

Abstract
GABA (γ-aminobutyric acid) is the primary inhibitory neurotransmitter in brain. The fast inhibitory effect of GABA is mediated through the GABAA receptor, a postsynaptic ligand-gated chloride channel. We propose that GABA can act as a ligand chaperone in the early secretory pathway to facilitate GABAA receptor cell surface expression. Forty-two hrs of GABA treatment increased the surface expression of recombinant receptors expressed in HEK 293 cells, an effect accompanied by an increase in GABA-gated chloride currents. In time-course experiments, a 1 hr GABA exposure, followed by a 5 hr incubation in GABA-free medium, was sufficient to increase receptor surface expression. A shorter GABA exposure could be used in HEK 293 cells stably transfected with the GABA transporter GAT-1. In rGAT-1HEK 293 cells, the GABA effect was blocked by the GAT-1 inhibitor NO-711, indicating that GABA was acting intracellularly. The effect of GABA was prevented by brefeldin A (BFA), an inhibitor of early secretory pathway trafficking. Coexpression of GABAA receptors with the GABA synthetic enzyme glutamic acid decarboxylase 67 (GAD67) also resulted in an increase in receptor surface levels. GABA treatment failed to promote the surface expression of GABA binding site mutant receptors, which themselves were poorly expressed at the surface. Consistent with an intracellular action of GABA, we show that GABA does not act by stabilizing surface receptors. Furthermore, GABA treatment rescued the surface expression of a receptor construct that was retained within the secretory pathway. Lastly, the lipophilic competitive antagonist (+)bicuculline promoted receptor surface expression, including the rescue of an secretory pathway-retained receptor. Our results indicate that a neurotransmitter can act as a ligand chaperone in the early secretory pathway to regulate the surface expression of its receptor. This effect appears to rely on binding site occupancy, rather than agonist-induced structural changes, since chaperoning is observed with both an agonist and a competitive antagonist.
Keywords: GABAA receptor, γ-aminobutyric acid, ligand chaperone, endoplasmic reticulum, secretory pathway, GABA transporter, glutamic acid decarboxylase
1. Introduction
The GABAA receptor is an inhibitory neurotransmitter receptor associated with a variety of neurological and psychiatric disorders and is the target of several classes of therapeutic agents [76]. It has been estimated that approximately 30% of synapses in the brain contain GABAA receptors [49]. The receptor mediates the fast inhibitory actions of the ubiquitous neurotransmitter γ-aminobutyric acid (GABA). Upon binding GABA, an integral chloride channel within the receptor is gated, allowing chloride influx and leading to membrane hyperpolarization.
GABAA receptors belong to the cys-loop ligand-gated ion channel family, whose other members include the nicotinic acetylcholine, glycine and 5-HT3 receptors [39]. Members of the cys-loop family of ligand-gated ion channels are pentameric in structure, with each subunit possessing a large extracellular N-terminus, four membrane spanning domains (M1-M4), a large cytoplasmic loop between M3 and M4, and an extracellular C-terminus. At least 17 GABAA receptor subunits have been identified. Assembly of the receptor is defined by discrete oligomerization residues that selectively limit the type of pentameric assemblages [7]. Although many receptor subtypes exist, the predominant receptor subtype in brain is composed of α1β2γ2 subunits, with two copies each of α1 and β2 subunits [43]. Two GABA binding sites are present on each pentamer, with the GABA binding pocket formed at the αβ subunit interface by the subunit N-terminal regions [1].
Dynamic cell surface expression of neurotransmitter receptors, including the GABAA receptor, has been intensely studied in recent years. It is now recognized that the regulation of receptor trafficking is an important mechanism for controlling synaptic efficacy and plasticity [12,28,41,48]. While receptor endocytosis and recycling are established determinants of receptor cell surface expression [37], the roles that receptor biogenesis and secretory pathway trafficking play in maintaining receptor surface populations are largely unexplored. It remains to be determined whether these processes are specifically regulated or are simply default processes governed by subunit translation rates and the endoplasmic reticulum (ER) quality control machinery.
It has been reported that mutation of agonist binding residues on glutamate receptors results in their ER-retention, leading to the speculation that agonist occupancy is required for glutamate receptor biogenesis [11,20,22,42,54,72]. The ER-retention of receptor ligand binding site mutants, however, may reflect terminal misfolding of the receptor subunits due to the mutation, rather than due to the absence of ligand binding. Thus, the hypothesis that neurotransmitter receptor biogenesis may be facilitated by, or require, the binding of neurotransmitter remains to be tested. Here, using recombinant GABAA receptors, we provide the first demonstration, to our knowledge, that a neurotransmitter can act as a ligand chaperone in the endoplasmic reticulum to promote the cell surface expression of its receptor.
2. Results
GABA treatment promotes surface expression of GABAA receptors composed of α1β2γ2LV5 subunits
α1β2γ2L receptors are the predominant receptor subtype in brain [41]. To test whether GABA might act as an ER chaperone to facilitate cell surface expression of this receptor subtype, HEK 293 cells were transiently transfected with α1β2γ2LV5 receptors and treated throughout the 42 hr transfection and expression period with GABA (100 μM). We reasoned that a high concentration of GABA and a long treatment time would aid in obtaining an effective intracellular concentration of GABA and allow time for receptor trafficking from the ER to the cell surface to occur. Three approaches were used to assess receptor surface expression. First, cell surface (nonpermeabilized conditions) and intracellular receptor (permeabilized conditions) populations were labeled using indirect immunofluorescence and visualized by confocal microscopy. Under control conditions (no GABA treatment), cell surface receptors were readily apparent (Fig. 1A). Cells treated with GABA, however, displayed more intense surface staining relative to untreated controls. A quantitative assessment of the GABA effect was conducted using flow cytometry (Table 1). In these experiments, cell surface receptors were detected by anti-V5 immunofluorescence in nonpermeabilized cells. For GABA-treated cells, receptor surface expression increased 75% relative to controls (no GABA treatment), confirming the results of the confocal microscopy experiments. Whole-cell patch-clamp experiments were then performed to provide a functional correlate to the microscopy and flow cytometry experiments. For these experiments, following the 42 hr GABA treatment period, cells were incubated in GABA-free medium for at least 2 hrs prior to electrophysiological recording to allow receptors to recover from any GABA-induced receptor desensitization that may have occurred during GABA treatment. Following this two hr GABA free period, GABA-gated chloride current peak amplitudes were recorded in response to a 2 sec application of 100 μM GABA (Fig. 1B). This high concentration of GABA was chosen to maximally activate the receptors and therefore allow comparison of peak current amplitudes among experimental treatments. In these experiments, cells expressing α1β2γ2LV5 receptors that had been treated with GABA for 42 hrs prior to electrophysiological recording displayed a greater than three fold increase in GABA peak amplitude response relative to untreated cells. These results are consistent with the confocal microscopy and flow cytometry findings that a 42 hr GABA treatment increases receptor cell surface expression.
Figure 1. GABA treatment promotes surface expression of GABAA receptors composed of α1β2γ2L subunits.

A) HEK 293 cells were transfected with α1, β2, and γ2LV5 subunit cDNAs and incubated throughout the transfection and expression period in the absence or presence of 100 μM GABA. Forty-two hrs post-transfection, cell surface and intracellular GABAA receptor populations were labeled by indirect immunofluorescence using an anti-V5 antibody and imaged by confocal microscopy (images representative of 20 independent experiments each performed in triplicate). B) The whole-cell patch-clamp technique was used to measure GABA-gated chloride currents. Following a 42 hr GABA incubation period, GABA-containing medium was removed from culture dishes and replaced with GABA-free medium for at least two hrs prior to electrophysiological recording to avoid receptor desensitization from prolonged GABA treatment. GABA-gated chloride current peak amplitudes were measured in response to GABA (100 μM) applied with a solenoid-controlled superfusion system. Asterisk indicates that GABA-gated chloride current peak amplitudes were significantly different between cells incubated in GABA for 42 hrs. vs. control (*, p ≤ 0.01, unpaired t-test).
