

Abstract
APOBEC-1 Complementation Factor (ACF) is an RNA-binding protein that interacts with apoB mRNA to support RNA editing. ACF traffics between the cytoplasm and nucleus. It is retained in the nucleus in response to elevated serum insulin levels where it supports enhanced apoB mRNA editing. In this report we tested whether ACF may have the ability to regulate nuclear export of apoB mRNA to the sites of translation in the cytoplasm. Using mouse models of obesity-induced, insulin resistance and primary hepatocyte cultures we demonstrated that both nuclear retention of ACF and apoB mRNA editing were reduced in the livers of hyperinsulinemic obese mice relative to lean controls. Coincident with an increase in the recovery of ACF in the cytoplasm was an increase in the proportion of total cellular apoB mRNA recovered in cytoplasmic extracts. Cytoplasmic ACF from both lean controls and obese mouse livers was enriched in endosomal fractions associated with apoB mRNA translation and ApoB lipoprotein assembly. Inhibition of ACF export to the cytoplasm resulted in nuclear retention of apoB mRNA and reduced both intracellular and secreted ApoB protein in primary hepatocytes. The importance of ACF for modulating ApoB was supported by the finding that RNAi knockdown of ACF reduced ApoB secretion. An additional discovery from this study was the finding that leptin is a suppressor ACF expression. Dyslipidemia is a common pathology associated with insulin resistance that is in part due to the loss of insulin controlled secretion of lipid in ApoB-containing very low density lipoproteins. The data from animal models suggested that loss of insulin regulated ACF trafficking and leptin regulated ACF expression may make an early contribution to the overall pathology associated with very low-density lipoprotein secretion from the liver in obese individuals.
Keywords: diabetes, insulin-resistance, APOBEC-1 Complementation Factor, apolipoprotein B, mRNA editing
1. Introduction
ACF (APOBEC-1 Complementation Factor) is a member of the ELAV/Hu family of RNA binding proteins, characterized by their multiple RNA Recognition Motifs (RRMs) [1]. This family of proteins functions in controlling the stability of AU-rich RNA [2]. ACF contains 3 RRMs in its N-terminus and has nanomolar affinity [3] for an 11 nucleotide AU-rich ‘mooring’ sequence 3′ of the edited cytidine in apoB mRNA [4; 5]. ACF is a required factor for apoB mRNA editing site recognition, [6; 7] and recruitment of APOBEC-1, a cytidine deaminase, to nucleotide C6666 for site-specific C to U editing [8]. This RNA modification enables two functional protein products to be expressed from one gene [9; 10] (ApoB100 and ApoB48) that serve as scaffold proteins for lipoprotein particle assembly and transport of lipid from the liver and small intestine. Editing of apoB mRNA is a characteristic of differentiated tissue types [11] and is regulated hormonally [12] and metabolically [13; 14; 15; 16; 17; 18].
While APOBEC-1 and apoB mRNA editing are not essential for life [19], ACF knockout was embryonically lethal [20]. ACF was essential at the pre-implantation stage prior to the developmental induction of apoB mRNA and apoB mRNA editing [21], and therefore ACF must have had a biological function other than the complementation of apoB mRNA editing. In this regard, ‘mooring’ sequences have been identified in numerous mRNAs, some of which encode proteins involved in signal transduction and cell proliferation [22]. Whether there is an alternative function of ACF has been an open question based on the finding that it is abundantly expressed in human liver even though APOBEC-1 and editing activity are not observed in this tissue.
ApoB mRNA editing occurred in the cell nucleus on spliced polyadenylated message [23; 24]. Despite compartmentalization of apoB editing, ACF is distributed throughout the cell and was recovered from cell extracts as inactive 60S cytoplasmic complexes and active 27S nuclear editosomes [25]. ACF trafficking from the cytoplasm into the nucleus was dependent on an NLS (Nuclear Localization Sequence) in the C-terminal half of the protein [26]. Increased nuclear abundance of ACF correlated with increased editing efficiency [12; 17; 27]. We recently reported that nuclear retention of ACF in response to metabolic stimulation correlated with phosphorylation of ACF’s serine residue(s) by insulin-stimulated protein kinase C [27; 28]. It was also observed that nuclear accumulation of ACF resulted from the inhibition of Protein Phosphatase 1 (PP1), suggesting that ACF export required de-phosphorylation. These findings were of interest as immuno-electron microscopy of rat liver thin sections suggested that cytoplasmic ACF was localized to the exterior surface of the endoplasmic reticulum, the site of apoB translation [25]. The localization of ACF with apoB translational complexes also was supported by the finding that ACF was required to protect edited apoB mRNA from Nonsense Mediated Decay (NMD) [29]. This led us to the hypothesis that, in addition to mRNA editing, ACF may have a function in the export of apoB mRNA from the nucleus to the cytoplasm and its continued association with apoB mRNA may modulate the availability of apoB mRNA for ApoB protein translation. Evidence will be presented from studies in animal models for obesity that the abundance and trafficking of ACF were regulated processes that affected ApoB protein expression.
