

Abstract
RP2 is a ubiquitously expressed protein encoded by a gene associated with X-linked retinitis pigmentosa (XLRP), a retinal degenerative disease that causes severe vision loss. Previous in vitro studies have shown that RP2 binds to ADP ribosylation factor-like 3 (Arl3) and activates its intrinsic GTPase activity, but the function of RP2 in the retina, and in particular photoreceptor cells, remains unclear. To begin to define the role of RP2 in the retina and XLRP, we have carried out biochemical studies to identify proteins in retinal cell extracts that interact with RP2. Here, we show that RP2 interacts with N-ethylmaleimide sensitive factor (NSF) in retinal cells as well as cultured embryonic kidney (HEK293) cells by mass spectrometry-based proteomics and biochemical analysis. This interaction is mediated by the N-terminal domain of NSF. The E138G and ΔI137 mutations of RP2 known to cause XLRP abolished the interaction of RP2 with the N-terminal domain of NSF. Immunofluorescence labeling studies further showed that RP2 co-localized with NSF in photoreceptors and other cells of the retina. Intense punctate staining of RP2 was observed close to the junction between the inner and outer segments beneath the connecting cilium, as well as within the synaptic region of rod and cone photoreceptors. Our studies indicate that RP2, in addition to serving as a regulator of Arl3, interacts with NSF and this complex may play an important role in membrane protein trafficking in photoreceptors and other cells of the retina.
Retinitis pigmentosa (RP) is a leading cause of inherited blindness with an incidence of 1 in 3500 individuals worldwide. It is a heterogeneous group of retinal degenerative diseases characterized by a reduction in visual field, night blindness and progressive loss of central vision often leading to complete blindness (1–3). RP can be inherited as an autosomal dominant, autosomal recessive or X-linked trait. To date, over 48 different genes have been implicated in the various forms of RP (http://www.sph.uth.tmc.edu/Retnet/) with most genes encoding proteins that are expressed in photoreceptor or retinal pigment epithelial (RPE) cells and are critical for photoreceptor cell structure, function and survival.
X-linked RP (XLRP) accounts for approximately 10–20% of RP cases. It is a particularly severe form of the disease, typically resulting in significant vision loss in the first decade and progressing to total blindness by the third or fourth decade of life (3–6). Approximately, 10–15% of the XLRP cases are caused by mutations in the RP2 gene. These include missense, splice-site, nonsense and frame shift mutations (7–11).
The RP2 gene encodes a ubiquitously expressed protein of 350 amino acids known as retinitis pigmentosa 2 protein or RP2 (8, 12, 13). Using polyclonal antibodies, Grayson et al. (14) first reported that RP2 is distributed throughout the human retina with immunoreactivity in photoreceptors extending from the tips of the outer segments to the synaptic terminals. RP2 is both myristoylated and palmitoylated at the N-terminus. This dual post-translational acylation is believed to target the protein to the plasma membrane of cells (12, 14). The N-terminal region of RP2 consisting of 151 amino acids (amino acid 42–192) shares a 30% sequence identity and partial functional conservation with the tubulin-specific chaperone protein (TBCC) (8, 15). In the presence of tubulin-specific co-factor D (TBCD), RP2 can substitute for TBCC by stimulating the GTPase activity of native tubulin (15). However, RP2 cannot replace TBCC in promoting the assembly of newly folded tubulin into heterodimers. The C-terminal region exhibits sequence and structural homology to nucleoside diphosphate (NDP) kinase, but the function of this domain remains to be determined. A high resolution structure of RP2 has been determined by X-ray crystallography (16). The N-terminal 228 amino acids fold into a β helix domain while the C-terminal domain (amino acid 229–350) forms a ferrredoxin-like α/β structure.
RP2 has been shown to bind to GTP bound form of ADP ribosylation factor-like 3 (Arl3), a member of the Arl subfamily of Ras-related GTP-binding proteins (15, 16). The high resolution structure of RP2 as a complex with Arl3-GppNHp and Arl3-GDP-AlF4 has been determined (16). The β-helix domain and a short upstream unstructured segment within the N-terminal region of RP2 serves as a high affinity binding site for Arl3 containing a bound GTP analogue (15–17). Myristoylation of RP2 weakens its interaction with Arl3 (15). Recently, Veltel et al. (17) have shown that RP2 is an efficient GTPase activating protein (GAP) for Arl3. The binding of RP2 to GTP-Arl3 resulted in a 90,000-fold stimulation of the intrinsic GTPase activity of Arl3.
Although structural studies of RP2 and its interaction with Arl3 have provided insight into the role of RP2 as a GAP protein for Arl3, the interaction of RP2 with Arl3 and other proteins in photoreceptor cells has not been investigated at a molecular level. We have generated a monoclonal antibody to RP2 and used this reagent to examine the distribution of RP2 in rodent and human photoreceptors and identify proteins that interact with RP2 in the retina. Here, we show that RP2 co-localizes and directly binds N-ethylmaleimide sensitive factor (NSF) in photoreceptor cells and cultured cell lines. Our data suggests that RP2 may have multiple functions in cells related to the trafficking of NSF to the cilium and synaptic region of photoreceptors in addition to its role as a GAP protein for Arl3.
EXPERIMENTAL PROCEDURES
Materials
Monoclonal and polyclonal antibodies against NSF and a polyclonal antibody against β-tubulin were purchased from Abcam (Cambridge, MA, USA); monoclonal antibody against α-tubulin was from Molecular Probes (Eugene, OR, USA); monoclonal antibody against acetylated a-tubulin was from Sigma-Aldrich; monoclonal antibody to actin was from Calbiochem (San Diego, CA, USA). CHAPS detergent was purchased from Anatrace (Maumee, OH, USA), and sucrose, NaCl, and Na2HPO4 were from Fisher Scientific International Inc. (Hampton, NH, USA). Sepharose 2B and Protein A 4 Fast Flow Sepharose beads were obtained from Amersham Biosciences (Uppsala, Sweden) and Complete protease inhibitor was from Roche (Roche Applied Science, Basel, Switzerland). Effectene transfection reagent was from Qiagen (Valencia, CA, USA). Unless otherwise stated all other chemicals were from Sigma-Aldrich.
Generation and characterization of a monoclonal antibody to RP2
Monoclonal antibody RP2-6H2 was generated from a hybridoma cell line produced by the fusion of NS-1 myeloma cells with lymphocytes from a mouse immunized with the human GST fusion proteins containing the N-terminal and C-terminal regions of RP2. A SPOTs kit (Sigma Genosys) was used to map the epitope for the RP2-6H2 antibody. This was achieved by synthesizing a series of peptides 8 amino acids in length on a solid support with an offset of 2 amino acids covering the N-terminal 90 amino acids. The immunoreactivity of RP2-6H2 to the peptides was determined by enhanced chemiluminescence (ECL). The RP2-6H2 antibody was purified from hybridoma culture fluid on a Protein G-Sepharose column.