Table 1.
Flow cytometry experiments in cells expressing α1β2γ2L receptors.
| Condition | Surface Receptors (% control) |
|---|---|
| Control | 100 |
| GABA | 175 ± 15* |
|
| |
| Control | 100 |
| GABA 1 hr | 163 ± 12* |
| 3 hrs | 190 ± 21* |
| 6 hrs | 347 ± 15** |
|
| |
| Control | 100 |
| GABA | 175 ± 14*,† |
| GABA + NO−711 | 79 ± 8 |
|
| |
| Control | 100 |
| GABA | 145 ± 4*,† |
| GABA + BFA | 84 ± 3 |
|
| |
| Control | 100 |
| GAD 67 cotransfection | 224 ± 44* |
Data are shown as the average receptor cell surface expression expressed as a percent of control ± SEM, n ≥ 3 for each experiment with each replicate conducted in triplicate. Data were compared via one-tailed, unpaired t-test, assuming unequal variances.
p ≤ 0.05 control vs. GABA
p ≤ 0.005 control vs. GABA
p ≤ 0.005 GABA vs. GABA + NO−711
GABA vs. GABA + BFA
Short incubation times are sufficient for the GABA effect
To determine if a shorter GABA incubation time would promote receptor cell surface expression (CSE), HEK 293 cells were transfected with α1β2γ2LV5 subunits and allowed to express receptor subunits in the absence of GABA for 42 hrs. Forty-two hrs post-transfection, cells were incubated in medium containing either vehicle or GABA (100 μM) for 1, 3 or 6 hr. The GABA containing medium was then replaced with GABA-free medium and the cells were returned to the incubator for 5, 3 or 0 hrs, respectively to allow the receptors time to traffic through the secretory pathway. The CSE of the receptor was then assessed. GABA treatments of increasing duration produced increasing levels of receptor surface expression (Fig. 2, Table 1). GABA treatment times of less than 1 hr did not result in an increase in receptor CSE (data not shown).
Figure 2. Time-course of GABA effect.

HEK 293 cells were transfected with α1β2γ2LV5 subunit cDNAs. At forty-two hrs post-transfection, the cells were incubated at 37°C in GABA (100 μM) for 1, 3 or 6 hrs. Following this incubation, the GABA-containing medium was removed and cells were incubated at 37°C in GABA-free medium for an additional 5, 3 or 0 hrs, respectively. Cells were then labeled for receptor surface and intracellular expression and visualized by confocal microscopy. Images are representative of 4 independent experiments each performed in triplicate.
Coexpression of GABAA receptors with the GABA transporter GAT-1
Because GABA treatment times of less than 1 hr did not promote receptor CSE, we speculatedthat GABA uptake into HEK 293 cells is inefficient since these cells do not contain high-affinity GABA transporters [66] and zwitterionic GABA would not readily cross the lipid bilayer. Thus, we expressed α1β2γ2LV5 receptors in HEK 293 cells stably expressing the GABA transporter rGAT-1 (rGAT-1HEK 293 cells). In these cells, a GABA incubation period of 15-30 min, followed by a GABA free incubation period of 5.5 hrs, was sufficient to produce an increase in receptor CSE (Fig 3, Table 1). In the rGAT-1HEK 293 cells, the GABA effect was blocked by the GAT-1 inhibitor NO-711, demonstrating that GABA uptake by a high-affinity GABA transporter is sufficient for the GABA effect to occur.
Figure 3. The GABA effect is prevented by the GAT-1 transporter blocker NO-711 in cells expressing rGAT-1.
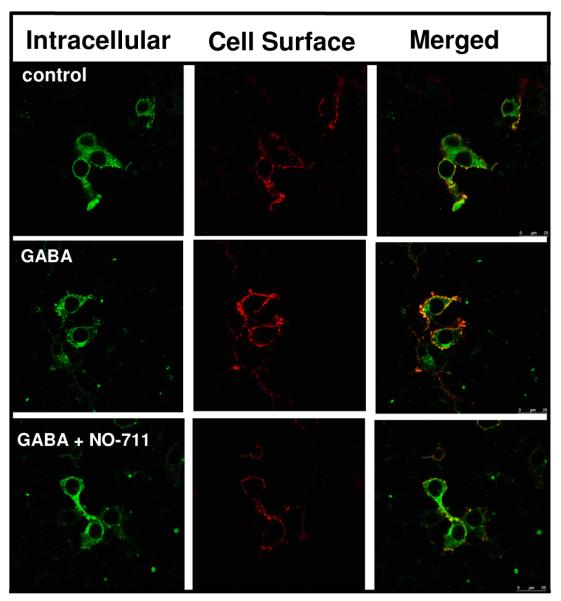
HEK 293 cells stably transfected with rat GAT-1 (rGAT HEK 293 cells) were transfected with α1β2γ2LV5 receptor subunit cDNAs. Forty-two hrs following transfection, rGAT HEK 293 cells were incubated in the absence or presence of GABA (100 μM) or GABA (100 μM) + NO-711 (100 μM) for 15-30 min. The medium was then replaced with drug-free medium and the cells were returned to the incubator for 5.5 hrs. Surface and intracellular receptor populations were then labeled using indirect immunofluorescence and visualized by confocal microscopy. Images are representative of 4 independent experiments each performed in triplicate.
The GABA effect is blocked by brefeldin A
To test our hypothesis that GABA acts in the endoplasmic reticulum (ER) to drive surface expression, we next determined if brefeldin A (BFA), which disrupts trafficking in the early secretory pathway [47], would prevent the GABA effect. Cells transfected with α1β2γ2LV5 subunits were allowed to express receptor in the absence of GABA for 42 hrs. At 42 hrs, cells were incubated at 37°C with either vehicle control (methanol. 0.01%), GABA (100 μM) + methanol or GABA (100 μM) + BFA (5 μg/ml) for six hrs (Fig. 4, Table 1). A six hr treatment with GABA significantly increased receptor CSE. This effect was blocked by BFA, suggesting that the GABA effect occurs in the early secretory pathway. The brefeldin experiments were conducted using a short GABA treatment period (6 hrs) since long incubations in BFA (i.e., >15 hrs) were cytotoxic.
Figure 4. The GABA effect is blocked by brefeldin A.
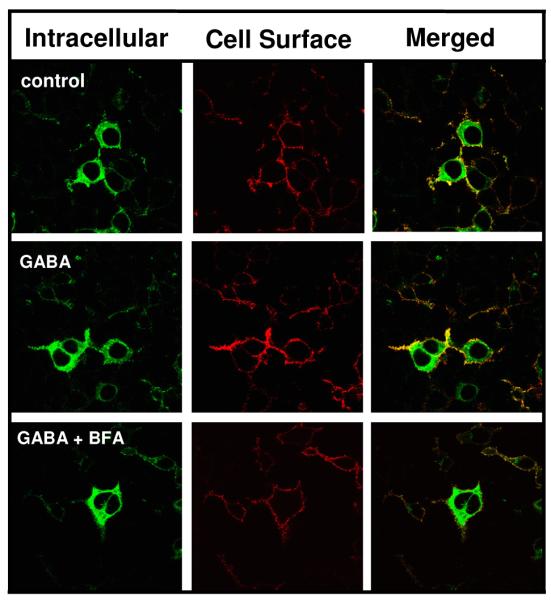
Forty-two hrs post-transfections, HEK 293 cells expressing α1β2γ2LV5 receptors were incubated at 37°C with either vehicle (methanol), GABA (100 μM) + methanol or GABA (100 μM) + BFA (5 μg/ml) for 6 hrs. Indirect immunofluorescence was then used to label cell surface and intracellular GABAA receptor populations using an anti-V5 antibody, and receptors were then imaged by confocal microscopy. Images are representative of 4 independent experiments each performed in triplicate.