2. Materials and Methods
2.1 Animal care and primary hepatocyte preparation
Male B6.V-Lepob/J, B6.Cg-m Leprdb/+ +/J and C57Bl/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME) and were used between 10 and 12 weeks of age, referred to herein as Ob/Ob, Db/Db, and cogenic lean control (respectively). Animals were housed in rooms with 12 hour light/dark cycles and fed normal rodent chow. Blood glucose levels were determined on blood drawn from the tail vein using a Bayer glucometer and Ascensia Contour Diabetic Test Strips. Fasting was conducted overnight for a 12 hour period. Insulin intraperitoneal injections were conducted with 1.5 IU/kg Novolin® N (Novo Nordisk, Princeton, NJ) and animals were sacrificed 4 hours following injection. Primary mouse hepatocytes were harvested as described previously [14]. Cells were plated in William’s E Media (Sigma, St Louis, MO) with 5% FBS overnight prior to treatment on BIOCOAT type I collagen coated dishes (Becton Dickinson Labware).
2.2 Subcellular fractionation
Whole liver extracts were prepared by Teflon to glass homogenization of liver in 0.25 STM (0.25 M sucrose, 50 mM Tris pH 7.0 and 5 mM MgCl2) supplemented with Complete Protease Inhibitors (Roche, Indianapolis, IN) and phosphatase inhibitors, PhosStop (Roche) then cleared at 3,000 X g for 10 min. Nuclear and cytoplasmic preparations were centrifuged as previously described [30] and the quality of the separation was assessed by western blotting extracts for cytoplasmic β-actin (Sigma) and nuclear histone H1 (Santa Cruz Biotechnology, Santa Cruz, CA). Sub-fractionation of cytoplasmic extract into HDM, LDM, and residual cytoplasmic fractions was conducted as previously described [31]. Subcellular fractionation of primary rat hepatocytes was conducted using the NE-PER kit (Thermo Scientific, Waltman, MA). Protein abundance in each fraction was assessed using the Bradford Assay (BioRad, Hercules, CA).
2.3 Immunological techniques and quantification
A polyclonal antibody recognizing the C-terminus of ACF [27] was used for the assessment of ACF abundance, while monoclonal antibodies recognizing either β-actin or histone H1 were used for normalization of protein input from the cytoplasmic or nuclear extracts (respectively). To assess total ACF, 40 μg of sample per lane were resolved on SDS-PAGE and western blots were sequentially probed for ACF and β-actin then visualized using ECL (Perkin Elmer Life Sciences, Waltham, MA). Non-saturated film exposures were scanned and the intensity of signal quantified using ImageJ (NIH) software. Total ACF in each fraction was determined as a relative value by calculating the ratio of the whole cell ACF western blot signal to that of β-actin. The ACF/ β-actin ratio for control lean animals was set to 1 in order to evaluate changes in ACF abundance that may have occurred in the experimental conditions.
The calculations used in quantifying ACF nuclear and cytoplasmic distributions have been reported [17]. Briefly, to calculate the subcellular distribution of ACF, densitometric scans of ACF western blots of cytoplasmic and nuclear extracts were normalized for β-actin and histone H1 signals and mathematically corrected to total amount of protein recovered in each extract. These values were expressed as the nuclear to cytoplasmic ratio (Supplemental, Table S1). To assess the cytoplasmic and nuclear distribution of ACF protein was quantified using the BioRad assay kit (BioRad Laboratories, CA) and 120 μg or 40 μg of extract (respectively) were resolved by SDS-PAGE and probed sequentially for ACF, β-actin and H1. The ACF western signal was normalized by dividing the ACF signals by β-actin or histone H1signals in the cytoplasmic and nuclear lanes, respectively. A relative N:C ratio was then calculated by dividing the corrected ACF band density from the nuclear lane by the corrected ACF band density in the cytoplasmic lane. The fold-change in ACF N:C ratio from that seen in the control animals was calculated by dividing the N:C ratio calculated for the insulin treated animal by the N:C ratio of the saline treated animal. Reported averages are from two trials.
2.4 ApoB Quantification
For western blotting, equivalent μg amounts of cytoplasmic extract from Ob/Ob and C57bl/6J mice were probed with an antibody recognizing the N-terminus of ApoB (Santa Cruz Biotechnology, Santa Cruz, CA) to assess the relative abundance of intracellular ApoB100 and ApoB48 proteins. For radioimmunoassay (RIA) assessment of ApoB, media was replaced at 24 post-infection of rat primary hepatocytes infected with virus expressing RNAi against acf. Media was harvested 24 hours following replacement and assessed by a quantitative RIA using a mouse monoclonal antibody raised against the N-terminus of rat ApoB [32]. ApoB secretion was calibrated using a standard curve prepared from rat VLDL. In addition, ApoB secreted in the media, as well as intracellular ApoB in whole cell lysates was quantified following 5 hr drug treatment of primary hepatocytes with at 5 μM Cantharidin. Cellular and media ApoB were normalized to whole cell protein content and results were reported as ng ApoB/mg cell protein as described above.
2.5 In vivo phosphorylation
In vivo 32Pi labeling was performed by intraperitoneal injection of mice with 0.2 mCi/g BW of orthophosphoric acid (NEN, Boston, MA) buffered with 50 mM HEPES, pH 7.0 and 150 mM NaCl. After 4 h, mice were sacrificed and hepatic cytoplasmic and nuclear extracts prepared as described above.