Plasmid construction and expression of RP2 and NSF
Full-length human RP2 was generated by PCR using human retina cDNA as a template. Constructs encoding the N-terminal domain of RP2 (RP2-N: 1–230 aa), the C-terminal domain of RP2 (RP2-C: 230–350 aa), and RP2 lacking the first 34 amino acids (RP2T34) were generated from the full-length RP2 by PCR. RP2 constructs were cloned into pGEX-4T1 using BamH1 and Xho1 sites to generate GST-RP2 fusion proteins and into pET-15b to produce His-tagged RP2. GST-RP2 disease-linked mutations (C86Y, P95L, Δ137, E138, C67Y, R118H, and R282W) were created by the Quick-Change site-directed mutagenesis kit (Stratagene, La Jolla, CA) following the manufacturer’s instructions. Chinese hamster ovary NSF constructs (NT-NSF: 1–205 aa; D1-NSF: 206–477 aa; and D2-NSF: 478-47 aa) in pQE-9 vector were kindly provided by James Rothman and cloned into pGEX-4T1 for expression as GST-fusion proteins. All of the constructs were verified by DNA sequencing.
The GST-fusion proteins and the His-tagged proteins were expressed in BL21 cells and purified on glutathione-Sepharose and Ni-NTA beads, respectively. In some experiments GST was cleaved from the fusion protein bound to glutathione-Sepharose by treatment with 20 units of thrombin overnight at 4°C.
Isolation of bovine retina tissue
Typically, 10 bovine retinas in 50% sucrose were homogenized in 6.5 ml of ice cold Buffer A (20 mM Tris-HCl, 0.1 M NaCl, 1 mM EDTA, 1 mM MgCl2, pH 7.4) containing Complete protease inhibitor in a glass homogenizer. The resulting mixture was further homogenized by passing the solution 8 times through a 28-gauge needle. The lysate was layered on 60% sucrose and centrifuged at 24,000 rpm for 60 min at 4°C in a swinging-bucket rotor TLS-55 (LE-80K; Beckman, Berkeley, CA). The fraction collected above the 60% sucrose solution was designated as the retinal extract. This extract was further separated into a membrane and soluble fraction as follows. One ml of retinal extract was pelleted at 13,000 rpm for 15 min at 4ºC in a SS-34 Sorvall rotor. The resulting pellet was resuspended with 1 ml of buffer A and stirred for 30 min. The suspension was centrifuged at 13,000 rpm for 15 min at 4°C in SS-34 rotor and membrane and supernatant (soluble) fractions were retained for analysis. The protein concentration was determined by BCA assay (Pierce, Rockford, IL).
Immunoprecipitation of RP2 and GST pull-down experiments
The purified RP2-6H2-antibody was coupled to CNBr-activated Sepharose 2B as described previously (18). Retinal membrane fraction was solubilized in 1% (vol/vol) Triton X-100 (TX-100) or 16 mM CHAPS containing Complete protease inhibitor for 30 min at 4 °C with continuous stirring. The solution was centrifuged at 30,000 × g for 15 min to remove residual non-solubilized material. An aliquot (0.4 ml) of the soluble fraction (Input) was incubated for 1 h with 50 μl of RP2-6H2-Sepharose pre-washed with column buffer (0.1% TX-100 or 10 mM CHAPS in Tris buffered saline, pH 7.4) in an Ultrafree filter unit (Millipore Corp., Billerica, MA). The unbound fraction was collected by low speed centrifugation and retained for analysis. The immunoaffinity matrix was subsequently washed extensively with column buffer prior to elution of the bound protein twice with 40 μl of 2% (w/v) SDS in TBS. The proteins were separated by SDS-PAGE for analysis by mass spectrometry and western blotting (see below).
For GST pull down experiments, 100 μl of 10 μg of GST alone, GST-N-NSF, GST-D1-NSF, or GST-D2-NSF were incubated with an equal volume of 10 μg of expressed and purified WT or mutant RP2 for 1 h at 4ºC. The mixture was then diluted to 500 μl PBS-Tween (phosphate buffered saline containing 0.5% Tween-20). After removing 50 μl, the remaining sample was added to 50 μl of glutathione-agarose pre-washed in PBS-Tween and gently stirred for 2 h at 4 ºC. The unbound fraction was collected. The affinity matrix was washed extensively in PBS-Tween and the bound protein was subsequently eluted with 4% SDS in PBS for analysis by SDS-PAGE and western blotting.
Construction and transfection of RP2-shRNA
Human RP2-targeting shRNAs 1 (target-position [nt] 361–379), 2 (nt 494–512), 3 (nt 901–921), and 4 (nt 1154–1174) were designed using the shRNA Sequence Designer (Clontech, Mountain View, CA). shRNA sequences were synthesized and cloned into pSIREN-DsRed-Express vector (Clontech). The shRNA tested to be most efficient was further subcloned into pcDNA3 for stable transfection of HeLa cells. A shRNA cassette containing human U6 promoter, shRNA3 and DsRed was further subcloned into pcDNA3 with BglII and HindIII, denoted RP2-shRNA3-pcDNA3. All constructs were confirmed by DNA sequencing before use.
RP2-sh3-pcDNA3 was stably transfected into HeLa cells with Effectene (Qiagen) and screened by administration of G418 (Invitrogen, Indianapolis, IN). After 48 h, cells were washed and total protein was extracted with lysis buffer (20 mM Tris, 150 mM NaCl, 1% TX-100). Protein expression was analyzed by SDS-PAGE and immunoblotted with antibodies to RP2. Detection and quantification were performed with anti-mouse Ig conjugated with Alexa 680 (dilution 1:20,000) and the LI-COR Odyssey system (Lincoln, NE).
