

Abstract
High spin oxoiron(IV) complexes have been proposed to be a key intermediate in numerous non-heme metalloenzymes. The successful detection of similar complexes has been reported for only two synthetic systems. A new synthetic high spin oxoiron(IV) complex is now reported that can be prepared from a well-characterized oxoiron(III) species. This new oxoiron(IV) complexes can also be prepared from a hydroxoiron(III) species via a proton-coupled electron transfer process—a first in synthetic chemistry. The oxoiron(IV) complexes has been characterized with a variety of spectroscopic methods: FTIR studies showed a feature associated with the Fe–O bond at ν(Fe16O) = 799 cm−1 that shifted to 772 cm−1 in the 18O complex; Mössbauer experiments show a signal with an δ = 0.02 mm/s and |ΔEQ | = 0.43 mm/s, electronic parameters consistent with a Fe(IV) center; and optical spectra had visible bands at λmax = 440 (εM = 3100), 550 (εM = 1900) and 808 (εM = 280) nm. In addition, the oxoiron(IV) complex gave the first observable EPR features in the parallel-mode EPR spectrum with g-values at 8.19 and 4.06. A simulation for an S = 2 species with D = 4.0(5) cm−1, E/D = 0.03, σE/D = 0.014, and gz = 2.04 generates a fit that accurately predicted the intensity, lineshape, and position of the observed signals. These results showed the EPR spectroscopy can be a useful method for determining the properties of high spin oxoiron(IV) complexes. The oxoiron(VI) complex was crystallized at −35°C and its structure was determined by X-ray diffraction methods. The complex has a trigonal bipyramidal coordination geometry with the Fe–O unit positioned within a hydrogen bonding cavity. The FeIV–O unit bond length is 1.680(1) Å, which is the longest distance yet reported for monomeric oxoiron(IV) complex.
Identification of intermediates in chemical catalysis is of fundamental interest in chemistry. This is exemplified in metalloenzymes that bind and activate dioxygen, in which mechanistic details are often surmised from spectroscopic measurements.1 Characterization of key intermediates, such as FeIV–O(H) species, has long been sought; recent findings on non-heme monooxygenases, halogenases and synthetic systems showed that FeIV–oxo complexes have detectable optical and vibrational properties.2 In this report, we demonstrate that oxidation of monomeric hydroxo- and oxoiron(III) complexes produce the same high-spin FeIV–oxo species. In addition, our results illustrate for the first time that EPR spectroscopy can be used to detect high-spin ferryl oxo species.3,4
Dioxygen activation by metal complexes is often proposed to proceed through monomeric MIV–oxo intermediates. With but a few exceptions, the ferryl intermediates in non-heme iron enzymes have eluded spectroscopic characterization. Of those characterized, the oxoiron(IV) species have all been high spin with an S = 2 spin ground state, whose primary coordination geometries are still uncertain.2f The vast majority of synthetic oxoiron(IV) complexes are prepared with oxidants other than dioxygen and have coordination geometries that support an intermediate spin state of S = 1.2e–g Two examples of synthetic high-spin oxoiron(IV) species have been described: one from Bakac using [FeII(H2O)6]2+ and ozone in water5a and the other from Que using [FeII(TMG3tren)(OTf)]+ and iodosylbenzene.5b,6
Our approach to high valent metal oxo chemistry utilizes tetradentate tripodal ligands with anionic nitrogen donors. We aimed to stabilize intermediates by incorporating intramolecular hydrogen bonds (H-bonds) between a coordinated exogenous ligand(s) and H-bond donors on the tripod.7 For instance, utilizing the urea-containing ligand, [H3buea]3−, we isolated the FeIII–O(H) complexes [FeIIIH3buea(OH)]− and [FeIIIH3buea(O)]2− whose oxygen ligands are derived from the deprotonation of H2O or O2 activation.7a,b
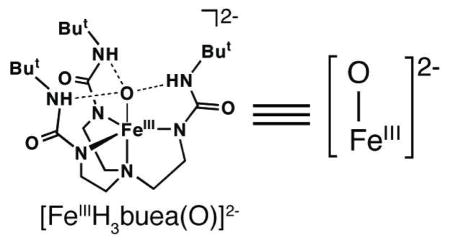
We reported previously that the oxomanganese(IV) complex, [MnIVH3buea(O)]− was prepared by the one-electron oxidation of a terminal MnIII–O complex, [MnIIIH3buea(O)]2−.8 This finding led us to reevaluate the redox properties of [FeIIIH3buea(O)]2− and we subsequently found that the FeIII/IV couple was significantly more negative (−0.90 V vs. [FeCp2]+/0, Figure S1A) than reported earlier (0.34 V).9 These new data prompted us to investigate the oxidation chemistry of [FeIIIH3buea(O)]2− and we found that an oxoiron(IV) species could be generated (Scheme 1).10 Spectroscopic studies showed that the high-spin complex, [FeIVH3buea(O)]− was produced (see below).