2.6 RNA analyses
Total hepatic abundance of editosome component (acf, apoB and apobec-1) mRNAs in each mouse model was determined, using RNA isolated from 100 mg of fresh tissue homogenized in TRI-Reagent (MRC, Cincinnati, OH) as per manufacturer’s instructions. cDNA was prepared by amplification of 2 μg of RNA primed by oligo dT and amplified using real time PCR. Real time reactions were monitored using TaqMan® (ABI, Foster City, CA) master mix with Exiqon mouse probes 60, 40, 59, 42, 26, 69 and 43 for apobec-1, apoB, acf, cugbp2, hnrnpa1, gryrbp and β-actin, respectively, and using the following primers: apobec-1, ACCTTCAGCTAACCTTCACACAGT and CGGACTTTGTTTCCTCAGGT; apoB, CCAGACAACCTCTTCCTAAAGACT and TCAATGTTTATTTTGTTCCTGTTCA; acf, TTTGCCTTTGTGGAATATGAGA and GGATGTCCCCACAACTGAAT; cugbp2, CACTGCCCACTTTGTACAGC and AGAGGTTTGCCCCCTCTG; hnrnpa1, GCTTTGGCGGTGGTAGTG and GCCCAAAATTGGAAGACTGA; gryrbp, GGGGACCAAAGTAGCAGACTC and TGTAGCCTGTTCTTTCCAAAAGT; and β-actin, AAGGCCAACCGTGAAAAGAT and GTGGTACGACCAGAGGCATAC. Primers designed for apoB annealed at nucleotides 3847-3870 and 3893-3917 for left and right primers, respectively, in the 3′ end of the transcript. Rat samples assessed by real time PCR used rat Exiqon probe numbers 70 and 18 for apoB and actin, respectively, and cDNA was amplified with the following primers: apoB, TCAATGTGAAGTATAACGAAGATGG and ATGTCCAGATGAGCCTCTCC; β-actin, CCCGCGAGTACAACCTTCT and CGTCATCCATGGCGAACT. Real time reactions were run on Applied Biosystems 7900HT Sequence Detection System cycler in 384 well format using cycling conditions: 95° C 10 minutes followed by 40 cycles of 95° C for 15 seconds and 60° C for 1 minute, and were analyzed using Applied Biosystems SDS2.2 software.
For evaluating the effect of hormone or metabolic treatment on the abundance of mRNAs encoding editosome components, 60 mm dishes of primary hepatocytes were rinsed twice in 1X PBS then harvested by scraping into TRI-Reagent (MRC). Two μg of RNA were used to amplify cDNA input for real time PCR. Subcellular fractionation of whole liver was carried out as described above with equivalent portions of cytoplasmic and nuclear extract placed in TRI-Reagent (MRC) for assessment of compartmental apoB abundance in Ob/Ob mice. Subcellular fractionation of primary rat hepatocytes following cantharidin treatment was carried out as described above. Total RNA recovered from the extraction was poly A purified using MicroPoly (A) Pursuit (Ambion, Austin, TX) and 150 ng were used as input for cDNA reaction to be used for real time PCR. β-actin was used to normalize for input in real time PCRs.
ApoB mRNA editing was quantified by the poisoned primer extension assay as previously described [33] on apoB cDNA amplified from 2 μg of total RNA isolated from the liver of Ob/Ob and C57Bl/6J mice as described above.
2.7 Treatments of primary hepatocytes
All hepatocytes were prepared as previously described and allowed to equilibrate overnight in fresh media [14]. Hormones used to treat mouse primary hepatocytes were: leptin, TNFα, IL-6, and resistin, were purchased from R&D systems (Minneapolis, MN) and the doses used in each treatment were based on recommended EC50 concentrations by the manufacturer: 0.01 ng/mL, 0.05 ng/mL, 0.02 ng/mL and 10 μg/mL, respectively. Hourly treatments for 4 hours at the suggested EC50, as well as 10-fold above and below the EC50 were tested. Resistin was only used at the provided EC50. Equal volumes of PBS were added as control treatments. Free fatty acid treatment was carried out for a period of 4 hours at two concentrations, 0.1 and 1 mM using a 2:1 ratio of oleate to palmitate [34].
2.8 RNAi Constructs and Treatment
Three non-conserved 22 nucleotide sequences were selected as targets of acf message, GenBank Accession Number NM_133400.1, using the RNAi Codex server at http://codex.cshl.edu. Perspective targets were PCR amplified from oligos; H2, TGCTGTTGACAGTGAGCGCGTTGTTGATGTCATTGTCTACTAGTGAAGCCACAGATGTAGTAGACAATGACATCAACAACTTGCCTACTGCCTCGGA, H3, TGCTGTTGACAGTGAGCGCACAGAGGCTATGCATTTGTAATAGTGAAGCCACAGATGTATTACAAATGCATAGCCTCTGTTTGCCTACTGCCTCGGA and W2, TGCTGTTGACAGTGAGCGAAATCCTGTACGTAAGGAACCTTTAGTGAAGCCACAGATGTAAAGGTTCCTTACGTACAGGATTTTGCCTACTGCCTCGGA containing the selected targets and mir-30 sequence context, including the hairpin loop structure, as previously described [35] using the primers 5′CAGAAGGGGATCCAAGGTATATTGCTGTTGACAGTGAGCG and 3′ CTAAAGTAGCCCCTTAAGCTTCCGAGGCAGTAGGCA. A nonspecific control sequence NS, TGCTGTTGACAGTGAGCGATCTCGCTTGGGCGAGAGTAAGTAGTGAAGCCACAGATGTACTTACTCTCGCCCAAGCGAGATTGCCTACTGCCTCGGA also was amplified. The resulting PCR products were cloned individually into pAd shRNAmU6 vector [36] using BamHI and HindIII restriction sites. Resulting vectors were recombined with pAdEasy-1 in the BJ5183-AD-1 E. coli strain and infective virus produced as per manufacturer’s instructions using the pAdEasy XL Adenoviral Vector System (Stratagene, La Jolla, CA). McArdle cells at 80% confluence were treated with 10 μL of virus for 2 hours in minimal media before replacing the media. Infectivity was monitored by fluorescence microscopy 24 hrs post infection prior to preparing whole cell lysates using Reporter Lysis Buffer (Promega, Madison, WI) per manufacture protocol. Primary rat hepatocytes were plated as previously described, treated with 30 μL of virus for 2 hrs in minimal media and then incubated with fresh media. Infectivity and harvest were conducted as described for McArdle cells at 48 hours post infection.
