

Abstract
Background
In order to identify biomarkers useful for the diagnosis of genetic white matter disorders we compared the metabolic profile of patients with leukodystrophies with a hypomyelinating or a non-hypomyelinating MRI pattern.
Methods
We used a non-a priori method of in vitro 1H-NMR spectroscopy on CSF samples of 74 patients with leukodystrophies.
Results
We found an elevation of CSF N-acetylaspartylglutamate (NAAG) in patients with Pelizaeus-Merzbacher disease (PMD) – PLP1 gene –, Pelizaeus-Merzbacher-like disease – GJC2 gene – and Canavan disease – ASPA gene. In the PMD group, NAAG was significantly elevated in the CSF of all patients with PLP1 duplication (19/19) but was strictly normal in 6 out of 7 patients with PLP1 point mutations. Additionally, we previously reported increased CSF NAAG in patients with SLC17A5 mutations.
Conclusions
Elevated CSF NAAG is a biomarker that suggests specific molecular diagnostic abnormalities in patients with white matter diseases. Our findings also point to unique pathological functions of the overexpressed PLP in PMD patients with duplication of this gene.
Keywords: NMR spectroscopy, N-acetylaspartylglutamate, biomarker, leukodystrophy, Pelizaeus-Merzbacher disease
Introduction
Elevation of CSF N-acetylaspartylglutamate (NAAG) in some leukodystrophies has been previously described but its relationship to disease is poorly understood, with the exception of Canavan disease where the elevation of N-acetylaspartate (NAA) may be directly implicated [1]. Using two capillary electrophoresis systems, NAAG has been shown to be elevated in the CSF of 29 out of 32 patients with a clinical presentation of Pelizaeus Merzbacher disease (PMD) [2]. This included 22 patients with PLP1 mutations and 7 patients with Pelizaeus-Merzbacher-like disease (PMLD). However, the nature of the mutations was not reported either in the PMD or the PMLD groups. An increase of NAAG was also observed in the CSF of one patient with Canavan disease [3], one PMLD patient harboring a homozygous deletion in the GJC2 gene [4] and two patients with a severe hypomyelination pattern of unknown cause [5]. In addition, we recently reported that CSF NAAG was increased in 6 patients with sialic acid storage disease (SASD) associated with mutations[6].
Therefore, we decided to use proton NMR spectroscopy (1H-NMRS) to determine whether the levels of CSF NAAG can facilitate the molecular diagnosis of patients with leukodystrophies with a hypomyelinating or a non-hypomyelinating pattern on brain MRI [7].
Material and Methods
Patients
Seventy-four children and adults from two referral centers for neurogenetics and neurometabolism were enrolled in clinical protocols approved by the ethics committees of the Hôpital de Clermont-Ferrand, France, and the National Institutes of Neurological Disorders and Stroke, Bethesda, MD, USA. Written informed consent was obtained from all patients or their legal guardians.
The cohort of patients with leukodystrophies with a hypomyelinating pattern consisted of (i) 26 male patients with PLP1 mutations presenting as PMD or X-linked spastic paraplegia type 2 [8] – PMD/SPG2 patients – , including 19 patients with PLP1 duplication and 7 patients with PLP1 missense or splice mutations (Table 1); (ii) 9 patients with PMLD including one patient with GJC2 mutations; and (iii) 10 patients with a leukodystrophy of unknown etiology despite extensive metabolic and genetic testing. The rest of the cohort consisted of 14 patients with leukodystrophies with a non-hypomyelinating pattern – including patients with Canavan disease, merosin deficiency, MLC1 mutation and complex I deficiency – and 15 diseased controls with complex neurological diseases but normal brain MRI.
Table 1.