Coexpression of glutamic acid decarboxylase 67 (GAD67) with GABAA receptors increases receptor cell surface expression
We next determined whether coexpression of α1β2γ2LV5 receptors with GAD67, a biosynthetic enzyme for GABA [68]would increase receptor surface expression (Fig. 5, Table 1). Cells coexpressing receptor and GAD67 displayed receptor cell surface expression that was more than twice that observed in controls (receptor + empty vector).
Figure 5. Coexpression of glutamic acid decarboxylase 67 (GAD67) with GABAA receptors increases receptor surface expression.
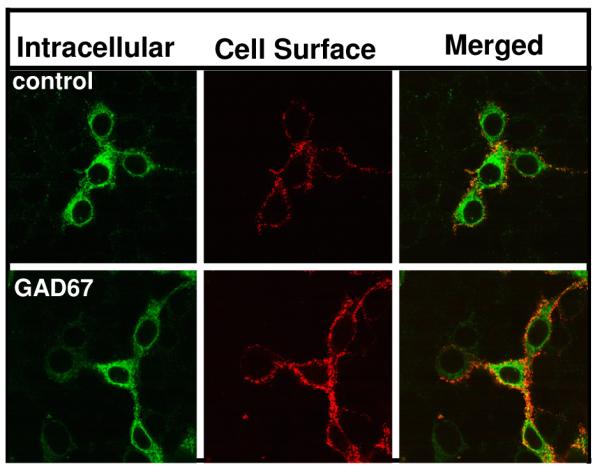
HEK 293 cells were transfected with α1β2γ2LV5 receptors alone or together with either empty vector or GAD67 cDNA. Forty-two hrs post-transfection, indirect immunofluorescence was used to label cell surface and intracellular GABAA receptor populations. Receptors were then visualized by confocal microscopy. Images are representative of 4 independent experiments each performed in triplicate.
The GABA effect is specific for GABAA receptors
To determine if the GABA effect is specific for GABAA receptors rather than a general effect on secretory pathway proteins, we tested whether GABA could act as a chaperone for 5-HT3a receptors, another member of the cys-loop ligand-gated ion channel family. We have previously examined the trafficking of V5-tagged 5-HT3 receptors [80]. Fig. 6 shows that 5-HT3a receptors are expressed, albeit weakly, at the cell surface 42 hrs post-transfection. Incubation of cells expressing these receptors with GABA (100μM) during the 42 hrs transfection/expression period did not result in an increase in receptor surface levels. Thus, GABA treatment does not non-specifically promote the maturation/forward trafficking of other secretory pathway proteins. Interestingly, a 42 hr serotonin (100 μM) treatment resulted in a robust increase in the surface expression of 5-HT3a receptors. While serotonin promoted surface expression of 5-HT3a receptors, it did not facilitate the surface expression of GABAA receptors (data not shown).
Figure 6. GABA does not promote the surface expression of 5-HT3 receptors.

HEK 293 cells were transiently transfected with V5-tagged mouse serotonin type 3a receptor (5-HT3a) cDNA and treated with GABA (100 μM) or serotonin (100 μM) throughout the 42 hr. transfection/expression period. Surface and intracellular 5-HT3a receptor populations were then labeled using indirect immunofluorescence and visualized by confocal microscopy. Images shown are representative of 3 independent experiments performed in triplicate.
Mutation of GABA binding residue α1F65 prevents the GABA effect
To determine if the effect of GABA requires an intact GABA binding site, receptors containing the GABA binding site mutant α1(F65A) were used. α1F65 is photoaffinity-labeled by the GABA agonist [3H]muscimol [67] and is critical for GABA activation of the receptor [65]. Cells expressing α1V5β2γ2L or α1(F65A)V5β2γ2L receptors were incubated throughout the transfection/expression period (42 hrs) in the absence or presence of GABA (100μM). Surface levels of wild type vs. mutant receptors were then assessed by immunofluorescence and flow cytometry. While GABA treatment increased surface expression of α1V5β2γ2L receptors, GABA had no effect on α1(F65A)V5β2γ2L receptors (Fig 7, Table 2). These results indicate that an intact GABA binding site is required for the GABA chaperoning effect. Of note, surface expression of α1(F65A)V5β2γ2L receptors was lower than that of wild type receptors (Table 2), indicating that mutation of an agonist binding site residue may affect receptor biogenesis, similar to findings for glutamate receptor agonist binding site mutants [11,20,42,54,72].
Figure 7. Mutation of the GABA binding residue α1(F65A) prevents the GABA effect.

HEK 293 cells were transfected with either α1V5β2γ2L or α1V5(F65A)β2γ2L subunit cDNAs. Cells were incubated throughout the transfection and expression period with or without 100 μM GABA. Forty-two hrs post-transfection, cell surface and intracellular GABAA receptor populations were labeled by indirect immunofluorescence and imaged by confocal microscopy. Images are representative of 5 independent experiments each performed in triplicate.
Table 2.
Mutation of GABA binding site residue α1(F65A) prevents GABA-induced increases in receptor cell surface expression.
| Condition | Surface Receptors (% of control α1β2γ2L receptors) |
|---|---|
| α 1 β 2 γ 2L | |
| Control | 100 |
| GABA | 146 ± 7* |
|
| |
| α1(F65A)β2γ2L | |
| Control | 14 ± 5† |
| GABA | 19 ± 6 |
Flow cytometry data are shown as the average receptor cell surface expression expressed as a percent of control ± SEM, n ≥ 3 for each experiment with each replicate conducted in triplicate. Data were compared via one-tailed, unpaired t-test, assuming unequal variances.
p ≤ 0.05 control vs. GABA for α1β2γ2L receptors
p ≤ 0.005 control α1β2γ2L vs. control α1(F65A)β2γ2L receptors
GABA promotes surface expression of secretory pathway-retained GABAA receptors
For our studies, we constructed extracellular C-terminal V5-tagged α1, β2 and γ2L subunits to label surface receptors in nonpermeabilized cells. The C-terminus was chosen to avoid residues involved in ligand binding, glycosylation and oligomerization present on the extracellular N-terminus. For the α1V5, β2V5 and γ2LV5 subunits, the trafficking/function of the subunits were normal in the context of α1β2γ2L receptors, regardless of which subunit had the V5 tag. However, we discovered that the β2V5 subunit in the context of α1β2V5 receptors, largely prevented surface expression of a receptor subtype (α1β2) that is otherwise readily expressed at the cell surface [14]. One possible explanation for this finding was that α1β2V5 receptors were surface competent but unstable at the cell surface, i.e., the V5 tag caused rapid endocytosis. To address this possibility, we took advantage of an endocytically-compromised receptor in which mutation of a dileucine endocytic signal on the receptor β2 subunit impairs receptor endocytosis [27]. Both confocal microscopy and flow cytometry experiments indicated that the surface expression of α1β2V5 receptors was not significantly different from α1β2(LL/AA)V5 receptors (Fig. 8, Table 3) indicating that rapid endocytosis did not account for the very low surface levels of α1β2V5 receptors. These experiments strongly suggested that low CSE of α1β2V5 receptors was due to secretory pathway retention. Since the surface expression of a variety of ER-retained receptors can be rescued by ligand chaperones [13], we examined whether GABA treatment could rescue the CSE of α1β2V5 receptors. GABA treatment was highly effective in promoting the surface expression of α1β2V5 receptors (Fig. 8, Table 3). GABA treatment also increased the surface expression of α1β2 (LL/AA)V5 receptors. The CSE of α1β2 (LL/AA)V5 receptors was increased by GABA nearly 19 fold (vs. 3 fold for α1β2V5 receptors), suggesting that once rescued from secretory pathway retention by GABA, the endocytically-compromised receptor mutant accumulates at the surface.