3. Results
3.1 Dysregulation of ACF in the genetically obese and insulin-resistant Ob/Ob mouse
Given that the normal hepatic response to insulin was to retain ACF in the nucleus, we evaluated whether this response was retained in insulin-resistant mice. We examined this question in the leptin-deficient Ob/Ob mice, a model for obesity-induced insulin-resistance. In Ob/Ob mice, hyperphagia, due to a lack of functional leptin, begins from birth and is responsible for the obese phenotype and leads to insulin-resistance in adult animals [37]. Ob/Ob mice were determined to be obese and diabetic, averaging twice the body weight of cogenic lean controls (40.1g vs. 21.4g) and characteristic of insulin-resistance, these mice had blood glucose levels approximately 2-fold higher than observed in lean mice (372 ± 70 mg/dL vs. 182 ± 30 mg/dL).
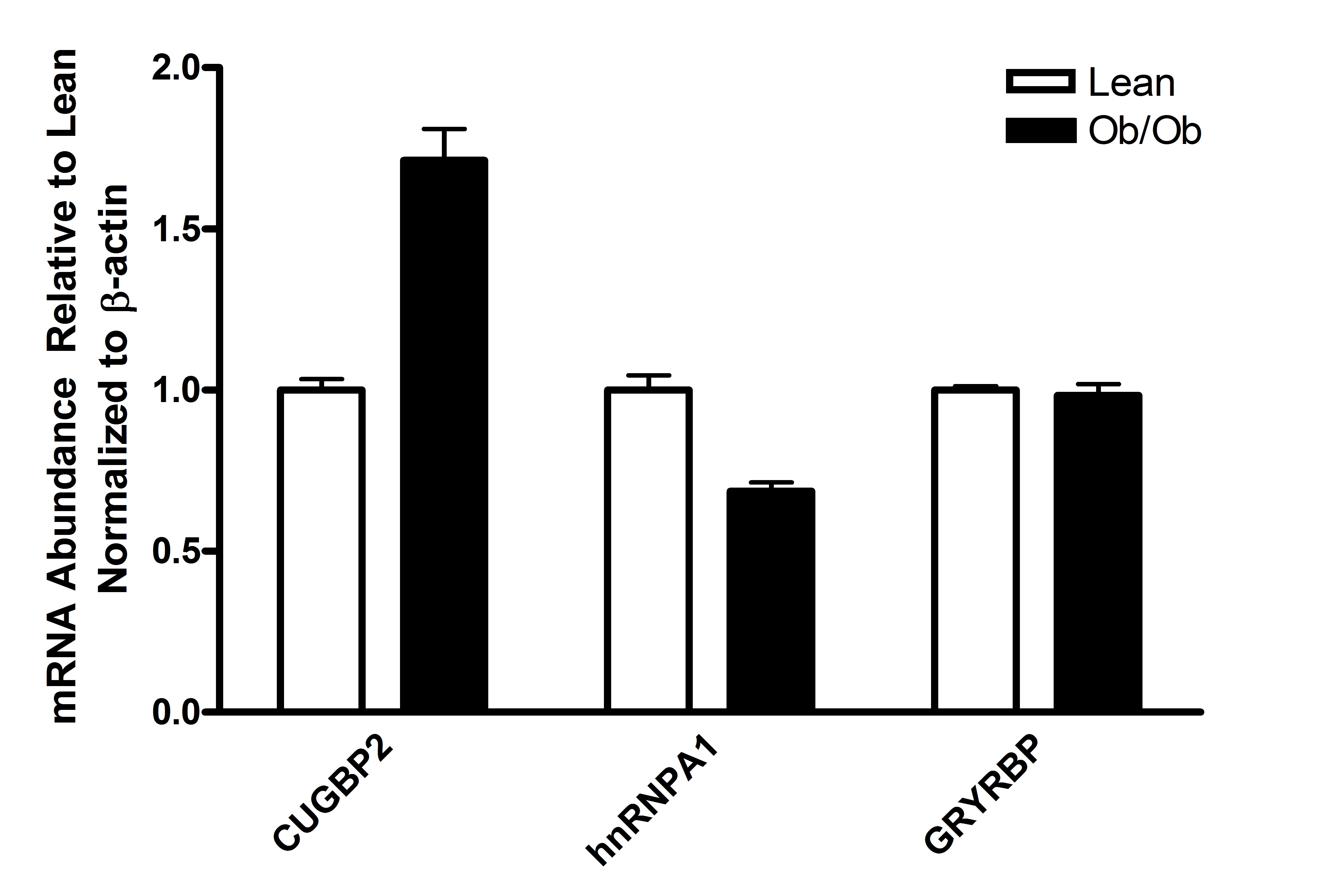
Using real-time-PCR, we examined the expression of hepatic acf, apobec-1 and apoB mRNAs using β-actin mRNA as a normalization control. While both apobec-1 and apoB mRNA appeared to be slightly more abundant in Ob/Ob mouse liver relative to cogenic lean control mice (normalized to β-actin mRNA abundance), these changes were not statistically significant with p≤0.29 and p≤0.17, respectively (Fig. 1A). Unexpectedly, the abundance of acf message was increased nearly 7-fold (p≤0.05) in Ob/Ob mouse liver. By western blotting, Ob/Ob total hepatic ACF protein abundance was increased approximately 5-fold (p≤0.01) relative to the abundance of hepatic ACF protein in lean control mice (normalized to β-actin protein abundance) (Fig. 1B). Previous studies showed that CUGBP2 [12; 38] and GRYRBP [39; 40] and hnRNPA1[40] are RNA-binding proteins that traffic between the nucleus and cytoplasm and whose interactions with apoB mRNA and ACF inhibit apoB mRNA editing. We evaluated whether the mRNAs encoding these proteins also were up regulated in Ob/Ob mouse liver. Real-time PCR showed that while cugbp2 mRNA was slightly elevated (1.7-fold, p≤0.4) relative to that quantified in lean controls, the abundance of hnrnpa1 and gryrbp mRNAs were not altered (Supplemental Fig S1). The data suggested that there was a significant difference in the magnitude of up regulation seen with ACF in Ob/Ob liver compared with other RNA binding proteins we evaluated. We therefore focused on ACF for the remainder of this study.