In-gel proteolysis and mass spectrometry
Identification of proteins in the bound, eluted fraction from the RP2-6H2 immunoaffinity matrix was obtained by liquid chromatography tandem mass spectrometry (LC/MS/MS) as follows: The bound fraction from the RP2-6H2 immunoaffinity column was eluted with 4% SDS and added to an equal volume of SDS cocktail (20 mM Tris-HCl, pH 6.8, 4% SDS, 20% sucrose, and 4% β-mercaptoethanol). The sample was applied to an 8% SDS-PAGE gel, and subjected to electrophoresis until the high molecular mass marker (~250 kDa) just entered the separating gel. The band was then cut and subjected to trypsin digestion as follows. The gel piece was washed with water several times and then a 1:1 mixture of 100 mM ammonium bicarbonate followed by rinsing 3 times with 100% acetonitrile. After shrinking and drying with 100% acetonitrile, the gel piece was incubated with 10 mM dithiotreitol followed by 55 mM iodoacetamide, washed with ammonium bicarbonate, and dried from 100% acetonitrile. The sample was then incubated with sequencing grade trypsin (Promega, Madison, WI) at a concentration of 40 ng trypsin/μl for 15 min at room temperature. The protease solution was removed, the sample was overlaid with 25 mM ammonium bicarbonate, and digested for 18 h at 37 °C. The solution was collected in a tube and the gel piece was re-extracted with 50 mM ammonium bicarbonate/60% acetonitrile/0.1% trifluoroacetic acid/5% formic acid. The samples were pooled and concentrated in a SpeedVac.
The peptides were separated and analyzed using an Agilent 1100 Series nanoflow HPLC coupled online to LTQ-FT as previously described (19). Fragment peak lists were generated by ExtractMSN (v3.2, ThermoFisher) using the default parameters and monoisotopic peak and charge state assignments were checked by DTA Supercharge, part of the MSQuant suite of software, and spectra were searched against the bovine IPI v3.50 database using Mascot Server and parameters as previously described (19).
SDS-PAGE and western blotting
Samples were denatured in 10 mM Tris-HCl, pH 6.8, 4% SDS, 20% sucrose, and 4% β-mercaptoethanol and subsequently separated by SDS-PAGE on 8–12 % NuPage or Lammeli resolving gels followed by electroblotting onto Immobilon-FL membranes (Millipore) for antibody labeling. Following blocking for 30 min at 22 °C in 0.5% skimmed milk in PBS, blots were incubated with specified antibodies for 1 h at 22 °C in 0.5% skimmed milk in PBS containing 0.05% Tween 20 (PBS-T) at the specified dilutions. Subsequently, the blots were labeled for 1 h at 22 °C with anti-mouse or anti-rabbit Ig conjugated with either Alexa 680 (dilution 1:20,000 in PBS and 0.5% skimmed milk; Molecular Probes) or LI-COR IRDye 800 (in 0.5% skimmed milk in PBST containing 0.02% SDS and dilution 1:10,000; Rockland, Gilbertsville, PA). Finally, the blots were rinsed three times for 20 min with PBS-T. Bands were visualized by infrared scanning using the LI-COR Odyssey® system (Lincoln, NE).
Immunofluorescence and confocal microscopy
Light-adapted Balb/c mouse retinas were used for immunocytochemical labeling studies. Whole mouse eyes were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (PB), pH 7.4 for 4 h and subsequently rinsed in 0.1 M phosphate buffer containing 10% sucrose as previously described. Cryosections were permeabilized and blocked with 0.1 M Na2HPO4 containing 0.2% TX-100 and 10% normal goat serum for 20 min and labeled overnight with the primary antibody diluted in 0.1 M PB containing 0.1% TX-100 and 2.5% normal goat serum. Sections were rinsed two times for 30 min each in PB and labeled for 1 h with either mouse or rabbit Cy3 (Molecular Probes). For double labeling experiments Alexa 568-conjugated goat anti-rabbit and Alexa 488 goat anti-mouse immunoglobulin were used (Molecular Probes). Labeled sections were also stained with DAPI nuclear stain (Molecular Probes). The stained sections were rinsed three times for 30 min each in PB and examined under a Zeiss LSM700 confocal microscope (Carl Zeiss AG, Jena, Germany) and processed with Zeiss Zen Image Browser.
RESULTS
Generation and characterization of anti-RP2 monoclonal antibody RP2-6H2
A monoclonal antibody against RP2 designated as RP2-6H2 was produced from a mouse immunized with the human N-terminal and C-terminal GST-RP2 fusion proteins. The specificity of the RP2-6H2 antibody is shown by western blotting in Fig. 1. RP2-6H2 specifically labeled a 40 kDa protein in a bovine retinal homogenate which closely co-migrated with His-tagged human RP2 expressed and purified from E. coli (Fig. 1A).
Figure 1.
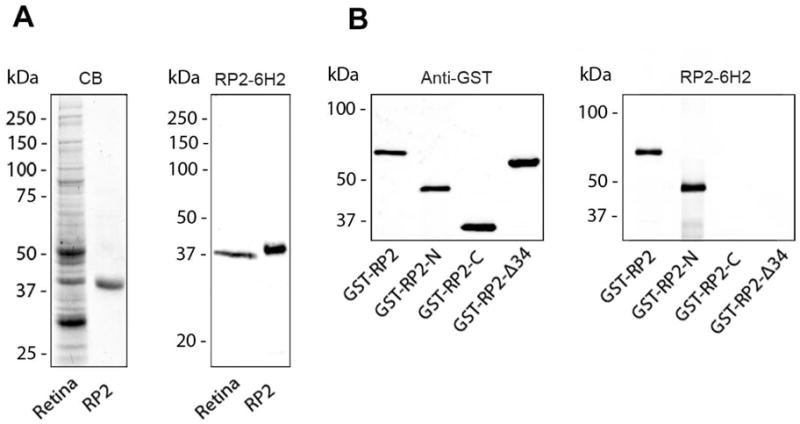
Characterization of RP2-6H2 antibody using bovine retinal extract and His-tagged human RP2. (A) Coomassie blue-stained gel (CB) and a western blot of a retinal extract (retina) and purified His-tagged RP2 (RP2) labeled with the RP2-6H2 antibody demonstrating the specificity of the antibody. (B) A series of GST-RP2 fusion proteins were used to map the epitope of RP2-6H2 antibody. The RP2-6H2 antibody labels the full length (GST-RP2) and the N-terminal fusion protein (GST-RP2-N) but not the C-terminal fusion protein (GST-RP2-C) or GST-RP2 lacking the first 34 amino acids (GST-RP2-Δ34) as shown in the right panel. The GST-fusion proteins were identified in the western blot labeled with an anti-GST antibody.
A series of GST-RP2 fusion proteins were used to localize the epitope for RP2-6H2. As shown in Fig. 1B, RP2-6H2 labeled the full-length and the N-terminal (aa 1 – 230) GST fusion proteins, but not the C-terminal domain (aa 230 – 350) protein or the full-length RP2 fusion protein lacking the N-terminal 34 amino acids indicating that the binding site for RP2-6H2 is within the N-terminal 34 amino acids of RP2. The epitope was further localized to a relatively conserved amino acid sequence YSWDQ (aa 27–31) using overlapping N-terminal synthetic peptides. RP2-6H2 generated against human RP2 cross-reacted with bovine, rat and mouse RP2 (data not shown).