Scheme 1.

Preparative routes to the FeIV–oxo complex.
During the course of our studies, we found that solutions containing less than 1 mM of the FeIII–O complex were consistently converted to [FeIIIH3buea(OH)]−, a result attributed to adventitious water and the high basicity of the oxo ligand in [FeIIIH3buea(O)]2− (pKa ~ 25 in DMSO).9 This observation led to the discovery that treating [FeIIIH3buea(OH)]− in DMF at −60°C with one equiv of [FeCp2]+ also produced [FeIVH3buea(O)]− (Scheme 1).11 Thus the same oxoiron(IV) species was obtained from the independent oxidation of either a monomeric FeIII–OH or FeIII–O complex. The formation of [FeIVH3buea(O)]− was followed optically and is shown in Figure 1 for the oxidation of [FeIIIH3buea(OH)]−. The strong absorption band of [FeIIIH3buea(OH)]− at λmax = 400 nm (εM = 5000) disappears with new features appearing at λmax = 350 (εM = 4200), 440 (εM = 3100), 550 (εM = 1900) and 808 (εM = 280) nm. The absorption band at λmax = 808 nm is suggestive of an oxoiron(IV) species2e,f and is not present in any other iron complexes that we have prepared. Moreover, the same optical band was found when [FeIVH3buea(O)]− was prepared from the oxidation of [FeIIIH3buea(O)]2− (Figure 1, inset). The bands between 300–600 nm are still under investigation but most likely arise from chargetransfer transitions. In comparison to other oxoiron(IV) species, these higher energy features are unique to [FeIVH3buea(O)]−: for instance, [FeIV(TMG3tren)(O)]2+ has a lone peak at 400 nm (εM = 9800 M−1cm−1) and the FeIV–oxo intermediate in taurine D dioxygenase has a single band at 318 nm.2a
Figure 1.

The oxidation of [FeIIIH3buea(OH)]− (0.20 mM) with [FeCp2]+ monitored spectrophotometrically at −60°C in DMF. The red spectrum was taken immediately after addition of [Cp2Fe]+ and subsequent spectra were recorded every minute for 12 min.12 The inset are spectra (600–1100 nm) for the products from the oxidation of [FeIIIH3buea(OH)]− (–) and [FeIIIH3buea(O)]2− ( ) (concentration, 2 mM).
) (concentration, 2 mM).
The isolation of [FeIVH3buea(O)]− was initially achieved by treating [FeIIIH3buea(OH)]− in acetonitrile at room temperature with [FeCp2]+ which caused the immediate precipitation of a brown solid. Redissolving the solid in DMSO afforded an optical spectrum nearly identical to that produced in Figure 1. The presence of the oxo ligand in [FeIVH3buea(O)]− was confirmed with isotopic labeling studies. The solid-state FTIR spectrum of the isolated product from the oxidation of [FeIIIH3buea(16OH)]− exhibited a new peak at 799 cm−1 that shifted to 768 cm−1 (sh) in the 18O-isotopomer (Figure 2A). Solution FTIR spectra (DMF) of [FeIVH3buea(O)]−, prepared from the oxidation of [FeIIIH3buea(O)]−, gave values for ν(Fe–16O) and ν(Fe–18O) of 798 and 765 cm−1 (Figure 2B). The observed shifts are expected based on a harmonic Fe–O oscillator model, which predicted a difference of 34 cm−1 between the 16O- and 18O-isotopomers. Note also that the frequency for the Fe–oxo bond measured for [FeIVH3buea(16O)]− is in good agreement to what Green predicted using Badger’s rule constructed for complexes with oxo–iron bonds.13 Furthermore, the negative-mode ESI-MS spectrum of [FeIVH3buea(16O)]− contained a strong ion peak at m/z = 512.2634 that shifted 2 mass-units in the 18O-isotopomer (Figure S2).14 These data and the calculated isotopic distribution support the formulation of a monomeric oxoiron(IV) species.
