

Abstract
Dendritic cells (DCs) appear in higher numbers within the CNS as a consequence of inflammation associated with autoimmune disorders, such as multiple sclerosis, but the contribution of these cells to the outcome of disease is not yet clear. Here, we show that stimulatory or tolerogenic functional states of intracerebral DCs regulate the systemic activation of neuroantigen-specific T cells, the recruitment of these cells into the CNS and the onset and progression of experimental autoimmune encephalomyelitis (EAE). Intracerebral microinjection of stimulatory DCs exacerbated the onset and clinical course of EAE, accompanied with an early T-cell infiltration and a decreased proportion of regulatory FoxP3-expressing cells in the brain. In contrast, the intracerebral microinjection of DCs modified by tumor necrosis factor α induced their tolerogenic functional state and delayed or prevented EAE onset. This triggered the generation of interleukin 10 (IL-10)-producing neuroantigen-specific lymphocytes in the periphery and restricted IL-17 production in the CNS. Our findings suggest that DCs are a rate-limiting factor for neuroinflammation.
Keywords: neuroimmunology, dendritic cells, myelin oligodendrocyte glycoprotein antigen, intracerebral injection, T-cell immune responses, neuroinflammation, autoimmunity
Introduction
Since the first discovery of dendritic cells (DCs) in the CNS (Matyszak and Perry, 1996), these cells have emerged as pivotal players in the development and maintenance of CNS autoimmunity and inflammation (for review, see Becher et al., 2006; McMahon et al., 2006). DCs are rarely detected in the healthy CNS, and when they are present, they localize to vascular-rich tissues including the meninges and choroid plexus (Matyszak and Perry, 1996; Hanly and Petito, 1998; McMenamin, 1999; Serot et al., 2000; Greenwood et al., 2003). Several studies have demonstrated a substantial accumulation of DCs in the brain and spinal cord in response to local inflammation induced by autoimmunity, infection, or trauma (Hanly and Petito, 1998; McMenamin, 1999; Suter et al., 2003; McMahon et al., 2005; Newman et al., 2005; Bailey et al., 2007). The mechanism(s) by which DCs accumulate in the CNS under inflammatory conditions are not well understood.
The concordance of (1) accumulation of DCs in the CSF during progression of experimental autoimmune encephalomyelitis (EAE), the animal model of multiple sclerosis (MS) (Fischer and Bielinsky, 1999; Aloisi et al., 2000; Fischer et al., 2000; Fischer and Reichmann, 2001; Juedes and Ruddle, 2001; Kivisäkk et al., 2004), (2) the localization of DCs at the proximity of inflamed microvessels in MS lesions (Serafini et al., 2006), and (3) the production of astrocyte-derived chemokines that promote recruitment of DCs into the CNS (Ambrosini et al., 2005) strongly suggest that brain microvessel endothelial cells regulate the recruitment of DCs into the CNS. We have previously demonstrated that the transmigration of DCs across brain microvessel endothelium is regulated by macrophage inflammatory protein-1α (MIP-1α), matrix metalloproteinases, and occludin perturbation. In addition, transmigration of DCs across brain microvessel endothelial cell monolayers contributed to the activation of antigen-specific T cells in vitro (Zozulya et al., 2007).
Conflicting data exist concerning the contribution of DCs to the outcome of CNS inflammation. It was proposed that DCs inhibit T-cell responses in the CNS (Suter et al., 2003), thus leading to protection from EAE (Kleindienst et al., 2005). However, other data suggests that DCs contribute to the induction and maintenance of neuroinflammation in EAE (Dittel et al., 1999; Weir et al., 2002). For example, increasing the number of DCs in the brain by systemic injection of FMS-like tyrosine kinase 3 ligand (Flt-3L) leads to a substantial increase in the severity of clinical EAE symptoms (Greter et al., 2005). Conversely, inhibition of Flt-3L signaling ameliorates EAE, providing further evidence that DC numbers in the brain correlate with the outcome of autoimmune responses (Whartenby et al., 2005). In addition, CNS-resident F4/80−CD11c+CD45high cells isolated from brains of animals experiencing relapsing EAE or Theiler's murine encephalomyelitis virus-induced demyelinating disease can efficiently present endogenous myelin proteolipid protein (PLP) antigen and activate naive PLP139–151-specific T cells in vitro (McMahon et al., 2005). Further supporting a stimulatory role for DCs in regulating CNS immune responses, DCs were recently shown to be the only CNS antigen-presenting cells (APC) population capable of inducing memory cytotoxic T-cell responses in lymphocytic choriomeningitis virus infection (Lauterbach et al., 2006).
Taking advantage of methods used to generate stimulatory or inhibitory tolerogenic DCs that can be injected intracerebrally, we addressed the role of functionally different DCs on the generation of neuroantigen-specific T-cell responses and clinical outcome of EAE. Our data demonstrate that the quantity and functional phenotypes of DCs in the brain regulate the onset and progression of EAE.
Materials and Methods
Animals, immunizations, and EAE scoring.
Four- to six-week-old female C57BL/6 mice were obtained from The Jackson Laboratory. 2D2 transgenic (Tg) mice were a gift from Dr. V. Kuchroo (Harvard Medical School, Boston, MA) (Bettelli et al., 2003). “Depletion of regulatory T-cell” (DEREG)-transgenic mice were a gift from Dr. T. Sparwasser (University of Muenchen, Munich, Germany) (Lahl et al., 2007). Experimental animals were housed in a pathogen-free facility at the University of Wisconsin, Medical School Animal Care Unit under guidelines of the National Institutes of Health or at the University of Wuerzburg Animal care facility according to German guidelines for animal care. Protocols for animal use were approved by the Animal Care and Use Committees of the University of Wisconsin–Madison and University of Wuerzburg (Regierung von Unterfranken).
For intracerebral injection, the mice were anesthetized by intraperitoneal injection of a ketamine (90 mg/kg)–xylazine (10 mg/kg) mixture. Dendritic cells (2.5 × 105) loaded or unloaded with myelin oligodendrocyte glycoprotein peptide 35–55 (MOG35–55) (10 μg/ml CyberSyn) in 20 μl of PBS, or an equal volume of PBS was injected into the right frontal lobe with an insulin syringe attached to a penetrating depth controller as described previously (Ling et al., 2003, 2006). The injection was restricted to the ventral-posterior region of the frontal lobe, and the penetrating depth of the syringe was 1.55 mm from the surface of the brain. For each intracerebral injection, the solution was injected slowly, and then the syringe was held in place for an additional minute to reduce backfilling of injected solution.
In some experiments, 5 × 106 2D2 transgenic T cells were adoptively transferred into mice (Sewell et al., 2003). Where indicated, DCs were incubated in the presence of 200 ng/ml pertussis toxin (PTX) (List Biological Laboratories) during antigen pulsing (Marriott et al., 1999). PTX-treated DCs were extensively washed before injection. No significant effect of PTX on DC phenotype, maturation and function was observed in vitro (Karman et al., 2004a) (data not shown).
For EAE induction, emulsion of equal volumes of complete Freund's adjuvant (CFA) (5 mg/ml) and 200 μg MOG35–55 supplemented with Mycobacterium tuberculosis (Strain H37Ra; Difco) were injected subcutaneously in the scapular region of each mouse. PTX (400 ng/mouse) was intraperitoneally injected on the days 0 and 2 relative to immunization. Clinical scores were monitored daily in a blind manner and recorded as follows: 0, no clinical disease; 1, flaccid tail; 2, gait disturbance or hind limb weakness; 3, hind limb paralysis and no weight bearing on hind limbs; 4, hind limb and forelimb paralysis and reduced ability to move around the cage; and 5, moribund or dead. The mean daily clinical score and SEM were calculated for each group. The significance of differences was calculated by Student's t and Wilcox tests as described by Fleming et al. (2005).
