

Abstract
SphK1 is an enzyme that converts sphingosine to bioactive sphingosine-1-phosphate (S1P). Recent in vitro data suggest a potential role of SphK1 in TNFα mediated inflammation. Our aims in this study were to determine the in vivo significance of SphK1 in TNFα mediated chronic inflammation and to define which pathogenic mechanisms induced by TNFα are SphK1 dependent. To pursue these aims, we studied the effect of SphK1 deficiency in an in vivo model of TNFα induced chronic inflammatory arthritis. Transgenic hTNFα mice, that develop spontaneous inflammatory erosive arthritis beginning at 14-16 weeks, were crossed with SphK1 null mice (SphK1-/-), on the C57BL6 genetic background. Beginning at 4 months of age, hTNF/SphK1-/- mice had significantly less severe clinically evident paw swelling and deformity, less synovial and periarticular inflammation and markedly decreased bone erosions as measured quantititatively through micro-CT images. Mechanistically, the mice lacking SphK1 had less articular COX-2 protein and fewer synovial Th17 cells than hTNF/SphK1+/+ littermates. Microarray analysis and real-time RT-PCR of the ankle synovial tissue demonstrated that hTNF/SphK1-/- mice had increased transcript levels of SOCS3 compared to hTNF/SphK1+/+ mice, likely also contributing to the decreased inflammation in the SphK1 deficient mice. Finally, significantly fewer mature osteoclasts were detected in the ankle joints of hTNF/SphK1-/- mice compared to hTNF/SphK1+/+ mice. These data indicate that SphK1 plays a key role in hTNFα induced inflammatory arthritis via impacting synovial inflammation and osteoclast number.
Introduction
Rheumatoid arthritis (RA) is a chronic, inflammatory, autoimmune disease characterized by synovial proliferation (pannus formation) which may eventually lead to bone erosions and joint deformity. The inciting agent that triggers the development of RA is unknown; however, it is clearly an inflammatory process as evidenced by consistently elevated levels of inflammatory mediators such as TNFα and IL-1β (1). The remarkable successes of anti-TNFα agents in the treatment of rheumatoid arthritis (2) indicate a key pathogenic role for TNFα in disease. Although much is known about the mechanisms of action of TNFα, a comprehensive understanding of the downstream mediators of disease in RA remains incomplete.
Recent in vitro data, from our group and others, implicate sphingosine kinase 1 (SphK1) derived sphingosine 1 phosphate (S1P) in the inflammatory cascade downstream of TNFα. TNFα induces TRAF2, which binds directly to SphK1, inducing translocation and tethering of SphK1 to the cell membrane, where it is exposed to its ligand sphingosine, leading to production and extracellular transport of S1P (3). Sphingosine, one of the products of sphingolipid metabolism, is derived from the deacylation of ceramide. While both SphK1 and 2 can phosphorylate sphingosine, SphK1 is the enzyme producing the majority of S1P in cells (4). Of importance to cancer biology, S1P enhances cell proliferation and survival, in contrast to ceramide and sphingosine, which are considered pro-apoptotic (4, 5). Recent data generated by a number of laboratories implied a role for S1P in the immune system (6). The most studied and clearest immune role of S1P is in lymphocyte trafficking. S1P acts as a chemoattractant agent via a concentration gradient from lymph node (low S1P) to lymph (high S1P) that induces lymphocyte egress from primary and secondary lymphoid structures (7, 8).
Additionally, in vitro data implicated S1P as a modulator of inflammatory responses following TNFα, IL1β or platelet-derived growth factor (PDGF) stimulation. The murine fibroblast cell line L929, treated with S1P, had increased cyclooxygenase 2 (COX-2) expression and prostaglandin E2 (PGE2) production compared to ceramide treated cells. These findings suggest a specific role for S1P in inflammation. The addition of TNFα to S1P further increased the production of PGE2 in both L929 and RA synovial fibroblasts (9, 10). Additional experiments showed that siRNA, specific for SphK1, reduced PGE2 and COX-2 production by L929 cells following stimulation with human TNFα. RA patients have increased expression of S1P1, a receptor for S1P, in the synovium (10) as well as increased expression of SphK1 compared to OA patients. These observations suggest a role for S1P in TNFα induced inflammation in rheumatoid arthritis.
To directly study the effect of SphK1 in TNFα induced arthritis, we initiated studies of SphK1 deficiency in hTNFα transgenic mice. This mouse model of inflammatory arthritis has a modified copy of human TNFα allowing for constitutive, deregulated expression. These mice develop an inflammatory arthritis, with swollen joints and joint deviation beginning at 4 months of age (11). This model is uniquely suited for our studies of SphK1 in TNFα induced inflammation, as the disease pathogenesis is directly due to TNFα overexpression. Furthermore, arthritis in these mice is a chronic process and the joints affected are similar to human RA (12). SphK1-deficient (SphK1-/-) mice allow for direct analysis and further clarification of SphK1's role in vivo in TNFα mediated arthritis. Exons 3, 4, 5, and the 5′ end of 6 of SphK1were replaced with a cassette for neomycin resistance and bred onto the C57Bl6 background. SphK1-/- mice exhibited no organ abnormalities, yet levels of S1P in serum and plasma were reduced compared to wildtype mice supporting SphK1 as the major producer of serum S1P (13). To define the role of SphK1 in TNFα induced inflammatory arthritis in vivo, we crossed the transgenic TNFα mice (hTNFα) with SphK1-/- mice (hTNF/SphK1-/-). Our results indicate that hTNF/SphK1-/- mice had significantly less inflammatory arthritis and erosive disease compared to hTNF/SphK1+/+ and hTNF/SphK1+/- mice. Furthermore, our data suggest that SphK1 deficiency impacts inflammatory arthritis via effects on COX-2, Th17 cells and SOCS3, as well as affecting erosions via impacting osteoclast generation in the affected joints.
Materials and Methods
Mice
hTNFα transgenic mice (Taconic) were crossed with SphK1 deficient (SphK1-/-) mice (13). All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC). Each mouse was screened for presence/absence of the hTNFα transgene and presence/absence of modified SphK1 using PCR amplification of tail DNA. The SphK1 activity deficient genotype results in a dysfunctional protein. mRNA for the defective gene is still produced. The primer sequences to detect the genotypes of interest were as follows: HTNF 1: 5′ CCT CAG CAA GGA CAG CAG 3′; HTNF 2: 5′ GCA GGC AGA AGA GCG TGG 3′; SK KO primer 1: 5′ TGT CAC CCA TGA ACC TGC TGT CCC TGC ACA3′; SK KO primer 2: 5′ AGA AGG CAC TGG CTC CTC CAG AGG AAC AAG 3′; SK KO primer neo; 5′TCG TGC TTT ACG GTA TCG CCG CTC CCG ATT3′. The PCR parameters for detection of hTNFα were as follows: 95°C for 15 min -1 cycle, 95°C for 45 sec, 60°C for 1 min, 72°C for 1 min - 30 cycles 72°C for 5 min - 1 cycle. The carrier has a 320bp DNA fragment, while in wild-types there is no detectable band. The PCR parameters for detection of SphK1 are as follows: 94°C for 1 min, 65°C for 1 min and 72°C for 1 min over 40 cycles. The wild-type allele yielded a DNA fragment of about 300bp, and the targeted allele yielded a fragment of 350bp.
Arthritis scoring
Mice were monitored weekly by a blinded observer and scored for signs of swelling and deviation beginning at 3 months of age. The scoring system graded the following: 0, 1+ (mild), or 2+ (moderate to severe) for swelling and 0, 1+, or 2+ for joint/paw deviation. Each front and hind paw was individually assigned a score for swelling and deviation, so that the highest score of 16 indicated the most severe disease effects. The scoring continued until the mice reached 6 months of age, after which they were sacrificed. The scores were compiled and sorted by genotype for analysis of arthritic changes that occur in each group over time.
Histological sectioning
After 3, 4, 5, and 6 months, mice were sacrificed and the hind joints were preserved in 10% formalin (Fisher, Atlanta, GA). The preserved sections were sent to AML Laboratory (Baltimore, MD) for paraffin embedding and sectioning at 7 microns. Sections were stained with H and E.
Pathological Scoring
H and E stained hind paw sections were sent to a blinded pathologist (PR) for scoring of degree of inflammation and synovial pathology found in the joints.
