



Abstract
Objective:
To assess the contribution of dementia-related neuropathologic lesions to age-related and disease-related change in cognitive function.
Methods:
A total of 354 Catholic nuns, priests, and brothers had annual clinical evaluations for up to 13 years, died, and underwent brain autopsy. The clinical evaluations included detailed testing of cognitive function from which previously established composite measures of global cognition and specific cognitive functions were derived. As part of a uniform neuropathologic evaluation, the density of neurofibrillary tangles was summarized in a composite measure and the presence of Lewy bodies and gross and microscopic cerebral infarction was noted.
Results:
During follow-up, rate of global cognitive decline was gradual at first and then more than quadrupled in the last 4 to 5 years of life consistent with the onset of progressive dementia. Neurofibrillary tangles, cerebral infarction, and neocortical Lewy bodies all contributed to gradual age-related cognitive decline and little age-related decline was evident in the absence of these lesions. Neurofibrillary tangles and neocortical Lewy bodies contributed to precipitous disease-related cognitive decline, but substantial disease-related decline was evident even in the absence of these lesions.
Conclusion:
Mild age-related decline in cognitive function is mainly due to the neuropathologic lesions traditionally associated with dementia.
GLOSSARY
- AD
= Alzheimer disease.
Much cognitive loss in old age is limited to very subtle decline evolving slowly over a period of years. The neurobiologic mechanisms underlying these changes are not known. Traditionally, this gradual age-related decline has been attributed to normative developmental processes and distinguished from the precipitous cognitive decline attributed to pathologic processes such as those underlying Alzheimer disease (AD) and other common dementias. However, few clinical–pathologic studies have examined the relation of pathologic indices to change in cognitive function over time,1,2 and none of these has separated slowly progressive age-related change from more rapid disease-related decline. Thus, knowledge about the contribution of common neurodegenerative lesions to cognitive aging is limited.
In the present study, we test the hypothesis that the neurodegenerative lesions associated with late-life dementia, including AD pathology, cerebrovascular disease, and Lewy bodies, contribute to age-related cognitive decline. The hypothesis is based on the observations that these neuropathologic lesions are commonly observed in the brains of old people who die with no or mild cognitive impairment3–8 and their cross-sectional correlation with level of cognitive function in persons without dementia is comparable to the correlation seen in persons with dementia.7 Data are from the Religious Orders Study. Participants are older Catholic nuns, priests, and brothers who completed up to 13 years of annual cognitive testing, died, and underwent brain autopsy. Summary measures of neurofibrillary tangles, cerebral infarction, and Lewy bodies were derived from a uniform postmortem examination of the brain. In analyses, we partitioned individual paths of cognitive change into a preterminal slowly progressive age-related component and a separate disease-related component. We then tested the association of postmortem measures of neurodegeneration with each component of change in cognitive function, first using a composite measure of global cognition and then measures of specific cognitive systems.
METHODS
Participants.
Clinical and pathologic data are from older Catholic nuns, priests, and brothers who participated in the Religious Orders Study.9 All subjects agreed to annual clinical evaluations and brain autopsy at the time of enrollment. The clinical evaluations began in 1994 and are continuing.
Eligibility for these analyses required a completed brain autopsy and longitudinal cognitive data. Of 508 study participants who died, 476 (93.7%) underwent brain autopsy. At the time of these analyses, 354 subjects had autopsy results available and longitudinal data. They had a mean age at death of 86.7 (SD 6.9), 59.0% were women, and 94.4% were white and non-Hispanic. They had participated in the Religious Orders Study a mean of 6.4 years (SD 3.2; range 0.9–13.1) with their final clinical evaluation a median of 6 months before death (interquartile range 3.0–9.5) and brain removal a median of 5.3 hours after death (interquartile range 3.8–9.8). At the last clinical evaluation, 47.5% met criteria for dementia.
Standard protocol approvals, registrations, and patient consents.
After a complete description of the study, written informed consent was obtained from all subjects. The study was approved by the Institutional Review Board of Rush University Medical Center.
Assessment of cognitive function.