Generation of different types of DCs.
DCs were generated as described previously (Inaba et al., 1992; Karman et al., 2004a). Briefly, bone marrow obtained from femurs and tibias of C57BL/6 mice was washed and plated in 24-well plates in RPMI 1640 with 10% FBS supplemented with 100 U/ml penicillin/streptomycin and 20 ng/ml granulocyte macrophage colony-stimulating factor (GM-CSF). GM-CSF was titrated from supernatants of the GM-CSF-secreting X63 cell line (a gift from Dr. A. Erdei, Eotvos University, Budapest, Hungary). Seven days after GM-CSF cultures, the nonadherent and loosely adherent cells were removed and replated in the absence of GM-CSF and cultured together for 4 h with or without MOG35–55 peptide (10 μg/ml). Nonadherent cells were collected for use as described previously (Karman et al., 2004a). To generate semimatured or fully matured DCs, cells were treated for 4 h with tumor necrosis factor (TNF)-α (500 U/ml; PeproTech) or lipopolysaccharide (LPS) (10 μg/ml), with or without MOG35–55 as described by Menges et al. (2002).
Isolation of spleen, lymph nodes, and CNS cells.
Spleen and lymph nodes were dissected, weighed, and transferred into cold HBSS (Cellgro). The isolated lymphocytes were washed with cold HBSS and resuspended either in staining buffer (PBS containing 1% BSA and 0.1% NaN3) for a direct cell-surface staining, or in culture medium (RPMI supplemented with 10% FBS) for an overnight cell culture followed by intracellular cytokine staining. For spleens, red blood cells were lysed with Tris-NH4Cl. Minced brain tissue was processed with Medicon inserts (BD Biosciences). Brain lymphocytes were isolated from the interface of a Percoll density gradient as described previously (Sewell et al., 2004). For some samples, the total number of isolated cells per gram of tissue was calculated.
Flow cytometry.
Single-cell suspensions were stained with saturating concentrations of antibodies at 4°C for 30 min. Monoclonal antibodies used for cell surface staining were purchased from BD Biosciences and included anti-CD8, anti-CD4, anti-vβ11, anti-LFA-1, anti-CD45, anti-CD11b, and anti-FoxP3. Nonspecific binding to cell surface Fc receptors was blocked with unlabeled FcγRII/FcγRIII-specific antibody (clone 2.4G2) as described previously (Karman et al., 2004a). For intracellular staining of interferon (IFN)-γ, single-cell suspensions from spleen (106 cells/ml) or brain lymphocyte preparation (105 cells/ml) were cultured for 12 h in 96-well plates in RPMI-10% FBS with or without MOG35–55 (10 μg/ml) in the presence of GolgiStop (1 μl/ml) (BD Biosciences) before being stained for extracellular ligands and intracellular cytokines. In some experiments, the supernatants of restimulated cells were assessed by cytokine bead array for measurement of IFN-γ, interleukin 17 (IL-17), and IL-10 according to the manufacturer's instruction (Bender MedSystems). Stained cells were acquired on a four-color FACSCalibur cytometer and were analyzed with FlowJo software (TreeStar) version 7.2.1.
Carboxyfluorescein succinimidyl ester staining.
Single-cell suspensions from the spleen of a 2D2 mouse were incubated with carboxyfluorescein succinimidyl ester (CFSE) (2.5 μm; Invitrogen) in HBSS for 5 min at 37°C. The reaction was quenched with 20% FBS. Cells were processed for adoptive transfer into C57BL/6 mice (5 × 106 cells in 100 μl of PBS per mouse).
Statistical analysis.
Differences between groups were determined with unpaired Mann–Whitney and Wilcox tests. p values <0.05 were considered to be significant.
Results
Neuroantigen-specific encephalitogenic T cells invade the CNS in response to intracerebral, but not systemic, injections of stimulatory dendritic cells
The accumulation of activated neuroantigen-specific T cells in the nervous tissue is a hallmark of autoimmune diseases of the CNS. Although the brain parenchyma is usually devoid of DCs, several recent studies have demonstrated that these cells are prominent components of CNS infiltrates in EAE and MS (Fischer and Reichmann, 2001; Suter et al., 2003; Greter et al., 2005; Bailey et al., 2007; Deshpande et al., 2007). To determine whether increasing the number of stimulatory DCs in the brain induces antigen-specific immune responses in the periphery, we injected MOG35–55 peptide-presenting DCs intracerebrally (intracerebral DCMOG) into C57BL/6 mice and subsequently analyzed the proliferation of MOG-specific (2D2) T cells in different organs 5 d after their adoptive transfer. We have previously shown that intracerebral microinjection of either ovalbumin (OVA) (Ling et al., 2006) or OVA-loaded DCs (Karman et al., 2004a) initiated antigen-specific T-cell responses in the cervical lymph nodes (CLN), spleen and the homing of OVA-specific T cells into the brain (Karman et al., 2004a). To extend these studies, we addressed whether the spatial localization of DCs in the brain is important for inducing neuroantigen-specific T-cell responses in the brain and CLNs. CFSE-labeled CD4+ T cells from 2D2 mice, expressing a Tg T-cell receptor specific for MOG35–55 peptide and defined by Vα3.2 and Vβ11+ subunit chains (Bettelli et al., 2003), were adoptively transferred into three groups of C57BL/6 animals (Fig. 1A, top). These groups included intracerebrally injected PBS and DCMOG mice and intravenously injected DCMOG mice (Fig. 1A, left, middle, and right of bottom panel, respectively). Five days after immunization, we analyzed the accumulation and activation of CFSE-labeled 2D2 Tg CD4+ T cells in the brain, CLNs and spleen. Our data show that intracerebral but not systemic injection of DCs resulted in antigen-specific T-cell accumulation in the brain. Based on high expression of LFA-1, antigen-specific CFSE-labeled T cells in the brain were highly activated (Fig. 1A, top center of bottom panel). As expected, MOG-specific T cells were stimulated in the CLN and spleen, where they underwent several cycles of proliferation before their accumulation in the brain (Fig. 1A, bottom of bottom panel, arrows). Only intracerebral injection of DCMOG induced the proliferation of MOG-specific T cells in the CLN and spleen and the accumulation of neuroantigen-specific T cells in the CNS. In contrast, intravenous injection of DCMOG resulted in 2D2 T-cell proliferation in the spleen but not in the CLN, and no accumulation of cells was found in the brain (Fig. 1A, most right column of bottom panel). As a control for injection-induced microtrauma, we injected the same volume of sterile PBS intracerebrally. Importantly, PBS injection alone did not induce proliferation of antigen-specific T cells in the periphery or the accumulation of these cells in the brain (Fig. 1A, far left plots). No MOG-specific T-cell accumulation in the CNS was observed after intracerebral injection of DCs pulsed with ovalbumin (intracerebral DCOVA) or other MHCII (major histocompatibility complex II)-restricted peptides (e.g., pigeon cytochrome c, PCC) (data not shown). Because we previously demonstrated that only the relevant antigen-expressing DCs induce antigen-specific T-cell accumulation in the CNS (Karman et al., 2004a, 2006), irrelevant peptides, such as OVA-expressing DCs, were not included in further studies. Analysis of the absolute number of tissue infiltrating CD4+Vβ11+LFA-1highCFSElow cells further confirmed that activated antigen-specific CD4+ T cells were accumulating in the brain when intracerebrally injected antigen-presenting DCs were present (Fig. 1B,C).
Figure 1.

