, the BioModels database [10], although it does use cross-referencing to controlled vocabularies, it does not yet include ontology information.
The BioPAX ontology was created from the beginning with the purpose of providing a pathway exchange format that aims to facilitate sharing of pathway information between databases and users. BioPAX is based on OWL (Web Ontology Language) that is designed for use by applications that need to process the content of information instead of just presenting information to humans. OWL provides a framework for controlled vocabulary along with a formal semantics. BioPAX concepts, unlike generic XML concepts, have relationships to each other that can be processed automatically (see [11, 12] for more information on using BioPAX vs. SBML). An automatic reasoner can infer that if B is a kind of A, then B inherits all of A’s property definitions. These relationships between different concepts are the key to merging or linking different sets of information from different sources. Additionally, each element of a BioPAX file is linked to an originating biological database, providing for a well-documented biological identification for each element of the model. These two features make the BioPAX standard a practical tool for reusable modeling modules. However, it has no support for all the critical information required for building a quantitative model and running simulations.
1.3. Challenges and solutions
Currently, there are multiple converters between the SBML and BioPAX standards. The BioModels database that stores curated models in SBML format can convert each model into the BioPAX standard. The Reactome database that stores curated pathways data in BioPAX format can generate an SBML file for each selection. However, these converters do not provide unique identification of species in SBML or physical entities in BioPAX; thus, they do not solve the problem of model reusability. The SBML output from the Reactome pathway database contains absolutely no additional information for Species and Reaction elements except names, and therefore these can not be easily identified in the context of several different models. Since BioPAX is an extensible format, one possible approach is to extend it to support kinetic data for simulations – but the number and complexity of the additional required abstractions is so large as to dwarf the entire existing format. A much more practical approach is to rely on the SBML format for kinetic models, and (i) implement some of the model-building operations at the level of the BioPAX files, before translation to SBML, and (ii) make use of existing SBML facilities to carry over the ontology and controlled vocabulary information during translation.
Primary challenges of converting of BioPAX data into a kinetic model include: (1) merging several BioPAX files through unique identification of BioPAX objects; (2) converting a BioPAX file into SBML format by deciding whether references to the same BioPAX entity have to be represented by the same or different SBML Species; (3) annotating the SBML model such that annotations would uniquely identify all Species and interactions across multiple datasets, and not just within the given model, and preserving the relationship information among model elements; (4) adding simulation-specific information such as kinetic laws and initial conditions. User intervention may be required to perform tasks (1) and (2), especially when data is coming from different sources. To reduce the user intervention to a minimum, if not eliminate it entirely, we employ a series of sophisticated tests based on a wide variety of attributes, including unique database identifiers, names and types of entities and interactions and relationships between them. Task (3) should be performed automatically, and the SBML file should ideally have a one-to one mapping with information from the BioPAX file. Task (4) is usually performed manually, but it could also be automated via retrieval of kinetic information from online sources. The implementation of mechanisms to facilitate tasks (3) and (4) fall beyond the scope of this manuscript, but are briefly discussed further where relevant.
2. Modeling using BioPAX
Creating a kinetic model using the data in BioPAX standard is a non-trivial problem. Most pathway databases are not model centric, and to build a particular model, one would typically select several elements from such a database (or several databases), i.e. several separate BioPAX files which should then be processed. Thus one important feature of the system is to have an algorithm for identifying molecular species and reactions that are common in the different BioPAX files and allow for easy merging of different pathway elements into a model. We have designed a BioPAX modeling framework intended to obtain, store, merge and complement data in BioPAX, thus facilitating generation of kinetic models, such as can be expressed in SBML. The BioPAX data in general lacks simulation-related information (such as concentrations, kinetic laws etc), but usually has a lot of auxiliary information which may be not essential for simulations (organisms, different names, linking molecular species to a variety of databases, etc). BioPAX model can be easily visualized more expressively than SBML models, by relying on this type of information – for example, by giving different BioPAX objects (proteins, small molecules, complexes etc) different representations (e.g. colors). Each object is linked to biological information from public databases. A modeler will need to add some information to convert a BioPAX model into a computable kinetic model in SBML format. Figure 1 illustrates a summary of possible workflows using the BioPAX modeling framework.
Fig. 1.

BioPAX modeling framework. Data from multiple sources in BioPAX format are imported into the BioPAX modeling framework, and can be converted into BioPAX-annotated reusable SBML, and further exported into the VCell modeling framework. The BioPAX modeling framework can be used to merge several BioPAX files. BioPAX-annotated SBML files can be stored locally or in the VCell database. Solid lines denote the implemented conversions. Dashed lines denote features to be implemented.