Immunohistochemistry
Unstained sections of paraffinized hind joints from 5 month old mice were de-paraffinized and stained using immunohistochemical techniques (Vectastain ABC kits, Vector Laboratories) to localize CD3ε (Santa Cruz, Santa Cruz, CA) and IL-23R (Abcam, Cambridge, MA) staining to indicate the location and number of T-cells and Th17 cells respectively.
FACS Analysis
Spleens were isolated at the time of sacrifice, weighed and spleen cells were isolated in single cell suspensions to quantify subsets of lymphocytes. Flow cytometry was performed using a FACSCalibur cytometer and Cell Quest plotting program. The cells were then harvested and stained with biotin-labeled Abs specific for CD3, CD4, CD8, or B220 (BD Biosciences, San Jose, CA). Isotype-matched mAbs were used as negative controls. A total of 106 cells were analyzed from each sample.
CT Imaging
All imaging was obtained using the Siemens Inveon Micro-CT/PET scanner at the small animal imaging facility in the Hollings Cancer Center at the Medical University of South Carolina. The hind paws were imaged at a spatial resolution of approximately 30 micrometers. For analysis of the erosion index, the gap found between the 4 and 5th distal tarsals, and the central and 3rd distal tarsal (Inter-Tarsal Diameter [mm] or ITSD) was measured. This value was corrected for bone density values (ITSD/Density). Following scanning, images were reconstructed at 30 micrometers (high resolution). Using this information, we determined the optimal protocol to scan both hindpaws.
Sphingolipid analysis
Serum samples
At 6 months of age, the mice were sacrificed and whole blood collected. For serum isolation, the samples were centrifuged at 7000 rpm for 5 min. and the serum layer was removed and placed in a fresh 1.5mL tube.
Tissue samples
Ankle joints of 6 month-old mice were isolated and homogenized in whole cell lysis buffer.
After serum and tissue collection, samples (100uL of serum and 1mg of protein) were sent to the MUSC's Lipidomics Core Facility, where advanced analyses of sphingosine and ceramide species were performed on a Thermo Finnigan TSQ 7000, triple-stage quadrupole mass spectrometer (Thermo Finnegan, Waltham, MA) operating in a multiple reaction monitoring (MRM) positive ionization mode, as described previously (14).
Preparation of total RNA
RNA from ankle joints from SphK1+/+, hTNF/SphK1+/+, and hTNF/SphK1-/- mice at 5 months of age was isolated by phase separation with TRIzol reagent (Invitrogen, Carlsbad, CA) and further purified using RNAeasy kit (Qiagen, Valencia, CA). After isolation, RNA purity and concentration was verified by spectrophotometry and by assessment with a Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA).
Microarray analysis
DNA microarray analysis was conducted on two independent ankle joint wildtype (SphK1+/+) RNA samples, three hTNF/SphK1+/+ samples and three hTNF/SphK1-/- samples. Samples were processed at the MUSC Proteogenomics Facility (http://proteogenomics.musc.edu) in accordance with Affymetrix protocols (Affymetrix, Santa Clara, CA). Synthesis of double stranded cDNA from total RNA, in vitro transcription of biotin-labeled cRNA targets and fragmentation of target cRNAs were performed following established Affymetrix procedures and as described previously (15, 16). Fragmented cRNA samples were hybridized overnight at 45°C to Affymetrix Mouse Genome 430 2.0 GeneChips®. Post hybridization washing, phycoerythrin-streptavidin staining and fluorescence scanning were performed using Affymetrix instrumentation in accordance with manufacturer protocols. The resulting raw data files were deposited in ArrayDB (12). Data are also deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/Accession #GSE20152). Hybridization data (CEL files) were normalized by RMA algorithm (17) using Affymetrix Expression Console software; detection calls were obtained by Affymetrix MAS5 algorithm. Expression data was compared using dChip software (18). Gene representations not receiving ‘present’ detection scores in ≥25% of all samples were excluded from further analysis. Genes differentially expressed between hTNF/SphK1+/+ and hTNF/SphK1-/- samples were detected by pairwise comparison using fold change and Student's unpaired t-test metrics. Significance thresholds for fold change and t-test metrics were chosen to maximize the number of genes discovered while maintaining an acceptable false discovery rate (FDR). Briefly, multiple comparisons were performed, each using a different threshold for fold change and t-test. For each comparison the number of genes discovered and the corresponding FDR were tabulated. Based on evaluation of these data, fold change >1.5 and p <0.05 yielded the optimum result, detecting 131 unique genes with estimated 20% FDR. Higher fold change thresholds and lower p value thresholds provided a modest reduction in the estimated FDR but were deemed overly restrictive for detection of differentially expressed genes, typically yielding fewer than 20-30 candidates (not shown).
Real-time Reverse Transcriptase PCR
RNA was isolated from the hind joints of 3 hTNF/SphK1+/+ and hTNF/SphK1-/- and 2 SphK1+/+ mice at 3 and 5 months of age independent of those used in the microarray study. 1ug of RNA was used to generate cDNA (SA Biosciences, Fredrick, MD) for reverse transcriptase real-time RT-PCR of COX-2, hTNFα, SphK2, mTNFα, SOCS3 and IL-6 (SA Biosciences, Fredrick, MD). The protocol followed for each primer was: Step 1: 95.0°C for 15:00 min. (1×), Step 2: 95.0°C for 00:15 min., Step 3: 60.0°C for 01:00 min. (40×), Step 4: 95.0°C for 01:00 min. and Step 5: 65.0°C for 02:00 (1×); Melting Curve: 65.0°C for 00:15 min., increase setpoint temperature after cycle 2 by 2.0°C (15×). The fold changes were calculated using the ΔΔCT method using SphK1+/+ mice as the control. Statistical analyses were used to determine if the differences in fold change calculated are significant.
Immunoblotting
Protein was isolated from the ankle joints of 2-3 mice in each group of similar age and genotype and homogenized in lysis buffer (20mM HEPES, 0.4 NaCl, 1.5mM MgCl, 0.1mM EGTA, 1mM dithiotheitol, 1mM EDTA, 20% glycerol, 0.1% non-Idet p-40) with phosphatase (Pierce, Rockford, IL) and protease (Invitrogen, Carlsbad, CA) inhibitors. Lysates were normalized using a Micro BCA protein Assay Kit (Pierce, Rockford, IL). Each sample (35-40ug) was resolved on a 4-20% polyacrylamide gel (SDS-PAGE) under denaturing conditions and transferred to a nitrocellulose membrane. After blocking with 100% Odyssey buffer (LI-COR, Lincoln, NE) for 1 hour at room temperature, the membranes were incubated with relevant primary antibodies (COX-2 (BD Biosciences, San Jose, CA) and Actin (Santa Cruz, Santa Cruz, CA)), diluted in Odyssey buffer and 0.1% Tween-20 overnight at 4°C. The blots were washed in PBS+0.1% Tween-20 and probed with appropriate infrared conjugated secondary antibodies. After washing, blots were imaged using an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE).
TRAP Staining
At the time of sacrifice, the hind joints of SphK1+/+, SphK1-/-, hTNF/SphK1+/+, and hTNF/SphK1-/- mice were isolated and preserved in 70% ethanol for 48 hours, changing the fixative after 12, 24, and 48 hours. The sections were sent to The Centre for Bone and Periodontal Research (Montreal, QC Canada) for TRAP staining. Sections were read by a blinded observer. The number and phenotype of TRAP positive cells were assessed.
Statistical Analysis
Statistical significance was set at p< 0.05 and determined using ANOVA (General linear model for unbalanced design); SAS 9.1 v. 3 and ANOVA with a Tukey's post-hoc test (GraphPad Prisim software, San Diego, CA).