Intracerebral but not systemic DCMOG injection results in MOG-specific 2D2 T-cell expansion in the periphery and the accumulation of these cells in the brain. A, Intracerebral PBS, intracerebral DCMOG, or intracerebral DCMOG injections were followed by an adoptive transfer of 5 × 106 CFSE-labeled 2D2 Tg T cells into C57BL/6 mice to track the migration of Vβ11+CD4+ T cells. The dot plots represent LFA-1 expression and CFSE proliferation of adoptively transferred CD4+Vβ11+ lymphocytes in the brain, CLN, and spleen of C57BL/6 mice. Arrows indicate cycles of cell proliferation. B, Flow cytometry dot plots of the accumulated CD4+Vβ11+ cells in the brain. Numbers in quadrants indicate percent positive cells. C, Absolute number of CD4+ Vβ11+LFA-1highCFSElow T cells in brain tissue. A total of six mice were analyzed in each experimental group. Each symbol represents a single mouse; small horizontal bars indicate the mean. ***p < 0.0001 (ANOVA).
It was shown previously that MOG-specific FoxP3+ regulatory T (Treg) cells accumulate in the CNS during EAE (Korn et al., 2007; O'Connor et al., 2007). To determine whether intracerebral DCs could also induce the accumulation of antigen-specific Treg cells in the brain, we intracerebrally injected 2D2 Tg mice with MOG-pulsed DCs and tracked the appearance of both MOG-specific and nonspecific Treg cells in the CNS and peripheral tissues (Fig. 2, top). We also confirmed our previous results showing that intracerebral DCMOG injection induces Vβ11+CD4+ MOG-specific T-cell accumulation in the brain (Fig. 2, top right panel of bottom panel). Intracerebral DCMOG injection resulted in approximately five times higher infiltration of MOG-specific Vβ11+CD4+ T cells in the brain compared with the intracerebral PBS injections (Fig. 2, top left). Unpulsed DCs were used as a control to test whether the observed effects depended on the accessibility of MOG antigen to DCs. Interestingly, we detected two times higher infiltration of MOG-specific Vβ11+CD4+ T cells in the brain after the injection of unpulsed DCs compared with intracerebral PBS injections, but this was clearly less compared with intracerebral DCMOG injection (Fig. 2, top center). This may indicate that DCs can uptake neuroantigens in the brain and induce the accumulation of antigen-specific T cells in the CNS. Regardless of the antigen, intracerebral DCs induced the presence of FoxP3-expressing CD4+ T cells with the prevalence of MOG-specific Vβ11+FoxP3+ Treg cells. Although ∼2% of MOG-specific Treg cells were found in the CLN in all groups of animals (Fig. 2, middle), no MOG-specific Treg cells were detected in the spleen after intracerebral injections (Fig. 2, bottom). These experiments demonstrate that intracerebral DCs elicit the accumulation of both encephalitogenic and regulatory T cells in the CNS. Altogether, these data suggest that antigen-carrying DCs in the brain contribute to the neuroantigen-specific T-cell accumulation in the CNS.
Figure 2.
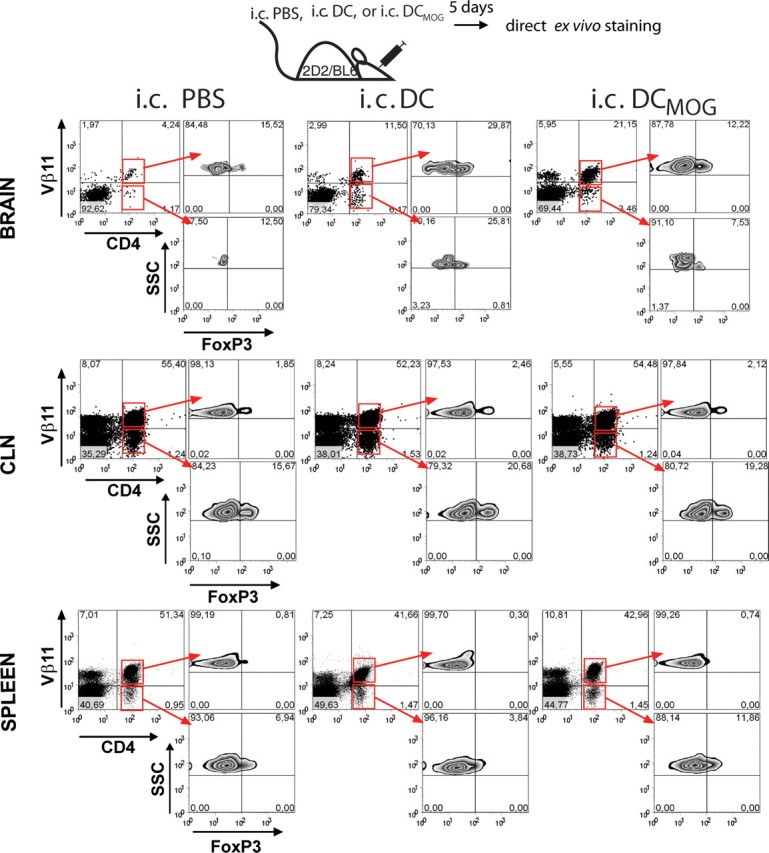
Intracerebral injection of DCMOG induces Vβ11+CD4+ MOG-specific T-cell accumulation in the brain with the prevalence of MOG over non-MOG-specific Vβ11+FoxP3+ Treg cells. 2D2/Bl6 mice were intracerebrally injected with PBS, unpulsed DCs, or DCs loaded with MOG (DCMOG). Five days after immunization, CNS, CLN, and spleen cells were analyzed by flow cytometry. Plots are gated for CD4+Vβ11+ (MOG-specific) and CD4+Vβ11− (non-MOG-specific) T cells (left) and analyzed for FoxP3 expression (right). Numbers in quadrants indicate the percentage of cells in each gate. Data plotted as dot plots of CD4+Vβ11+, CD4+Vβ11− and as contour plots of FoxP3 distribution in different organs in response to intracerebral injections. Data are from two experiments with three mice per group.
When we used transgenic mice termed DEREG mice (Lahl et al., 2007), in which FoxP3+ cells can easily be detected and followed for tissue distribution based on GFP expression regardless of their antigen specificity, we did not observe any differences in the fraction of GFP+CD4+FoxP3-expressing Treg cells in response to different types of DCs or PBS injections in the periphery (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, higher percentages of CNS-derived GFP+ cells were detectable in intracerebrally injected animals with no differences between intracerebrally injected PBS and DCMOG groups compared with intravenous DCMOG. This suggests that microtrauma induced by intracerebral injection of PBS or DCs was not sufficient to induce the accumulation of antigen-specific T cells in the brain but attracts Treg cells that can be found in the brain 5 d after injection (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Clinical and immunological signs of EAE are exacerbated in response to intracerebral DCMOG at different phases of EAE
In Figure 1, we demonstrated that intracerebrally injected DCs that carry MOG antigen contribute to the activation of MOG-specific T cells in the periphery and the accumulation of these cells in the CNS. From our previous studies, we also learned that in addition to presenting antigen in the CNS, DCs can deliver antigens from inflamed CNS and induce homing of peripheral antigen-specific T cells into nervous tissue (Karman et al., 2004b; Ling et al., 2006). To further understand the in vivo significance of these results, we extended our work to analyze whether intracerebrally injected DCs loaded with MOG peptide would influence the clinical course or onset of EAE. In these experiments, we also used DCs that were pulsed with MHC class II-specific OVA peptide (DCOVA) in parallel with DCMOG. Thus, DCMOG, DCOVA, or equal volume of sterile PBS was intracerebrally injected into the CNS, and 5 d later, EAE was actively induced by immunization with MOG35–55 antigen (Fig. 3A, top). Mice intracerebrally injected with DCMOG experienced a significantly accelerated EAE onset and a more progressive course of the disease compared with intracerebrally injected PBS and DCOVA animals (Fig. 3A, bottom; Table 1). Accordingly, the frequency of CD4+LFA-1+ double-positive IFN-γ-producing cells in the brain of intracerebrally injected DCOVA mice (3.6%) was significantly lower at 7 d after EAE induction compared with intracerebrally injected DCMOG (16.7%) mice (Fig. 3B). Also, a higher percentage of activated LFA-1+CD4+ T cells was observed in intracerebrally injected DCMOG mice (∼80%) compared with intracerebrally injected DCOVA (37%) and PBS (23.5%) mice. Still, modest acceleration of EAE clinical scores (Fig. 3A) and cellular infiltration (Fig. 3B) was observed in response to intracerebrally DCOVA injections. This might suggest that OVA-pulsed and intracerebrally injected DCs could amplify neuroinflammation by contributing to CNS antigen-specific T-cell activation. In support of this, myeloid DCs were recently described as a superior CNS cell population in EAE mice, capable of inducing naive CD4+ T-cell proliferation and cytokine production with endogenous peptides (Bailey et al., 2007) (for review, see Miller et al., 2007b).