Figure. 1.
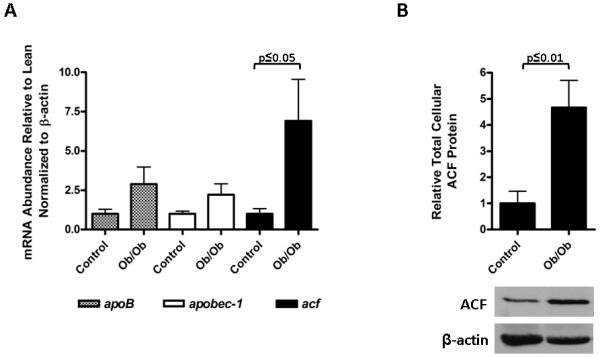
Increased ACF mRNA and protein expression in Ob/Ob liver. A. The abundance of Ob/Ob hepatic apoB, apobec-1 and acf mRNA relative to that quantified from lean control liver was assessed by real time PCR and normalized to β-actin mRNA. Lean animal values were set to 1. Results and standard deviations were the average of the analysis of 3 livers and statistical significance was calculated using a student’s t-test. B. Graphic representation of total hepatic ACF protein abundance normalized to β-actin. Scanning densitometry of western blots probed for ACF and β-actin using ImageJ (NIH) was used to create an ACF to β-actin ratio. For lean animals this ratio was set to one. A representative western blot of ACF and β-actin is shown below the graph. The results are representative of 3 liver preparations and statistical significance was calculated using a student’s t-test.
To determine what was responsible for the elevated expression of ACF mRNA and protein, we considered that the Ob/Ob mice lacked leptin and that this may have indirectly or directly contributed to our findings. In addition to leptin however, obesity is associated with increased levels of other hormones such as the adipokine resistin and pro-inflammatory cytokines Il-6 and TNFα [41; 42; 43]. To evaluate the effect of these factors on acf mRNA expression under defined in vitro conditions, primary hepatocytes from lean mice were treated with either resistin, Il-6, TNFα or free fatty acids [34; 44]. All of these treatments failed to modulate acf mRNA abundance (data not shown). However, treatment of primary hepatocytes with leptin demonstrated a dose-dependent suppression of acf mRNA (p≤0.05) (Fig. 2). Consequently, the ability of leptin to alter acf mRNA abundance was unique to the agents we tested. Although ACF protein levels were not significantly suppressed by leptin during this 4 hour time course (data not shown), our findings are consistent with the possibility that the absence of leptin during the lifespan of the Ob/Ob mouse contributed to the over expression of ACF. However, these data did not rule out the possibility that insulin resistance in the Ob/Ob mouse liver also may have contributed to the observed changes in ACF abundance.
Figure. 2.
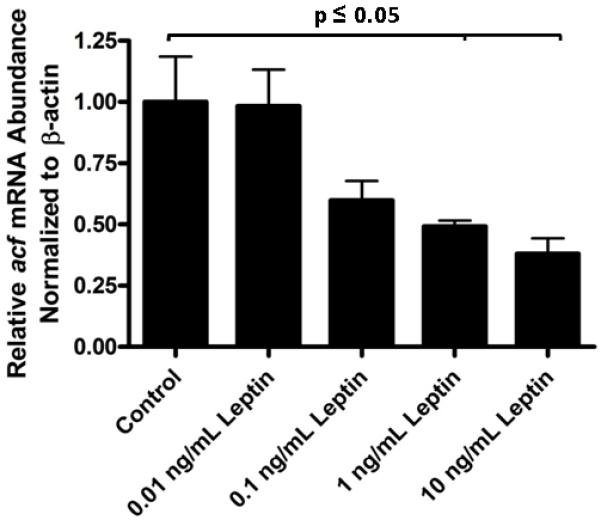
Acute leptin treatment suppresses acf mRNA abundance. The relative abundance of acf mRNA in primary mouse hepatocytes, normalized to β-actin mRNA abundance was determined by real time PCR following 4 hours of treatment with increasing concentrations of leptin. Control acf mRNA to β-actin mRNA ratio was set to one. Reduction in acf mRNA to β-actin mRNA ratios at both 1 and 10 ng/mL leptin treatment were statistically significant (p≤0.05 and n=3, where n is the average of one treated 6 mm plate of hepatocytes).