Figure 8. GABA promotes surface expression of GABAA receptors composed of α1β2 and α1β2(LL/AA) subunits.

HEK 293 cells were transfected with α1β2V5 or α1β2V5(LL/AA) subunit cDNAs. Cells were incubated throughout the transfection and expression period in the absence or presence of 100 μM GABA. Forty-two hrs post-transfection, cell surface and intracellular GABAA receptor populations were labeled by indirect immunofluorescence using an anti-V5 antibody and imaged by confocal microscopy. Images representative of at least 5 independent experiments each performed in triplicate.
Table 3.
GABA treatment promotes cell surface expression of α1β2 and α1β2 (LL/AA) receptors.
| Condition | Surface Receptors (% of control α1β2 receptors) |
|---|---|
| α 1 β 2 | |
| Control | 100 |
| GABA | 311 ± 26* |
|
| |
| α1β2(LL/AA) | |
| Control | 98 ± 1 |
| GABA | 1888 ± 294† |
Flow cytometry data are shown as the average receptor cell surface expression expressed as a percent of control ± SEM, n ≥ 3 for each experiment with each replicate conducted in triplicate. Data were compared via one-tailed, unpaired t-test, assuming unequal variances.
p ≤ 0.01 control vs. GABA for α1β2 receptors
p ≤ 0.05 GABA-treated α1β2 (LL/AA) vs. GABA-treated α1β2 receptors
The lipophilic antagonist (+)bicuculline promotes surface expression of GABAA receptors
For other receptors, membrane permeant antagonist drugs have been reported to have ligand chaperone activity [79], thus we next performed experiments with the lipophilic competitive antagonist (+)bicuculline on α1β2γ2LV5 and α1β2V5 receptors. (+)Bicuculline has an octanol/water partition coefficient (indicator of hydrophobicity) of 2.62 (calculated at www.molinspiration.com), which is similar to drugs such as benzodiazepines that are highly lipophilic and readily cross cell membranes. For these experiments (+)bicuculline was dissolved in DMSO, a solvent known to act as a chemical chaperone for other receptors [51]. In these experiments, we observed that 0.01% DMSO itself promoted a small but significant increase in α1β2γ2LV5 receptor CSE when compared to control (Fig. 9; Table 4), thus, DMSO may act as a chemical chaperone on GABAA receptors. Treatment of cells with (+)bicuculline increased surface expression when compared to DMSO treatment (Fig. 9, Table 4) indicating that (+)bicuculline promotes receptor surface expression. Similar results were observed using α1β2V5 receptors. Because the α1β2V5 receptors are likely retained in the secretory pathway under control conditions (see above), these results indicate that (+)bicuculline acts as a ligand chaperone and suggests that occupancy of the ligand binding site, and not structural movements associated with agonist binding, underlie ligand chaperoning at the GABAA receptor.
Figure 9. The lipophilic competitive antagonist (+)bicuculline promotes surface expression of GABAA receptors.

HEK 293 cells expressing α1β2V5 or α1β2γ2LV5 receptors were incubated throughout the transfection and expression period in the absence or presence of either DMSO (0.1%) or (+)bicuculline (100 μM) dissolved in DMSO. Forty-two hrs post-transfection, indirect immunofluorescence was used to label cell surface and intracellular GABAA receptor populations using an anti-V5 antibody and the cell were then imaged by confocal microscopy. Images are representative of 4 independent experiments each performed in triplicate.
Table 4.
(+)Bicuculline treatment promotes receptor cell surface expression of α1β2 and α1β2γ2L receptors.
| Condition | Surface Receptors (% control) |
|---|---|
| α 1 β 2 | |
| Control | 100 |
| DMSO | 142 ± 9* |
| (+)Bicuculline | 308 ± 4** |
|
| |
| α 1 β 2 γ 2L | |
| Control | 100 |
| DMSO | 115 ± 1* |
| (+)Bicuculline | 164 ± 6** |
Flow cytometry data are shown as the average receptor cell surface expression expressed as a percent of control ± SEM, n ≥ 3 for each experiment with each replicate conducted in triplicate. Data were compared via one-tailed, unpaired t-test, assuming unequal variances.
p ≤ 0.05 DMSO vs. control α1β2 receptors
DMSO vs. control α1β2γ2L receptors
p ≤ 0.005 (+)Bicuculline vs. DMSO α1β2 receptors
(+)Bicuculline vs. DMSO α1β2γ2L receptors
3. Discussion
In this study GABA treatment of HEK 293 cells expressing GABAA receptors resulted in an increase in receptor surface expression. Several lines of evidence indicate that this effect is due to GABA acting as a ligand chaperone. First, the GABA effect is blocked by brefeldin A, a compound that inhibits trafficking in the early secretory pathway. Second, the GABA effect is facilitated by expression of the GABA transporter GAT-1 and is blocked by the GAT inhibitor NO-711 showing that GABA uptake into the cell is important for the GABA effect. Third, coexpression of the receptor with the GABA synthetic enzyme GAD67 results in an increase in receptor surface expression experiments. Lastly, we show that GABA can rescue the surface expression of a receptor construct that is retained in the secretory pathway. The chaperoning action of GABA is specific for GABAA receptors and not a general effect on secretory pathway proteins since GABA does not chaperone 5-HT3a receptors. Furthermore, GABA fails to promote the surface expression of GABAA receptors in which a key GABA binding residue is mutated.
Evidence that receptor ligands can act in the ER to facilitate receptor biogenesis comes from studies of membrane permeant drugs called “pharmacological chaperones”, “pharmacochaperones” or “pharmacoperones” [3,4,13,29,75] which can facilitate the biogenesis and surface expression of a variety of receptors and ion channels including δ opioid [52], β1-adrenergic [33], dopamine D4 [73], and nicotinic acetylcholine (nACh) receptors [35,59]). In the latter case, drugs that chaperone nACh receptors have been termed SePhaChARNS for “selective pharmacological chaperoning of acetylcholine receptor number and stoichiometry” [40]. In the future, it will be important to examine whether the neurotransmitters which activate these receptors can also act as ligand chaperones, as this may represent a physiological mechanism to post-translationally regulate receptor number.
It remains to be determined whether GABA acts as a ligand chaperone in neurons. This would require a cytoplasmic GABA pool near the ER where receptor biogenesis occurs. GABA is synthesized by GAD65 and GAD67 isoforms, which produce “transmitter” and “metabolic” GABA, respectively [53]. While GAD65 has a restricted synaptic distribution, GAD67 is present throughout both dendrites and soma, and would therefore be favorably positioned for the production of “chaperone” GABA. Because GABA synthesis and metabolism are linked to the Krebs cycle, a particularly exciting possibility is that metabolic activity may affect GABAA receptor biogenesis through a GABA chaperone mechanism. Alternatively, “chaperone” GABA could derive from synaptically-released GABA. In this regard, GAT-2 is localized to both dendritic plasma and ER membranes in extrasynaptic regions [15,82] and GABAA receptor mRNA is in found in dendrites [18] raising the possibility that GABAA receptors may be synthesized dendritically with their biogenesis regulated by GAT-2 mediated uptake of synaptically-released GABA.
For GABA to function as a ligand chaperone to increase receptor biogenesis, an ER pool of excess receptor subunits would need to exist. The synthesis of ion channels in the ER, including cys-loop receptors, is inefficient, with only ~ 30% of synthesized subunits incorporated into mature, surface competent receptors/channels [23,44,61]. Thus, 70% of synthesized subunits are degraded without apparent use. It is unclear whether these subunits are degraded due to terminal misfolding, or are properly folded but for unknown reasons fail to be incorporated into pentamers. The observation that proteasome inhibitors increase nACh receptor assembly and surface expression [10] suggests that some percentage of subunits that would otherwise be degraded are structurally competent and not terminally misfolded. Thus, the ER may act as a reservoir for structurally competent receptor subunits or higher order oligomers. We propose that GABA acts as a ligand chaperone in the ER, facilitating the use of “excess” subunits in a reserve pool. Because different GABAA receptor subtypes exist in a single neuron (synaptic vs. extrasynaptic), it is possible that GABA chaperoning may favor the biogenesis of certain receptor subtypes. Such receptor subtype selectivity has been demonstrated for nicotine chaperoning of nACh receptors [71,74].