A BioPAX model can be converted into SBML for use with different simulation software tools, and can be exported into the Virtual Cell software framework (VCell) (http://vcell.org, [13]) for running kinetic simulations and further model development. BioPAX models along with easily reusable, BioPAX-annotated SBML models will be stored in the VCell database. Several BioPAX models can be merged into a larger BioPAX model. The merged model can be compactly visualized as a set of modules, where all elements of the same BioPAX model are compressed into a single container node.
3. Modeling framework design
The BioPAX modeling framework prototype implementation is designed to be a part of the VCell software. To handle the BioPAX ontology classes, we use Jena. Jena is a Java application programming interface that provides support for handling RDF (Resource Description Framework) documents, including OWL documents, such as BioPAX files. In the description below, we will use the following notations:
By p-interactions we denote the elements of the BioPAX class type physicalInteraction, as well as of those of all of its subclasses (for example, biochemicalReaction).
By p-entities we denote the elements of the BioPAX class physicalEntities, as well as of those of all of its subclasses (for example, protein).
By p-participants we denote the elements of the BioPAX utility class physicalEntityParticipant, and of all of its subclasses (sequenceParticipant).
Generally, in BioPAX pathways, there is one p-participant for each participation of a p-entity in a p-interaction, and a set of p-participants will be mapped onto Species in SBML. A p-entity corresponds to a reacting entity independent of location and modifications (such as phosphorylation of proteins), and thus in many cases corresponds to speciesType in SBML.
3.1. Features
As the framework is based on Jena, which allows handling of generic OWL files, it is not tied to the current version of BioPAX and can support BioPAX extensions and future versions of BioPAX. The framework can use data taken from databases offering BioPAX web interfaces, from BioPAX files provided by the user, and from the VCell repository of BioPAX modeling projects (which are described in more detail in section 3.2). Some of this data, such as data taken from the VCell repository of BioPAX files and BioPAX-annotated SBML files, might already be in a state that allows a straight-forward translation into kinetic simulation models; while in general, data available as BioPAX ontology documents, such as from most public pathway databases, will require nontrivial processing and the addition of information. Processing primarily includes:
Merging BioPAX files, including identification and linking of p-entities referring to the same resource from different files. For example, two files brought into the framework might each have an entry for the same protein, possibly with a slightly different name.
Mapping of BioPAX objects onto sets of Species and Reactions. For example, several objects that refer to the same p-entity can translate into one or more Species due to modifications, such as the phosphorylated or unphosphorylated state of a protein.
Converting a BioPAX model into a fully annotated SBML model. Each Species and Reaction in the SBML model has an Annotation element that uniquely identifies this element, in the same way as BioPAX does.
This processing is explained in more detail below.
3.2. BioPAX to model conversion
3.2.1 Merging BioPAX files and identification of unique resources
OWL provides means for one file to refer to another (via owl:import) and to link objects as identical (via owl:sameAs). When several files are coming from the same source, it may be enough to simply compare unifXREF. When the files are coming from different sources, the complexity of the problem varies widely because information from different BioPAX files can be very different. In the best case scenario, any p-entity may still have the complete specification, including a type (for example rna, protein, etc), properties specific for the type (for example, sequence for a protein, chemical-formula for a smallMolecule), and one or more references to a database identifier. In the worst case scenario, each Species may just be called a physicalEntity with no additional detail (an example is when the user brings in a BioPAX file converted from SBML by the BioModels database sbml2biopax tool). To identify a list of unique Species, we use a series of tests that provide either a certain decision, or give a likelihood score that two p-entities refer to the same resource. See Figure 2 for additional details about the algorithm. A similar algorithm for identifying Species will be described in the next subsection in more detail.
Fig. 2.

Flowchart of algorithm for identification of unique p-entities. The first “else” stands for the case when either one or both p-entities have no unifXREF. Individual types tests include comparing sequence property for protein and rna, chemical-formula for smallMolecule, etc. The user sets negative and positive thresholds for the score function. If the score is above a positive threshold, p-entities are declared to be identical, if below negative threshold – distinct, otherwise the user has to make the decision.
3.2.2 Identification of Species and Reactions
A crucial question to be answered when converting from BioPAX to SBML is whether two p-participants should correspond to one Species or two different Species, which will be decided by an algorithm similar to the one described in the previous subsection. Here, we describe the most important steps.