Figure 3.

Intracerebral DCMOG injection induces an earlier onset of EAE, altering the severity and duration of the disease. Mice intracerebrally injected with PBS, DCOVA, or DCMOG were induced with MOG-EAE 5 d after injections and followed for EAE clinical scores. A, Daily mean clinical EAE score for each group of mice is plotted. B, Intracellular IFN-γ staining on CNS-purified and MOG35–55 peptide restimulated leukocytes. Dot plots are gated on CD4+ T cells, and numbers demonstrate the percentage of IFN-γ-positive cells on LFA-1+ activated cells. Data acquired from brain tissue 7 d after EAE induction. C, Graphical representation of EAE day onset for intracerebral DCMOG-immunized animals compared with intracerebrally injected PBS and DCOVA mice. (Student's t test, ***p = 0.0003 for intracerebrally injected PBS group compared with intracerebrally injected DCMOG group; **p = 0.005 for intracerebral DCOVA compared with intracerebral DCMOG.) D, Graphical representation of the disease severity that summarizes the number of days with EAE clinical scores ≥2.5 for intracerebral DCMOG compared with intracerebrally injected PBS and DCOVA mice. (Student's t test, ***p = 0.0005 for intracerebral PBS compared with intracerebral DCMOG; *p = 0.01 for intracerebral PBS compared with intracerebral DCOVA and intracerebral DCOVA compared with intracerebral DCMOG.)
Table 1.
EAE induction after intracerebral immunizations
| Immunization | Incidence(%) | No.of mice | Meanday onseta | Meanmaximal scoreb |
|---|---|---|---|---|
| Intracerebral PBS + subcutaneous CFA-MOG | 100 | 8 of 8 | 11.5 ± 1.2 | 2.26 ± 0.3 |
| Intracerebral DCMOG + subcutaneous CFA-MOG | 100 | 12 of 12 | 8.2 ± 1.0***c | 2.62 ± 0.35*c |
| Intracerebral DCOVA + subcutaneous CFA-MOG | 100 | 9 of 9 | 10.4 ± 0.5**d | 2.6 ± 0.25*e |
| Intracerebral DCMOG + subcutaneous CFA | 0 | 0 of 6 |
aMean day of onset was calculated for the mice which developed EAE. Data represent mean ± SE.
bMean maximal score was calculated for all mice in the group. Data represent mean ± SE.
cp values of intracerebral DCMOG compared with intracerebral PBS group.
dp values of intracerebral DCOVA compared with intracerebral DCMOG.
ep values of intracerebral DCOVA compared with intracerebral PBS group.
*p < 0.05,
**p < 0.01,
***p < 0.001; Student's t test.
To test whether the induction of neuroantigen-specific T cells by intracerebral DCMOG in conjunction with CFA is sufficient to induce clinical EAE, one group of intracerebrally injected animals (intracerebral DCMOG) received only subcutaneous CFA and no MOG antigen during EAE induction (Table 1). These mice did not develop clinical signs of EAE, indicating that subcutaneously injected MOG antigen is still required for full susceptibility to clinical disease (Table 1).
Graphical representation of EAE day onset for intracerebrally DCMOG-immunized animals compared with intracerebrally injected DCOVA and PBS mice confirms an earlier onset of disease (Fig. 3C). Likewise, graphical representation of the disease severity in number of days with EAE clinical scores ≥2.5 from all animals involved in EAE experiments (Table 1) shows a higher number of mice with EAE score of 2.5 or above in intracerebrally injected DCMOG groups compared with intracerebrally injected PBS and DCOVA groups (Fig. 3D). A significantly increased disease severity was also observed between intracerebrally injected PBS and DCOVA animals, proving DC contribution to CNS-triggered neuroinflammation (Fig. 3D). Immunohistopathology was performed in addition to clinical EAE scoring, and cellular infiltration was analyzed in the animal cohorts described above. On day 17 after EAE induction, immunohistopathology directly correlated with the EAE development in all experimental groups and was followed by a higher cellular infiltration and demyelination degree at lesion sites in the optic nerves (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), brain and spinal cord tissues (not shown) in intracerebrally injected DCMOG and DCOVA animals compared with intracerebrally injected PBS animals. These data show that injected DCs contribute to cellular infiltration and demyelination in the CNS.
The intracerebral DCOVA group in the last set of experiments has helped us conclude that antigen-specific T cells do not accumulate in the CNS in the absence of their specific antigen and neuroinflammation, and that intracerebral injections of PBS or DCs (pulsed or not pulsed with antigen) induces the same level of microtrauma in the brain that does not result in the accumulation of antigen-specific cells in this tissue.
We next wanted to determine whether the accumulation of T cells in the brain after intracerebral DCMOG injection and EAE induction correlates with clinical disease (Fig. 4, experimental scheme). Our data show that intracerebral DCMOG injection resulted in earlier accumulation of CD4+ and also CD8+ T cells in the brain (preclinical phase) (Fig. 4A). At day 14 after EAE induction (peak of disease), differences in T-cell accumulation between intracerebral PBS and intracerebral DCMOG were not statistically significant (Fig. 4B). These data indicate that increasing the number of MOG-presenting DCs in the CNS induces early immune responses in the periphery, and the accelerated accumulation of T cells in the brain contributes to the amplification of neuroinflammation during early stages of the EAE clinical course.
Figure 4.

Intracerebral DCMOG injection results in increased infiltration of T cells into the CNS before EAE onset. Intracerebral DCMOG or intracerebrally injected PBS mice were induced with EAE 5 d after injection and used for T-cell analysis before EAE onset (day 7 after EAE induction) and at the peak of the disease (day 14 after EAE induction). A, B, The dot plots represent the percentage of CD4+ and CD8+ CNS infiltrating T cells (top) and absolute cell number per gram of CNS tissue (bottom) 7 d (A) and 14 d (B) after EAE immunization. Dot plots presented are representative analyses of unpooled samples from five separate experiments, each with three to four mice per group. [Two-tailed Student's t test, p = 0.01 for CD4+ T cells (gray bars) and p = 0.05 for CD8+ T cells (white bars).]