A variety of studies have investigated the mechanism(s) by which ligand chaperoning occurs but detailed structural mechanisms are lacking. It is generally acknowledged that ligand chaperones act as “maturational enhancers” that facilitate protein folding, thus stabilizing native conformations [5,16,45,46,52,56]. It has been speculated that this occurs when ligand binding decreases the energy of activation required for native folding [9] and/or stabilizes conformations within the hydrophobic core [52]. Such chaperone-induced stabilization can release receptors from ER quality control proteins [38,56] and promote the assembly of heteromeric receptors [46]. While the mechanism of chaperoning remains to be understood at the structural level, homology modeling of liganded vasopressin V2 receptor transmembrane helix mutants suggests that certain antagonists can restore proper transmembrane helix-helix interactions [79]. For GnRH receptor mutants, a ligand-mediated bridge is proposed to substitute for a salt bridge that is present in wild type but not mutant receptors [29].
Regarding GABAA receptors, GABA chaperoning must occur subsequent to the formation of the αβ subunit interface, since the GABA binding pocket is formed at this interface [1]. Thus, both α and β subunits must each achieve a folding competency that favors αβ subunit oligomerization prior to any chaperoning step. The binding of GABA to the αβ subunit interface could promote subsequent oligomerization steps leading to pentamerization. Alternatively, GABA could bind to existing pentamers releasing them from the ER quality control system or exposing export signals. Whatever the mechanism, GABA chaperoning does not require structural changes association with receptor activation since we demonstrate that the lipophilic competitive antagonist (+)bicuculline acts as a ligand chaperone.
Our data in rGAT-1HEK 293 cells indicate that active transport of GABA into the cell can mediate the chaperone effect. We further propose that GABA is actively transported into the ER lumen. To our knowledge, it is not known whether neurotransmitter transporters can transport neurotransmitters into the ER, however, membrane vesicles prepared from cells expressing ER-retained GAT-1 mutants show that ER-retained transporters can transport [3H]GABA with Km and Vmax values similar to wild type surface GAT-1 transporters [62]. The transport of GABA into the ER lumen would require that the transporter work in reverse a manner. It is well-known that GATs at the plasma membrane can operate in a reverse mode to release GABA into the extracellular space [2,55,66,77,78]. Based on an ER luminal membrane potential of −85 [8] and estimated cytoplasmic concentrations of 15 mM sodium, 10 mM chloride and 2 mM GABA [50,57,58,63,81], we have used the GABA transporter reversal potential calculator of Sepehr Eskandari (California State Polytechnic University, Pomona) http://www.csupomona.edu/ ~seskandari/physiology/physiological_calculators/gat_v_rev.html to determine if electrical and chemical driving forces would be sufficient for GAT to reverse transport GABA into the ER lumen. Even at osmotically untenable intraluminal sodium and chloride concentrations of 150 mM, the intraluminal concentration of GABA is calculated to be 32 μM, a concentration within the range of binding to even low affinity GABAA receptors. Thus, our findings suggest that neurotransmitter transporters may have a heretofore unrecognized functional role in the ER. Although we conclude that expression of GAT-1 increases GABAA receptor cell surface expression via a GABA chaperoning mechanism, it could be argued that a protein/protein interaction between GAT-1 and the GABAA receptor facilitates receptor surface expression. In this regard, a direct interaction between the dopamine transporter (DAT) and the dopamine D2 receptor increases DAT surface expression [36]. Several lines of evidence argue against GAT-1 directly facilitating GABAA receptor expression through a protein/protein interaction. First, coexpression of GABAA receptors and GAT-1 does not result in an increase in receptor surface expression unless GABA is present. While it is possible that the presence of GABA could promote such an interaction we consider this to be unlikely and note that the DAT/D2 interaction is agonist independent [36]. Second, the GAT-1 blocker NO-711 prevents the GABA effect indicating that transport of GABA appears necessary for the GABA effect.
Ligand chaperones have therapeutic potential for treating a variety of diseases such as cystic fibrosis, retinitis pigmentosa, and long QT syndrome for which recognized mutations in channels and receptors cause ER retention [4]. For example, mutations in the vasopressin type 2 (V2) receptor are causative for approximately 90% of nephrogenic diabetes insipidus cases. A subset of these mutants complete translation but are misfolded and remain within the ER. Membrane permeant V2 receptor ligands access the ER lumen, bind to mutant V2 receptors, presumably stabilizing a favorable tertiary protein conformation, thus promoting receptor surface expression [5,30]. Interestingly, once at the cell surface these mutant receptors display normal functional properties. Most recently, nicotine has been shown to act as a ligand chaperone to normalize nACh receptor subunit stoichiometry for nACh receptors that carry mutations linked to autosomal dominant nocturnal frontal lobe epilepsy [69]. For the GABAA receptor, several epilepsy-associated subunit missense and truncation mutants are retained within the ER including a γ2 subunit R43Q missense mutation [6,24,31,60], γ2 subunit truncation mutation Q351 [25], δ subunit susceptibility variants E177A and R220H [17,19,21,34] and α1 subunit mutant A332D [17,21,34]. These GABAA receptor channelopathies are due to protein misfolding and are potential targets for rescue by ligand chaperones. In light of our finding that GAD67 expression increases receptor surface levels, a potential therapeutic approach for the rescue of epilepsy-associated receptor mutants might be gene therapy with AAV-GAD67, which is currently in clinical trials for the treatment of Parkinson’s disease [32].
In conclusion, we have demonstrated that the neurotransmitter GABA acts as a ligand chaperone in the early secretory pathway to promote GABAA receptor cell surface expression. The ability of a neurotransmitter to act as ligand chaperone to regulate the biogenesis of its own receptor may provide a receptor-specific, post-translational mechanism for controlling the surface expression of newly-synthesized receptors.
4. Experimental Procedures
Materials
Human GABAA receptor subunits in pcDNA3.1 were a gift from Paul Whiting (Merck, Sharp and Dohme, Essex). α1, β2, γ2L subunits were tagged at their C-termini with the V5 epitope (GKPIPNPLLGLDST) by in-frame insertion of subunit coding sequence into the multiple cloning site of pcDNA3.1V5-His A using the restriction sites Xho/XbaI, EcoRI/XbaI, HindIII/BamHI, respectively. Following insertion, all subunit clones were sequenced in their entirety. Glutamic acid decarboxylase in pCMV6 and pEGFP-N1 were obtained from Origene (Rockville, MD) and Clontech (Mountain View, CA), respectively. HEK 293 cells were purchased from ATCC (Manassas, VA). rGAT-1HEK cells were a gift from Harald Sitte (Medical University of Vienna, Austria). Mouse anti-V5 antibody and goat anti-mouse secondary antibodies conjugated to Alexa 488 and Alexa 555 were obtained from Invitrogen (Carlsbad, CA). The anti-V5 antibody recognizes an epitope (shown above) in the P and V proteins of the paramyxovirus SV5 [70]
HEK 293 cell culture and transfection
HEK 293 and rGAT-1HEK cells were plated (1 × 105) on either 35 mm poly-L-lysine coated glass bottom insert dishes (MatTek Corp., Ashland, MA) for imaging experiments or plastic dishes for flow cytometry and incubated overnight at 37°C in a humidified incubator with 5% CO2/95% air. Cells were then transfected with a total of 0.6 μg/dish of plasmid DNA using the calcium phosphate method as previously described [26].