Distribution of RP2 in rodent and human retina by immunofluorescence microscopy
The distribution of RP2 in cryosections of rodent and human retina labeled with the RP2-6H2 antibody was examined by confocal scanning microscopy. Intense immunofluorescence staining was observed in the inner segment and outer plexiform layers and more moderate staining was seen in the outer nuclear layer of photoreceptors from mouse retina (Fig. 2). In addition there was strong labeling of the inner nuclear layer (INL) and inner plexiform layer (IPL) of the inner retina as well as the ganglion cell layer (GCL; see Fig 5) consistent with the ubiquitous expression of RP2. Intense punctate staining was observed in the outer plexiform layer as well as in the ciliary region at the junction between the inner and outer segments (Fig 2 insert) possibly corresponding to the basal body and/or centriole labeling. The latter punctuate staining, however, was not always evident (see Fig 5) suggesting that this labeling may be dependent on the state of the tissue. At the level of immunoflurorescence microscopy, it was not possible to unambiguously determine whether RP2 was in the cytoplasmic fraction or associated with the membrane, although the punctuate labeling pattern observed in the synaptic layers and inner segment was consistent with the association of RP2 with membrane vesicles. A similar pattern of RP2 immunolabeling was observed for rat retina (data not shown) and human retina (Fig. 2). In contrast to an earlier report (14), little, if any, immunoreactivity was observed in the outer segment layer of either human or rodent retinas.
Figure 2.
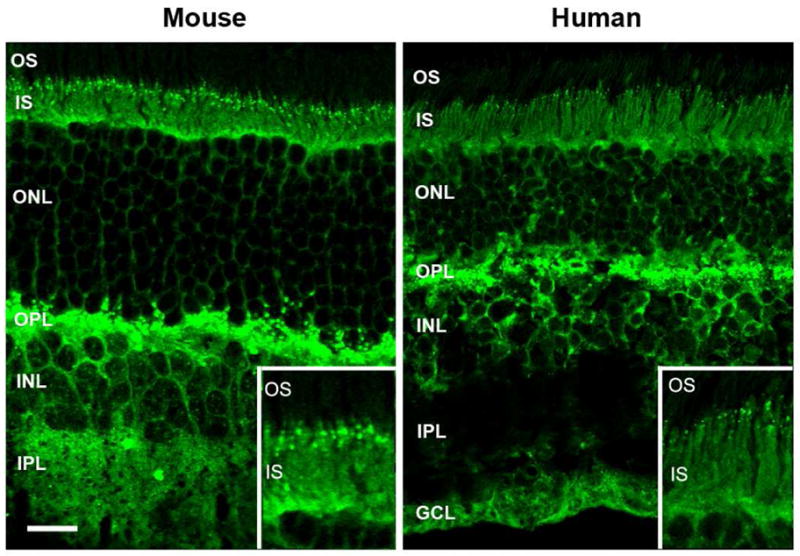
Immunofluorescence microscopy of mouse and human retinal cryosections labeled with the RP2-6H2 antibody. RP2 in both mouse and human retina is present throughout most of the retina, including intense labeling in the inner segment and outer plexiform layer of the outer retina and the inner nuclear layer, inner plexiform layer, and ganglion cell layer of the inner retina; labeling is absent in the outer segment layer. Inset: An enlarged image of the junction between the inner and outer segments showing punctate labeling of RP2. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Bar = 20 μm.
Figure 5.

Confocal scanning microscopy of mouse retina double labeled for RP2 and NSF. (A) Labeling of NSF with an anti-NSF polyclonal antibody; (B) labeling of RP2 with the RP2-6H2 monoclonal antibody; (C) merged image showing co-localization of RP2 and NSF. More intense co-staining was observed for both NSF and RP2 near the junction of the inner and outer segments and outer plexiform layers (D–F). In a number of samples, punctuate staining was observed for RP2 (H) and not NSF (G). Merged image (I) showing RP2 and NSF are not co-localized at these punctations. Bar = 20 μm. Retinal layers are defined in Figure 2.
Co-immunoprecipitation of NSF with RP2 from retinal and HEK293 membrane extracts
Co-immunoprecipitation studies were carried out to identify candidate proteins that interact with RP2. A detergent-solubilized bovine retinal extract was applied to an immunoaffinity matrix consisting of the RP2-6H2 monoclonal antibody directly coupled to Sepharose beads. After extensive washing to remove unbound proteins, the bound RP2 and candidate interacting proteins were eluted from the matrix with SDS and resolved by SDS-PAGE. A control sample which did not contain the retinal extract was run in parallel to identify immunoglobulin protein bands that dissociate from the immunoaffinity matrix under denaturing conditions. As shown in the Coomassie blue stained gels in Fig. 3A, the co-immunoprecipitated fraction contained a number of proteins including proteins with apparent molecular masses of 80 kDa, 55 kDa, 40 kDa, 37 kDa, 27 kDa and a 22 kDa. The 22 kDa and 50–55 kDa proteins which were present in the control samples correspond to the IgG light and heavy chains of the RP2-6H2 antibody, respectively.
Figure 3.