To further explore the potential suppressive effects of leptin on acf mRNA we quantified acf, apoB and apobec-1 mRNAs in the livers from the Db/Db genetic mouse model for obesity-induced insulin resistance. In these mice, leptin is expressed, but knockout of the long form of the leptin receptor (Ob-Rb), expressed primarily in the brain, induces hyperphagia leading to obesity and insulin resistance. Important to this study, despite knockout of Ob-Rb, leptin signaling in the liver is intact due to a short form of the leptin receptor (Ob-Ra) expressed in the liver [45; 46]. Both apoB and apobec-1 mRNAs were suppressed in Db/Db liver to statistically significant levels relative to that in the livers of cogenic littermates (p≤0.01 and p≤0.05, respectively). However, acf mRNA levels were not significantly different from that seen in control livers (Supplemental Fig. S2, left panel). Total ACF protein levels in the Db/Db mouse liver also were not significantly different from that quantified in control animals (Supplemental Fig. S2, right panel). The data suggested that leptin signaling in Db/Db and lean control mouse liver maintained comparable levels of ACF mRNA and protein expression. Taken together, these findings supported a novel role for leptin in regulating hepatic ACF abundance but that insulin resistance did not make a significant contribution to the regulation of ACF abundance.
3.2 ACF trafficking defect in Ob/Ob mouse liver
Given the large increase in ACF expression in the liver of Ob/Ob mice together with their insulin resistance, it was unclear whether these animals retained the capacity for insulin-dependent regulation of ACF nuclear retention. Prior to liver harvest and subcellular fractionation, animals were fasted and received either control saline or 1.5 IU insulin/kg body weight by intraperitoneal injection to promote ACF’s nuclear retention. Consistent with previous published findings in rats [25], fasting of control mice resulted in a marked increase in the proportion of total cellular ACF in the cytoplasm and insulin treatment produced a 4-fold increase in nuclear ACF abundance within 4 hours following injection (Fig. 3A and relevant quantification in Supplemental Table S1). The calculated proportion of ACF that traffics in response to insulin represents only a small fraction of the total cellular ACF. Nonetheless, these data demonstrated that the previously characterized insulin-dependent nuclear retention of ACF in rats [17; 25] was conserved in mice.
Figure. 3.

Ob/Ob and Db/Db mouse hepatic ACF does not accumulate in the nucleus following insulin administration. Ob/Ob (A) and Db/Db (B) mice were fasted for 12 hours and then injected intraperitoneally with 1.5 IU insulin or saline 4 hours prior to harvesting liver for extract preparation. Subcellular fractionation was performed to generate nuclear and cytoplasmic extracts of which 40 and 120 μg (respectively) were resolved by SDS-PAGE and transferred to nitrocellulose. Western blots were probed with ACF peptide-specific antibody and for β-actin and histone H1 as fraction-specific loading controls for the cytoplasm and nucleus (respectively). The nuclear to cytoplasmic ratio (N:C) of ACF signal was calculated as described in Materials and Methods and Supplemental Table 1. The fold change in N:C ratio was calculated (as the relative N:C ratio insulin-treated mice divided by the relative N:C ratio measured in saline-treated mice). The calculations are the average of two trials.
In contrast to the lean control mice, Ob/Ob hepatic ACF was not responsive to fasting. ACF failed to redistribute to the cytoplasm (Fig. 3A). Moreover, insulin administration did not promote nuclear accumulation of ACF in Ob/Ob mouse liver as evidenced by the calculated ‘no difference’ (1-fold) in the relative nuclear to cytoplasmic ratio of ACF in insulin-treated versus saline-treated animals (Fig. 3A). Similar analyses were conducted using Db/Db animals to evaluate whether these animals also lacked an insulin response in ACF trafficking in the absence of the ACF over expression observed in Ob/Ob mice. Insulin injection did not stimulate nuclear retention of ACF in Db/Db mice (Fig. 3B) suggesting that the lack of a robust response was a general characteristic of the obesity-induced insulin-resistance state and was not simply due to ACF over expression characteristic of Ob/Ob mice.
3.3 ApoB mRNA Editing is resistant to insulin stimulation in Ob/Ob liver
Our laboratory [17; 25] and others [15; 47] have shown that apoB mRNA editing is stimulated by insulin. Given that Ob/Ob mice are hyperinsulinemic [48] we had anticipated a greater amount of nuclear retention of hepatic ACF. Because this was not observed, we questioned whether editing activity itself had become insulin-resistant. The proportion of total hepatic apoB mRNA edited in fed Ob/Ob and Db/Db mice was quantified by the poisoned primer extension assay (Fig. 4A). Ob/Ob and Db/Db liver editing was 67% +/− 3% (n=3) and 56% +/− 5% (n=3) respectively and therefore lower than that quantified in fed lean control animals 73% +/− 1%. These findings suggested that apoB mRNA editing was not responsive to the elevated insulin levels that were present in fed obese mice.
Figure. 4.