Results
Arthritis Scores
In the single gene hTNFα model of inflammatory arthritis, the front and hind paws are primarily affected. To assess arthritis development and the impact of deficiency of SphK1, beginning at 3 months of age, the front and hind paws were monitored weekly for paw swelling and deformity by a blinded observer. Figure 1A pictorially compares hTNF/SphK1+/+ and hTNF/SphK1-/- 6 month old littermates. The amount of swelling and deviation in the front and hind paws was noticeably less severe in the hTNF/SphK1-/- mouse than in the hTNF/SphK1+/+. At the end of each month, from 3-6 months, the arthritis scores were compiled to assess differences in arthritis scores in the different groups (Figure 1B). As expected, SphK1+/+, SphK1+/-, and SphK1-/-mice, lacking the hTNF transgene, had no signs of disease; accordingly, these mice had an arthritis score of 0 for all months measured. hTNF/SphK1+/+ and hTNF/SphK1+/- mice began to show signs of joint swelling and deviation beginning at 4 months of age. Overtime, the arthritis score increased as the mice aged indicating progressive disease severity. hTNF/SphK1+/+ and hTNF/SphK1+/- mice had similar disease onset and severity indicating that only one copy of SphK1 is required for progression of disease. Arthritis in the hTNF/SphK1-/-mice was also first detectable at 4 months of age; however, progression and severity of disease was significantly decreased as indicated by arthritis scores at months 5 and 6 compared to hTNF/SphK1+/+ and hTNF/SphK1+/- mice.
Figure 1. Arthritis score in mice varying in hTNFα and SphK1 status.
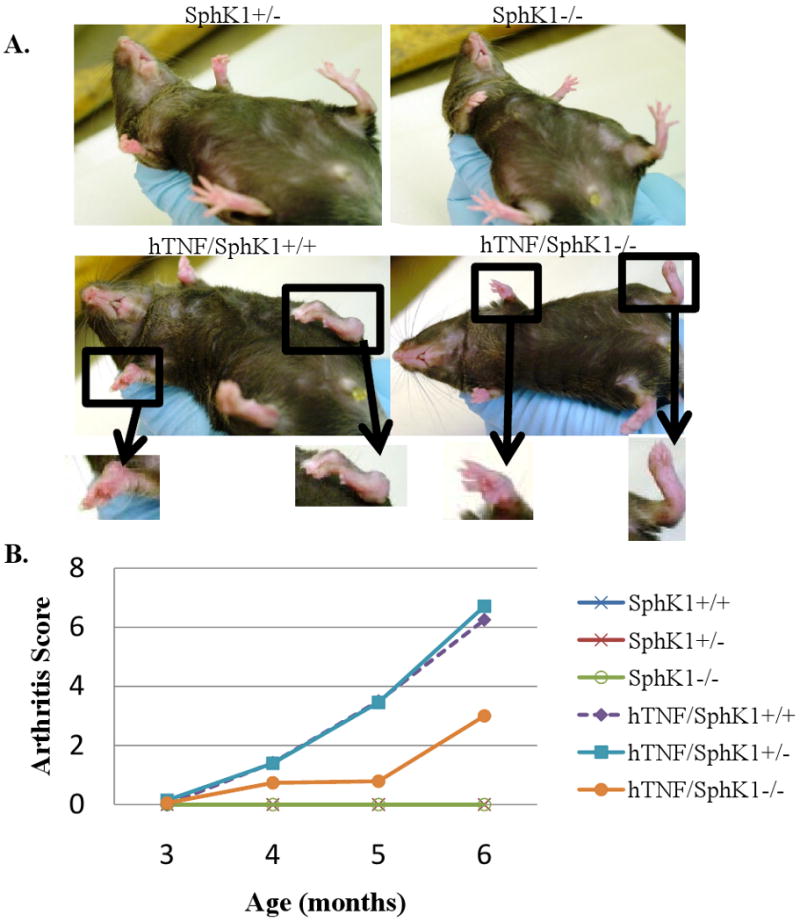
7 month old littermates of hTNF/SphK1-/-show decreased severity and progression of arthritis. Less disease noted in the hTNF/SphK1-/- mice compared to hTNF/SphK1+/+ mice (A). Observed arthritis scores in groups varying in SphK1 and TNFα genotype from 3 months to 6 months of age (B). Decreased severity and disease progression is observed in hTNF/SphK1-/- mice compared to hTNF/SphK1+/+ mice. Data are presented as mean +/- SEM, n>16 mice/group, p< 0.05, 0.01 by ANOVA.
Synovial Pathology
After imaging, the mice were sacrificed and the hind paws were collected, fixed in 10% formalin, and H and E stained. As expected, there was no detectable synovial proliferation, joint inflammation, or periarticular inflammation detected in SphK1+/+ and SphK1-/- sections as they do not express the hTNFα transgene (data not shown). At all time points, hTNF/SphK1-/- mice had significantly less periarticular inflammation and less joint inflammation compared to age-matched hTNF/SphK1+/+ mice (Figures 2B and C). In contrast, no significant difference was detected in synovial proliferation between hTNF/SphK1+/+ and hTNF/SphK1-/- mice at any time-point from 3-6 months of age (data not shown), although there was a trend towards less synovial proliferation in the 3 month old hTNF/SphK1-/- mice. Surprisingly, at 3 months of age, the hTNFα expressing mice already had significant synovial proliferation that was not evident as paw swelling or deformity. The synovial proliferation did not progress as the mice aged despite increasing inflammation and joint destruction.
Figure 2. Ankle and foot joint pathology scores at 3 and 6 months.
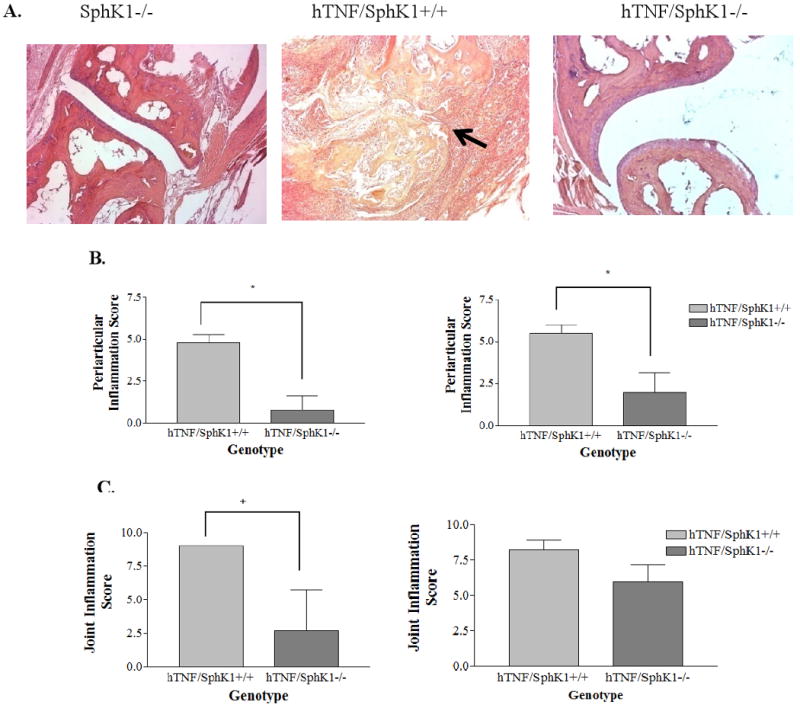
Representative H and E sections of ankles and feet of 5 month old mice after micro-CT imaging (A). Notice definable joint spaces in SphK1-/- and hTNF/SphK1-/- mice compared to hTNF/SphK1+/+ mice. n=4-5/group, 100×. Pathology scoring of periarticular inflammation (B), and joint inflammation (C) of H and E sections from hTNF/SphK+/+ and hTNF/SphK1-/- mice at 3 and 6 months of age. n=4-5 mice. Data represents mean pathology score +/- SEM. *p<0.05, + p<0.01 by Student's t-test
Erosion Index
The key clinical endpoint in inflammatory arthritis is the appearance of erosive disease. To detect and quantitate erosions in the hind paws, SphK1+/+, SphK1-/-, hTNF/SphK1+/+ and hTNF/SphK1-/- mice were scanned using a micro CT/PET scanner at 3, 4, 5 and 6 months. Due to the amount of radiation required for the procedure, which might impact disease expression, we did not sequentially follow the same mice overtime. SphK1+/+ and SphK1-/- mice never showed signs of bone erosion. At 3 months of age (Figure 3B), the hTNF/SphK1+/+ and hTNF/SphK1-/- mice began developing bone erosions. At 5 (Figures 3C) and 6 months of age (data not shown), the hTNF/SphK1-/- mice had markedly lower erosion indices as compared to hTNF/SphK1+/+. These differences were easily detectable by CT scan as shown in Figure 3A. The index provides a quantitative assessment of the differences between groups. hTNF/SphK1+/- mice were not scanned due to their being no difference in joint scores from the hTNF/SphK+/+ mice.