Genetic, biochemical and clinical characteristics of PMD/SPG2 patients.
| PLP1 Mutation | NAAG (μmol/L)* | Age | Clinical Score |
|---|---|---|---|
| p.Cys109TrpfsX11 | ND | 3 | 3 |
| p.Asp103ThrfsX12 | ND | 17 | 3 |
| p.Val219Phe | ND | 38 | 3 |
| c.IVS4+1G>A / p.Val209GlufsX27 | ND | 9 | 3 |
| p.Leu223Phe | 38 | 10 | 0 |
| p.Cys220Tyr | ND | 10 | 3 |
| p.Asp103ThrfsX12 | ND | 18 | 4 |
| Duplication | 30 | 2 | 1 |
| Duplication | 36 | 6 | 3 |
| Duplication | 40 | 2 | 2 |
| Duplication | 65 | 6 | 3 |
| Duplication | 89 | 5 | 3 |
| Duplication | 32 | 17 | 2 |
| Duplication | 84 | 8 | 2 |
| Duplication | 60 | 11 | 3 |
| Duplication | 47 | 4 | 3 |
| Duplication | 106 | 13 | 2 |
| Duplication | 36 | 8 | 3 |
| Duplication | 56 | 5 | 2 |
| Duplication | 205 | 8 | 1 |
| Duplication | 88 | 9 | 1 |
| Duplication | 55 | 3 | 1 |
| Duplication | 32 | 5 | 2 |
| Duplication | 23 | 3 | 1 |
| Duplication | 53 | 5 | 3 |
| Duplication | 52 | 7 | 2 |
Diseased controls, mean= 5 ±1 μmol/L (n=15). ND: below detection limit (<3 μmol/L).
Methods
In order to assess clinical severity, we used a score previously validated in patients with PLP1 mutations [9]. Briefly, form 0 referred to absence of motor achievement; form 1 – head control between 2 and 4 years; form 2 – sitting position between 2 and 5 years; form 3 – sitting position between 1 and 2 years, walk with support at a mean age of 3.5 ±1.5 years (range 2–6); form 4 – autonomous walking.
Frozen CSF – stored at −80°C – prepared for 1H-NMRS with minimal handling [5, 10]. First, CSF samples were deproteinized using a 10kDa filter (Nanosep, Omega) to avoid interference from high molecular weight species such as lipoproteins. Before use, the filter was washed twice with water by centrifugation to remove glycerol. A 100μl aliquot of 3.89 mM [trimethylsilyl]-2,2,3,3-tetradeuteropropionic acid in 2H2O (TSP-2H2O, Aldrich) was then added to 500 μl of the ultrafiltrate, providing a chemical shift reference (δ=0.00ppm), a concentration reference and a deuterium lock signal. The pH of the ultrafiltrate was adjusted to 2.50± 0.05 with concentrated HCl. Finally, 500μl of the sample was placed in a 5 mm NMR tube (Wilmad Royal Imperial). The proton NMR spectra were determined on an Avance-500 SB spectrometer (Bruker, France) equipped with a 5 mm BBI (broadband inverse) probe; samples were not spun. Spectra were collected at 25°C and consisted in 32K data points with a spectral width of 6,000 Hz and a total acquisition times of 27 min. A 90° radiofrequency pulse, following a water signal presaturation of 10s, was used for each 128 scans. Shimming of the sample was performed on the deuterium signal until the resonance line width for TSP was <1Hz. Before a Fourier transformation into 64K data points, a sine window multiplication (sine bell shift of 90°) was used to reduce noise. The phase and the baseline were corrected manually using the spectrometer software (X-Win NMR 3.5, Bruker, France). Two-dimensional proton NMRS, 1H-1H correlation spectroscopy (COSY), was performed to confirm the identification of metabolites of interest in patients’ body fluids. The spectra were recorded at 500 MHz using 4k datapoints in F2 and a spectral width of 6002 Hz. 256 Increments and 16 scans per increment were used. The TR was 1.5 s, during which the water resonance was presaturated. Prior to Fourier transformation, a sine window multiplication was applied in both time domains. The values of NAAG were calculated by comparison with the known concentration of the internal standard (TSP).
A one-way between groups analysis of covariance was conducted to compare the levels of CSF NAAG between PMD/SPG2 patients with PLP1 duplication and PLP1 point mutations using age and clinical score as covariates. Preliminary checks were conducted to ensure that there was no violation of the assumptions of normality, linearity, homogeneity of variances and homogeneity of regression slopes.
Results
Elevated CSF NAAG in patients with PLP1, GJC2 and ASPA mutations
In a 1-dimensional 1H-NMR spectrum (pH=2.5), NAAG presents as a prominent singlet resonance at 2.04 ppm and smaller resonances at 2.01 and 2.23 (multiplets), 2.47 (triplet), 2.81 and 2.91 (doublet of doublets), 4.32 (doublet of doublets), and 4.72 ppm (doublet of doublets) [11]. Characteristic cross peaks in 2-dimensional COSY 1H-NMRS are shown in Figure 1.