Two p-participants are the same Species if they have the same location and the same chemical identity. The same chemical identity can be assumed if only if they refer to the same p-entity with the same modifications. If the p-entity is a complex, all components have to be the same. A smallMolecule with modifications is a different object. To find out whether two rnas, dnas, or proteins have the same modifications, we evaluate sequenceFeatures. This usually gives a definite answer in the case of p-participants of the same Reactions, but often only likelihoods for p-participants of different Reactions. The lack of definite answers in the latter case stems from problems with sequenceFeature recommended usage, such as using sequenceFeatures both for chemical modifications (e.g. phosphorylations) and for description of non-modified features (e.g. binding sites), mentioning only sequenceFeatures “relevant to the interaction”, and defining the same sequenceFeature separately for each interaction. In some, but not all, cases, these issues can be resolved by evaluating a sequenceFeature’s featureType (e.g. phosphorylation site) and featureLocation, if it is specific enough. The BioPAX standard notes that sequence features might be replaced in future versions by states, which we expect to be a great improvement (for our purposes). If proper information on sequence features is absent, we might still infer chemical modifications by analyzing other properties, for example similarity of names or the occurrence of phrases like “phospho”. Since reactions listed in the same file usually form one network, is very likely that at least one p-participant per reaction is the same Species as a p-participant in another reaction. Similarly, it is somewhat likely that not all p-participants referring to a p-entity are a different Species. All of these likelihoods contribute to computing a score, which will be compared to a tunable threshold for automatic or manual decision-making.
The score for each test roughly corresponds to the negative logarithm of the probability that a statement (e.g. two p-entities being the same species) is false although tests are positive. If the tests have a low probability of false positives and little correlation to each other, each positive test result lowers the probability that the statement is false by a factor roughly equal to the probability of a false positive divided by the probability of a correct positive. If two tests have high degree of correlation or high false positive probabilities, they may be combined and treated as one test. The total score will roughly estimate the negative logarithm of the probability that the statement is wrong in spite of one or more positive test results.
For example, we can estimate the probability that two p-entities are the same species if they have similar names. Similarity can be measured by edit distances (such as Damerau-Levenshtein [14] or Jaro-Winkler [15] distance). For the case of Damerau-Levenshtein we need to divide it by the length of the longer name to get a number between zero and one. We might say, for example, that if the normalized distance is less than 0.2, there is only a five percent chance that they are not the same species, and then the score would be the negative logarithm of 0.05.
3.2.3 Extension of BioPAX
BioPAX is based on OWL, which provides a standard to extend any ontology easily by adding new properties to existing classes. There are two main issues that we deal with:
To store in a BioPAX file the information necessary to enable immediate and fully automatic conversion to SBML, we introduce to BioPAX two new properties, speciesID and speciesTypeID. These correspond to SBML’s Species and SpeciesType elements, and can be assigned to p-participants and p-entities. A Species in SBML is an entity which can be quantified in a kinetic simulation, and a SpeciesType is a set of Species differing only in location. Therefore, in SBML, two entities are the same Species if they are of the same SpeciesType and at the same location, and they are different, if the location or the SpeciesType is different. It is optional for a Species to belong to a SpeciesType. In many cases, a p-entity in BioPAX corresponds to a SpeciesType in SBML, and in this case, all referring p-participants with the same location correspond to the same Species. Therefore, this is our default for a conversion if no speciesID or speciesTypeID is given. Exceptions are primarily rnas, dnas and proteins with modifications, such as phosphorylations, which cause the same p-entity to correspond to different SpeciesTypes and different Species. The properties speciesID and speciesTypeID are introduced to mark these exceptions. A speciesID or speciesTypeID can be added to any p-participant or p-entity to control to which Species or SpeciesType it corresponds in SBML. Adding a speciesID or speciesTypeID to a p-entity is the same as adding it to all referring p-participants which do not have this property already assigned.