Figure 2.

FTIR spectra of [FeIVH3buea(16O)]− (•••) and [FeIVH3buea(18O)]− (–) measured at room temperature. (A) Solid-state samples (Nujol) prepared from the oxidation of [FeIIIH3buea(16/18OH)]− and (B) solution samples (DMF) prepared from the oxidation of [FeIIIH3buea(16/18O)]2−.
Parallel-mode X-band EPR spectra of the FeIV–oxo complex showed a sharp resonance at g = 8.19 and a broad valley at g = 4.06 (Figure 3A).15 The g = 8.19 signal is indicative of a transition from the |2±〉 doublet of an S = 2 spin manifold (inset of Figure 3A).10 Temperature-dependent EPR studies showed that this is an excited state signal originating from a positive value of the zero-field splitting. A simulation for an S = 2 species with D = 4.0(5) cm−1, E/D = 0.03, σE/D = 0.014,16 and gz = 2.040(5) generates a fit that accurately predicted the intensity, lineshape, and position of the observed signals. The simulation also predicted the broad feature at g = 4.06, arising from the lower |1±〉 doublet of the same S = 2 manifold. Our analysis of the EPR data further showed a spin concentration of 20 mM that was in good agreement with the starting concentration of the Fe(III) precursor (~25 mM). The Mössbauer spectrum of the complex (powder or in DMF) showed a doublet with δ = 0.02 mm/s and |ΔEQ | = 0.43 mm/s (Figure 3B).17 The near zero isomer shift is indicative of an Fe(IV) oxidation state. EPR and Mössbauer spectra of [FeIVH3buea(O)]− prepared from [FeIIIH3buea(OH)]− showed the same species. Thus, the Mössbauer and EPR data together indicate a high-spin (S = 2) iron(IV) assignment for the oxidation product of [FeIIIH3buea(O)]2− or [FeIIIH3buea(OH)]−.
Figure 3.

(A) X-band EPR spectrum of 20 mM [FeIVH3buea(O)]− in DMF (–) and simulation ( ); experimental conditions: B1||B; temperature, 10 K; microwaves, 2 mW at 9.28 GHz. The signals at g = 4.3 and 1.99 are impurities, observed due to B1||B misalignment. Inset: energy levels of the S = 2 manifold as a function of magnetic field. (B) Mössbauer spectrum of [FeIVH3buea(O)]− in Nujol at 4.2 K. The solid line is a least square fit of the experimental data.
); experimental conditions: B1||B; temperature, 10 K; microwaves, 2 mW at 9.28 GHz. The signals at g = 4.3 and 1.99 are impurities, observed due to B1||B misalignment. Inset: energy levels of the S = 2 manifold as a function of magnetic field. (B) Mössbauer spectrum of [FeIVH3buea(O)]− in Nujol at 4.2 K. The solid line is a least square fit of the experimental data.
The electronic parameters (D, E/D) of [FeIVH3buea(O)]− are comparable to those of other S = 2 oxoiron(IV) species (Table S1).2a,5 All the complexes have nearly axial symmetry (i.e., small E/D), and since the g = 8 signal intensity is proportional to (E/D)4, weak EPR signal intensities are predicted.10 It is unclear why only [FeIVH3buea(O)]− has produced observable EPR signals. Such signals should be detectable for oxoiron(IV) species with less symmetric ligand fields,3 such as those often found within protein active sites. Interestingly, the shift of the gz-value from 2.00 (spin only) to 2.040(5) is significant, suggesting dominant orbital angular momentum contributions from a ligand (possibly an oxygen atom) rather than metal-based orbitals, which would cause negative g-shifts.3
The oxoiron(IV) complex is sufficiently stable at −35°C to allow formation of single crystals that were suitable for analysis by X-ray diffraction methods.10,18 The molecular structure of [FeIVH3buea(O)]− has the expected trigonal bipyramidal coordination geometry with the terminal oxo ligand (O1) positioned trans to the apical N1 atom (Figure 4). The Fe1–O1 bond length is 1.680(1) Å and the O1–Fe1–N1 angle is 179.50(4)°. The value for the Fe–O bond distance is slightly (but statistically significant) longer than values found for other synthetic non-heme oxoiron(IV) complexes.2e,5c,19 For example, the recently reported molecular structure of [FeIV(TMG3tren)(O)]2+ has an Fe–O bond distance of 1.661(2) Å.5c Moreover, the Fe–N bond lengths between these two high spin oxoiron(IV) complexes differ significantly. In [FeIVH3buea(O)]−, the Fe1–N1 and average Fe–Neq bond lengths are 2.057(1) Å and 1.983(1) Å, whereas in [FeIV(TMG3tren)(O)]2+ these bond distances are 2.112(3) and 2.005 Å, respectively. These differences could reflect the highly anionic character of the [H3buea]3− ligand and the H-bonding network that surrounds the FeIV–O unit, both of which are absent in other systems.