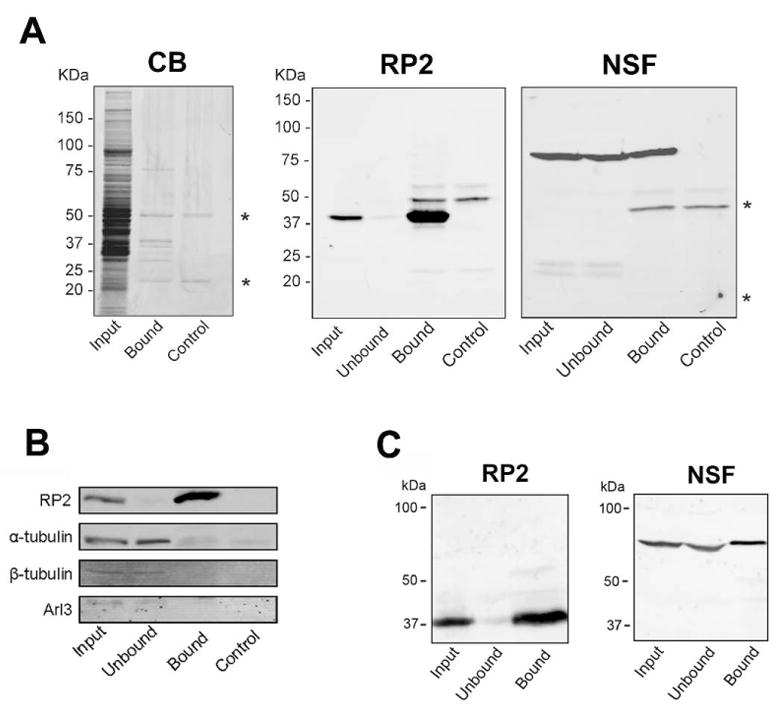
Immunoprecipitation of retinal and HEK293 cell membrane extracts on a RP2-6H2 immunoaffinity matrix. (A) The solubilized retinal membranes (Input) and unbound (Unbound) and SDS-eluted (Bound) fractions were analyzed on SDS gels stained with Coomassie blue (CB) and western blots labeled with the RP2-6H2 antibody (RP2). Quantitative immunoprecipitation was observed for RP2. The SDS-eluted fraction from a control sample (Control) without retinal membranes revealed the immunoglobulin protein bands (*) that dissociated from the immunoaffinity matrix under denaturing conditions. The western blot labeled with the NSF antibody (NSF) confirmed co-immunoprecipitation of NSF with RP2 from retinal extract. (B) Western blots of retinal extracts labeled with antibodies against a and β tubulin and Arl3 showed that little if any of these proteins co-immunoprecipitated with RP2. (C) Endogenous NSF from HEK293 cell homogenate also co-immunoprecipitated with endogenous RP2.
Mass spectrometry was used to identify proteins that co-immunoprecipitated with RP2 on a RP2-6H2 immunoaffinity matrix. The bound fraction was digested with trypsin as outlined in the Experimental section and the resulting peptides were analyzed by LC-MS/MS. NSF (Mass 86,140 daltons) was identified as a major protein along with RP2 (Mass 40,290 daltons). In addition, α and β tubulin and several other proteins were found in the immunoprecipitated fraction (Supplemental Table S1).
The co-immunoprecipitated fraction from the RP2-6H2 immunoaffinity matrix was further analyzed by western blotting to confirm the identity of the Coomassie blue stained proteins that migrated with apparent molecular masses of 40 kDa and 80 kDa. As shown in Fig. 3A, the RP2-6H2 antibody labeled the 40 kDa RP2 protein and an antibody against NSF intensely labeled the 80 kDa protein in the retinal extract and bound fractions from the immunoaffinity matrix indicating that a significant fraction of NSF is associated with RP2. Western blots were also labeled with α and β tubulin and Arl3 antibodies to determine if these proteins co-immunoprecipitated with RP2 from retina homogenates. Tubulins and Arl3 were detected in the retinal membrane extract and unbound fractions, but were not present in significant amounts in the bound fraction suggesting that the tubulins detected by LC-MS/MS were present in low amounts, possibly representing contaminating proteins (Fig. 3B).
Since both RP2 and NSF are ubiquitously expressed in cells, we investigated whether RP2 associates with NSF in a mammalian culture cell line. HEK293 cells were homogenized and the detergent solubilized extract was applied to a RP2-6H2-immunoaffinity matrix. The extract together with the unbound and bound fractions were analyzed by western blotting. As shown in Fig. 3C, the bound fraction contained NSF indicating that a significant fraction of NSF is associated with RP2 in HEK 293 cells as well as retinal extracts.
We considered the possibility that the RP2-6H2 antibody nonspecifically interacts with NSF in the co-immunoprecipitation experiments. To test this we applied a bovine retinal extract to the RP2-6H2 immunoaffinity matrix, and the resulting unbound fraction depleted of RP2, but containing NSF, was reapplied to another fresh RP2-6H2 immunoaffinity matrix. NSF from the unbound fraction depleted of RP2 did not bind to the RP2-6H2 immunoaffinity matrix (data not shown). To further confirm that the RP2-6H2 antibody does not interact with NSF nonspecifically, a cell line with a reduced endogenous RP2 level mediated by RNAi was used to repeat the immunoprecipitation study. The efficiency of knockdown was quantified by immunoblotting the homogenized cell lysate. The RP2-targeted shRNA markedly decreased RP2 expression by approximately 55%. When the cell lysate was applied to the RP2-6H2 immunoaffinity matrix, the amount of NSF co-immunoprecipitated with RP2 was proportionally less, indicating that NSF does not bind to RP2-6H2 antibody nonspecifically (data not shown).
RP2 is associated with the membrane fraction of retinal extracts
RP2 is devoid of transmembrane segments, but contains both myristoylation and palmitoylation post-translational modifications (12). To determine if RP2 and interacting proteins are preferentially present in the soluble or membrane fractions, a retinal extract was separated into soluble and membrane fractions by high speed centrifugation and the fractions were analyzed by western blotting. Figure 4 shows that RP2 was preferentially present in the membrane fraction while NSF was present in both fractions. In contrast, Arl3 was primarily in the soluble fraction.
Figure 4.
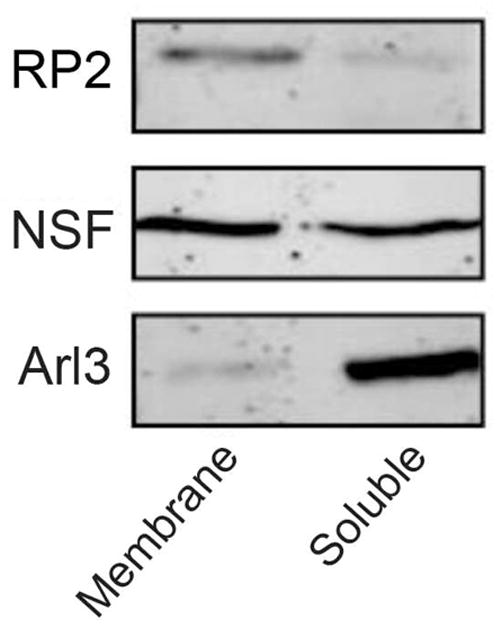
RP2 is associated with the membrane fraction of a retinal extract. Western blots of retinal extracts with RP2-6H2 antibody and antibodies against NSF and Arl3 show that RP2 is preferentially present in the membrane fraction while NSF is present in both fractions and Arl3 is primarily in the soluble fraction.