Figure 3. Measure of bone erosion in test mice due to SphK1.
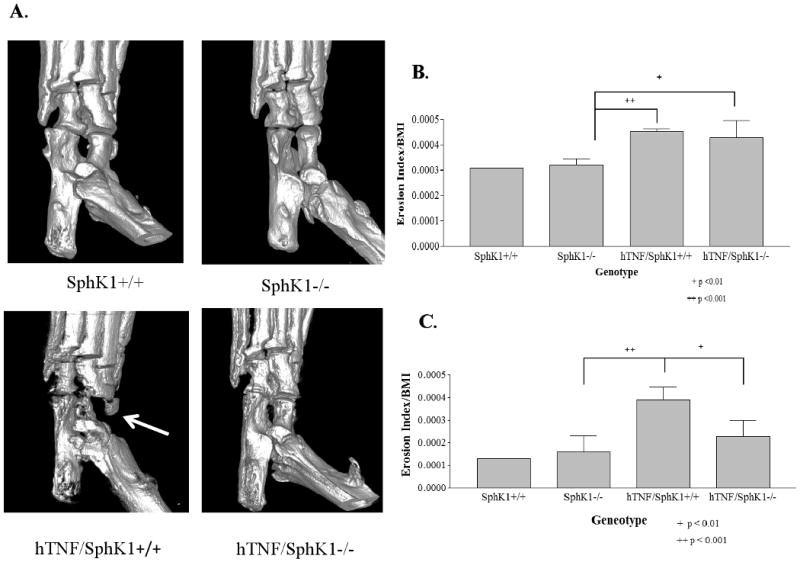
Representative micro-CT images of ankle joints in hTNF/SphK1-/- mice show less erosions than hTNF/SphK1+/+ mice as indicated by the arrow (A). Erosion index of mice 3 months of age (B) and 5 months of age (C). hTNF/SphK1-/- and hTNF/SphK1+/+ mice show signs of erosion as early as 3 months of age (p <0.01). At 5 months of age, significantly less erosion seen in hTNF/SphK1-/-mice than hTNF/SphK1+/+ mice. Correction for bone mineral density precludes comparing erosion scores from one month to the next. Data are presented as mean +/- SD, n = 3-5/group, + p< 0.01, ++ p< 0.001 by ANOVA.
Lack of mature osteoclasts detected in hTNF/SphK1-/- mouse joints
Based on the decreased erosions detected in the joints of the hTNF/SphK1-/- compared to hTNF/SphK1+/+ mice, we investigated the role of osteoclasts in this model. S1P is involved in bone homeostasis and osteoclastogenesis (19). Furthermore, osteoclasts are required for erosions that occur in the hTNFα model of arthritis (20). Therefore, we assessed the number of osteoclasts in the hind joints of hTNF/SphK1+/+ vs. hTNF/SphK1-/- mice. At 6 months of age, the hind joints were isolated and prepared for TRAP staining (Figure 4). Evaluation of the joint sections showed that SphK1+/+ and SphK1-/- mice had fewer mature osteoclasts compared to hTNF/SphK1+/+ mice. hTNF/SphK1-/- had fewer multinucleated TRAP+ osteoclasts in their joints compared to hTNF/SphK1+/+ mice. Further analysis revealed that the location of multinucleated TRAP+ cells differed in the hTNF/SphK1+/+ mice compared to hTNF/SphK1-/-mice. Multinucleated TRAP+ cells were seen actively resorbing bone and more osteoclast activity was present around the chondrocytes and cartilage in the hTNF/SphK1+/+ mice. In comparison, TRAP activity in the hTNF/SphK1-/- mice was present in the inflammatory infiltrate away from the bone margin. Therefore, hTNF/SphK1+/+ mice not only had more multinucleated TRAP+ cells, but these cells appeared more activated and bone resorbing compared to cells from hTNF/SphK1-/- mice.
Figure 4. TRAP staining in test mice with/without SphK1.
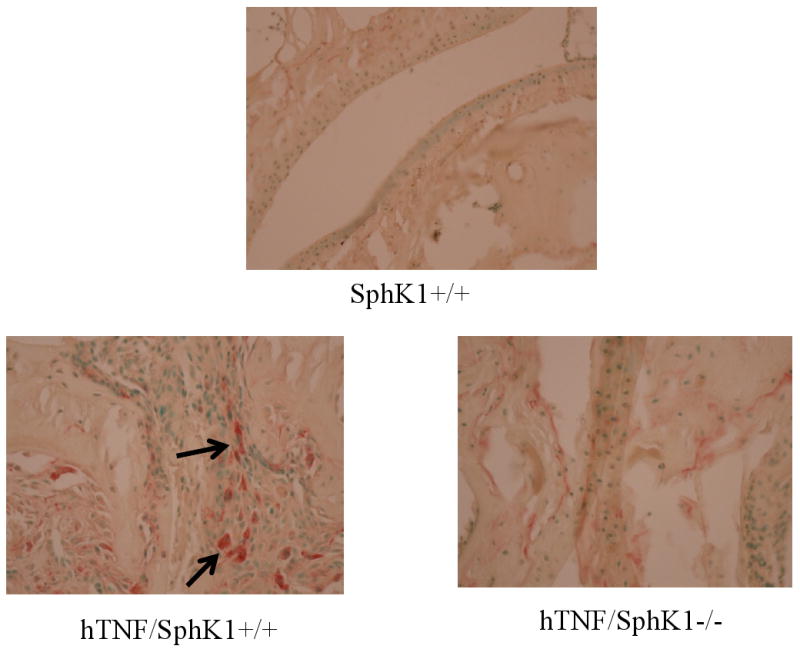
Representative TRAP staining of hTNF/SphK1+/+ mice (n=8) compared to hTNF/SphK1-/- mice (n=3). Arrows indicate TRAP positive cells. All images are at 400× magnification.
Spleen weights and cellular subsets
Previous data showed that lymphocyte egress from secondary lymphoid organs occurs in response to the S1P gradient through the S1P1R (8, 21, 22). To determine the effect of the lack of SphK1 on lymphocyte egress in this model of inflammation, the spleens were isolated at the time of sacrifice and analyzed. hTNF/SphK1-/- mice had significantly increased spleen weights compared to hTNF/SphK1+/+ mice (data not shown). To determine if there were differences in the subsets of lymphocytes or overall lymphocyte number, we isolated and sorted spleen cells using flow cytometry. There was a significant increase in the number of B220 positive, B-cells, CD3, CD4 and CD8 positive T-cells in spleens of hTNF/SphK1+/+ mice compared to hTNF/SphK1-/- mice (Table 1) indicating an effect of the SphK1 knockout on splenic retention of all major lymphocyte subsets in this inflammatory arthritis model.
Peripheral Blood Analysis
Based on differences detected in lymphocyte populations in the spleen, we collected whole blood to analyze the levels of circulating PBMCs. hTNF/SphK1+/+ and hTNF/SphK1-/- mice had decreased circulating lymphocytes compared to SphK1+/+ mice, a known effect of TNFα (23). However, there was no significant difference in circulating lymphocytes in hTNF/SphK1+/+ mice vs. hTNF/SphK1-/- mice (Figure 5).
Figure 5. Peripheral blood analysis in test mice.
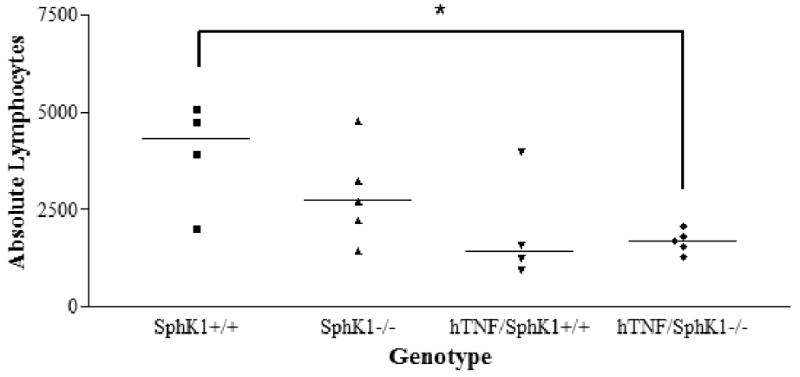
Peripheral blood analysis showed a significant decrease in absolute circulating lmphocytes in hTNF expressing mice compared to wildtype mice regardless of SphK1 genotype. No difference was detected in hTNF/SphK1+/+ mice compared to hTNF/SphK1-/- mice. Data presented as individual mice with line denoting mean of each group. n=4-5 mice/group, *p<0.05 by ANOVA with Tukey's multiple comparison correction.