-
To be able to use tools which support BioPAX, but do not tolerate extensions, we enclose each nonstandard property within a comment tag. Thus, standard tools will simply see a comment, which is a standard BioPAX property and which does not mandate processing. Our framework will read comments searching for new properties. For example, speciesID is declared through:
<bp:COMMENT rdf:datatype="…XMLSchema#string">
<bp:SPECIES-ID
rdf:datatype="…XMLSchema#string">
speciesID</bp:SPECIES-ID> </bp:COMMENT>
3.2.4 Mapping BioPAX files onto SBML files annotated with ontology and controlled vocabulary information
The SBML language specification has recently introduced two features that facilitate and standardize the inclusion of additional information that is not required for the numerical interpretation of the model, but which can help describe the model and relate model and model elements to each other, both within the same file or between files from different sources. The most flexible approach is the use of the standardized syntax for the Annotation element, which can decorate any class such as Species or Reaction. Additional XML namespaces defined at arbitrary URIs can be included, using a restricted form of the Dublin Core (http://dublincore.org) embedded in RDF. This generalized syntax allows one to add any valid RDF/OWL fragments to SBML elements, effectively allowing us to directly add the BioPAX data (such as the corresponding p-entity information for a particular SBML Species). This is not quite trivial, though, especially for the case of molecular complexes and modifications, where references to p-participants need to be added, and additional “dummy” SBML Species are being created. Additionally, the standardization of the Relation element, a subtype of Annotation, allows direct inclusion of unifXREF information using the bqbiol:is, bqbiol:isVersionOf and bqbiol:isDescribedBy qualifiers.
The detailed description of this process is outside the scope of this paper. Furthermore, potentially the most effective and powerful approach to create a mapping of all BioPAX information into corresponding SBML files is the use of the sboTerm attribute which is included in SBase and thus inherited essentially by all relevant SBML classes. SBO is designed to be more comprehensive ontology framework than BioPAX, including definitions and relationship hierarchies for concepts that are specific to quantitative modeling (in the modeling framework, quantitative parameter, mathematical expression, and event branches). The scope of BioPAX discussed here (p-entity and p-particpant) is mainly covered by the participant type branch composed of the participant functional type and participant physical type sub-branches, applied to the Species, SpeciesType, and SpeciesReference elements. However, since SBO is relatively new, and still expanding and evolving quite rapidly, we chose to relegate this approach to future versions of the software framework.
3.3. Data organization: BioPAX modeling project
When the user imports a BioPAX file into the framework, he has an option to create a new BioPAX modeling project or to add the file to an existing project. Projects are stored as a collection of BioPAX files either locally or in the VCell database. The project consists of: (1) the data-source: BioPAX files imported into the framework that are designated read-only and stay unmodified; and (2) BioPAX models and BioPAX-annotated SBML models. BioPAX models have new properties added to store a log of files added to the project. If several files are added to the project, the BioPAX model imports the source files and marks identity relationships. Most ontology-processing tools like Jena and Protégé that can handle any OWL file can process the composite model.
To create a model ready for simulation, the BioPAX model can be imported into the VCell software or converted to SBML format. It can also be exported to a BioPAX file without extensions. This is useful if the user intends to process the file with tools which expect BioPAX but do not tolerate extensions. In this case new data is included as comments.
3.4. System Architecture
The software is organized into several layers: (1) initialization and configuration; (2) graphical user interface; (3) specification of actions initiated by the user or by other events; (4) organization of background threads; (5) representation of the BioPAX data; and (6) general utilities. Calls are made only within layers or from a higher to a lower layer. Lower layers communicate to higher layers through message passing based on call-backs. The data is stored in RDF format using the Jena API. On top of the RDF model is a layer of model components, where there is a component for each node or statement of the model, as well as components representing groups of smaller components. Each object that is a model component can be visualized as an element of a graph.
3.5. Model visualization
To facilitate the organization of the data and to make selections, the framework provides a graphical representation to view the source data as well as the data needed to create kinetic models ready for simulation. This graphical interface can handle any OWL model but specifically supports the BioPAX ontology. Inspired by the graphical user interface of the VCell “physiology” editor, it displays a graph consisting of p-interactions among p-entities in the VCell style (see Figure 3). This is a bipartite graph in its fully flattened form (with nodes for both entities and interactions), using for each of the p-entity and p-interaction classes a separate symbol. Each p-interaction is connected by an edge with the p-entities participating in it. Complexes are displayed in a way that alludes to their components. All other objects are hidden, until they are properties of an object which becomes selected.
Fig. 3.