A battery of cognitive tests was administered at baseline and repeated annually until death. It included the following measures of episodic memory: Word List Memory, Recall, and Recognition and immediate and delayed recall of the East Boston Story and Story A from Logical Memory. Semantic memory was assessed with a 20-item Boston Naming Test, 15-item version of Extended Range Vocabulary, 20-item form of the National Adult Reading Test, and Verbal Fluency. Digit Span Forward, Digit Span Backward, Digit Ordering, and Alpha Span were used to assess working memory. Perceptual speed was measured with Number Comparison and the oral form of the Symbol Digit Modalities Test.
Initial analyses used a previously established composite measure of global cognition based on all 19 tests. We did this because composite measures of this sort are able to accommodate a much wider range of performance than individual tests, thereby minimizing floor and ceiling artifacts and other forms of measurement error. This composite measure has yielded remarkably similar estimates of terminal cognitive decline in separate cohorts,10,11 likely a reflection of its sound metric properties. To calculate the composite measure, raw scores on each test were converted to z scores, using the baseline mean and SD in the entire cohort, and then averaged. In addition, because prior research has shown that terminal changes occur in multiple cognitive domains,10–12 we also formed composite measures of specific cognitive functions. Further information on the individual tests and the derivation of the composite scores is contained in previous publications.9,13
Neuropathologic assessment.
A standard protocol was followed for brain removal (at Rush and 11 predetermined sites across the United States), tissue sectioning and preservation, and quantification of pathologic findings, as described in more detail elsewhere.14–17 Each brain was cut coronally using a Plexiglas jig into 1-cm slabs which were examined for cerebral infractions after being fixed in 4% paraformaldehyde. The age, volume, and location of each infarction was noted. For these analyses, we counted old cortical and subcortical gray and white matter infarctions including ischemic lesions with small amounts of hemorrhage. We excluded infarctions that occurred within 6 months of death or in the brainstem or cerebellum. In addition, we used hematoxylin and eosin stain to document chronic microscopic infarcts. In primary analyses, gross and microscopic infarcts were treated as present or absent. In secondary analyses of each type of infarction, a no infarct reference group was contrasted with 1 and multiple infarct subgroups.
We used tangle density as the measure of AD pathology because prior work found that it was most strongly associated with level of cognition and the association of amyloid with cognition was not significant when tangles were included in the analysis.18 The density of tau-immunoreactive neurofibrillary tangles in 6 brain regions was quantified with immunohistochemistry and computer-assisted sampling: entorhinal cortex, CA1/subiculum, dorsolateral prefrontal cortex or midfrontal gyrus (Brodmann area 46/9), inferior temporal cortex (Brodmann area 20), inferior parietal cortex or angular/supramarginal gyrus (Brodmann area 39/40), and primary visual cortex (Brodmann area 17). Regional tangle density scores were standardized and averaged to yield a composite measure.
Lewy bodies were identified with α-synuclein immunohistochemistry (Zymed Laboratories, South San Francisco, CA; 1:100) using the avidin-biotin method with alkaline phosphatase as the color developer, as previously described.14,15 Six brain regions were examined: substantia nigra, entorhinal cortex, cingulate cortex, midfrontal cortex, middle temporal cortex, and inferior parietal cortex. In analyses, a no Lewy body reference group was contrasted with those with any neocortical Lewy bodies (i.e., in midfrontal, middle temporal, or inferior parietal areas) and those with Lewy bodies confined to the substantia nigra or limbic regions.
Data analysis.
The first analytic goal was to partition cognitive paths into slow change and rapid change components. Previous research in this10 and other11,12,19 cohorts indicates that cognitive decline tends to markedly accelerate in the last few years of life. To identify this point, we constructed a series of mixed-effect models20 that allowed rate of cognitive decline to change in the last n months before death. We tested values of n ranging from 48 to 72 based on prior research10,11 and the distribution of follow-up time in this cohort. We selected the analysis with the highest log likelihood value, indicating the best model fit. We did not use clinical diagnostic data to dichotomize cognitive trajectories given evidence that years of accelerated cognitive decline usually precede incident diagnoses of mild cognitive impairment21 and dementia.22,23
The second analytic goal was to assess the contribution of postmortem variables to cognitive change. In separate analyses for each pathologic index, we repeated the core model with terms for the interactions of the pathologic index with time before the change point (to estimate the association with age-related decline) and with time after the change point (to estimate the association with disease-related decline). A final model included terms for all pathologic indices. A composite measure of global cognition was the primary outcome measure. Analyses were subsequently repeated using measures of specific cognitive functions in place of the global cognitive measure.