RP2 and NSF co-localize in photoreceptors and other retinal cells
Co-localization of RP2 and NSF in retina tissue was investigated by immunofluorescence microscopy using a rabbit polyclonal antibody to NSF and the mouse RP2-6H2 monoclonal antibody (Fig. 5). NSF showed a similar pattern of labeling as RP2 with intense labeling in the inner segments, outer and inner plexiform layers, and inner nuclear layer with fainter labeling in the outer nuclear layer. No significant labeling of the outer segment layer was observed for either NSF or RP2. Merged image of NSF and RP2 labeling confirmed the co-localization of RP2 and NSF over the length of the photoreceptor cells (Fig. 5C).
At the junction of the inner and outer segments distinct patterns of labeling were observed for different mouse retinal sections. In some cases, intense diffuse, but not punctuate, co-labeling of NSF and RP2 was evident suggesting that the RP2-NSF complex accumulated in this region of the photoreceptor cell (Fig 5A–F). In many samples (Fig 5G–I), pronounced punctate staining was observed for RP2 similar to that shown in Fig 2. Interestingly, NSF did not co-localize with RP2 within these structures (Fig. 5G–I). In all cases, however, RP2 and NSF colocalized throughout the rest of the retina. The junction of the inner and outer segments of photoreceptors, known as the ciliary region, consists of the basal body at the base, a transitional connecting cilium, and an axoneme projecting into the outer segment. To further define the localization of RP2 and NSF within the ciliary region, co-localization studies were performed with a monoclonal antibody to acetylated a-tubulin that has been shown to label stabilized microtubules such as the axoneme and the connecting cilium, but not the basal body of photoreceptors (20, 21). NSF and acetylated a-tubulin signals were observed in distinct regions, with NSF present proximally in the ciliary region and acetylated a-tubulin found distally in the region (Figure S1). Co-localization of RP2 and acetylated a-tubulin could not be performed since both our antibodies against these two proteins are of mouse monoclonal origin. Nonetheless, the presence of NSF at the base of the cilium together with the co-localization of NSF and RP2 suggests that RP2 is also densely localized at the base of the photoreceptor connecting cilium, possibly within basal body and/or centriole, consistent with a recent report (22). In a control study, no labeling was observed when the primary antibody was omitted or substituted with an irrelevant antibody confirming the specificity of labeling (data not shown).
RP2 binds to the N-terminal domain of NSF
NSF consists of three domains: the N-terminal domain (aa 1–205), D1 domain (aa 206–477) and D2 domain (aa 478–744) (23). In order to further characterize the interaction of RP2 with NSF, a series of GST fusion proteins corresponding to these domains were expressed in bacteria and purified by glutathione affinity chromatography (Fig. 6A). The fusion proteins were then immobilized on glutathione-Sepharose for analysis of their binding to RP2. As shown by western blotting in Fig. 6B, a significant amount of RP2 is pulled-down with the N-terminal fusion protein (GST-NTNSF) relative to that found for D1 (GST-D1NSF) and D2 (GST-D2NSF) fusion proteins or GST as a control, suggesting that RP2 preferentially binds to the N-terminal region of NSF.
Figure 6.

Binding of RP2 to the N-terminal domain of NSF. (A) Coomassie Blue-stained gel showing the purified N-terminal (GST-NTNSF), D1 (GST-D1NSF), and D2 (GST-D2NSF) fusion proteins and the purified recombinant wild-type (WTRP2), and disease-causing mutant (E138G and L188P) RP2 proteins. (B) Western blot labeled with the RP2-6H2 antibody shows that RP2 interacts primarily with the fusion protein containing the N-terminal domain of NSF. GST protein was used as a control.
The binding of disease-linked mutants of RP2 to the N-terminal domain of NSF
The binding of purified RP2 containing disease-causing mutations to the N-terminal domain of NSF was determined by an in vitro binding assay and western blotting. The R118H, L188P, R282W RP2 mutants retained the ability to bind to the N-terminal domain of NSF similar WT-RP2 (Fig. 7). In contrast, no significant binding of ΔI137 and E138G RP2 disease-associated mutants was observed.
Figure 7.
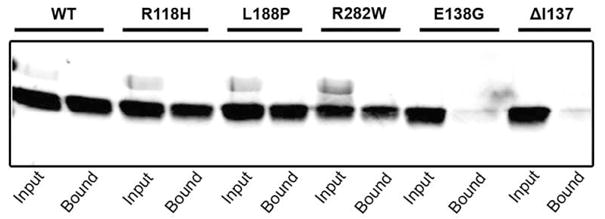
Binding of disease-linked mutants of RP2 to N-terminal domain of NSF. The binding of wild-type (WT) and mutant RP2 proteins to immobilized GST fusion protein containing the N-terminal domain of NSF. Binding was detected on western blots labeled with the RP2-6H2 antibody. The binding of RP2 to the N-terminal NSF domain is essentially abolished for the E138G and Δ137 RP2 disease mutations.
DISCUSSION
In previous studies RP2 has been shown to bind to ADP-ribosylation factor-like 3 (Arl3) in a GTP-dependent manner (15, 16), and in vitro RP2 serves as a GTPase-activating protein (GAP) protein for Arl3 (17). However, the physiological significance of this activity and its existence in photoreceptor cells has not been explored. In order to gain further insight into the role of RP2 in photoreceptors and how mutations in RP2 contribute to retinal degeneration, we have investigated RP2 interacting proteins by immunoprecipitation and mass spectrometry. Our studies indicate that RP2 specifically associates with NSF in photoreceptors and other cells of the retina as well as cultured cell lines. Evidence for this interaction is based on experiments employing a highly specific monoclonal antibody (RP2-6H2) with immunoprecipitation and immunofluorescence localization.
A RP2 immunoaffinity matrix that binds essentially all RP2 proteins coprecipitated NSF from detergent-solubilized retinal tissue and HEK293 cell extracts as determined by mass spectrometry-based proteomic analysis and confirmed by western blotting. Arl3 is a known binding partner for RP2 but it was not pulled down with RP2 using our RP2-6H2 antibody. This is most likely due to the fact that Arl3 and the RP2-6H2 antibody share a similar binding site on RP2. The RP2-6H2 antibody recognizes the amino acid sequence YSWDQ (aa 27–31) of RP2 as determined by epitope mapping, and this is part of the N-terminal domain that has been shown to serve as a binding site for Arl3-GTP (16). Thus if a subfraction of RP2 is already bound to Arl3-GTP, it would not be accessible to the RP2-6H2 antibody.
Control experiments support the specificity of interaction between RP2 and NSF. First, when a retinal membrane extract depleted of RP2 was applied to the RP2-6H2 immunoaffinity matrix, NSF was not detected in the SDS-eluted fraction. Second, we used shRNA to knockdown the endogenous RP2 expression in HeLa cells. When an extract from RP2-shRNA-stably transfected HeLa cells was applied to the antibody matrix, the NSF detected in the SDS-eluted fraction decreased proportionally.