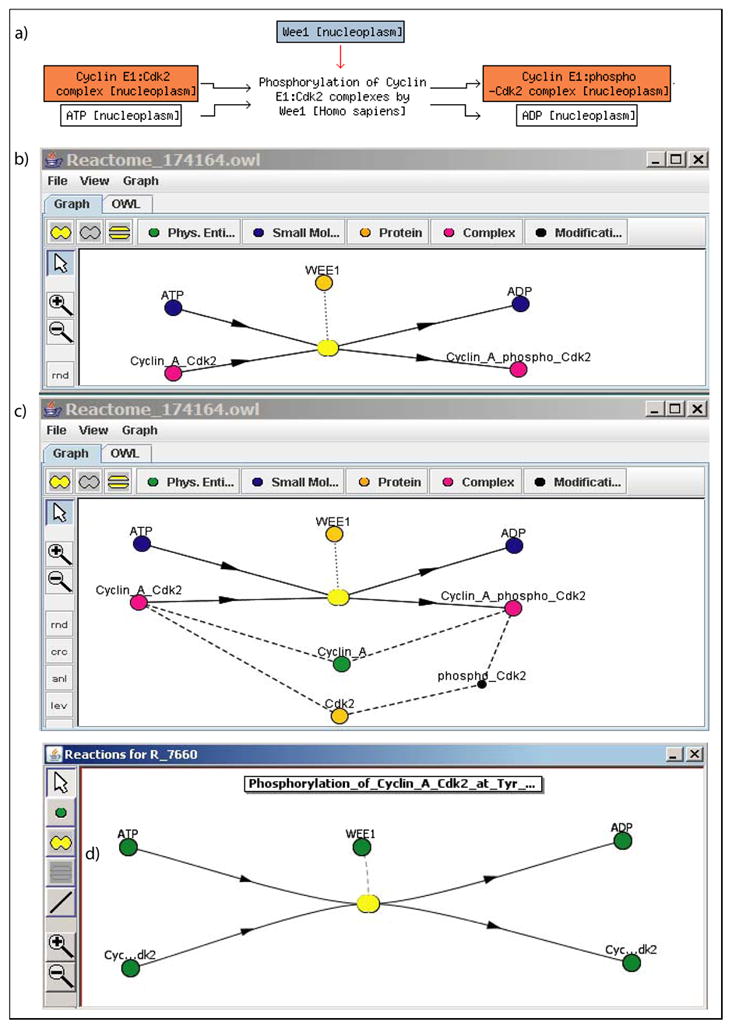
Screenshots of BioPAX modeling user interface prototype. (a) Phosphorylation of Cyclin A:Cdk2 complexes by Wee1 as displayed by Reactome. (b) A compact representation: only species and reactions are shown in the form compliant with the VCell GUI. (c) An extended view, where a user gains an understanding that both complexes contain the same components Cyclin_A and Cdk2; Cdk2 is a protein, which participates in one of these complexes in phosphorylated form. (d) The model exported into VCell, kinetic parameters and rate constants added within VCell framework.
4. Conclusions and future directions
We have introduced a modeling framework that is based on the BioPax ontology. Ontologies provide a great deal of flexibility in data representation, analysis and visualization. Users have the option of modifying or adding new tests for identification of elements of kinetic model and tweak corresponding score functions to address files coming from different sources. The tests themselves and the appropriate models can be validated against known data. Visualization of ontologies can be made very flexible, allowing the user to select which resources are visible and which are hidden, similar to the CytoScape visualization framework. Arbitrary sets of objects can be collapsed and expanded again. The user can decide which kinds of property relationships are represented by graph edges. Currently, project files are stored in a local directory. When such a file-based repository becomes large, searching will be inefficient and one needs to provide a BioPAX-compatible database. This can be accommodated by extending the VCell database schema. Another possible option is to use BioWarehouse [16], which provides specific interfaces for different data sources.
The framework provides explicit specification of each and every molecular species and interactions. However, the data may allow multiple interpretations, such as a given interaction can be applied to several phosphoforms of a protein. An intelligent framework can be used to generate kinetic rules for interactions [17]. These efforts should be concurrent with development of the next level of BioPAX ontology that describes protein modifications.
A highly desirable feature is automated data retrieval and verification using external web-resources. After the user picks elements to be included in a model, the framework should try to infer additional elements (interactions, modifications, kinetic constants) to make a kinetic model complete, and then request this information from external web resources. For example, if the user develops a model involving interactions between proteins A and B, the framework should search local files, the VCell repository and external databases for all data that affect these interactions. Qualitative information, such as reaction catalysts, can be requested from databases like Reactome that provide an API for querying and retrieving BioPAX data over the web. Quantitative information, such as kinetic constants, can be requested from emerging databases of reaction kinetics, such as SABIO-RK (http://sabio.villabosch.de/SABIORK).