Immunofluorescence labeling/confocal microscopy
To label cell surface receptors, living HEK 293 cells were incubated on ice in 100 μl of HEPES buffer (HEPES 25 mM, NaCl 140 mM, KCl 5.4 mM, CaCl2 1.8 mM, glucose 15 mM, pH=7.4) containing mouse anti-V5 antibody (1:100) for 1 hr, followed by a 1 hr incubation on ice with 1 ml of buffer containing an Alexa 555-conjugated goat anti-mouse antibody (1:1000). Cells were then fixed with 4% paraformaldehyde, permeabilized with Triton X-100 (0.25%) and blocked with fetal bovine serum and bovine serum albumin for 1 hr at room temperature. Intracellular receptors were then labeled with the anti-V5 antibody (1:100) for 1 hr, followed by a 1 hr incubation at room temperature with 1 ml of buffer containing an Alexa 488-conjugated rabbit anti-mouse antibody (1:1000). For fluorescence confocal microscopy, a Bio-Rad MRC-1024 laser scanning system with an argon/krypton laser was used (Bio-Rad, Hercules, CA). Alexa 488 was excited at 488 nm and the emission light collected using a 522/30 emission filter. For Alexa 555 detection, a 543 nm excitation wavelength was used and the emission light collected at 600/25 nm. A 60X objective was used to collect images. Laser intensity, photomultiplier gain, and iris were optimized for each set of experiments but kept constant within each experiment. Images were captured using LaserSharp MRC-1024 software. Each experiment was performed in triplicate.
Flow cytometry
Cell surface receptors in HEK 293 cells were labeled as describe above. Cells were then gently lifted from the dishes using PBS containing EDTA (5 mM), 500 μl of cell suspension was added to 200 μl of 4% paraformaldehyde [64], and fluorescence was analyzed by flow cytometry. Data were collected using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) made available through the Research Core Facility at the Louisiana State University Health Sciences Center-Shreveport. The FACSCalibur has an argon 488nm laser as well as a 635nm red diode laser. Data analysis was performed using CELLQuest software (BD Biosciences). Dead cells were excluded from analysis by forward and 90 degree laser light scatter properties, and a minimum of 10,000 events were collected for each sample. Spectral overlap between the fluorochromes was compensated electronically based on single-color control samples. Each experiment was performed in triplicate.
Whole-cell patch-clamp experiments
Whole-cell patch-clamp recordings were performed on HEK 293 cells expressing recombinant GABAA receptors. Patch electrodes were made from 1.5-mm outer diameter thin-walled borosilicate glass (World Precision Instruments, Sarasota, FL) on a Flaming/Brown micropipette puller (Sutter Instruments, Novato, CA) to yield a tip resistance of 2-4 MΩ. Coverslips containing HEK 293 cells were placed in a recording chamber (1ml volume) on the stage of a Nikon Diaphot inverted microscope and superfused with external solution at a rate of 6 ml/min (gravity flow). GABA-gated currents were recorded at room temperature (20-25°C) at a holding potential of 0 mV in a bath containing (in mM): NaCl 140, KCl 5, CaCl2 1, MgCl2 1, HEPES 10 and glucose 24, pH 7.4, 320 mOsm/kg H2O. The pipet solution contained (in mM): K-gluconate 80, N-methyl-D-glucamine chloride 40, Cs4BAPTA 5 and Mg-ATP 5. The ATP regeneration system Tris phosphocreatine (20 mM) and creatine kinase were added to minimize GABA rundown. GABA application (100 μM) was accomplished with a solenoid-controlled superfusion system positioned within 50 microns of the cell that released GABA for 2 sec. Currents were recorded using an Axopatch 1D amplifier (Axon Instruments) filtered at 2 kHz (four-pole Bessel filter) and detected at 10 kHz using pClamp 5.51 acquisition software (Axon Instruments, Foster City, CA).
Acknowledgements
The authors wish to thank Harald Sitte (Medical University of Vienna) for the gift of the rGAT-1HEK 293 cells, as well as George Richerson (Yale University), Aurelio Galli (Vanderbilt University) and Harald Sitte for helpful discussions. This work was supported by National Institute of Mental Health grant R01MH062640 to NJL.
Footnotes
Classification terms: GABA receptors, GABA, ligand-gated ion channels
Section: Cellular and Molecular Biology of Nervous Systems
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Amin J, Weiss DS. GABAA receptor needs two homologous domains of the beta-subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–9. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- [2].Attwell D, Barbour B, Szatkowski M. Nonvesicular release of neurotransmitter. Neuron. 1993;11:401–7. doi: 10.1016/0896-6273(93)90145-h. [DOI] [PubMed] [Google Scholar]
- [3].Bernier V, Bichet DG, Bouvier M. Pharmacological chaperone action on G-protein-coupled receptors. Curr Opin Pharmacol. 2004;4:528–33. doi: 10.1016/j.coph.2004.08.001. [DOI] [PubMed] [Google Scholar]
- [4].Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004;15:222–8. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- [5].Bernier V, Lagace M, Lonergan M, Arthus MF, Bichet DG, Bouvier M. Functional rescue of the constitutively internalized V2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059. Mol Endocrinol. 2004;18:2074–84. doi: 10.1210/me.2004-0080. [DOI] [PubMed] [Google Scholar]
- [6].Bianchi MT, Song L, Zhang H, Macdonald RL. Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci. 2002;22:5321–7. doi: 10.1523/JNEUROSCI.22-13-05321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bollan K, Robertson LA, Tang H, Connolly CN. Multiple assembly signals in gamma-aminobutyric acid (type A) receptor subunits combine to drive receptor construction and composition. Biochem Soc Trans. 2003;31:875–9. doi: 10.1042/bst0310875. [DOI] [PubMed] [Google Scholar]
- [8].Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium. 2005;38:303–10. doi: 10.1016/j.ceca.2005.06.010. [DOI] [PubMed] [Google Scholar]
- [9].Chen Y, Liu-Chen LY. Chaperone-like effects of cell-permeant ligands on opioid receptors. Front Biosci. 2009;14:634–43. doi: 10.2741/3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Christianson JC, Green WN. Regulation of nicotinic receptor expression by the ubiquitin-proteasome system. Embo J. 2004;23:4156–65. doi: 10.1038/sj.emboj.7600436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Coleman SK, Moykkynen T, Jouppila A, Koskelainen S, Rivera C, Korpi ER, Keinanen K. Agonist occupancy is essential for forward trafficking of AMPA receptors. J Neurosci. 2009;29:303–12. doi: 10.1523/JNEUROSCI.3953-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–62. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- [13].Conn PM, Ulloa-Aguirre A. Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol Metab. 2009;21:190–7. doi: 10.1016/j.tem.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ. Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J Biol Chem. 1996;271:89–96. doi: 10.1074/jbc.271.1.89. [DOI] [PubMed] [Google Scholar]
- [15].Conti F, Minelli A, Melone M. GABA transporters in the mammalian cerebral cortex: localization, development and pathological implications. Brain Res Brain Res Rev. 2004;45:196–212. doi: 10.1016/j.brainresrev.2004.03.003. [DOI] [PubMed] [Google Scholar]
- [16].Corringer PJ, Sallette J, Changeux JP. Nicotine enhances intracellular nicotinic receptor maturation: a novel mechanism of neural plasticity? J Physiol Paris. 2006;99:162–71. doi: 10.1016/j.jphysparis.2005.12.012. [DOI] [PubMed] [Google Scholar]