Figure 4.

Thermal ellipsoid diagram of [FeIVH3buea(O)]−. The ellipsoids are drawn at the 50% probability level and non-urea hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angle (deg): Fe1–O1, 1.680(1); Fe1–N1, 2.064(1); Fe1–N2, 1.988(1), Fe1–N3, 1.997(1); Fe1–N4, 1.981(1); O1–Fe1–N1, 179.50(4); N2–Fe1–N3, 116.46(4); N2–Fe1–N4, 122.50(4), N3–Fe1–N4, 115.89(4).
The characterization of [FeIVH3buea(O)]− allowed us to compare for the first time the structural properties of two monomeric oxoiron complexes that differ by only one electron. We have previously described the molecular structure of [FeIIIH3buea(O)]2−, which also has trigonal bipyramidal coordination geometry with the oxo ligand nestled within a Hbonding cavity created by the [H3buea]3− ligand.7a There are significant bond length contractions upon oxidation of the FeIII–oxo complex to the oxoiron(IV) species (Figure 5): the Fe–O and Fe–N1 bond lengths decrease by 0.133 and 0.212 Å, and the change in the average Fe–Neq bond distances is −0.076 Å. In fact, the FeIV–O unit is situated closer to the equatorial plane as indicated by the comparatively small displacement of the iron center from the plane (0.262 Å). In addition, the oxo ligand is further removed (by 0.206 Å) from the plane formed by urea nitrogen atoms, N5, N6, and N7. This displacement of the oxo ligand in [FeIVH3buea(O)]− caused a decrease in the N–H–O angles to approximately 150°, possibly leading to diminished Hbonding interactions as predicted by DFT theory.20 Further supports for this premise comes from FTIR studies that showed the bands associated with the NH groups in [FeIVH3buea(O)]− are substantially sharper and at higher energy than those found in [FeIIIH3buea(O)]2− (Figure S3).
Figure 5.

Comparison of key metrical parameters between [FeIVH3buea(O)]− and [FeIIIH3buea(O)]2−. Key: d[Fe–Neq] is displacement of the iron centers from the plane formed by N2, N3, and N4; d[O–NH] is the displacement of oxo ligand (O1) from the plane formed by N5, N6, and N7 (see Figure 4 for atom labels).
At room temperature [FeIVH3buea(O)]− converts to [FeIIIH3buea(OH)]− (Figure S4) with a half life of 2.2 h,10 a process we suggest was caused by the homolytic cleavage of a C–H bond from DMF.21 This process occurred as well when [FeIVH3buea(O)]− was treated with diphenylhydrazine, forming azobenzene in nearly quantitative yield.
The formation of a monomeric oxoiron(IV) complex via the proton-coupled oxidation of an FeIII–OH species has never been observed and is ascribed to the increased acidity of the hydroxo ligand upon formation of an FeIV center.22 The loss of a proton from the complex is likely facilitated by the H-bonding network surrounding the iron center. Our results with FeIII–OH species are reminiscent of those found in ruthenium chemistry, in which the oxidation of RuIII–OH complexes produced RuIV–oxo species in aqueous media.23 Together with our findings it suggests that FeIV–oxo complexes may be more stable than their hydroxo analogs in certain non-heme systems, hindering the detection of FeIV–OH species. Finally, the observed EPR signals for a highspin ferryl oxo species provide an additional quantitative means for probing mechanisms in other non-heme systems.
Supplementary Material
Acknowledgments
ACKNOWLEDGMENT is made to the NIH (GM050781 to ASB; GM77387 to MPH) for financial support. We thank R. Zarkesh collecting the X-ray data.