Synovial T cells
We next assessed the cellular makeup of the synovial inflammation in the hTNF groups differing in SphK1 expression in the 5 month age group by immunohistochemistry. We did not detect differences in overall number of synovial CD3 expressing cells (data not shown). Significantly increased numbers of IL23r+ cells, presumably Th17 cells, however, were detected in hTNF/SphK1+/+ synovium as compared to SphK1+/+ synovium, with Th17 cells significantly decreased to absent in the hTNF/SphK1-/- mice (Figure 6). We were not able to achieve satisfactory staining by immunohistochemistry using B cell specific antibodies to assess differences in synovial B cells. These findings of decreased numbers of Th17 cells were consistent with the decrease in joint swelling, articular inflammation and periarticular inflammation in the hTNF/SphK1-/- mice and the reported role of S1P in Th17 formation (24).
Figure 6. Ankle joint Th17 quantification in test mice.
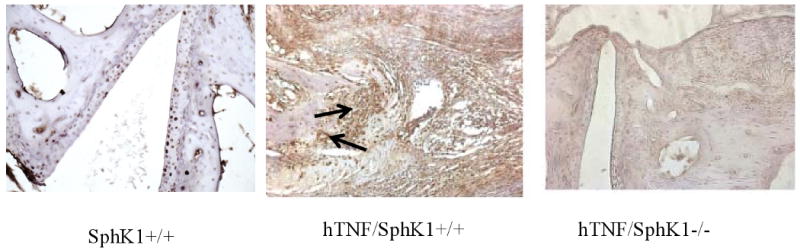
Representative immunohistochemistry (IHC) of ankle joints staining for IL-23R-positive (Th17) cells. Less Th17cells (brown staining) were detected in hTNF/SphK1-/- mice compared to hTNF/SphK1+/+ mice as indicated by the arrow. n=3-5 in each group
SphK1-/- and hTNF/SphK1-/- mice had significantly less detectable levels of serum S1P
Since lymphocyte egress is influenced by S1P levels, we measured serum sphingolipid levels at the time of sacrifice (Figure 7). SphK1-/- mice had significantly less detectable S1P in serum than SphK1+/+ or SphK1+/- mice. Similarly, hTNF/SphK1-/- mice had significantly less S1P in serum as compared to hTNF/SphK1+/+ or hTNF/SphK1+/- mice. The levels of S1P in SphK1-/- and hTNF/SphK1-/- did not differ significantly; therefore, the presence of hTNFα did not significantly increase the levels of serum S1P in the study mice. This finding likely reflects the localized nature of disease in this arthritis model.
Figure 7. Serum S1P levels in hTNF and SphK1 mice.
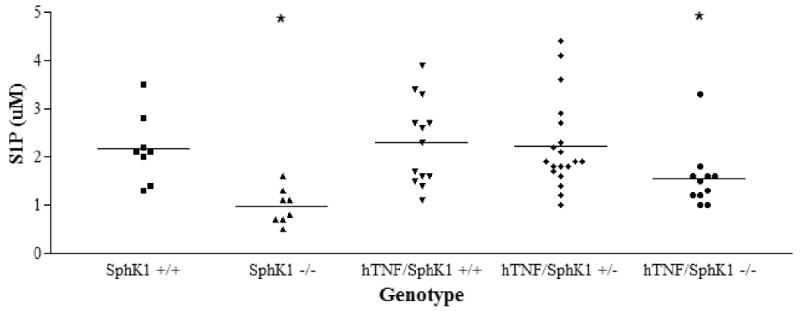
Less S1P was detected in SphK1-/- mice and hTNF/SphK1-/- mice compared to other groups of mice. n ≥8, *p<0.05 by ANOVA
Sphingolipid levels in the synovium
To assess the effect of hTNFα expression and SphK1 genotype on joint sphingolipid levels, we measured sphingolipid levels in the ankle joints of SphK1+/+, SphK1-/-, hTNF/SphK1+/+, and hTNF/SphK1-/- mice. Significantly increased levels of total ceramide were present in the joints of the hTNF/SphK1+/+ compared to SphK1+/+ mice, due to TNFα induced neutral sphingomyelinase activity (25). There was a trend towards further increased joint tissue ceramide levels in hTNF/SphK1-/- mice compared to hTNF/SphK+/+ mice (Figure 8A). Sphingosine levels were significantly increased in the hTNF/SphK1-/- mice likely secondary to a backup of ceramide and sphingosine due to the lack of SphK1 (Figure 8B). We did not detect a significant difference in S1P levels between the groups in the ankle joints of the mice. The lack of differences in tissue S1P levels in SphK1 deficient mice is attributed to the rapid turnover of S1P by S1P lyase and phosphatases present in the tissue, as previously reported (13). These data do indicate that the TNFα and the genetic knockout of SphK1 impact the local sphingolipid makeup of the joint in this model of inflammatory arthritis.
Figure 8. Tissue ceramide and sphignosine levels in mice of varying genotype.
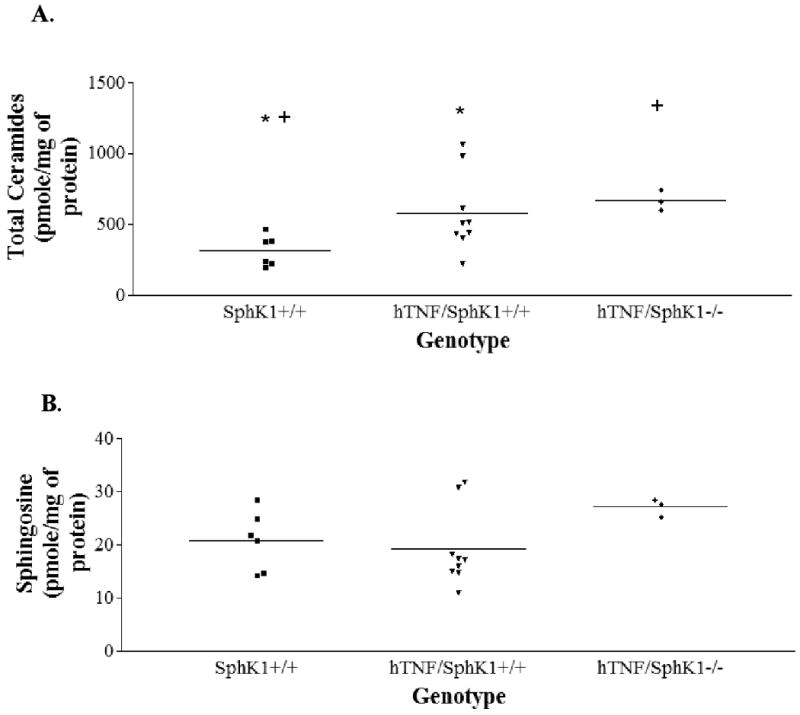
Significantly higher levels of ceramide were detected in hTNF/SphK1+/+ and hTNF/SphK1-/- mice compared to SphK1+/+ and SphK1-/- mice (A). SphK1+/+ vs. hTNF/SphK1+/+ *p<0.01 ; SphK1+/+ vs. hTNF/SphK1-/- +p<0.001; (B). Higher levels of sphingosine detected in hTNF/SphK1-/- mice. Center lines represent means and each shape represents one mouse.