The model which is augmented with such quantitative data for simulation purposes will be encoded in SBML format. A critical capability is the ability to preserve the ontology information from the BioPAX format in the SBML format. Initially, this is being implemented by extending the SBML model with all the relevant ancillary Species (that otherwise may not be necessary for performing the actual numerical simulations), and by using the SBML Annotation element (that can encode arbitrary RDF-type data) to hold the BioPAX-specific information. This improved level of BioPAX/SBML translation allows for algorithms and operations specific to ontology-processing tools to also be performed on the SBML files, thus enhancing the reusability of the generated SBML files, as well as allowing reverse generation of qualitative models from quantitative models that were further processed. Ideally, in the long term, a more sophisticated translation mechanism would be highly desirable, by using direct ontology-level mapping to the newly developed SBO framework.
When fully implemented, such capabilities will provide an intelligent data-driven modeling framework that can exploit the growing number of systems biology resources, such as pathway and model repositories, and experimental data repositories.
Contributor Information
Oliver Ruebenacker, Email: oruebenacker@exchange.uchc.edu.
Ion. I. Moraru, Email: moraru@exchange.uchc.edu.
James C. Schaff, Email: schaff@exchange.uchc.edu.
Michael L. Blinov, Email: blinov@exchange.uchc.edu.
References
- 1.Luciano JS. PAX of mind for pathway researchers. Drug Discov Today. 2005;10:937–42. doi: 10.1016/S1359-6446(05)03501-4. [DOI] [PubMed] [Google Scholar]
- 2.Luciano JS, Stevens RD. e-Science and biological pathway semantics. BMC Bioinformatics. 2007;8(Suppl 3):S3. doi: 10.1186/1471-2105-8-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vastrik I, et al. Reactome: a knowledgebase of biological pathways and processes. Genome Biol. 2007:8. [Google Scholar]
- 4.Krummenacker M, Paley S, Mueller L, Yan T, Karp PD. Querying and computing with BioCyc databases. Bioinformatics. 2005;21:3454–5. doi: 10.1093/bioinformatics/bti546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerami EG, Bader GD, Gross BE, Sander C. cPath: open source software for collecting, storing, and querying biological pathways. BMC Bioinformatics. 2006;7:497. doi: 10.1186/1471-2105-7-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Z, Mellor J, Wu J, Yamada T, Holloway D, Delisi C. VisANT: data-integrating visual framework for biological networks and modules. Nucleic Acids Res. 2005;33:W352–7. doi: 10.1093/nar/gki431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hucka M, et al. The Systems Biology Markup Language (SBML): A Medium for Representation and Exchange of Biochemical Network Models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 9.Lloyd CM, Halstead MD, Nielsen PF. CellML: its future, present and past. Progress in Biophys and Mol Biol. 2004;85:433–450. doi: 10.1016/j.pbiomolbio.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Le Novere N, et al. BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006;34:D689–91. doi: 10.1093/nar/gkj092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stromback L, Lambrix P. Representations of molecular pathways: an evaluation of SBML, PSI MI and BioPAX. Bioinformatics. 2005;21:4401–7. doi: 10.1093/bioinformatics/bti718. [DOI] [PubMed] [Google Scholar]
- 12.Stromback L, Jakoniene V, Tan H, Lambrix P. Representing, storing and accessing molecular interaction data: a review of models and tools. Brief Bioinform. 2006;7:331–8. doi: 10.1093/bib/bbl039. [DOI] [PubMed] [Google Scholar]
- 13.Moraru II, Schaff JC, Slepchenko BM, Loew LM. The Virtual Cell: An integrated modeling environment for experimental and computational cell biology. Ann NY Acad Sci. 2002;971:595–6. doi: 10.1111/j.1749-6632.2002.tb04535.x. [DOI] [PubMed] [Google Scholar]
- 14.Damerau FJ. A technique for computer detection and correction of spelling errors. Communications of the ACM. 1964;7:171–176. [Google Scholar]
- 15.Jaro MA. Probabilistic linkage of large public health data files. Stat Med. 1995;14:491–8. doi: 10.1002/sim.4780140510. [DOI] [PubMed] [Google Scholar]
- 16.Lee TJ, Pouliot Y, Wagner V, Gupta P, Stringer-Calvert DW, Tenenbaum JD, Karp PD. BioWarehouse: a bioinformatics database warehouse toolkit. BMC Bioinformatics. 2006;7:170. doi: 10.1186/1471-2105-7-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hlavacek WS, Faeder JR, Blinov ML, Posner RG, Hucka M, Fontana W. Rules for modeling signal-transduction systems. Sci STKE. 2006:re6. doi: 10.1126/stke.3442006re6. [DOI] [PubMed] [Google Scholar]