To address the question whether the migration of intracerebrally injected DCs out of the brain would be required to influence EAE disease onset, we inhibited the migratory capacity of DCs with PTX, blocking chemokine receptor signaling in vitro. After in vitro treatment, PTX pretreated DCs were injected into the brain. In Figure 5, we show that pretreatment of DCMOG before their intracerebral injection does not exacerbate the onset of EAE (no significant differences between intracerebral PBS vs intracerebral DCMOG(PTX)) (Fig. 5A,B). The intracerebral DCMOG injection led to a significantly accelerated EAE clinical onset compared with both intracerebral PBS and intracerebral DCMOG(PTX) groups (Fig. 5A,B). Furthermore, the disease severity was significantly enhanced if intracerebral PBS and intracerebral DCMOG groups were compared; however, we did not find statistical differences between intracerebral DCMOG and intracerebral DCMOG(PTX) groups (Fig. 5C).
Figure 5.

Pretreatment of DCMOG cells with PTX before their intracerebral injection does not exacerbate the onset of EAE. A, Mice were injected intracerebrally with PBS, DCMOG, or DCMOG pretreated with PTX 5 d before EAE induction. Daily mean clinical EAE scores are shown. B, Graphical representation of EAE day onset for intracerebral DCMOG immunized animals compared with intracerebrally injected PBS and DCMOG(PTX) mice. (Student's t test, **p = 0.008 for intracerebral PBS compared with intracerebral DCMOG; ***p = 0.0005 for intracerebral DCMOG compared with intracerebral DCMOG(PTX).) C, Graphical representation of the disease severity that summarizes the number of days with EAE clinical scores ≥2.5 for intracerebral DCMOG compared with intracerebrally injected PBS and DCMOG(PTX) mice. (Student's t test, **p = 0.006.)
Intracerebral DCMOG injection induces earlier occurrence of IFN-γ-producing MOG-specific CD4+ T cells in the spleen during EAE
To further define the mechanisms by which intracerebral DCs regulate the early development and severity of clinical EAE symptoms, we studied the kinetics and frequency of IFN-γ-producing, MOG-specific peripheral T cells. Intracerebral delivery of DCMOG induced significantly higher numbers of IFN-γ-secreting, MOG-reactive CD4+ T cells in the peripheral lymphoid organs (spleen) (Fig. 6) (lymph nodes) (not shown). These cells were detected earlier in the course of EAE compared with intracerebrally injected PBS animals (as measured at day 7 after EAE induction) (Fig. 6A) and also remained significantly higher at the peak of EAE (day 14) (Fig. 6B). We also detected the frequency of IL-17-producing CD4+ T cells in the peripheral immune organs, which appeared to be slightly elevated in the intracerebral DCMOG compared with the intracerebral PBS group at earlier time points of EAE (day 7) (Fig. 6A) and significantly higher at later time points (day 14) (Fig. 6B). Interestingly, IFN-γ-producing CD4+ T cells from cultured splenocytes isolated at different time points after EAE induction (day 7 and day 14) could also be detected in media without MOG restimulation from intracerebral DCMOG, but not from intracerebrally injected PBS animals. In the clinical phase of EAE (day 14), double positive IFN-γ+IL-17+ CD4+ T cells could also be detected in intracerebral DCMOG but not in intracerebral PBS group (Fig. 6B).
Figure 6.

Exacerbation of EAE during intracerebral DCMOG injection is associated with increased numbers of MOG-specific IFN-γ- and IL-17-producing CD4+ T cells in the peripheral immune organs. A, B, Dot plots demonstrate IFN-γ and IL-17-producing CD4+ T cells in media or after MOG35–55 peptide restimulation 7 (A) and 14 d (B) after EAE induction. Data shown are representative of seven independent experiments, each with three to four mice per group with similar outcome.
The intracerebral injection of DCMOG decreases the proportion of CD4+FoxP3-expressing cells in the brain during EAE
Growing evidence indicates a bidirectional interaction between Treg cells and DCs (Tang and Bluestone, 2006; Tang et al., 2006). We therefore speculated that the impact of intracerebral DC modulation on CNS autoinflammation might be reflected in a change in the absolute numbers or in the ratio between encephalitogenic and regulatory CD4+ T cells in the CNS. Lymphocytes isolated from noninflamed (naive) brains contained only a few CD4+ T cells that predominantly exhibited the FoxP3high regulatory phenotype (Fig. 7A). The expression of FoxP3 on CD4+CD25high cells was clearly decreased in the brain of intracerebrally injected DCMOG animals (Fig. 7B, bottom) compared with intracerebrally injected PBS control group (Fig. 6B, top). This data show that in parallel with early EAE onset and higher accumulation of MOG-specific T cells after intracerebral DCMOG injection, the frequency of FoxP3high cells in the brain is decreased, and intracerebral DCMOG alters the ratio between encephalitogenic and regulatory T cells.
Figure 7.

Intracerebral DCMOG injection before EAE induction results in increased numbers of CD4+ T cells in the CNS, downregulating the number of regulatory T cells at the expense of effector cells. A, B, The dot plots represent the percentage of CD4+CD25high T cells (left) and the percentage of FoxP3+ regulatory T cells (right) within CD25high population gated on CNS lymphocytes isolated from naive (A) and intracerebrally injected animals (B and experimental scheme). Data presented are representative analyses of three separate experiments, each with two mice per group.
Intracerebral injection of TNF-α treated, semimature DCs ameliorates EAE by inducing peripheral IL-10-secreting cells and restricting the CNS from IL-17-producing CD4+ T cells
It has been proposed that CNS DCs have a dual role during EAE, as they could provide stimulatory or suppressive signals at different stages of disease (Deshpande et al., 2007). Modifying DC phenotypes with TNF-α elicits the generation of IL-10-producing T cells and leads to antigen-specific prevention of EAE (Menges et al., 2002) (for review, see Steinman et al., 2003). We therefore asked the question whether the functional state of intracerebral DCs is decisive for the qualitative and quantitative outcome of experimental CNS inflammation. We injected MOG-pulsed and TNF-α- or LPS-treated (DCTNF-α or DCLPS) semimature DCs intracerebrally and subsequently induced EAE. Among other microbial products, LPS treatment results in an upregulation of maturation markers and an increase in the ability of DCs to stimulate T cells (for review, see Reis e Sousa, 2006). Intracerebrally injected DCLPS significantly exacerbated EAE onset and increased EAE severity compared with intracerebrally injected PBS animals (Fig. 8; Table 2). Intracerebral injection of DCTNF-α beneficially modulated EAE compared with the intracerebrally injected DCLPS group. Protection ranged from delaying disease onset to full prevention of clinical symptoms (only 35% disease incidence) (Fig. 8; Table 2). Onset of EAE clinical symptoms was on average 3–4 d later in intracerebrally injected DCTNF-α and PBS mice compared with intracerebrally injected DCLPS animals. Also, the severity of disease after onset was lower in intracerebrally injected DCTNF-α groups compared with intracerebrally injected DCLPS groups. To assess the mechanism of how intracerebral DCTNF-α injections promoted EAE protection, we measured the frequency and function of peripheral and CNS-infiltrating immune cells in intracerebrally injected DCTNF-α, DCLPS, and PBS mice. Although clinical disease was delayed or EAE was ameliorated (Fig. 8; Table 2), intracerebrally injected DCTNF-α EAE mice did not have decreased numbers of CNS infiltrating CD4+ and CD8+ T cells at any time points (Fig. 9A,B). The ratio of encephalitogenic to regulatory T cells was higher in intracerebrally injected DCLPS and DCTNF-α groups compared with intracerebrally injected PBS mice, similar to that observed between intracerebrally injected DCMOG and PBS groups (Fig. 7) (data not shown). We observed a higher accumulation of CD45+CD11bhigh macrophages in intracerebrally injected DCLPS mice and similar subsets of CD45+CD11bhigh macrophages and CD45+CD11blow microglia in intracerebrally injected PBS and DCTNF-α groups (Fig. 9A). Similar to intracerebrally injected DCMOG animals (Fig. 4), CD4+ T cells persisted at later stages of EAE (day 17) in intracerebrally injected DCTNF-α and DCLPS groups compared with intracerebral PBS controls (Fig. 9B). To find factors that may be responsible for earlier EAE onset (intracerebral DCLPS) or a delay in EAE onset (intracerebral DCTNF-α) at earlier time points of disease, we measured the cytokines produced both in the CNS and periphery at different stages of EAE. Notably, lymphocytes isolated from the CNS of intracerebrally injected DCTNF-α and DCLPS mice produced comparable levels of IL-10 at earlier (day 7) and later (day 17) time points, which were higher compared with intracerebrally injected PBS animals (Fig. 9C,D, left panels). Although there was no IFN-γ signal detected in the CNS for intracerebrally injected PBS animals, 962 pg/ml of IFN-γ was detected in intracerebral DCLPS group, which was 10-fold higher than the concentration of IFN-γ in intracerebral DCTNF-α mice (95 pg/ml) before EAE onset (day 7 after EAE induction). At this point, intracerebral DCTNF-α completely protected both the CNS compartment and peripheral immune organs from IL-17-producing cells (26 pg/ml and 263 pg/ml, respectively) compared with the intracerebrally injected DCLPS group (1235 pg/ml and 10910 pg/ml, respectively) (Fig. 9C). At later time points of EAE (day 17), the intracerebrally injected DCLPS group contained cells with a phenotype polarized toward Th-17 profile with 1350 pg/ml of IL-17 in the CNS and 7841 pg/ml of IL-17-producing cells in the periphery (spleen), which was twofold higher than IL-17 production detected in intracerebrally injected DCTNF-α and PBS mice in the CNS and periphery (Fig. 9D). However, a great majority of CNS lymphocytes in intracerebrally injected DCTNF-α mice included IFN-γ-producing cells (2468 pg/ml of IFN-γ in intracerebral DCTNF-α compared with 962 pg/ml in intracerebral DCLPS mice), suggesting a Th1 phenotype in this group at later time points of EAE (day 17). At the same time, intracerebral DCTNF-α injections induced a higher amount of peripheral IL-10 production after both 7 and 17 d after EAE induction compared with the intracerebral DCLPS group (Fig. 9C,D).