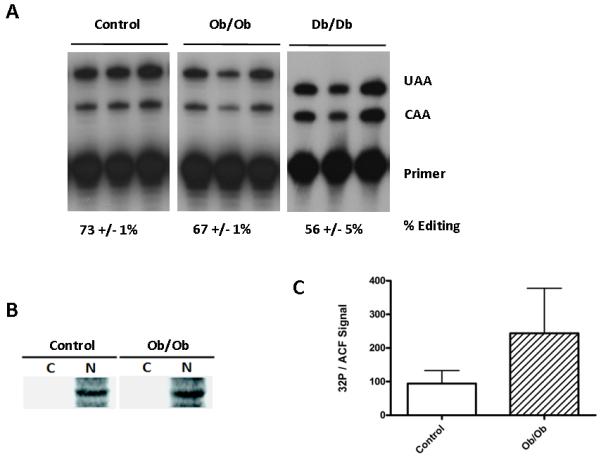
ACF phosphorylation and apoB editing are uncoupled in Ob/Ob mice. A. ApoB mRNA was amplified from 2 μg of total RNA and apoB mRNA editing efficiency was evaluated by poisoned primer extension as previously described [50]. Shown is the editing activity of in the liver of three different animals. The percent editing activity is the average of the editing activity determined in each animal and was calculated as a quotient of the signal from the primer extension product of edited apoB mRNA divided by the signals from primer extension products from the edited and unedited mRNAs multiplied by 100. B. Fed mice were injected with 32Pi buffered orthophosphoric acid 4 hours prior to liver harvest and subcellular fractionation. Cytoplasmic and nuclear extracts were immunoprecipitated with ACF C-terminal specific polyclonal antibody, resolved by SDS PAGE, transferred to PVDF membrane and autoradiographed. C. The specific activity of ACF was calculated as the PhosphorImager quantified 32P signal divided by ACF western signal. The calculated specific activity and error are the average of a determination performed on an extract from each of three animals.
We have previously shown that elevated insulin levels correlated with an enhanced abundance of phosphoACF in the nucleus of hepatocytes [27]. To evaluate whether Ob/Ob hepatocytes retained the ability to phosphorylated ACF, hepatic extracts were prepared from fed control and Ob/Ob mice that had been injected intraperitoneally with 32Pi. Nuclear and cytoplasmic extracts were immunoprecipitated with the C-terminal ACF polyclonal antibody and the ACF recovered analyzed by SDS PAGE and autoradiographed. ACF was determined to be phosphorylated and phosphoACF was properly compartmentalized to the nucleus in both lean controls and Ob/Ob mouse livers (Fig. 4B). Quantification of the 32P signal recovered with nuclear ACF per μg of protein suggested that Ob/Ob nuclear ACF had twice the specific labeling of ACF compared to lean control ACF (Fig. 4C). Although phosphorylation of ACF is not required for apoB mRNA binding [27; 28], the apparent stimulation of nuclear ACF phosphorylation without a commensurate stimulation in apoB mRNA editing suggested some insulin regulation of protein kinase C activity on ACF remained intact but that editosome assembly and/or activity was insulin-resistant in the Ob/Ob hepatocyte. The data suggest an uncoupling of the previously observed role of ACF phosphorylation in the stimulation of apoB mRNA editing
3.4 Increased cytoplasmic ACF correlates with increased cytoplasmic apoB mRNA and enhanced cytoplasmic ApoB protein
Given the localization of cytoplasmic ACF to the exterior surface of the endoplasmic reticulum (ER), enhanced ACF expression may have facilitated apoB mRNA translation. To evaluate whether the enhanced cytoplasmic levels of ACF had an impact on apoB mRNA, poly A+ mRNA was isolated from nuclear and cytoplasmic extracts of Ob/Ob and C57Bl/J6 mice and apoB and β-actin sequences amplified by real time PCR amplification. Despite the finding that total hepatic cellular apoB mRNA was not significantly increased in Ob/Ob mice relative to lean control mice (Fig. 1), a 2.5-fold increase (p≤0.01) in cytoplasmic apoB mRNA abundance was observed in Ob/Ob mice relative to that quantified in the cytoplasm of lean controls (Fig. 5A). The result was commensurate with a reduction in the relative abundance of nuclear apoB mRNA (p≤0.01) in Ob/Ob animals (Fig. 5A). Interestingly, within hepatic cytoplasmic extracts of both mice and rats, ACF was localized to the low density microsomal fraction (LDM) (Supplementary Fig. S3). This cytoplasmic fraction in rat hepatocytes has been described to contain approximately 75% of cytoplasmic apoB message and apoB translation complexes [31]. Cytoplasmic ACF in the Ob/Ob mice was recovered in both LDM and high density microsomal (HDM) fractions suggesting that a preponderance of ACF that is over expressed in Ob/Ob mice may retain its association with apoB mRNA in the cytoplasm.
Figure. 5.
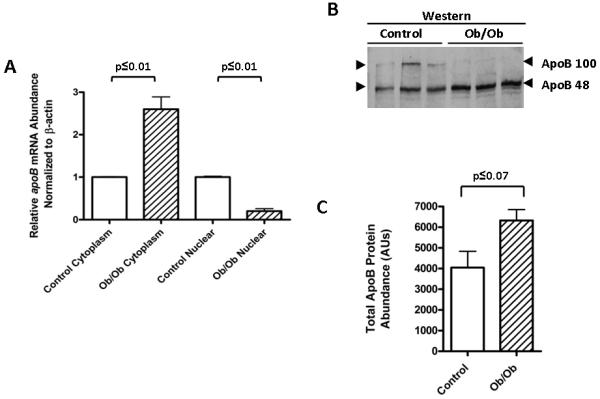
Enhanced ACF abundance supports increases in intracellular ApoB protein. A. Real time PCR was performed on poly A+ purified RNA from cytoplasmic and nuclear subcellular extracts of Ob/Ob and cogenic control animal liver. β-actin mRNA was quantified as a normalization control for RNA input. The relative nuclear and cytoplasmic abundance of apoB mRNA in control mice was set to one. The statistical significance was determined to be p≤0.01 for both cytoplasmic and nuclear differences using a student’s t-test with n=3. B. Hepatic cytoplasmic extracts from 3 different lean and Ob/Ob mice were resolved by SDS-PAGE on 5% gels and probed by western blotting using an anti-ApoB antibody reactive with the N-terminus. Arrowheads denote the position of both ApoB100 and ApoB48. C. Total protein abundance from ‘B’ represented graphically. Statistical significance was calculated using a student t-test (n=3).