RESULTS
Global cognitive decline.
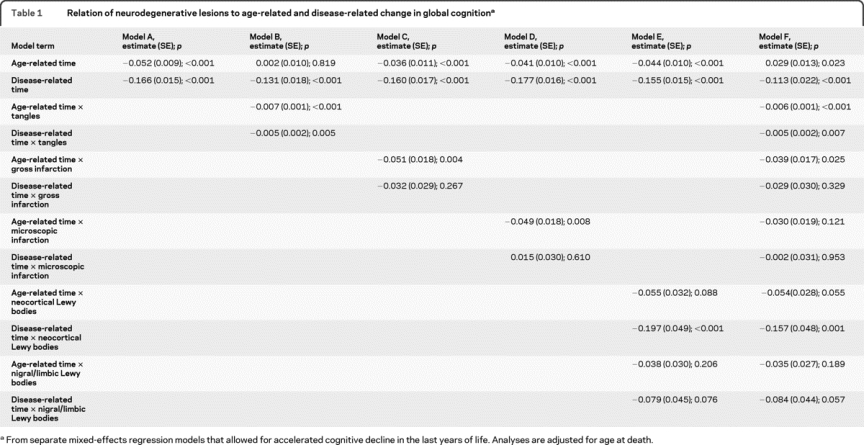
To make use of all cognitive data, primary analyses were conducted with a composite measure of global cognition based on all individual tests. At study onset, it ranged from −3.4 to 1.4 (mean −0.3, SD 0.7), with higher values indicating better performance. We fit a series of models that allowed rate of change in global cognition to shift before death. Optimal results were obtained with change points ranging from about 50 to 55 months before death with a change point of 52 months providing the best fit (figure e-1 on the Neurology® Web site at www.neurology.org). In this analysis (table 1, model A), global cognition declined at a mean of 0.052 unit per year before this point (age-related change). During the last 52 months of life, this rate increased more than 4-fold to 0.218 unit per year (disease-related change). Baseline level of cognitive function had a positive correlation with age-related rate of cognitive change (r = 0.32, p = 0.002) but not with disease-related cognitive change (r = 0.05, p = 0.575). Further, age-related cognitive change was not associated with disease-related cognitive change (r = 0.06, p = 0.648).
Table 1 Relation of neurodegenerative lesions to age-related and disease-related change in global cognition
Neuropathologic lesions and global cognitive decline.
We began analyses with a previously established composite measure of the density of tau-immunoreactive neurofibrillary tangles. Scores ranged from 0 to 65.7 (median 4.4; interquartile range 1.4–10.1) with higher values indicating more pathology. In a mixed-effects analysis (table 1, model B), higher tangle density was associated with more rapid age-related and disease-related decline in global cognition. Further, the effect of tangles was roughly equivalent for both age-related and disease-related change. As shown in figure 1A, which is based on this analysis, virtually no age-related change in global cognition occurred at low levels of tangles (25th percentile, green line) compared to substantial decline at high levels (75th percentile, red line). By contrast, much disease-related global cognitive decline occurred despite low levels of tangles, suggesting the involvement of other pathologic factors.

Figure 1 Neuropathologic lesions and global cognitive decline
Predicted path of global cognitive decline at high (75th percentile, red line) and low (25th percentile, green line) levels of neurofibrillary tangles (A); with chronic gross (B) and microscopic (C) infarction present (red line) or absent (green line); with Lewy bodies present (neocortical, red line; nigral/limbic, blue line) or absent (green line) (D).
One or more chronic gross cerebral infarctions were found at autopsy in 36.8% of subjects and 31.2% had 1 or more microinfarctions. As shown in table 1, both gross (model C) and microscopic (model D) infarction were associated with a more than 2-fold increase in rate of age-related global cognitive decline. By contrast, neither gross nor microscopic infarction was associated with disease-related cognitive decline. These effects are portrayed in figure 1, B and C. Results were comparable when gross and microscopic infarction analyses were repeated with a no-infarction reference group contrasted with 1 infarction and multiple infarction subgroups.