Footnotes
Supporting Information Available: Experimental details for all chemical reactions and figures for all spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–350. doi: 10.1021/cr9900275. and references therein. [DOI] [PubMed] [Google Scholar]
- 2.(a) Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]; (b) Hausinger RP. Critical Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]; (c) Krebs C, Fujimori DG, Walsh CT, Bollinger JM. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]; (e) Rohde JW, In JH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]; (f) Nam W. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (g) Que L., Jr Acc Chem Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]; (h) Sinnecker S, Svensen N, Barr EW, Ye S, Bollinger JM, Jr, Neese F, Krebs C. J Am Chem Soc. 2007;129:6168–6179. doi: 10.1021/ja067899q. [DOI] [PubMed] [Google Scholar]
- 3.Kostka KL, Fox BF, Hendrich MP, Collins TJ, Rickard CEF, Wright LJ, Münck E. J Am Chem Soc. 1993;115:6746–6757. [Google Scholar]
- 4.Krzystek J, England J, Ray K, Ozarowski A, Smirnov D, Que L, Jr, Telser J. Inorg Chem. 2008;47:3483–3485. doi: 10.1021/ic800411c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Pestovsky O, Stoian S, Bominaar EL, Shan X, Münck E, Que L, Jr, Bakac A. Angew Chem, Int Ed. 2005;44:6871–6874. doi: 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]; (b) England J, Martinho M, Farquhar ER, Frisch JR, Bominaar EL, Münck E, Que L., Jr Angew Chem Int Ed. 2009;48:3622–3626. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) England J, Guo Y, Farquhar ER, Young VG, Jr, Münck E, Que L., Jr J Am Chem Soc. 2010;132:8635–8644. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbreviations: TMG3tren, tris(tetramethylguanidino)tren; [H3 buea]3−, tris(tert-butylureaylethylene)aminato.
- 7.(a) MacBeth CE, Golombek AP, Young VG, Jr, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]; (b) Borovik AS. Acc Chem Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]; (c) Shook RL, Borovik AS. Chem Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shook RL, Borovik AS. Inorg Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsell TH, Yang MY, Borovik AS. J Am Chem Soc. 2009;131:2762–2763. doi: 10.1021/ja8100825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta R, Borovik AS. J Am Chem Soc. 2003;125:13234–13242. doi: 10.1021/ja030149l. [DOI] [PubMed] [Google Scholar]
- 10.See supporting information for experimental details.
- 11.A nearly irreversible one-electron oxidative process was found for [FeIIIH3buea(OH)]− at a potential of −0.033 V vs [Cp2Fe]+/0 (Figure S1B).
- 12.The small peak in the initial spectrum (red) at λmax ~ 600 nm is from [Cp2Fe]+ (Figure S7).
- 13.Green MT. J Am Chem Soc. 2006;128:1902–1906. doi: 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- 14.16O-isotopomer calc’d: 512.2648. 18O-isotopomer: found, 514.2676; calc’d: 514.2691.
- 15.The perpendicular-mode spectrum of the sample used for Figure 1B had signals from [FeIIIH3buea(OH)]− (g = 9.3) and adventitious iron (g = 4.3) that each accounted for 0.5 mM Fe; a residual amount (0.01 mM) of [FeCp2]+ (g = 2) was also present (Figure S5).
- 16.σE/D is one standard deviation in the mean value for E/D, which dominates the broadening of EPR signals.
- 17.Mössbauer measurements of multiple samples gave yields of [FeIVH3buea(O)]− between 50–90% based on the amount of FeIICp2 present.
- 18.Single crystals were grown from a DMF/ether solution of K[FeIVH3buea(O)] at −35°C in the dark. Crystal growth took months.
- 19.Klinker EJ, Kaizer J, Brennessel WW, Woodrum NL, Cramer CJ, Que L., Jr Angew Chem, Int Ed. 2005;44:3690–3694. doi: 10.1002/anie.200500485. [DOI] [PubMed] [Google Scholar]
- 20.Dey A, Hocking RK, Larsen PL, Borovik AS, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2006;128:9825–9833. doi: 10.1021/ja061618x. [DOI] [PubMed] [Google Scholar]
- 21.BDEC–H (DMF) = 82 kcal/mol; Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies; CRC Press; Taylor and Francis Group: New York (2007), 119.
- 22.A pKa ~ 11 is estimated for a putative [FeIVH3buea(OH)].
- 23.(a) Moyer BA, Meyer TJ. J Am Chem Soc. 1978;100:3601–3603. [Google Scholar]; (b) Moyer BA, Meyer TJ. Inorg Chem. 1981;20:436–444. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.