Figure 1.

One-dimensional and 2-dimensional (COSY) 1H-NMR spectra of NAAG in CSF. The upper part shows the methyl signal (CH3 of NAAG) in the 1D spectrum (2.04 ppm). The lower part shows the correlations between the two diastereotopic protons of CH2 (H17) – 2 doublets of doublets at 2.91 and 2.85 ppm – and the correlations between the two diastereotopic protons of CH2 (H8) – multiplets at 1.99 and 2.23 ppm – and the two protons of CH2 (H9) – triplet at 2.43 ppm – in the 2D spectrum. The signals of protons of CH2 (H8) and CH2 (H9) are not visible on the 1D spectrum because of their small intensities and of overlapping with glutamate and methyl signals.
Compared to our group of disease controls, in vitro 1H-NMRS of CSF showed a significant elevation of NAAG in the CSF of PMD/SPG2 patients compared to controls (47 ±44 μmol/L vs 5 ±1 μmol/L, p<0.001). The mean values of NAAG were normal in (i) patients with PMLD, (ii) patients with other leukodystrophies with a hypomyelinating MRI pattern, and (iii) patients with leukodystrophies with a non-hypomyelinating pattern (Figure 2). However, 1H-NMRS revealed a mild to moderate elevation in the CSF of one PMLD patient associated with GJC2 compound heterozygous mutations (14 μmol/L), one patient with Canavan disease associated with ASPA mutations (17 μmol/L), and two patients presenting with a hypomyelinating leukodystrophy of unknown etiology (33 and 60 μmol/L respectively) (Figure 2). Of note, we did not observe any CSF elevation of NAA except in the CSF of the patient with Canavan disease (98 μmol/L, Figure 3).
Figure 2.
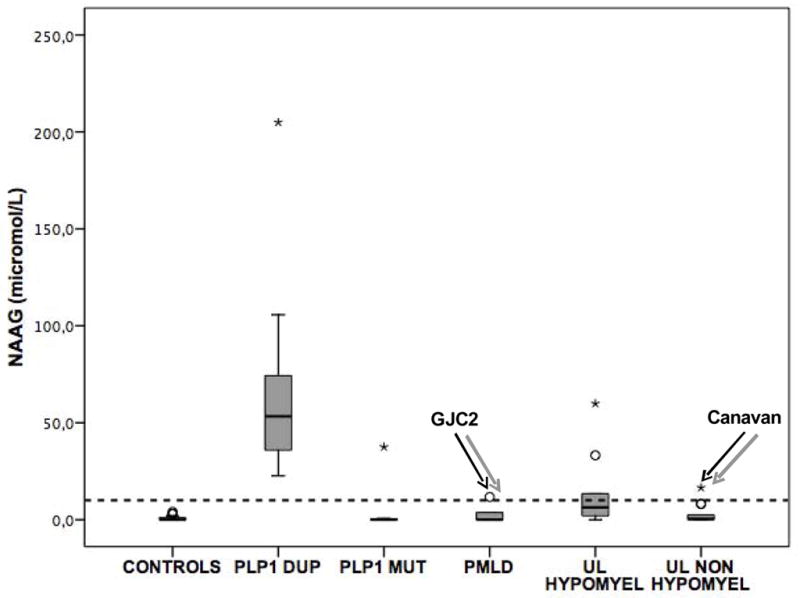
Values of CSF NAAG in the cohort of 74 patients. The dotted line represents the upper limit of normal values of CSF NAAG (12 μmol/L [5]). DUP: duplication; MUT: point mutation; PMLD: Pelizaeus-Merzbacher-like disease; UL: unknown leukodystrophy. Circles and stars denote outliers that are farther than 1.5 and 3 interquartile ranges respectively from the nearer edge of the box.
Figure 3.

1H-NMR spectra of CSF from 2.0 to 2.2 ppm showing the signals of NAA (singlet at 2.033 ppm), N-acetylneuraminic acid (NANA; singlet at 2.047 ppm), NAAG (singlet at 2.040 ppm) and and glutamic acid (multiplet at 2.16 ppm). CSF of a patient with (a) Canavan disease showing a marked elevation of NAA (98 μmol/L) and a moderate elevation of NAAG (17 μmol/L): (b) PLP1 duplication showing elevated NAAG (55 μmol/L), compared to (c) normal CSF spectrum.