At autopsy, 1 or more Lewy bodies were present in the neocortex of 9.6% and confined to the substantia nigra or limbic regions in another 10.5%. The presence of neocortical Lewy bodies (red line, figure 1D) was associated with an approximate doubling of disease-related decline relative to those without Lewy bodies (green line, figure 1D) and a nearly significant effect on age-related decline (table 1). By contrast, nigral/limbic Lewy bodies (blue line, figure 1D) were not associated with either age-related or disease-related decline in global cognition.
We constructed an additional model that included all pathologic measures (table 1, model E). In the absence of pathologic findings, there was a slight age-related improvement in global cognition though substantial disease-related decline remained, as shown by the terms for time. Gross cerebral infarction was associated with age-related decline in global cognition; neocortical Lewy bodies were associated with disease-related decline; and neurofibrillary tangles were associated with both age-related and disease-related decline.
Neuropathologic lesions and decline in cognitive systems.
To determine whether the association of pathologic burden with cognitive decline varied across cognitive systems, we repeated the previous set of analyses for each of 4 composite measures of specific cognitive functions. Higher tangle density was associated with more rapid age-related and disease-related decline in all cognitive systems (first 2 rows of table 2). This global effect of neurofibrillary tangles was evident in the predicted cognitive paths associated with relatively high (75th percentile, red line) vs relatively low (25th percentile, green line) tangle density in figure 2. By contrast, cerebral infarction (third through sixth rows of table 2) and Lewy bodies (last 4 rows of table 2) had selective effects across time and cognitive systems. Gross cerebral infarction was associated with more rapid age-related decline in episodic and working memory and more rapid disease-related decline in semantic memory (figure e-2). Microscopic infarction was associated with increased age-related decline in semantic memory (figure e-3). As shown in figure 3, neocortical Lewy bodies (red line) were associated with accelerated age-related decline in perceptual speed and disease-related decline in episodic, semantic, and working memory, whereas Lewy bodies in nigral or limbic regions (blue line) were not associated with decline. With all pathologic measures in the same model, tangles continued to be associated with age-related and disease-related decline in multiple cognitive systems; gross infarction was associated with age-related working memory decline; and neocortical Lewy bodies were associated with age-related perceptual speed decline and disease-related decline in episodic and semantic memory (table e-1).
Table 2 Relation of neurodegenerative lesions to age-related and disease-related change in specific cognitive functions
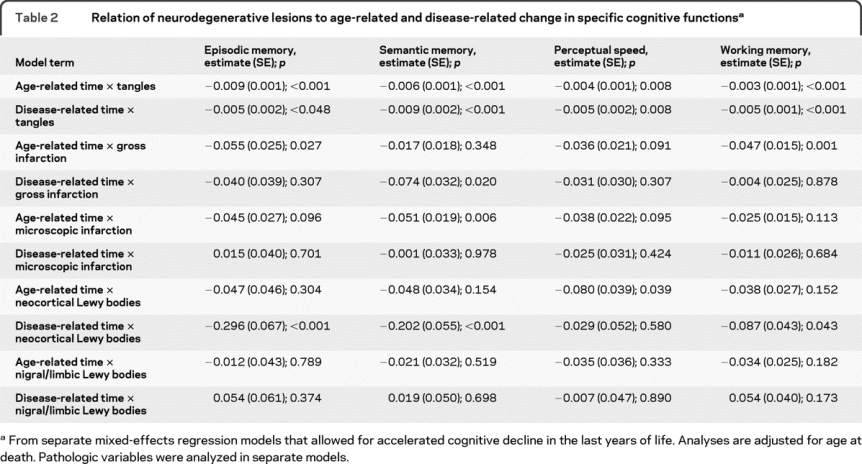

Figure 2 Neurofibrillary tangles and decline in specific cognitive domains
Relation of high (75th percentile, red line) and low (25th percentile, green line) levels of neurofibrillary tangles to decline in episodic memory (A), semantic memory (B), working memory (C), and perceptual speed (D).

Figure 3 Lewy bodies and decline in specific cognitive domains
Relation of Lewy bodies (neocortical, red line; nigral/limbic, blue line; absent, green line) to decline in episodic memory (A), semantic memory (B), working memory (C), and perceptual speed (D).