Synovial SphK2, COX-2, hTNFα, and mTNFα Expression
To assess mechanisms for the beneficial effect of SphK1 deficiency on TNFα induced inflammatory arthritis, RNA was isolated from the joints of 5 month SphK1+/+, hTNF/SphK1+/+, and hTNF/SphK1-/- mice and used in real-time RT-PCR assays to quantitate expression of genes known to be important in inflammatory pathways affected by S1P. Five months was chosen as the time point due to the arthritis being active, but not advanced. Message levels of SphK2 were assessed to determine whether SphK2 was upregulated to compensate for the lack of functional SphK1. However, no difference was detected in message levels of SphK2 in hTNF/SphK1+/+ vs. hTNF/SphK1-/- joints (data not shown); therefore, little or no compensatory increase of SphK2 occurred. TNFα is known to increase COX-2 protein expression. COX-2 message was upregulated in both hTNF/SphK1-/- and hTNF/SphK1+/+ joints compared to SphK1+/+ joints, however, no difference in COX-2 message expression was detected between hTNF/SphK1+/+ mice and hTNF/SphK1-/- mice (data not shown). When message levels of hTNFα were measured, there was a 100-fold increase in both hTNF/SphK1+/+ and hTNF/SphK1-/- joints compared to SphK1+/+ joints (data not shown). Therefore, the presence or absence of SphK1 did not affect the transcription of the transgene derived hTNFα. mTNFα was upregulated in both hTNF/SphK1+/+ and hTNF/SphK1-/- mice showing that hTNFα leads to activation of the murine inflammatory pathway and transcription of other pro-inflammatory cytokines. However, there was a trend towards less mTNFα in hTNF/SphK1-/-mice than hTNF/SphK1+/+ mice (data not shown), although the difference did not reach statistical significance.
COX-2 Immunoblots
To confirm the real-time RT-PCR results at the protein level, immunoblots were performed using joint protein extracts isolated from SphK1+/+, hTNF/SphK1+/+, and hTNF/SphK1-/- mice. COX-2 was elevated in 5 month old- hTNF/SphK1+/+ compared to SphK1+/+ joints. COX-2 protein levels were significantly decreased in hTNF/SphK1-/- compared to hTNF/SphK1+/+ joints (Figure 9). Thus, although COX-2 message levels were the same in hTNF/SphK1+/+ and hTNF/SphK1-/- joints, protein expression of COX-2 was decreased in hTNF/SphK1-/- joints consistent with prior in vitro reports of SphK1 impacting COX-2 expression (9). At 3 months, no differences in COX-2 expression were detected between SphK1+/+, hTNF/SphK1+/+ and hTNF/SphK1-/- joints (data not shown).
Figure 9. Immunoblot of COX-2 in test mice in the presence/absence of SphK1.
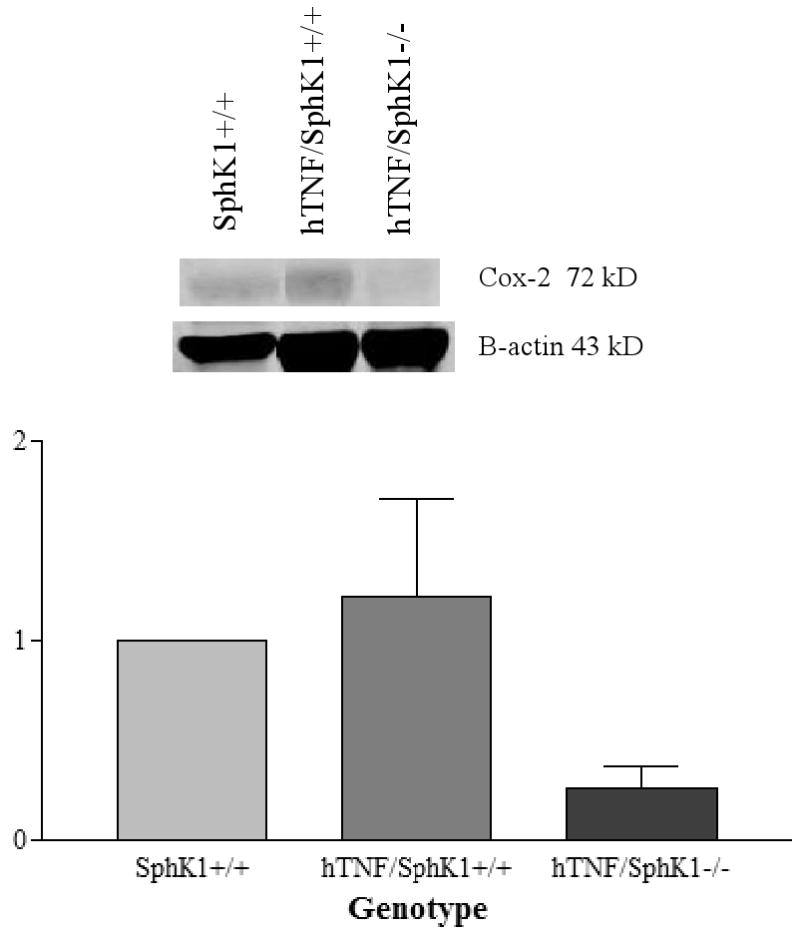
Synovial protein expression of COX-2 from the ankles of 5 month old mice of varying genotype. Data presented are representative samples. p-value not statistically significant.
Differential expression of mRNA in joints
Microarray analysis was performed on RNA isolated from the ankle joints of 5 month old hTNF/SphK1+/+ and hTNF/SphK1-/- mice with SphK1+/+ mice used as the control. Differential expression analysis revealed that thousands of transcripts were upregulated in the hTNF/SphK1+/+ mice compared to SphK1+/+ mice, including SphK1 (not shown). However, only 131 were identified as differentially expressed (39 upregulated, 92 downregulated) in hTNF/SphK1-/- compared to hTNF/SphK1+/+ joints (Figure 10A). Of these differentially regulated genes, based on their known roles in inflammation, their known interactions, and their having the highest differential expression, we selected three, SphK1, IL-6 and SOCS3, for further confirmation and study. Differential expression of all 3 genes was confirmed using real time RT-PCR (Figure 10B and C) of joint tissue. Complement factor D (CFD), which was not upregulated in the microarray analysis, was used as a control comparator and also did not show differential expression by real time RT-PCR. The upregulation of SphK1 occurs due to the genetic alteration in the SphK1-/- mice. Overproduction of the non-functional protein leads to disruption of the negative feedback in this pathway causing upregulation of the SphK1 transcript. In light of the increased levels of IL-6, as well as SOCS3, we believe that SOCS3, a natural regulator of IL-6 signaling, is impacting the signaling of IL-6, leading to decreased pro-inflammatory signaling.
Figure 10. Differential gene expression in the ankle joints of mice in the presence/absence of SphK1.
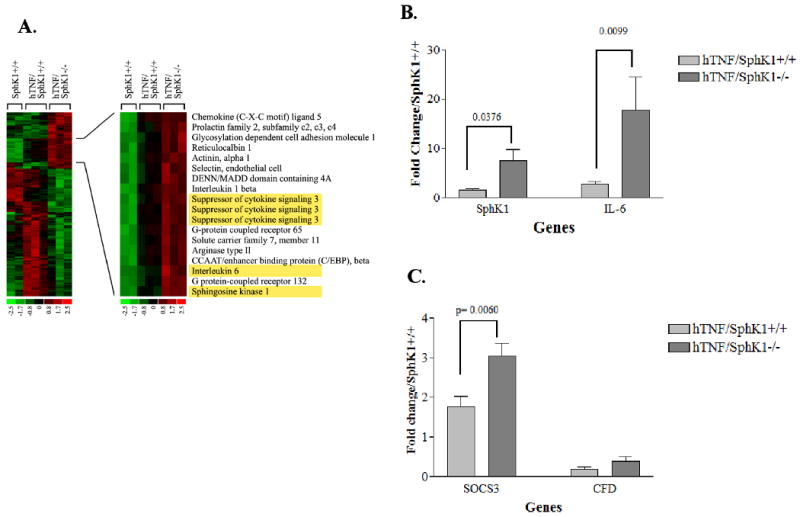
Heat map of microarray expression data for (A) from SphK1+/+ (n=2), hTNF/SphK1+/+ (n=3) and hTNF/SphK1-/- (n=3) mice. Left panel shows profiles for all genes differentially expressed. Right panel shows a subset that is upregulated. Yellow boxes represent genes further investigated in these mice (A). Real time RT- PCR confirmation of microarray analysis of SphK1 and IL-6 (B), SOCS3 and complement factor D (CFD) (C). CFD was used as a no difference control. Each gene measured in triplicate; p-values (Student's t test) are noted above the brackets.