The association of RP2 with NSF as determined by immunoprecipitation is consistent with the colocalization of these proteins in the retina. We found that both NSF and RP2 co-localized in photoreceptors and other cells of the mouse and human retina. Intense labeling was observed in the inner segments, synaptic layers and inner nuclear layers, fainter labeling in the outer nuclear layer, and essentially no labeling over most of the outer segment layer. In an earlier study, Grayson et al. (14) showed RP2 labeling throughout the photoreceptor cells extending from the tips of outer segments to the synaptic terminals. The reason for the difference in labeling with respect to the outer segment layer is unclear, although it could reflect either differences in the state of the retina tissue used in the labeling experiments or the epitopes for the antibodies. The monoclonal antibody in our study was generated against the N-terminus of RP2, while the polyclonal antibody used in their study was directed against the C-terminus of RP2.
Our study showing the co-localization of RP2 and NSF supports the biochemical studies showing that the two proteins interact. Both proteins colocalized throughout the retina. Within the photoreceptors, RP2 and NSF co-localized in the synaptic region, outer nuclear layer and inner segments. Within the ciliary region of the photoreceptors, however, the degree of immunostaining varied from intense yet diffuse to pronounced and punctuate labeling. The intense, diffuse labeling pattern may represent an accumulation of the RP2 protein in the ciliary region which could lead to an intense punctation pattern at a later time point. The observed punctuate staining of RP2 observed in both human and mouse photoreceptors may represent a stage in which NSF has dissociated from RP2 thereby enabling Arl3 to bind suggesting a dynamic interplay between RP2, NSF and Arl3 in the ciliary region. Hence, it is possible that RP2 together with NSF and Arl3 may play a crucial role in the vesicle trafficking of proteins to the photoreceptor outer segments as well as in the synaptic region of photoreceptors. Additional studies employing different RP2 antibodies together with Arl3 antibodies are needed to further define the molecular interactions and dynamics in this region of the photoreceptors. Despite the variation of RP2 and NSF labeling at the ciliary region, the two proteins co-localized in the remaining parts of the retina in all samples examined.
The functional significance of RP2 and NSF interaction is further strengthened by our finding that XLRP-causing mutations of RP2 abolished the binding to NSF. We have demonstrated that the N-terminus (amino acid 1–205) of NSF is essential in the interaction with RP2. This domain of NSF has been shown to be necessary, but not sufficient for the binding and disassembly activities of NSF (24). We also found that the disease-causing ΔI137 and E138G mutations in RP2 abolish binding to the N-terminal domain of NSF. RP2-ΔI137 has been shown to cause destabilization of the β helix of the protein whereas RP2-E138G associates with Arl3-GTP with reduced affinity (16). Further experiments are needed to refine the region of RP2 that interacts with NSF.
This newly-described interaction between RP2 and NSF is intriguing as NSF is a protein that promotes vesicle/membrane fusion by associating with the SNARE complex to facilitate the disassembly of the SNARE complex in an ATP hydrolysis-dependent manner, thereby recycling the complex for another round of fusion. The fact that RP2 did not co-precipitate with any of the SNARE complex implies that RP2 interacts with the population of NSF that is not associated with the SNARE complex. Thus RP2 may regulate the availability of NSF in a negative way by sequestering it or in a positive way by concentrating the local levels of NSF. When ATP becomes available, NSF would be ready to interact with the SNARE complex nearby. On the contrary, it is conceivable that NSF could act with RP2 outside the context of SNARE complex. Recent studies have suggested that NSF regulates the function of the AMPA glutamate receptor located in the postsynaptic density, independent of the SNARE complex (25). NSF may restrict the point of contact between RP2 and Arl3 or other interacting partners to the ciliary region, as this is the only place where RP2 and NSF do not co-localize on occasions. NSF may restrict the point of contact between RP2 and Arl3 or other interacting partners to the ciliary region, as this is the only place where RP2 and NSF do not co-localize on occasions.
The role of RP2 in rod photoreceptors remains to be firmly established. Its function as a GAP protein for Arl3 relating to its interaction with NSF and the mechanism regulating the shift in binding of RP2 between Arl3 and NSF needs to be defined. In a preliminary study, we investigated the possible formation of a RP2-NSF-Arl3 complex and we observed that the three proteins do not form a complex. RP2 appears to bind to only one protein at a time. In addition, our Arl3 monoclonal antibody labeled photoreceptors specifically at the connecting cilium (results not published), consistent with an earlier report (14). It is tempting to speculate that since all three proteins are found either at or near the connecting cilium, this could serve as a potential site for convergence of pathway for Arl3, RP2 and NSF. In summary, our studies show that RP2 co-localizes and directly binds NSF in the retina, specifically near the region of connecting cilium and synaptic regions in photoreceptor cells. The significance of this interaction awaits functional knockdown studies.
Supplementary Material
Acknowledgments
We thank Andrew Ho for technical support and other members of the Molday lab for helpful discussions.
ABBREVIATIONS
- NSF
N-ethylmaleimide sensitive factor
- RP
retinitis pigmentosa
- XLRP
X-linked retinitis pigmentosa
- GAP
GTPase activating protein
- RPE
retinal pigment epithelium
- SDS
sodium dodecyl sulfate
- LC-MS/MS
liquid chromatography tandem mass spectroscopy
- CHAPS
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate
- TX-100
Triton X-100
- PBS
phosphate buffered saline
Footnotes
SUPPORTING INFORMATION AVAILABLE
Double labeling of NSF and acetylated a-tubulin (Figure S1) and a listing of proteins found in the co-precipitated fraction from the RP2-6H2 immunoaffinity matrix (Table S1). This material is available free of charge via Internet at http://pubs.acs.org.
Supported by Grants from the National Institutes of Health (EY 02422); Canadian Institutes for Health Research (CIHR RMF-92101);MT 5822), the Macula Vision Research Foundation; and the Osk. Huttunen Foundation and Sigrid Juselius Foundation. C.L.C. was supported by a NSERC Postdoctoral Fellowship; F.M.D was supported on a Foundation Fighting Blindness – Canada Postdoctoral Fellowship and an Arthur and June Willms Postdoctoral Fellowship. J.C. was supported on a NSERC studentship. R.S.M. holds a Canada Research Chair in Vision and Macular Degeneration.