DISCUSSION
In this study, more than 350 older people completed up to 13 years of annual cognitive testing, died, and underwent brain autopsy. Individual paths of change in cognitive function were separated into gradual age-related and more precipitous disease-related components. Age-related cognitive decline was associated with neurofibrillary tangles, cerebral infarction, and Lewy bodies and was not evident in the absence of these lesions. This indicates that the neurodegenerative lesions traditionally associated with dementia are principally responsible for the gradual age-related cognitive decline that precedes dementia and that AD and related disorders have a much greater impact on late-life cognitive functioning than previously recognized.
Gradual cognitive decline in old age has mainly been thought to reflect normative age-related developmental processes.19,24 In this cohort, however, there was no age-related cognitive decline absent postmortem evidence of neurodegenerative disease, and multiple pathologic lesions were associated with rate of age-related cognitive decline. These data challenge the concept of normative cognitive aging and suggest instead that neurodegenerative disease plays a role in virtually all late-life cognitive decline. Yet the correlation between postmortem markers and cognitive decline is far from perfect. Understanding individual differences in the cognitive consequences of these early neurodegenerative changes may suggest strategies for delaying the onset of cognitive symptoms in dementia, which epidemiologic research has identified as a key to reducing its public health burden.25,26
Many individuals in this cohort experienced more rapid cognitive decline in their last 4 to 5 years of life. This terminal acceleration in cognitive decline is well-recognized.10–12,19 Although its neurobiological basis is not well-understood, clinical studies have suggested that dementia disorders may be involved. Consistent with this idea, neurofibrillary tangles and neocortical Lewy bodies, postmortem markers of AD and Lewy body dementia, were each associated with precipitous cognitive decline. By contrast, neither gross nor microscopic cerebral infarctions were associated with terminal change in cognition. Previous research has shown that cerebral infarction contributes to risk of dementia but is rarely its sole cause.14–17 The present data suggest that this may be because cerebral infarction mainly contributes to early mild cognitive decline rather than late rapid decline.
Although controlling for all postmortem pathologic measures virtually eliminated age-related global cognitive decline, substantial disease-related decline remained. This suggests that the factors responsible for precipitous cognitive decline in the last few years of life substantially differ from the factors responsible for cognitive change preceding this period. This is consistent with the independence of age-related and disease-related cognitive change observed in this study and may explain why most risk factors for dementia do not predict dementia progression. The results also indicate that factors other than tangles and neocortical Lewy bodies are contributing to variability in disease-related cognitive decline. This could include other pathologic features such as the TAR DNA-binding protein 43.27,28 In addition, neurodegeneration in the form of loss of neurons and synapses may be the most proximate cause of precipitous cognitive decline,29,30 leaving less variability to be accounted for by more distal contributors to neurodegeneration such as tangles and Lewy bodies.
The relation of pathology to cognitive decline was complex, varying not only by lesion type but also by the portion of the late-life cognitive trajectory studied and the cognitive domain assessed. Higher tangle density adversely affected all forms of cognition at all trajectory points whereas the effects of cerebral infarction and Lewy bodies varied across time and domains of cognitive function. It is noteworthy that Lewy bodies were associated with decline in episodic memory, a defining characteristic of AD, and that all forms of pathology contributed to age-related decline in working memory, a change often attributed to normal aging. Although most late-life cognitive decline appears to have a neurodegenerative component, inferring the type of neurodegeneration from cognitive data alone is difficult.
This study has several strengths. Psychometrically sound composite measures of cognition were used in analyses, minimizing floor and ceiling artifacts and other forms of measurement error. The availability of up to 14 evenly spaced observations per individual allowed us to accommodate a within-person shift in rate of change in cognitive function. The high rates of participation in follow-up and autopsy reduce the likelihood that missing data affected results.
Study limitations should also be noted. The findings are based on a selected cohort so their generalizability remains to be determined. In addition, use of a fixed cutpoint to separate age-related decline from disease-related decline did not allow us to capture individual differences in the onset of accelerated cognitive decline, and this may have affected results.
AUTHOR CONTRIBUTIONS
Statistical analysis was supervised by Dr. Leurgans. Dr. Wilson had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of data analysis.