Discussion
S1P is implicated in mouse models of inflammation, including DSS induced colitis (26), and collagen induced arthritis (CIA) (27). These murine models of inflammatory disease demonstrated a significant decrease in disease development in the setting of decreased S1P, due to the genetic lack of SphK1 (DSS) or RNA knockdown of enzyme (CIA). There are multiple factors critical for disease development in most inflammatory disease models, making it difficult to define the finite mechanisms impacted by SphK1 deficiency. The mechanistic advantage of the hTNFα model of inflammatory arthritis is that the causative factor is known (TNFα). Thus, this model allows for direct assessment of the role of SphK1 in mediating TNFα mediated inflammatory diseases. Based on the efficacy of agents targeting TNFα in rheumatoid arthritis (2), understanding the mechanisms underlying their effectiveness is critical. Such research may define mechanisms for failure to respond to anti-TNFα therapy, as well as perhaps identify therapies with similar or improved efficacy with fewer or less severe toxicities.
Although an interaction between TNFα and SphK1 appears clear, the pathways linking these factors are not fully identified. In vitro data published previously, and confirmed in vivo in this manuscript, indicate that blocking SphK1 decreases TNFα-induced PGE2 and COX-2 production. One identified molecular interaction between SphK1 and TNFα is that SphK1 contains a TNFα receptor activating factor (TRAF2) binding site (28). This interaction of SphK1 with TRAF2 is necessary for nuclear factor kappa B (NFκB) activation after stimulation with TNFα in L929 fibroblasts (29). Therefore, the TRAF2 binding site in SphK1 is one proven link between TNFα, SphK1 and NFκB, a known transcription factor that upregulates inflammatory mediators such as COX-2, leading to the production of PGE2. Theorized models conceptualize co-cellular localization of TNFα, SphK1 and NFκB (30, 31).
Our findings indicate that, in vivo, the lack of SphK1 impacted two aspects of inflammatory arthritis. The first was articular/periarticular inflammation that was significantly decreased in the joints of SphK1 deficient hTNFα transgenic mice. The second was joint erosions, which were markedly decreased in the hTNF/SphK1-/- mice as revealed through micro-CT scanning. These improvements in disease parameters are in contrast to the apparent minimal effect of SphK1 deficiency on synovial proliferation, indicating there are different TNFα induced pathways underlying TNFα induced synovial inflammation versus synovial proliferation, the latter of which is not SphK1 dependent. Our findings also indicate that synovial proliferation alone does not directly parallel development of bone erosions. An interesting feature of disease in this mouse model is that the development of erosions at 3 months of age preceded the onset of clinically evident joint swelling or joint deformity (32). Synovial proliferation was evident pathologically at 3 months, regardless of SphK1 expression, and did not increase significantly from 3-6 months despite marked increases in joint swelling and deformity. Thus, in this model, synovial proliferation does not appear to play a major role in joint swelling, joint deviation or bone erosion. We found that both hTNF/SphK1+/+ and hTNF/SphK1-/- mice expressed equal levels of hTNFα as assessed by real-time PCR. Therefore, the lack of disease progression in the hTNF/SphK1-/- was not due to decreased expression of the hTNFα transgene in the SphK1 deficient mice, though it may explain the equal development of synovial proliferation.
Our studies thus far identify two potential mechanisms for decreased articular inflammation in SphK1 deficient mice. The first is the role of SphK1 and S1P in the development of Th17 T cells. Th17 cells are increased in the target organs of autoimmune diseases, most prominently rheumatoid arthritis (33). They are postulated to play a key role in the pathogenesis of disease in both human rheumatoid arthritis and animal models of this disease (33, 34). Prior in vitro studies implicate S1P in Th17 development. The addition of S1P to splenic CD4+ T cells led to the secretion of IL-17. S1P enhanced in vitro development of Th17 cells and mice treated with FTY-720, a S1P receptor antagonist, had decreased percentages of Th17 cells (24). Our data demonstrated decreased numbers of IL23r+ cells in the joints of hTNF/SphK1-/- mice compared to hTNF/SphK1+/+ mice suggesting that the prior in vitro findings of the impact of S1P on Th17 T cell development was also true in vivo in inflammatory diseases. We believe that the decreased number of articular Th17 cells, in the SphK1 deficient mice, is mechanistically implicated in the decreased inflammation seen in the hTNFα SphK1 deficient mice. Linking the two primary findings we observed in the SphK1 mice, i.e. decreased inflammation and decreased erosions, IL-17 is known to induce osteoclastogenesis in vitro and in vivo (35). It is possible, therefore, that through inhibition of Th17 development, SphK1 deficiency has a dual linked effect on inflammation and bone destruction in this model.
Our investigations in the hTNFα transgenic mice have implicated a second inflammatory pathway impacted by SphK1 and possible mechanism to explain the decreased inflammation observed. The microarray results revealed that SOCS3 was consistently and significantly upregulated in the joints of hTNF/SphK1-/- mice. However, hTNFα induced increased IL-6 production in the joints of the study mice regardless of SphK1 expression. IL-6's role in inflammation is established, though recent investigations suggest that IL-6 can also be anti-inflammatory, depending on the biologic setting in which it is expressed (36). SOCS3 functions as a negative regulator in the JAK/STAT pathway utilized by IL-6 receptor signaling. SOCS3 transcription is increased in the presence of IL-6 signaling with an end effect of SOCS3 inhibiting prolonged STAT3 phosphorylation and IL-6 downstream inflammatory effects (37).
Increased SOCS3 transcription levels, in parallel with increased IL-6 transcription, suggest that the pro-inflammatory effects of IL-6 signaling were inhibited by the increased SOCS3 expression in the SphK1 deficient mice. We are not aware of any previous publications implicating an interaction between SphK1 and SOCS3. Previous in vitro studies suggested, however, that increased levels of ceramide induced increased SOCS3 expression (38). Due to the equilibrium between sphingosine and ceramide demonstrated in vitro, in the setting of SphK1 deficiency, increased ceramide levels would be expected. Indeed, a trend towards increased levels of ceramide isomers was present in the joints of the hTNFα SphK1 deficient mice, possibly increasing SOCS3 expression. We propose that the increased expression of SOCS3, induced dually by IL-6 and ceramide, resulted in decreased IL-6 induced inflammation in the SphK1 deficient mice. Further cluster analysis of the genes up/down regulated in the hTNFα/SphK1-/- mice indicated that 29 of these genes are impacted by SOCS3 expression (data not shown) further implicating SOCS3 in the phenotypic differences observed.
SOCS3 expression may also explain the marked difference in erosions seen in the study mice. SOCS3 was previously shown to be important in osteoclastogenesis. In an IL-1 induced inflammatory arthritis model, mice lacking functional copies of SOCS3, had increased numbers of articular osteoclasts measured through TRAP staining (39) and significantly worse arthritis. Mice overexpressing SOCS3 had significantly less arthritis in the collagen induced model (40). Therefore, we believe that the increased levels of SOCS3 found in the hTNF/SphK1-/- mice may inhibit osteoclastogenesis explaining our micro-CT results. Furthermore, in a recent publication, it was shown that the lack of S1P affected osteoclast precursor migration (19). Thus, it is possible that in our model, the lack of S1P directly led to the observed decreased number of osteoclasts in the ankle joint.
We assessed other alternative explanations for decreased erosions, including effects on RANKL expression (no difference in expression in the joints of the SphK1 deficient vs. wildtype mice, data not shown) and effects on MMP9 expression (also no difference based on SphK1 genotype). Thus, although effects on RANKL or MMP expression cannot be totally excluded based on these findings, we believe it is more likely that the impact of SphK1 deficiency on the development of erosions is due either to indirect IL-17 and SOCS3 effects or direct S1P effects on osteoclastogenesis.
Limitations of this study are that the effects we are studying are minimally dependent on the adaptive immune system, which is clearly important in rheumatoid arthritis pathogenesis (41, 42). Furthermore, the disease in these mice is isolated to the small joints as opposed to human disease, which may involve larger joints and is often systemic in its effects. Additionally, S1P levels in the serum were not elevated in the diseased mice we studied, despite a significant increase in SphK1 expression in the joints of the hTNFα transgenic mice compared to wildtype mice, again emphasizing the localized effect in this model. Despite these limitations, we believe these experimental results are informative as we have defined a clear role of SphK1 in TNFα induced arthritis and identified distinct pathways operative in TNFα induced inflammation that appear dependent on SphK1 expression. We believe that the protective effect of SphK1 deficiency in vivo is mediated via multiple co-dependent mechanisms including decreased COX2 expression, decreased articular Th17 cells, increased SOCS3 expression, and decreased osteoclastogenesis. These findings further suggest that SphK1, ceramide and/or S1P are potential therapeutic targets in human disease for blocking inflammation and bone loss in TNFα induced arthritis.