References
- 1.Berson EL, Sandberg MA, Rosner B, Birch DG, Hanson AH. Natural course of retinitis pigmentosa over a three-year interval. Am J Ophthalmol. 1985;99:240–251. doi: 10.1016/0002-9394(85)90351-4. [DOI] [PubMed] [Google Scholar]
- 2.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Fishman GA. Retinitis pigmentosa. Genetic percentages. Arch Ophthalmol. 1978;96:822–826. doi: 10.1001/archopht.1978.03910050428005. [DOI] [PubMed] [Google Scholar]
- 4.Bird AC. X-linked retinitis pigmentosa. Br J Ophthalmol. 1975;59:177–199. doi: 10.1136/bjo.59.4.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jay M. On the heredity of retinitis pigmentosa. Br J Ophthalmol. 1982;66:405–416. doi: 10.1136/bjo.66.7.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fishman GA, Farber MD, Derlacki DJ. X-linked retinitis pigmentosa. Profile of clinical findings. Arch Ophthalmol. 1988;106:369–375. doi: 10.1001/archopht.1988.01060130395029. [DOI] [PubMed] [Google Scholar]
- 7.Breuer DK, Yashar BM, Filippova E, Hiriyanna S, Lyons RH, Mears AJ, Asaye B, Acar C, Vervoort R, Wright AF, Musarella MA, Wheeler P, MacDonald I, Iannaccone A, Birch D, Hoffman DR, Fishman GA, Heckenlively JR, Jacobson SG, Sieving PA, Swaroop A. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545–1554. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AA, Rosenberg T, Pinckers AJ, Fundele R, Rosenthal A, Cremers FP, Ropers HH, Berger W. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19:327–332. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- 9.Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73:1131–1146. doi: 10.1086/379379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelletier V, Jambou M, Delphin N, Zinovieva E, Stum M, Gigarel N, Dollfus H, Hamel C, Toutain A, Dufier JL, Roche O, Munnich A, Bonnefont JP, Kaplan J, Rozet JM. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype-phenotype correlations and impact on genetic counseling. Hum Mutat. 2007;28:81–91. doi: 10.1002/humu.20417. [DOI] [PubMed] [Google Scholar]
- 11.Miano MG, Testa F, Filippini F, Trujillo M, Conte I, Lanzara C, Millan JM, De Bernardo C, Grammatico B, Mangino M, Torrente I, Carrozzo R, Simonelli F, Rinaldi E, Ventruto V, D'Urso M, Ayuso C, Ciccodicola A. Identification of novel RP2 mutations in a subset of X-linked retinitis pigmentosa families and prediction of new domains. Hum Mutat. 2001;18:109–119. doi: 10.1002/humu.1160. [DOI] [PubMed] [Google Scholar]
- 12.Chapple JP, Hardcastle AJ, Grayson C, Spackman LA, Willison KR, Cheetham ME. Mutations in the N-terminus of the X-linked retinitis pigmentosa protein RP2 interfere with the normal targeting of the protein to the plasma membrane. Hum Mol Genet. 2000;9:1919–1926. doi: 10.1093/hmg/9.13.1919. [DOI] [PubMed] [Google Scholar]
- 13.Evans RJ, Hardcastle AJ, Cheetham ME. Focus on molecules: X-linked Retinitis Pigmentosa 2 protein, RP2. Exp Eye Res. 2006;82:543–544. doi: 10.1016/j.exer.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 14.Grayson C, Bartolini F, Chapple JP, Willison KR, Bhamidipati A, Lewis SA, Luthert PJ, Hardcastle AJ, Cowan NJ, Cheetham ME. Localization in the human retina of the X-linked retinitis pigmentosa protein RP2, its homologue cofactor C and the RP2 interacting protein Arl3. Hum Mol Genet. 2002;11:3065–3074. doi: 10.1093/hmg/11.24.3065. [DOI] [PubMed] [Google Scholar]
- 15.Bartolini F, Bhamidipati A, Thomas S, Schwahn U, Lewis SA, Cowan NJ. Functional overlap between retinitis pigmentosa 2 protein and the tubulin-specific chaperone cofactor C. J Biol Chem. 2002;277:14629–14634. doi: 10.1074/jbc.M200128200. [DOI] [PubMed] [Google Scholar]
- 16.Kuhnel K, Veltel S, Schlichting I, Wittinghofer A. Crystal structure of the human retinitis pigmentosa 2 protein and its interaction with Arl3. Structure. 2006;14:367–378. doi: 10.1016/j.str.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Veltel S, Gasper R, Eisenacher E, Wittinghofer A. The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat Struct Mol Biol. 2008;15:373–380. doi: 10.1038/nsmb.1396. [DOI] [PubMed] [Google Scholar]
- 18.Connell G, Bascom R, Molday L, Reid D, McInnes RR, Molday RS. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc Natl Acad Sci U S A. 1991;88:723–726. doi: 10.1073/pnas.88.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwok MC, Holopainen JM, Molday LL, Foster LJ, Molday RS. Proteomics of photoreceptor outer segments identifies a subset of SNARE and Rab proteins implicated in membrane vesicle trafficking and fusion. Mol Cell Proteomics. 2008;7:1053–1066. doi: 10.1074/mcp.M700571-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sale WS, Besharse JC, Piperno G. Distribution of acetylated alpha-tubulin in retina and in vitro-assembled microtubules. Cell Motil Cytoskeleton. 1988;9:243–253. doi: 10.1002/cm.970090306. [DOI] [PubMed] [Google Scholar]
- 21.Liu Q, Lyubarsky A, Skalet JH, Pugh EN, Jr, Pierce EA. RP1 is required for the correct stacking of outer segment discs. Invest Ophthalmol Vis Sci. 2003;44:4171–4183. doi: 10.1167/iovs.03-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans RJ, Schwarz N, Nagel-Wolfrum K, Wolfrum U, Hardcastle AJ, Cheetham ME. The retinitis pigmentosa protein RP2 links pericentriolar vesicle transport between the Golgi and the primary cilium. Hum Mol Genet. 2010;19:1358–1367. doi: 10.1093/hmg/ddq012. [DOI] [PubMed] [Google Scholar]
- 23.Tagaya M, Wilson DW, Brunner M, Arango N, Rothman JE. Domain structure of an N-ethylmaleimide-sensitive fusion protein involved in vesicular transport. J Biol Chem. 1993;268:2662–2666. [PubMed] [Google Scholar]
- 24.Nagiec EE, Bernstein A, Whiteheart SW. Each domain of the N-ethylmaleimide-sensitive fusion protein contributes to its transport activity. J Biol Chem. 1995;270:29182–29188. doi: 10.1074/jbc.270.49.29182. [DOI] [PubMed] [Google Scholar]
- 25.Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.