- [17].Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, Saint-Hilaire JM, Carmant L, Verner A, Lu WY, Wang YT, Rouleau GA. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–9. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- [18].Costa E, Auta J, Grayson DR, Matsumoto K, Pappas GD, Zhang X, Guidotti A. GABAA receptors and benzodiazepines: a role for dendritic resident subunit mRNAs. Neuropharmacology. 2002;43:925–37. doi: 10.1016/s0028-3908(02)00199-5. [DOI] [PubMed] [Google Scholar]
- [19].Feng HJ, Kang JQ, Song L, Dibbens L, Mulley J, Macdonald RL. Delta subunit susceptibility variants E177A and R220H associated with complex epilepsy alter channel gating and surface expression of alpha4beta2delta GABAA receptors. J Neurosci. 2006;26:1499–506. doi: 10.1523/JNEUROSCI.2913-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fleck MW. Glutamate receptors and endoplasmic reticulum quality control: looking beneath the surface. Neuroscientist. 2006;12:232–44. doi: 10.1177/1073858405283828. [DOI] [PubMed] [Google Scholar]
- [21].Gallagher MJ, Shen W, Song L, Macdonald RL. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the alpha1 subunit. J Biol Chem. 2005;280:37995–8004. doi: 10.1074/jbc.M508305200. [DOI] [PubMed] [Google Scholar]
- [22].Gill MB, Vivithanaporn P, Swanson GT. Glutamate binding and conformational flexibility of ligand-binding domains are critical early determinants of efficient kainate receptor biogenesis. J Biol Chem. 2009;284:14503–12. doi: 10.1074/jbc.M900510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gorrie GH, Vallis Y, Stephenson A, Whitfield J, Browning B, Smart TG, Moss SJ. Assembly of GABAA receptors composed of alpha1 and beta2 subunits in both cultured neurons and fibroblasts. J Neurosci. 1997;17:6587–96. doi: 10.1523/JNEUROSCI.17-17-06587.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hales TG, Tang H, Bollan KA, Johnson SJ, King DP, McDonald NA, Cheng A, Connolly CN. The epilepsy mutation, gamma2(R43Q) disrupts a highly conserved inter-subunit contact site, perturbing the biogenesis of GABAA receptors. Mol Cell Neurosci. 2005;29:120–7. doi: 10.1016/j.mcn.2005.01.002. [DOI] [PubMed] [Google Scholar]
- [25].Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH, Richards MC, Williams DA, Mulley JC, Berkovic SF, Scheffer IE, Petrou S. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–6. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Herring D, Huang R, Singh M, Dillon GH, Leidenheimer NJ. PKC modulation of GABAA receptor endocytosis and function is inhibited by mutation of a dileucine motif within the receptor beta 2 subunit. Neuropharmacology. 2005;48:181–94. doi: 10.1016/j.neuropharm.2004.09.015. [DOI] [PubMed] [Google Scholar]
- [27].Herring D, Huang R, Singh M, Robinson LC, Dillon GH, Leidenheimer NJ. Constitutive GABAA receptor endocytosis is dynamin-mediated and dependent on a dileucine AP2 adaptin-binding motif within the beta 2 subunit of the receptor. J Biol Chem. 2003;278:24046–52. doi: 10.1074/jbc.M301420200. [DOI] [PubMed] [Google Scholar]
- [28].Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–43. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Janovick JA, Patny A, Mosley R, Goulet MT, Altman MD, Rush TS, 3rd, Cornea A, Conn PM. Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Mol Endocrinol. 2009;23:157–68. doi: 10.1210/me.2008-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jean-Alphonse F, Perkovska S, Frantz MC, Durroux T, Mejean C, Morin D, Loison S, Bonnet D, Hibert M, Mouillac B, Mendre C. Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J Am Soc Nephrol. 2009;20:2190–203. doi: 10.1681/ASN.2008121289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kang JQ, Macdonald RL. The GABAA receptor gamma2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of alpha1beta2gamma2S receptors in the endoplasmic reticulum. J Neurosci. 2004;24:8672–7. doi: 10.1523/JNEUROSCI.2717-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D, Strybing K, Eidelberg D, During MJ. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369:2097–105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- [33].Kobayashi H, Ogawa K, Yao R, Lichtarge O, Bouvier M. Functional rescue of beta-adrenoceptor dimerization and trafficking by pharmacological chaperones. Traffic. 2009;10:1019–33. doi: 10.1111/j.1600-0854.2009.00932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Krampfl K, Maljevic S, Cossette P, Ziegler E, Rouleau GA, Lerche H, Bufler J. Molecular analysis of the A322D mutation in the GABA receptor alpha-subunit causing juvenile myoclonic epilepsy. Eur J Neurosci. 2005;22:10–20. doi: 10.1111/j.1460-9568.2005.04168.x. [DOI] [PubMed] [Google Scholar]
- [35].Kuryatov A, Luo J, Cooper J, Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human alpha4beta2 acetylcholine receptors. Mol Pharmacol. 2005;68:1839–51. doi: 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- [36].Lee FJ, Pei L, Moszczynska A, Vukusic B, Fletcher PJ, Liu F. Dopamine transporter cell surface localization facilitated by a direct interaction with the dopamine D2 receptor. Embo J. 2007;26:2127–36. doi: 10.1038/sj.emboj.7601656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Leidenheimer NJ. Regulation of excitation by GABA(A) receptor internalization. Results Probl Cell Differ. 2008;44:1–28. doi: 10.1007/400_2007_039. [DOI] [PubMed] [Google Scholar]
- [38].Leskela TT, Markkanen PM, Pietila EM, Tuusa JT, Petaja-Repo UE. Opioid receptor pharmacological chaperones act by binding and stabilizing newly synthesized receptors in the endoplasmic reticulum. J Biol Chem. 2007;282:23171–83. doi: 10.1074/jbc.M610896200. [DOI] [PubMed] [Google Scholar]
- [39].Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA. Cys-loop receptors: new twists and turns. Trends Neurosci. 2004;27:329–36. doi: 10.1016/j.tins.2004.04.002. [DOI] [PubMed] [Google Scholar]
- [40].Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R, Banghart MR, Dougherty DA, Goate AM, Wang JC. Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. Aaps J. 2009;11:167–77. doi: 10.1208/s12248-009-9090-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Luscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. 2004;102:195–221. doi: 10.1016/j.pharmthera.2004.04.003. [DOI] [PubMed] [Google Scholar]
- [42].Mah SJ, Cornell E, Mitchell NA, Fleck MW. Glutamate receptor trafficking: endoplasmic reticulum quality control involves ligand binding and receptor function. J Neurosci. 2005;25:2215–25. doi: 10.1523/JNEUROSCI.4573-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–43. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- [44].Merlie JP, Lindstrom J. Assembly in vivo of mouse muscle acetylcholine receptor: identification of an alpha subunit species that may be an assembly intermediate. Cell. 1983;34:747–57. doi: 10.1016/0092-8674(83)90531-7. [DOI] [PubMed] [Google Scholar]
- [45].Morello JP, Salahpour A, Laperriere A, Bernier V, Arthus MF, Lonergan M, Petaja-Repo U, Angers S, Morin D, Bichet DG, Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–95. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, Lester HA. Assembly of alpha4beta2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci. 2003;23:11554–67. doi: 10.1523/JNEUROSCI.23-37-11554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nebenfuhr A, Ritzenthaler C, Robinson DG. Brefeldin A: deciphering an enigmatic inhibitor of secretion. Plant Physiol. 2002;130:1102–8. doi: 10.1104/pp.011569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Newpher TM, Ehlers MD. Glutamate receptor dynamics in dendritic microdomains. Neuron. 2008;58:472–97. doi: 10.1016/j.neuron.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nutt D. GABAA receptors: subtypes, regional distribution, and function. J Clin Sleep Med. 2006;2:S7–11. [PubMed] [Google Scholar]