Figure 8.

Intracerebral injection of TNF-α treated, semimature DCs (DCTNF-α) ameliorates EAE. DCs modified by TNF-α (DCTNF-α) or LPS (DCLPS) were pulsed with MOG and intracerebrally injected into mice before EAE induction. The intracerebral PBS, intracerebral DCTNF-α, or intracerebral DCLPS injections were followed with active EAE induction 5 d after intracerebral injections. The mice were observed for EAE clinical scores over time. Graphical representation shows the proportion of mice in three groups on each day experiencing no onset or no clinical symptoms (gray portion of bar), mild EAE (black portion of bar), or severe EAE (red portion of bar).
Table 2.
Effect of tolerogenic and stimulatory intracerebral immunizations of DC on EAE induction
| Immunization | Incidence(%) | No.of mice | Meanday onset | Meanmaximal score |
|---|---|---|---|---|
| Intracerebral PBS + subcutaneous CFA-MOG | 77 | 17 of 22 | 14.0 ± 3 | 2.52 ± 1.0 |
| Intracerebral DCTNF-α + subcutaneous CFA-MOG | 35 | 6 of 17 | 14.0 ± 2 | 2.17 ± 1.1a |
| Intracerebral DCLPS + subcutaneous CFA-MOG | 100 | 9 of 9 | 10.0 ± 2* | 2.88 ± 0.5*a |
ap values are compared with intracerebral PBS and intracerebral DCTNF-α[ρ].
*p < 0.05; Student's t test.
Figure 9.

MOG-loaded and intracerebrally injected DCLPS and DCTNF-α induce similar infiltration of leukocytes into the brain but result in different cytokine profiles in the CNS and periphery. A, B, Intracerebrally injected PBS, DCLPS, or DCTNF-α groups of injected mice were analyzed for leukocyte infiltration before EAE onset (A) and at the later phase of EAE (B). Plots are gated for infiltration of CD4+ and CD8+ T cells (left) and CD45+CD11bhigh macrophages and CD45+CD11blow microglia (right). C, D, Cytokine bead array of secreted cytokines in culture supernatants of CNS lymphocytes and splenocytes collected after 48 h of restimulation with MOG35–55 peptide. The amount of IFN-γ, IL-17 and IL-10-producing cells in response to MOG restimulation at day 7 (C) and day 17 (D) after EAE induction. Data collected are from two separate experiments with CNS cells isolated from two to four mice and pooled.
These data indicate that TNF-α-treated, MOG-pulsed DCs injection into the brain induces a predominantly IL-10 dominated peripheral immune response that restrains Th17-mediated pathology during EAE.
Discussion
DCs have emerged as pivotal regulators of CNS autoimmune and inflammatory responses. Taking advantage of the targeted intracerebral microinjection technology, combined with methods to generate stimulatory or inhibitory tolerogenic DCs, we have analyzed the role of functionally different DCs on the generation of neuroantigen-specific immune responses and their relevance for modulating experimental neuroinflammation using the mouse model of MOG35–55-induced EAE.
Our results demonstrate that immunogenic DCs pulsed with MOG antigen and injected into the brain (intracerebral DCMOG) activated naive antigen-specific T cells in peripheral immune organs and promoted CNS invasion of neuroantigen-specific CD4+ T cells. However, the intracerebral DCMOG did not induce EAE if injected without active immunization. This implies that peripheral induction for the disease is necessary. In this model, a mixture of MOG and CFA needs to be delivered subcutaneously to induce EAE, and in this case, the intracerebral DCMOG accelerated EAE initiation. Together, this may suggest that peripheral antigen presentation and immunization are necessary and sufficient to induce EAE and that intracerebral DCMOG can only modify the kinetics of disease. Thus, the main role of DCs in the brain appears to be in the effector phase of the immune response, rather than in the priming phase. The increasing number of MOG-loaded stimulatory DCs in the brain also accelerated the onset of EAE disease and resulted in exacerbated clinical severity and the extent of the disease. This was accompanied by an early infiltration of IFN-γ-producing MOG-reactive CD4+ T cells from the periphery into the CNS and a decrease in the proportion of FoxP3-expressing CD4+ regulatory T cells, suggesting a shift toward the enrichment of encephalitogenic T cells. In contrast, intracerebral injection of TNF-α-treated and MOG-loaded tolerogenic dendritic cells (intracerebral DCTNF-α) before EAE induction prevented clinical signs of EAE disease (35% disease incidence) or delayed EAE onset, followed by decreased IL-17 production in the CNS and increased level of IL-10-producing peripheral and CNS CD4+ T cells.