ACKNOWLEDGMENT
The authors thank the hundreds of Catholic nuns, priests, and brothers who have participated in the Religious Orders Study; Julie Bach, MSW, and Traci Colvin, MPH, for study coordination; John Gibbons, MS, and Greg Klein, MS, for data management; Woojeong Bang, MS, for statistical programming; and Priya Patel for word processing.
DISCLOSURE
Dr. Wilson serves as a Consulting Editor for Aging, Neuropsychology, and Cognition and Psychology and Aging; and receives research support from the NIH/NIA (R01AG024871 [principal investigator], P30AG10161 [coinvestigator], R01AG11101 [coinvestigator], R01AG15819 [coinvestigator], R01AG021972 [coinvestigator], U24AG026395 [coinvestigator], R01AG017917 [coinvestigator], R01AG009966 [coinvestigator], and U01AG016979 [coinvestigator]) and the NIH/NIEHS (ES10902 [coinvestigator]). Dr. Leurgans receives/has received research support from the NIH (NINDS R01NS28127 [biostatistician], NINDS R01NS40902 [biostatistician], NIA P01AG009466 [statistics core leader], P30AG010161 [data core leader], NIA R01AG024480 [biostatistician], NIA P01AG014449 [statistics core leader], R01AG033678 [biostatistician], NIA R01AG017917 [biostatistician]), NIA R01AG024871 [biostatistician], NIA R01AG22018 [biostatistician], and NIA R01 AG15819 [biostatistician]) and the Michael J. Fox Foundation. Dr. Boyle receives research support from the NIH (R01AG034374 [principal investigator] and R01AG033678 [principal investigator]). Dr. Schneider serves as an Associate Editor of the Journal of Alzheimer's Disease; serves as a consultant for Avid Radiopharmaceuticals, Inc.; and receives research support from the NIH (R01AG024480 [neuropathologist], P01AG009466 [neuropathologist], P01AG014449 [neuropathologist], R01AG015819 [neuropathologist], R01AG017917 [neuropathologist], R01AG031553 [neuropathologist], R21AG030346 [neuropathologist], R01AG034374 [neuropathologist], R01AG033678 [neuropathologist], R01AG036042 [neuropathologist], RC2AG036547 [neuropathologist], R01HL096944 [neuropathologist], and P30AG010161 [pathology core leader]). Dr. Bennett serves on the editorial board of Neurology®; has served as a consultant to Schering-Plough Corp., Medivation, Inc., and the Gerson Lehrman Group; and receives research support from Danone Inc., the NIH (R01AG017917 [principal investigator], R01AG015819 [principal investigator], R01AG036042 [principal investigator], RC2AG036547 [principal investigator], U01AG032984 [co-principal investigator, leader of epidemiologic cohort studies], R01AG024480 [coinvestigator], R01AG024871 [Co-Investigator], P01AG009466 [coinvestigator], U24AG026395 [coinvestigator], R01AG030142 [coinvestigator], P01AG01449 [coinvestigator], R01HL096944 [coinvestigator], R01AG033678 [coinvestigator], R01AG034374 [coinvestigator], R01AG032755 [coinvestigator], and P30AG010161 [principal investigator–administrative core leader, Religious Orders Study core leader]), and the Illinois Department of Public Health.
Address correspondence and reprint requests to Dr. Robert S. Wilson, Rush Alzheimer's Disease Center, Rush University Medical Center, 600 South Paulina Ave., Suite 1038, Chicago, IL 60612 rwilson@rush.edu
Supplemental data at www.neurology.org
e-Pub ahead of print on September 15, 2010, at www.neurology.org.
Study funding: Supported by the NIH/NIA (R01AG015819, P30AG010161, and R01AG034374). The sponsors had no role in the design and conduct of the study; in the collection, management, analysis, or interpretation of the data; or in the preparation, review, or approval of the manuscript.
Disclosure: Author disclosures are provided at the end of the article.
Received February 11, 2010. Accepted in final form May 17, 2010.