Acknowledgments
We would like to acknowledge the following people for their contributions to manuscript: Dr. Michael Rosol for his expertise in imaging, Victor Fresco for microarray hybridization, Dr. Phillip Ruiz for pathological scoring, Ivan Molano for flow cytometry analysis, Renée Bernatchez and the Center for Bone and Periodontal Research for TRAP staining, AML labs for histology, MUSC lipidomics core facility for lipid analysis, Dr. Srinivasan Shanmugarajan and Dr. Sakamuri V. Reddy for TRAP analysis and Dr. Ashley Snider for technical help.
Footnotes
This work was supported by the American College of Rheumatology Research and Education Foundation's Within Our Reach: Finding a Cure for Arthritis campaign and U.S. National Institutes of Health (NIH) grants R01GM062887 and 1F31AR057307-01.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc.(AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third pasty. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Saxne T, Palladino MA, Jr, Heinegard D, Talal N, Wollheim FA. Detection of tumor necrosis factor alpha but not tumor necrosis factor beta in rheumatoid arthritis synovial fluid and serum. Arthritis Rheum. 1988;31:1041–1045. doi: 10.1002/art.1780310816. [DOI] [PubMed] [Google Scholar]
- 2.Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, Bijl H, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 3.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 4.Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. Molecular cloning and functional characterization of murine sphingosine kinase. J Biol Chem. 1998;273:23722–23728. doi: 10.1074/jbc.273.37.23722. [DOI] [PubMed] [Google Scholar]
- 5.Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim Biophys Acta. 2002;1585:193–201. doi: 10.1016/s1388-1981(02)00341-4. [DOI] [PubMed] [Google Scholar]
- 6.El Alwani M, Wu BX, Obeid LM, Hannun YA. Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol Ther. 2006;112:171–183. doi: 10.1016/j.pharmthera.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Graeler M, Goetzl EJ. Activation-regulated expression and chemotactic function of sphingosine 1-phosphate receptors in mouse splenic T cells. Faseb J. 2002;16:1874–1878. doi: 10.1096/fj.02-0548com. [DOI] [PubMed] [Google Scholar]
- 8.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 9.Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, Chalfant CE, Obeid LM, Hannun YA. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. Faseb J. 2003;17:1411–1421. doi: 10.1096/fj.02-1038com. [DOI] [PubMed] [Google Scholar]
- 10.Kitano M, Hla T, Sekiguchi M, Kawahito Y, Yoshimura R, Miyazawa K, Iwasaki T, Sano H, Saba JD, Tam YY. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis Rheum. 2006;54:742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- 11.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. Embo J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Argraves GL, Barth JL, Argraves WS. The MUSC DNA Microarray Database. Bioinformatics. 2003;19:2473–2474. doi: 10.1093/bioinformatics/btg325. [DOI] [PubMed] [Google Scholar]
- 13.Allende ML, Sasaki T, Kawai H, Olivera A, Mi Y, van Echten-Deckert G, Hajdu R, Rosenbach M, Keohane CA, Mandala S, Spiegel S, Proia RL. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52487–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- 14.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Song W, Barth JL, Yu Y, Lu K, Dashti A, Huang Y, Gittinger CK, Argraves WS, Lyons TJ. Effects of oxidized and glycated LDL on gene expression in human retinal capillary pericytes. Invest Ophthalmol Vis Sci. 2005;46:2974–2982. doi: 10.1167/iovs.04-1505. [DOI] [PubMed] [Google Scholar]
- 16.Hammad SM, Twal WO, Barth JL, Smith KJ, Saad AF, Virella G, Argraves WS, Lopes-Virella MF. Oxidized LDL immune complexes and oxidized LDL differentially affect the expression of genes involved with inflammation and survival in human U937 monocytic cells. Atherosclerosis. 2009;202:394–404. doi: 10.1016/j.atherosclerosis.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishii M, Egen JG, Klauschen F, Meier-Schellersheim M, Saeki Y, Vacher J, Proia RL, Germain RN. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature. 2009;458:524–528. doi: 10.1038/nature07713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M, Lang S, Turk B, Pietschmann P, Woloszczuk W, Haralambous S, Kollias G, Steiner G, Smolen JS, Schett G. Tumor necrosis factor alpha-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum. 2002;46:785–792. doi: 10.1002/art.10097. [DOI] [PubMed] [Google Scholar]
- 21.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 22.Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115:84–105. doi: 10.1016/j.pharmthera.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Ishida H, Shimizu M, Suemura M, McCloskey R, Kishimoto T. Long-term follow-up of the changes in circulating cytokines, soluble cytokine receptors, and white blood cell subset counts in patients with rheumatoid arthritis (RA) after monoclonal anti-TNF alpha antibody therapy. J Clin Immunol. 1999;19:305–313. doi: 10.1023/a:1020543625282. [DOI] [PubMed] [Google Scholar]
- 24.Liao JJ, Huang MC, Goetzl EJ. Cutting edge: Alternative signaling of Th17 cell development by sphingosine 1-phosphate. J Immunol. 2007;178:5425–5428. doi: 10.4049/jimmunol.178.9.5425. [DOI] [PubMed] [Google Scholar]
- 25.Hayakawa M, Jayadev S, Tsujimoto M, Hannun YA, Ito F. Role of ceramide in stimulation of the transcription of cytosolic phospholipase A2 and cyclooxygenase 2. Biochem Biophys Res Commun. 1996;220:681–686. doi: 10.1006/bbrc.1996.0464. [DOI] [PubMed] [Google Scholar]
- 26.Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, Obeid LM. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. Faseb J. 2009;23:143–152. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai WQ, Irwan AW, Goh HH, Howe HS, Yu DT, Valle-Onate R, McInnes IB, Melendez AJ, Leung BP. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J Immunol. 2008;181:8010–8017. doi: 10.4049/jimmunol.181.11.8010. [DOI] [PubMed] [Google Scholar]
- 28.Taha TA, Hannun YA, Obeid LM. Sphingosine kinase: biochemical and cellular regulation and role in disease. J Biochem Mol Biol. 2006;39:113–131. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- 29.Xia P, Wang L, Moretti PA, Albanese N, Chai F, Pitson SM, D'Andrea RJ, Gamble JR, Vadas MA. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem. 2002;277:7996–8003. doi: 10.1074/jbc.M111423200. [DOI] [PubMed] [Google Scholar]
- 30.Pettus BJ, Chalfant CE, Hannun YA. Sphingolipids in inflammation: roles and implications. Curr Mol Med. 2004;4:405–418. doi: 10.2174/1566524043360573. [DOI] [PubMed] [Google Scholar]
- 31.Chalfant CE, Spiegel S. Sphingosine 1-phosphate and ceramide 1-phosphate: expanding roles in cell signaling. J Cell Sci. 2005;118:4605–4612. doi: 10.1242/jcs.02637. [DOI] [PubMed] [Google Scholar]
- 32.Hayer S, Redlich K, Korb A, Hermann S, Smolen J, Schett G. Tenosynovitis and osteoclast formation as the initial preclinical changes in a murine model of inflammatory arthritis. Arthritis Rheum. 2007;56:79–88. doi: 10.1002/art.22313. [DOI] [PubMed] [Google Scholar]
- 33.Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–970. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 34.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 37.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 38.Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2009;297:E211–224. doi: 10.1152/ajpendo.91014.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong PK, Egan PJ, Croker BA, O'Donnell K, Sims NA, Drake S, Kiu H, McManus EJ, Alexander WS, Roberts AW, Wicks IP. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest. 2006;116:1571–1581. doi: 10.1172/JCI25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, Nagata K, Yoshimura A. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest. 2001;108:1781–1788. doi: 10.1172/JCI13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kollias G, Douni E, Kassiotis G, Kontoyiannis D. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol Rev. 1999;169:175–194. doi: 10.1111/j.1600-065x.1999.tb01315.x. [DOI] [PubMed] [Google Scholar]
- 42.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]