Recently, several studies have contributed to the notion that antigen-presenting cells in the brain play a key role in determining the outcome of CNS inflammation (Greter et al., 2005; McMahon et al., 2005; Bailey et al., 2007). Under inflammatory conditions (e.g., autoimmunity and infectious disease), DCs accumulate in the CNS parenchyma, suggesting that CNS DCs are critical for the initiation, regulation, and/or maintenance of immune responses in the CNS. Although numerous studies unambiguously emphasize the potential relevance of DCs for CNS immune surveillance or autoimmune reactions, the true contribution of DCs in the initiation and perpetuation of neuroantigen-specific T-cell responses remains elusive. Our work tested the relevance and impact of CNS-derived DCs by taking advantage of a DC delivery method into the CNS. We found that the activation and CNS accumulation of neuroantigen-specific T cells critically depended on the route of MOG antigen delivery by DCs and the site of DC origin. Local brain DCs are likely to provide critical signals for attracting antigen-specific CD4+ and CD8+T cells into the inflamed CNS (Carson et al., 1999; Tang and Cyster, 1999). A combination of different cytokines, chemokines and inflammatory mediators might be responsible for the regulation of local inflammatory responses via intracerebral DCs. At the same time, several chemokines such as MIP-1α (CCL3), MIP-3β (CCL20), MCP-1 (CCL2), and RANTES (CCL5) are produced in the CNS during acute and chronic EAE (Serafini et al., 2000; Bailey et al., 2007). Defined by chemokine responsiveness, DC populations can differentially migrate to CNS compartments (Bailey et al., 2007). Myeloid DCs were recently shown to preferentially accumulate in the perivascular inflammatory foci of the spinal cord and cerebellum, clustering there with T cells at the peak of EAE (Bailey et al., 2007). The establishment of such DC-T and DC-B cell clusters in the CNS may be necessary to sustain intrathecal clonal expansion of T cells and production of anti-myelin-specific antibodies as detected in EAE and MS lesions.
Spatial CNS localization of MOG antigen-pulsed DCs is critical in the accumulation of antigen-specific T cells in the brain. To further strengthen the importance of specific antigen presentation by intracerebral DCs, we performed experiments directly in 2D2 mice. We show that intracerebral DCMOG injection significantly increased MOG-specific CD4+Vβ11+ T-cell accumulation in the CNS (4.2 vs 21.15%). Unpulsed DC injection into the brain induced a detectable accumulation of neuroantigen-specific cells in the brain under these experimental conditions, which might be attributable to the intrinsic ability of intracerebral DCs to pick up local CNS antigen and present it to naive T cells, resulting in their expansion.
Severity of EAE as well as the number of MS plaques seem to correlate with the presence and function of DCs (Pashenkov et al., 2001; Greter et al., 2005; Serafini et al., 2006). One of the most important observations from our studies is that the amount of DCs in the brain could be a limiting factor in CNS autoimmune diseases. Our data show that in the early phase of EAE autoimmune disease, the number of DCs in the brain is critical, and this is a rate-limiting factor for the development of the disease. This and other studies (Greter et al., 2005; McMahon et al., 2005) collectively suggest that CNS-associated DCs are capable of inducing pathogenetically relevant T-cell responses locally in the brain.
Some data indicate that regulatory T cells are critical in the maintenance of immune privilege status of the CNS (Korn et al., 2007; O'Connor et al., 2007). We investigated the influence of intracerebral DCMOG injections on FoxP3 Treg accumulation in the CNS. Although the naive brain per se contained a very low number of CD4+ T cells, the majority of these cells expressed high levels of FoxP3. This might indicate that the high level of FoxP3 cells in the brain could be a part of the mechanisms maintaining an “immunological privileged” milieu of the CNS. Our experiments show that intracerebral DC injections induce the CNS accumulation of FoxP3 negative neuroantigen-specific T cells, which occurs at the expense of FoxP3 Treg cells. Thus, immunogenic DCs clearly alter the ratio of encephalitogenic to regulatory T cells. Whether regulatory T cells play a critical role in modifying CNS autoimmunity needs to be further studied (Zozulya and Wiendl, 2008).
DCs with regulatory or tolerogenic properties are capable of attenuating EAE (for review, see Miller et al., 2007a). It was previously demonstrated that DCMOG matured with TNF-α and systemically injected into mice before EAE induction induced antigen-specific protection from EAE in mice (Menges et al., 2002). Here, we show that semimature DCs have a protective effect on subsequent CNS-directed autoimmune responses, emphasizing the notion that the functional state of DCs has a clear impact on the quantity and quality of CNS-directed immune responses. The mechanism of EAE-attenuation by tolerogenic TNF-α-matured DCs is noteworthy. Strikingly, absolute numbers of CNS-infiltrating immune cells are equally high in animals receiving intracerebral injections of DCTNF-α or DCLPS. Although we did not test the production of cytokines by CNS DCs in this study, it is well known that multiple immune mediators can be produced by DCs. It was proposed that DCs can polarize CD4+ T cells to become either Th17 or Th1 cells, producing IL-17 or IFN-γ, respectively. Tolerogenic or stimulatory DCs influence this polarization differently (Shortman and Liu, 2002; Shortman and Naik, 2007). Thus, IL-17 polarized CNS T cells with pathological function in the relapsing EAE model, and their clustering with myeloid DCs in the CNS indicate the latter as the only CNS APC population that biased antigen-specific T cells toward a Th17 profile (Bailey et al., 2007). Endogenously collecting CNS antigen and highly producing IL-6 and TGF-β, DCs polarized Th17 responses which could be an intrinsic property of myeloid DCs or the CNS environment (Bailey et al., 2007; Miller et al., 2007b). Our data show that intracerebral injection of DCTNF-α completely protects the CNS from infiltration of IL-17-producing cells but promotes IL-10 cells both in the CNS and in the periphery at early time points of EAE. This supports the idea that CNS DCs, depending on their functional state, can accelerate IL-17 appearance, leading to earlier EAE onset or restrict IL-17 generation in the target organ, thus delaying the disease.
In summary, our study shows that intracerebral DCs are capable of mounting CNS-specific T-cell responses and, in contrast to peripheral DCs, of inducing specific accumulation of neuroantigen-specific T cells in the brain. The quantity of stimulatory DCs in the CNS is a rate-limiting factor for the onset of subsequent EAE. Furthermore, the functional state of intracerebral DCs is decisive for the outcome of a subsequent autoimmune CNS inflammation: the presence of tolerogenic DCs in the brain protects from early development of EAE clinical signs by inducing IL-10 and restricting CNS-infiltrating IL-17 cells. Intracerebral DCs can therefore be considered as a crucial immune cell population during CNS-specific immune responses and can change the outcome of autoimmune inflammatory CNS disorders. This has clear implications for understanding the pathogenesis of MS and considering DCs as potential targets for future therapies.
Footnotes
This work was supported by National Institutes of Health Grant RO1-NS 37570-01A2 (Z.F.), German Federal Ministry of Education and Research (BMBF, to H.W.), and German Research Foundation (SFB 581, TP A8, to H.W.). We thank Dr. Dana C. Baiu, Melissa G. Harris, and Heidi A. Schreiber for helpful discussions and critical review of this manuscript, and K. Macvilay and B. Reuter for technical assistance.