Association of elevated CSF NAAG with the nature of the PLP1 mutation rather than clinical severity
1H-NMRS revealed an elevation of NAAG in the CSF of all 19 patients with PLP1 duplication (Table 1 and Figure 2). Conversely, CSF NAAG was strictly normal in 6 out of 7 patients with PLP1 missense, nonsense or splice mutations (Table 1 and Figure 2). The only patient with PLP1 mutations who displayed a significant elevation of CSF NAAG presented with the most severe clinical presentation – clinical score of 0 (Table 1). However, there was no correlation between the levels of CSF NAAG and clinical severity among the cohort of PMD/SPG2 patients, or among patients with PLP1 duplication only. There was no correlation either between CSF NAAG and age. Moreover, when adjusting for both age and clinical severity, we found a significant elevation of NAAG in patients with PLP1 duplication versus PLP1 point mutations (p=0.012, partial eta squared=0.265). Therefore, the elevation of NAAG in the CSF of PMD/SPG2 patients strongly suggests the nature of the mutation – i.e. duplication in the PLP1 gene – rather than clinical severity.
Discussion
In this study, using in vitro 1H-NMRS we showed that NAAG is always elevated in the CSF of patients with PLP1 duplication. We also confirmed increased CSF NAAG in patients with GJC2 mutations and Canavan disease [2, 3]. Despite the elevation of CSF NAAG in one patient with a PLP1 point mutation displaying a very severe form of the disease, we did not find any correlation between the levels of CSF NAAG and clinical severity in PMD/SPG2 patients, which is consistent with our recent findings in SASD patients [6]. Since we had relatively few patients with mutated PLP1 and severe clinical score (0–1), we cannot completely exclude that some patients with severe missense mutation also have elevated CSF NAAG. Nevertheless, there was a marked difference in the levels of CSF NAAG between the patients with PLP1 duplication and those with PLP1 point mutations, even when adjusting for clinical severity. Therefore, we suggest that quantifying NAAG levels in CSF of patients with white matter diseases can be a useful and simple tool to orient molecular diagnostic testing, especially in hypomyelinating leukodystrophies. Of note, in vivo 1H-MRI spectroscopy is unable to provide similar information since the technique cannot discriminate between the NAA and NAAG peaks unlike in vitro 1H-NMRS (Figure 3). Consequently, the relatively high NAA concentrations in brain tissues prevent the measurement of NAAG in vivo. The detection limit of 1H-NMRS for CSF NAAG is higher than of techniques such as two capillary electrophoresis [2] or mass spectrometry (personal observation). However, contrary to these techniques, 1H-NMRS requires no derivatization or extraction. It can therefore simultaneously detect compounds of different nature which allows the non-a priori identification of new metabolic abnormalities in patients with uncharacterized or known genetic diseases [6, 12].
The absence of correlation between CSF NAAG and clinical severity in PMD/SPG2 and SASD patients, as well as the observed neuroprotective effect of increased extracellular NAAG during GCPII inhibition [13], suggest that NAAG is an unlikely contributor to the hypomyelination phenotype observed in these patients. NAAG undergoes a tricellular metabolic sequence wherein it is synthesized in neurons from N-acetylaspartate (NAA) and L-glutamate, hydrolyzed to NAA and glutamate by astrocytes and further hydrolyzed to L-aspartate and acetate by oligodendrocytes [14]. NAA was also shown to provide acetyl groups for myelin synthesis [15]. It is therefore possible that the elevation of CSF NAAG in some hypomyelinating disorders reflects a compensatory mechanism for the altered maturation of oligodendrocytes in an effort to enhance myelin synthesis.