REFERENCES
- 1.Green MS, Kaye JA, Ball MJ. The Oregon Brain Aging Study: neuropathology accompanying healthy aging in the oldest old. Neurology 2000;54:105–113. [DOI] [PubMed] [Google Scholar]
- 2.Galvin JE, Powlishta KK, Wilkins K, et al. Predictors of preclinical Alzheimer's disease and dementia: a clinicopathologic study. Arch Neurol 2005;62:758–765. [DOI] [PubMed] [Google Scholar]
- 3.Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol 2003;60:729–736. [DOI] [PubMed] [Google Scholar]
- 4.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 2006;63:665–672. [DOI] [PubMed] [Google Scholar]
- 5.Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol 2006;62:38–46. [DOI] [PubMed] [Google Scholar]
- 6.Driscoll I, Resnick SM, Troncoso JC, An Y, O'Brien R, Zonderman AB. Impact of Alzheimer's pathology on cognitive trajectories in nondemented elderly. Ann Neurol 2006;60:688–695. [DOI] [PubMed] [Google Scholar]
- 7.Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer's disease pathology and cerebral infarctions. Neurology 2005;64:834–841. [DOI] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 9.Wilson RS, Bienias JL, Evans DA, Bennett DA. Religious Orders Study: overview and change in cognitive and motor speed. Aging Neuropsychol Cogn 2004;11:280–303. [Google Scholar]
- 10.Wilson RS, Beckett LA, Bienias JL, Evans DA, Bennett DA. Terminal decline in cognitive function. Neurology 2003;60:1782–1787. [DOI] [PubMed] [Google Scholar]
- 11.Wilson RS, Beck TL, Bienias JL, Bennett DA. Terminal cognitive decline: accelerated loss of cognition in the last years of life. Psychosom Med 2007;69:131–137. [DOI] [PubMed] [Google Scholar]
- 12.Thorvaldsson V, Hofer SM, Berg S, Skoog I, Sacuiu S, Johansson B. Onset of terminal decline in cognitive abilities in individuals without dementia. Neurology 2008;71:882–887. [DOI] [PubMed] [Google Scholar]
- 13.Wilson RS, Beckett LA, Barnes LL, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 2002;17:179–193. [PubMed] [Google Scholar]
- 14.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009;66:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69:2197–2204. [DOI] [PubMed] [Google Scholar]
- 16.Schneider JA, Wilson RS, Cochran EJ, et al. Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology 2003;60:1082–1088. [DOI] [PubMed] [Google Scholar]
- 17.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 2004;62:1148–1155. [DOI] [PubMed] [Google Scholar]
- 18.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 2004;61:378–384. [DOI] [PubMed] [Google Scholar]
- 19.Sliwinski MJ, Strawski RS, Hall CB, Katz M, Verghese J, Lipton R. Distinguishing preterminal and terminal cognitive decline. Eur Psychol 2006;11:172–181. [Google Scholar]
- 20.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics 1982;38:963–974. [PubMed] [Google Scholar]
- 21.Howieson DB, Carlson NE, Moore MM, et al. Trajectory of mild cognitive impairment onset. J Int Neuropsychol Soc 2008;14:192–198. [DOI] [PubMed] [Google Scholar]
- 22.Amieva H, Jacqmin-Gadda H, Orgogozo JM, et al. The 9 year cognitive decline before dementia of Alzheimer type: a prospective population-based study. Brain 2005;128:1093–1101. [DOI] [PubMed] [Google Scholar]
- 23.Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer's disease. J Int Neuropsychol Soc 2008;14:266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baltes P, Nesserlroade J. History and rationale of longitudinal research. In: Nesserlroade J, Baltes P, eds. Longitudinal Research in the study of Behavior or Development. Sand Diego, CA: Academic Press; 1979:1–39. [Google Scholar]
- 25.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health 1998;88:1337–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sloane PD, Zimmerman S, Suchindran C, et al. The public health impact of Alzheimer's disease, 2000–2050: potential implication of treatment advances. Annu Rev Public Health 2002;23:213–231. [DOI] [PubMed] [Google Scholar]
- 27.Amador-Ortiz C, Lin WL, Ahmed Z, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol 2007;61:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Josephs KA, Whitewell JL, Knopman DS, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinico-pathologic and radiologic phenotype. Neurology 2008;70:1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991;30:572–580. [DOI] [PubMed] [Google Scholar]
- 30.Scheff SW, Price DA, Schmitt FA, Dekosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007;68:1505–1508. [DOI] [PubMed] [Google Scholar]