References
- Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol Today. 2000;21:141–147. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, Aloisi F, Coccia EM. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol. 2005;64:706–715. doi: 10.1097/01.jnen.0000173893.01929.fc. [DOI] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- Becher B, Bechmann I, Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med. 2006;84:532–543. doi: 10.1007/s00109-006-0065-1. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Disproportionate recruitment of CD8+ T cells into the central nervous system by professional antigen-presenting cells. Am J Pathol. 1999;154:481–494. doi: 10.1016/S0002-9440(10)65294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande P, King IL, Segal BM. Cutting edge: CNS CD11c+ cells from mice with encephalomyelitis polarize Th17 cells and support CD25+CD4+ T cell-mediated immunosuppression, suggesting dual roles in the disease process. J Immunol. 2007;178:6695–6699. doi: 10.4049/jimmunol.178.11.6695. [DOI] [PubMed] [Google Scholar]
- Dittel BN, Visintin I, Merchant RM, Janeway CA., Jr Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. J Immunol. 1999;163:32–39. [PubMed] [Google Scholar]
- Fischer HG, Bielinsky AK. Antigen presentation function of brain-derived dendriform cells depends on astrocyte help. Int Immunol. 1999;11:1265–1274. doi: 10.1093/intimm/11.8.1265. [DOI] [PubMed] [Google Scholar]
- Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol. 2001;166:2717–2726. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- Fischer HG, Bonifas U, Reichmann G. Phenotype and functions of brain dendritic cells emerging during chronic infection of mice with Toxoplasma gondii. J Immunol. 2000;164:4826–4834. doi: 10.4049/jimmunol.164.9.4826. [DOI] [PubMed] [Google Scholar]
- Fleming KK, Bovaird JA, Mosier MC, Emerson MR, LeVine SM, Marquis JG. Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;170:71–84. doi: 10.1016/j.jneuroim.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Greenwood J, Amos CL, Walters CE, Couraud PO, Lyck R, Engelhardt B, Adamson P. Intracellular domain of brain endothelial intercellular adhesion molecule-1 is essential for T lymphocyte-mediated signaling and migration. J Immunol. 2003;171:2099–2108. doi: 10.4049/jimmunol.171.4.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- Hanly A, Petito CK. HLA-DR-positive dendritic cells of the normal human choroid plexus: a potential reservoir of HIV in the central nervous system. Hum Pathol. 1998;29:88–93. doi: 10.1016/s0046-8177(98)90395-1. [DOI] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juedes AE, Ruddle NH. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J Immunol. 2001;166:5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- Karman J, Ling C, Sandor M, Fabry Z. Initiation of immune responses in brain is promoted by local dendritic cells. J Immunol. 2004a;173:2353–2361. doi: 10.4049/jimmunol.173.4.2353. [DOI] [PubMed] [Google Scholar]
- Karman J, Ling C, Sandor M, Fabry Z. Dendritic cells in the initiation of immune responses against central nervous system-derived antigens. Immunol Lett. 2004b;92:107–115. doi: 10.1016/j.imlet.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Karman J, Chu HH, Co DO, Seroogy CM, Sandor M, Fabry Z. Dendritic cells amplify T cell-mediated immune responses in the central nervous system. J Immunol. 2006;177:7750–7760. doi: 10.4049/jimmunol.177.11.7750. [DOI] [PubMed] [Google Scholar]
- Kivisäkk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B, Wujek J, Ravid R, Staugaitis SM, Lassmann H, Ransohoff RM. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- Kleindienst P, Wiethe C, Lutz MB, Brocker T. Simultaneous induction of CD4 T cell tolerance and CD8 T cell immunity by semimature dendritic cells. J Immunol. 2005;174:3941–3947. doi: 10.4049/jimmunol.174.7.3941. [DOI] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Bäckström BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach H, Zuniga EI, Truong P, Oldstone MB, McGavern DB. Adoptive immunotherapy induces CNS dendritic cell recruitment and antigen presentation during clearance of a persistent viral infection. J Exp Med. 2006;203:1963–1975. doi: 10.1084/jem.20060039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Sandor M, Fabry Z. In situ processing and distribution of intracerebrally injected OVA in the CNS. J Neuroimmunol. 2003;141:90–98. doi: 10.1016/s0165-5728(03)00249-2. [DOI] [PubMed] [Google Scholar]
- Ling C, Sandor M, Suresh M, Fabry Z. Traumatic injury and the presence of antigen differentially contribute to T-cell recruitment in the CNS. J Neurosci. 2006;26:731–741. doi: 10.1523/JNEUROSCI.3502-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott I, Inscho EW, Bost KL. Extracellular uridine nucleotides initiate cytokine production by murine dendritic cells. Cell Immunol. 1999;195:147–156. doi: 10.1006/cimm.1999.1531. [DOI] [PubMed] [Google Scholar]
- Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74:599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- McMahon EJ, Bailey SL, Miller SD. CNS dendritic cells: critical participants in CNS inflammation? Neurochem Int. 2006;49:195–203. doi: 10.1016/j.neuint.2006.04.004. [DOI] [PubMed] [Google Scholar]
- McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol. 1999;405:553–562. [PubMed] [Google Scholar]
- Menges M, Rössner S, Voigtländer C, Schindler H, Kukutsch NA, Bogdan C, Erb K, Schuler G, Lutz MB. Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J Exp Med. 2002;195:15–21. doi: 10.1084/jem.20011341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol. 2007a;7:665–677. doi: 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. 2007b;1103:179–191. doi: 10.1196/annals.1394.023. [DOI] [PubMed] [Google Scholar]
- Newman TA, Galea I, van Rooijen N, Perry VH. Blood-derived dendritic cells in an acute brain injury. J Neuroimmunol. 2005;166:167–172. doi: 10.1016/j.jneuroim.2005.04.026. [DOI] [PubMed] [Google Scholar]
- O'Connor RA, Malpass KH, Anderton SM. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J Immunol. 2007;179:958–966. doi: 10.4049/jimmunol.179.2.958. [DOI] [PubMed] [Google Scholar]
- Pashenkov M, Huang YM, Kostulas V, Haglund M, Söderström M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001;124:480–492. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157:1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, Aloisi F. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. 2006;65:124–141. doi: 10.1097/01.jnen.0000199572.96472.1c. [DOI] [PubMed] [Google Scholar]
- Serot JM, Béné MC, Foliguet B, Faure GC. Monocyte-derived IL-10-secreting dendritic cells in choroid plexus epithelium. J Neuroimmunol. 2000;105:115–119. doi: 10.1016/s0165-5728(99)00240-4. [DOI] [PubMed] [Google Scholar]
- Sewell DL, Reinke EK, Co DO, Hogan LH, Fritz RB, Sandor M, Fabry Z. Infection with Mycobacterium bovis BCG diverts traffic of myelin oligodendroglial glycoprotein autoantigen-specific T cells away from the central nervous system and ameliorates experimental autoimmune encephalomyelitis. Clin Diagn Lab Immunol. 2003;10:564–572. doi: 10.1128/CDLI.10.4.564-572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, Sandor M, Fabry Z. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol. 2004;155:55–63. doi: 10.1016/j.jneuroim.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- Suter T, Biollaz G, Gatto D, Bernasconi L, Herren T, Reith W, Fontana A. The brain as an immune privileged site: dendritic cells of the central nervous system inhibit T cell activation. Eur J Immunol. 2003;33:2998–3006. doi: 10.1002/eji.200323611. [DOI] [PubMed] [Google Scholar]
- Tang HL, Cyster JG. Chemokine up-regulation and activated T cell attraction by maturing dendritic cells. Science. 1999;284:819–822. doi: 10.1126/science.284.5415.819. [DOI] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA. Plasmacytoid DCs and T(reg) cells: casual acquaintance or monogamous relationship? Nat Immunol. 2006;7:551–553. doi: 10.1038/ni0606-551. [DOI] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir CR, Nicolson K, Bäckström BT. Experimental autoimmune encephalomyelitis induction in naive mice by dendritic cells presenting a self-peptide. Immunol Cell Biol. 2002;80:14–20. doi: 10.1046/j.1440-1711.2002.01056.x. [DOI] [PubMed] [Google Scholar]
- Whartenby KA, Calabresi PA, McCadden E, Nguyen B, Kardian D, Wang T, Mosse C, Pardoll DM, Small D. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:16741–16746. doi: 10.1073/pnas.0506088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zozulya AL, Wiendl H. The role of regulatory T cells in multiple sclerosis. Nat Clin Pract Neurol. 2008;4:384–398. doi: 10.1038/ncpneuro0832. [DOI] [PubMed] [Google Scholar]
- Zozulya AL, Reinke E, Baiu DC, Karman J, Sandor M, Fabry Z. Dendritic cell transmigration through brain microvessel endothelium is regulated by MIP-1alpha chemokine and matrix metalloproteinases. J Immunol. 2007;178:520–529. doi: 10.4049/jimmunol.178.1.520. [DOI] [PMC free article] [PubMed] [Google Scholar]