The elevation of CSF NAAG in patients with PLP1 duplication, but usually not in patients with point mutations, favors a specific effect of the over-expressed wild-type PLP1 gene as supported by in vitro and preclinical in vivo studies [16, 17]. Overexpression of wild-type PLP in the CNS may affect the integrity of neurons as suggested by the increased mortality of neurons when cultured in the presence of conditioned media from PLP overexpressing cells but not from DM20 – a splice variant of PLP – overexpressing cells. This effect of conditioned media may be mediated by a negative pH shift elicited by PLP [18] and could be related to the similarity of PLP and its four hydrophobic domain motif to proteins that are known to function as pores or channels [19]. Interestingly, abnormal connexin 47 associated with GJC2 gene mutations was reported to alter the function of intercellular channels [20]. The sialin transporter, which was recently found to also function as an aspartate transporter [21], may have a role in neuronal secretory processes due to its additional non-lysosomal localization [22]. Therefore, the implication of PLP1, connexin 47 and sialin in the formation of ion channels/transporters in membranes suggests increased excretion of NAAG into the extracellular compartment. The lack of concurrent elevation of NAA further supports the hypothesis that NAAG elevation in CSF of patients with PLP1 duplication results from excessive neuronal release of NAAG rather than from its overproduction. We found two patients presenting with hypomyelination on brain MRI and increased CSF NAAG but without mutations of PLP1, GJC2 or SLC17A5. A similar entity has been previously reported [5]. In these cases, a candidate genes approach should focus on genes encoding for proteins related to the structure and function of ion channels or transporters.
Acknowledgments
This work was supported by the Assistance Publique des Hôpitaux de Paris (CRC 05169, FM), the ELA Foundation (OBT) and by the Intramural Program of the NIH/NINDS (RS). The authors are grateful to Emile Van Schaftingen for his critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arun P, Madhavarao CN, Moffett JR, Namboodiri MA. Regulation of N-acetylaspartate and N-acetylaspartylglutamate biosynthesis by protein kinase activators. J Neurochem. 2006;98:2034–2042. doi: 10.1111/j.1471-4159.2006.04068.x. [DOI] [PubMed] [Google Scholar]
- 2.Burlina AP, Ferrari V, Burlina AB, Ermani M, Boespflug-Tanguy O, Bertini E. N-acetylaspartylglutamate (NAAG) in Pelizaeus-Merzbacher disease. Adv Exp Med Biol. 2006;576:353–359. doi: 10.1007/0-387-30172-0_26. discussion 361–353. [DOI] [PubMed] [Google Scholar]
- 3.Burlina AP, Ferrari V, Divry P, Gradowska W, Jakobs C, Bennett MJ, Sewell AC, Dionisi-Vici C, Burlina AB. N-acetylaspartylglutamate in Canavan disease: an adverse effector? Eur J Pediatr. 1999;158:406–409. doi: 10.1007/s004310051102. [DOI] [PubMed] [Google Scholar]
- 4.Sartori S, Burlina AB, Salviati L, Trevisson E, Toldo I, Laverda AM, Burlina AP. Increased level of N-acetylaspartylglutamate (NAAG) in the CSF of a patient with Pelizaeus-Merzbacher-like disease due to mutation in the GJA12 gene. Eur J Paediatr Neurol. 2008;12:348–350. doi: 10.1016/j.ejpn.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 5.Wolf NI, Willemsen MA, Engelke UF, van der Knaap MS, Pouwels PJ, Harting I, Zschocke J, Sistermans EA, Rating D, Wevers RA. Severe hypomyelination associated with increased levels of N-acetylaspartylglutamate in CSF. Neurology. 2004;62:1503–1508. doi: 10.1212/01.wnl.0000123094.13406.20. [DOI] [PubMed] [Google Scholar]
- 6.Mochel F, Engelke UF, Barritault J, Yang B, McNeill NH, Thompson JN, Vanderver A, Wolf NI, Willemsen MA, Verheijen FW, Seguin F, Wevers RA, Schiffmann R. Elevated CSF N-acetylaspartylglutamate in patients with free sialic acid storage diseases. Neurology. 2010;74:302–305. doi: 10.1212/WNL.0b013e3181cbcdc4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750–759. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saugier-Veber P, Munnich A, Bonneau D, Rozet JM, Le Merrer M, Gil R, Boespflug-Tanguy O. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet. 1994;6:257–262. doi: 10.1038/ng0394-257. [DOI] [PubMed] [Google Scholar]