- [50].Otsuka M, Obata K, Miyata Y, Tanaka Y. Measurement of gamma-aminobutyric acid in isolated nerve cells of cat central nervous system. J Neurochem. 1971;18:287–95. doi: 10.1111/j.1471-4159.1971.tb00567.x. [DOI] [PubMed] [Google Scholar]
- [51].Papp E, Csermely P. Chemical chaperones: mechanisms of action and potential use. Handb Exp Pharmacol. 2006:405–16. doi: 10.1007/3-540-29717-0_16. [DOI] [PubMed] [Google Scholar]
- [52].Petaja-Repo UE, Hogue M, Bhalla S, Laperriere A, Morello JP, Bouvier M. Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. Embo J. 2002;21:1628–37. doi: 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pinal CS, Tobin AJ. Uniqueness and redundancy in GABA production. Perspect Dev Neurobiol. 1998;5:109–18. [PubMed] [Google Scholar]
- [54].Priel A, Selak S, Lerma J, Stern-Bach Y. Block of kainate receptor desensitization uncovers a key trafficking checkpoint. Neuron. 2006;52:1037–46. doi: 10.1016/j.neuron.2006.12.006. [DOI] [PubMed] [Google Scholar]
- [55].Richerson GB, Wu Y. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol. 2003;90:1363–74. doi: 10.1152/jn.00317.2003. [DOI] [PubMed] [Google Scholar]
- [56].Robert J, Auzan C, Ventura MA, Clauser E. Mechanisms of cell-surface rerouting of an endoplasmic reticulum-retained mutant of the vasopressin V1b/V3 receptor by a pharmacological chaperone. J Biol Chem. 2005;280:42198–206. doi: 10.1074/jbc.M510180200. [DOI] [PubMed] [Google Scholar]
- [57].Rose CR. Na+ signals at central synapses. Neuroscientist. 2002;8:532–9. doi: 10.1177/1073858402238512. [DOI] [PubMed] [Google Scholar]
- [58].Rose CR, Ransom BR. Regulation of intracellular sodium in cultured rat hippocampal neurones. J Physiol. 1997;499(Pt 3):573–87. doi: 10.1113/jphysiol.1997.sp021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sallette J, Pons S, Devillers-Thiery A, Soudant M, de Carvalho L. Prado, Changeux JP, Corringer PJ. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46:595–607. doi: 10.1016/j.neuron.2005.03.029. [DOI] [PubMed] [Google Scholar]
- [60].Sancar F, Czajkowski C. A GABAA receptor mutation linked to human epilepsy (gamma2R43Q) impairs cell surface expression of alphabetagamma receptors. J Biol Chem. 2004;279:47034–9. doi: 10.1074/jbc.M403388200. [DOI] [PubMed] [Google Scholar]
- [61].Schmidt J, Rossie S, Catterall WA. A large intracellular pool of inactive Na channel alpha subunits in developing rat brain. Proc Natl Acad Sci U S A. 1985;82:4847–51. doi: 10.1073/pnas.82.14.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Scholze P, Freissmuth M, Sitte HH. Mutations within an intramembrane leucine heptad repeat disrupt oligomer formation of the rat GABA transporter 1. J Biol Chem. 2002;277:43682–90. doi: 10.1074/jbc.M205602200. [DOI] [PubMed] [Google Scholar]
- [63].Schwartz EA. Depolarization without calcium can release gamma-aminobutyric acid from a retinal neuron. Science. 1987;238:350–5. doi: 10.1126/science.2443977. [DOI] [PubMed] [Google Scholar]
- [64].Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci. 2001;21:3063–72. doi: 10.1523/JNEUROSCI.21-09-03063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sigel E, Baur R, Kellenberger S, Malherbe P. Point mutations affecting antagonist affinity and agonist dependent gating of GABAA receptor channels. Embo J. 1992;11:2017–23. doi: 10.1002/j.1460-2075.1992.tb05258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sitte HH, Singer EA, Scholze P. Bi-directional transport of GABA in human embryonic kidney (HEK-293) cells stably expressing the rat GABA transporter GAT-1. Br J Pharmacol. 2002;135:93–102. doi: 10.1038/sj.bjp.0704446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Smith GB, Olsen RW. Identification of a [3H]muscimol photoaffinity substrate in the bovine gamma-aminobutyric acidA receptor alpha subunit. J Biol Chem. 1994;269:20380–7. [PubMed] [Google Scholar]
- [68].Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19:500–5. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- [69].Son CD, Moss FJ, Cohen BN, Lester HA. Nicotine Normalizes Intracellular Subunit Stoichiometry of Nicotinic Receptors Carrying Mutations linked to Autosomal Dominant Nocturnal Frontal Lobe Epilepsy. Mol Pharmacol. 2009 doi: 10.1124/mol.108.054494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Southern JA, Young DF, Heaney F, Baumgartner WK, Randall RE. Identification of an epitope on the P and V proteins of simian virus 5 that distinguishes between two isolates with different biological characteristics. J Gen Virol. 1991;72(Pt 7):1551–7. doi: 10.1099/0022-1317-72-7-1551. [DOI] [PubMed] [Google Scholar]
- [71].Tumkosit P, Kuryatov A, Luo J, Lindstrom J. Beta3 subunits promote expression and nicotine-induced up-regulation of human nicotinic alpha6* nicotinic acetylcholine receptors expressed in transfected cell lines. Mol Pharmacol. 2006;70:1358–68. doi: 10.1124/mol.106.027326. [DOI] [PubMed] [Google Scholar]
- [72].Valluru L, Xu J, Zhu Y, Yan S, Contractor A, Swanson GT. Ligand binding is a critical requirement for plasma membrane expression of heteromeric kainate receptors. J Biol Chem. 2005;280:6085–93. doi: 10.1074/jbc.M411549200. [DOI] [PubMed] [Google Scholar]
- [73].Van Craenenbroeck K, Clark SD, Cox MJ, Oak JN, Liu F, Van Tol HH. Folding efficiency is rate-limiting in dopamine D4 receptor biogenesis. J Biol Chem. 2005;280:19350–7. doi: 10.1074/jbc.M414043200. [DOI] [PubMed] [Google Scholar]
- [74].Walsh H, Govind AP, Mastro R, Hoda JC, Bertrand D, Vallejo Y, Green WN. Up-regulation of nicotinic receptors by nicotine varies with receptor subtype. J Biol Chem. 2008;283:6022–32. doi: 10.1074/jbc.M703432200. [DOI] [PubMed] [Google Scholar]
- [75].White E, McKenna J, Cavanaugh A, Breitwieser GE. Pharmacochaperone-mediated rescue of calcium-sensing receptor loss-of-function mutants. Mol Endocrinol. 2009;23:1115–23. doi: 10.1210/me.2009-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Whiting PJ. GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discov Today. 2003;8:445–50. doi: 10.1016/s1359-6446(03)02703-x. [DOI] [PubMed] [Google Scholar]
- [77].Wu Y, Wang W, Diez-Sampedro A, Richerson GB. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron. 2007;56:851–65. doi: 10.1016/j.neuron.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wu Y, Wang W, Richerson GB. Vigabatrin induces tonic inhibition via GABA transporter reversal without increasing vesicular GABA release. J Neurophysiol. 2003;89:2021–34. doi: 10.1152/jn.00856.2002. [DOI] [PubMed] [Google Scholar]
- [79].Wuller S, Wiesner B, Loffler A, Furkert J, Krause G, Hermosilla R, Schaefer M, Schulein R, Rosenthal W, Oksche A. Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J Biol Chem. 2004;279:47254–63. doi: 10.1074/jbc.M408154200. [DOI] [PubMed] [Google Scholar]
- [80].Zhang R, Wen X, Militante J, Hester B, Rhubottom HE, Sun H, Leidenheimer NJ, Yan D, White MM, Machu TK. The role of loop F residues in determining differential d-tubocurarine potencies in mouse and human 5-hydroxytryptamine 3A receptors. Biochemistry. 2007;46:1194–204. doi: 10.1021/bi0616100. [DOI] [PubMed] [Google Scholar]
- [81].Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–68. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
- [82].Zhu XM, Ong WY. A light and electron microscopic study of betaine/GABA transporter distribution in the monkey cerebral neocortex and hippocampus. J Neurocytol. 2004;33:233–40. doi: 10.1023/b:neur.0000030698.66675.90. [DOI] [PubMed] [Google Scholar]