- 9.Cailloux F, Gauthier-Barichard F, Mimault C, Isabelle V, Courtois V, Giraud G, Dastugue B, Boespflug-Tanguy O. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet. 2000;8:837–845. doi: 10.1038/sj.ejhg.5200537. [DOI] [PubMed] [Google Scholar]
- 10.Mochel F, Barritault J, Boldieu N, Eugene M, Sedel F, Durr A, Seguin F. Contribution of in vitro NMR spectroscopy to metabolic and neurodegenerative disorders. Rev Neurol (Paris) 2007;163:960–965. doi: 10.1016/s0035-3787(07)92640-5. [DOI] [PubMed] [Google Scholar]
- 11.Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, Mandal R, Sinelnikov I, Xia J, Jia L, Cruz JA, Lim E, Sobsey CA, Shrivastava S, Huang P, Liu P, Fang L, Peng J, Fradette R, Cheng D, Tzur D, Clements M, Lewis A, De Souza A, Zuniga A, Dawe M, Xiong Y, Clive D, Greiner R, Nazyrova A, Shaykhutdinov R, Li L, Vogel HJ, Forsythe I. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37:D603–610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mochel F, Sedel F, Vanderver A, Engelke UF, Barritault J, Yang BZ, Kulkarni B, Adams DR, Clot F, Ding JH, Kaneski CR, Verheijen FW, Smits BW, Seguin F, Brice A, Vanier MT, Huizing M, Schiffmann R, Durr A, Wevers RA. Cerebellar ataxia with elevated cerebrospinal free sialic acid (CAFSA) Brain. 2009;132:801–809. doi: 10.1093/brain/awn355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Post JP, de Visser SJ, de Kam ML, Woelfler M, Hilt DC, Vornov J, Burak ES, Bortey E, Slusher BS, Limsakun T, Cohen AF, van Gerven JM. The central nervous system effects, pharmacokinetics and safety of the NAALADase-inhibitor GPI 5693. Br J Clin Pharmacol. 2005;60:128–136. doi: 10.1111/j.1365-2125.2005.02396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baslow MH. Functions of N-acetyl-L-aspartate and N-acetyl-L-aspartylglutamate in the vertebrate brain: role in glial cell-specific signaling. J Neurochem. 2000;75:453–459. doi: 10.1046/j.1471-4159.2000.0750453.x. [DOI] [PubMed] [Google Scholar]
- 15.Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen RW. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem. 2001;78:736–745. doi: 10.1046/j.1471-4159.2001.00456.x. [DOI] [PubMed] [Google Scholar]
- 16.Regis S, Grossi S, Corsolini F, Biancheri R, Filocamo M. PLP1 gene duplication causes overexpression and alteration of the PLP/DM20 splicing balance in fibroblasts from Pelizaeus-Merzbacher disease patients. Biochim Biophys Acta. 2009;1792:548–554. doi: 10.1016/j.bbadis.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Dhaunchak AS, Nave KA. A common mechanism of PLP/DM20 misfolding causes cysteine-mediated endoplasmic reticulum retention in oligodendrocytes and Pelizaeus-Merzbacher disease. Proc Natl Acad Sci U S A. 2007;104:17813–17818. doi: 10.1073/pnas.0704975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boucher SE, Cypher MA, Carlock LR, Skoff RP. Proteolipid protein gene modulates viability and phenotype of neurons. J Neurosci. 2002;22:1772–1783. doi: 10.1523/JNEUROSCI.22-05-01772.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitagawa K, Sinoway MP, Yang C, Gould RM, Colman DR. A proteolipid protein gene family: expression in sharks and rays and possible evolution from an ancestral gene encoding a pore-forming polypeptide. Neuron. 1993;11:433–448. doi: 10.1016/0896-6273(93)90148-k. [DOI] [PubMed] [Google Scholar]
- 20.Kamasawa N, Sik A, Morita M, Yasumura T, Davidson KG, Nagy JI, Rash JE. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: implications for ionic homeostasis and potassium siphoning. Neuroscience. 2005;136:65–86. doi: 10.1016/j.neuroscience.2005.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyaji T, Echigo N, Hiasa M, Senoh S, Omote H, Moriyama Y. Identification of a vesicular aspartate transporter. Proc Natl Acad Sci U S A. 2008;105:11720–11724. doi: 10.1073/pnas.0804015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aula N, Kopra O, Jalanko A, Peltonen L. Sialin expression in the CNS implicates extralysosomal function in neurons. Neurobiol Dis. 2004;15:251–261. doi: 10.1016/j.nbd.2003.11.017. [DOI] [PubMed] [Google Scholar]