From these data we predicted that elevated ACF in the LDM fraction in Ob/Ob livers may stabilize apoB mRNA translation complexes associated with the ER. Moreover, the expression of ApoB48 from edited apoB mRNA might be enhanced due to the ability of the additional ACF to more effectively protect edited mRNA from NMD [29]. Analysis of the western blots of cytoplasmic fractions revealed a 60% increase in total ApoB (p≤0.07) (Fig. 5D) with the majority of the increase in the abundance of ApoB48 (increased 2-fold in Ob/Ob animal p≤0.01) (Fig. 5C). These findings are strong support for our hypothesis that alterations in the regulation of ACF can affect downstream pathways involved in ApoB protein expression.
3.5 ACF Localization Affects ApoB Secretion
Our hypothesis predicts that nuclear export of ACF could affect apoB mRNA nuclear export to the cytoplasm and its translation. To further assess this prediction we returned to our previously characterized nuclear retention of ACF in rat primary hepatocytes treated with cantharidin to inhibit PP1 [27]. Cantharidin treatments of primary rat hepatocytes at 5 μM, 10X the IC50 of PP1, resulted in a marked increase in nuclear ACF, coordinate with a reduction of cytoplasmic ACF (Fig. 6A). In parallel, intracellular and secreted ApoB protein were assessed by radioimmunoassay [32] and normalized for protein input. A reduction in both intracellular (p≤0.02) and secreted (p≤0.001) ApoB was observed relative to DMSO control treatment (Fig. 6B). While the most robust inhibition was observed in ApoB secretion, the reduction of intracellular ApoB was also significant. The data suggested a requirement for PP1 activity for proper packaging and secretion of nascent ApoB VLDL as well as possibility that PP1 activity may have been requirement for the translation or stability of ApoB itself. Real time PCR demonstrated that while apoB mRNA was slightly elevated in the cytoplasmic fraction of cantharidin treated hepatocytes the result was not statistically significant (p≤0.32) (Fig. 6C). However, the ~2-fold increase in the nuclear abundance of apoB mRNA was statistically significant (p≤0.05). These findings supported our hypothesis that ACF nuclear export was consequential for the amount of ApoB protein that was expressed and secreted however we cannot discount from this experiment that cantharidin treatment may have affected other processes in hepatocytes that were also responsible for the reduction in ApoB secretion.
Fig. 6.
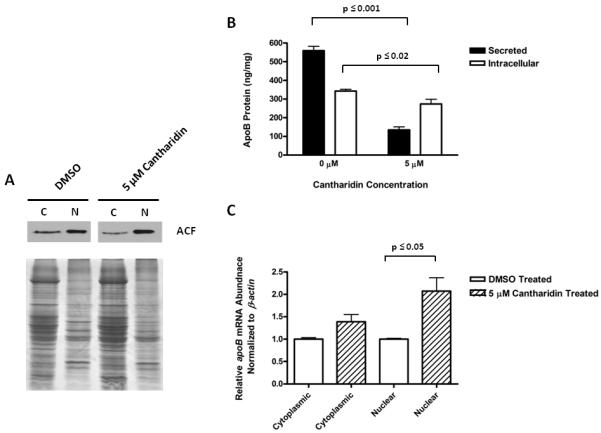
Nuclear Retention of ACF Results in Nuclear retention of apoB mRNA and a Reduction of Intracellular and Secreted ApoB Protein. A. Western blot analysis of ACF distribution between the cytoplasm and nucleus following treatment with 5.0 μM cantharidin (10X the IC50 of PP1) or the solvent, DMSO, used for dissolve the compound. B. The amount of intracellular ApoB protein (ng) per mg of total intracellular protein (open bars) and secreted ApoB protein per mg total protein in the media (closed bars), are shown following treatment of primary rat hepatocytes with cantharidin as in A. ApoB protein was assessed by RIA as previously described. Results and standard deviations are reported where n= 5 (for intracellular) or 8 (for secreted). C. Real time PCR was used to assess the relative abundance of apoB mRNA in the cytoplasm and nucleus, normalized to β-actin, with cantharidin treatment as in parts A & B. The results are representative of 3 trials and statistical significance was calculated using a student t-test.
3.6 ACF Abundance Affects ApoB Secretion
To demonstrate that ApoB expression was indeed modulated by ACF availability and abundance, an RNAi knockdown analysis was conducted in rat primary hepatocytes. Constructs were developed in the pAdEasy system to target rat acf mRNA. Each of three adenoviruses targeting one of 3 unique sites within acf mRNA were assessed in McArdle cells, a rat hepatic cell line, for their efficacy along with virus encoding a nonspecific and random sequence as a control ‘NS’ (Fig. 7A). The target site designated ‘H2’ resulted in the most efficient reduction of ACF protein and was subsequently used in treatment of primary rat hepatocytes where it demonstrated an MOI-dependent reduction of ACF protein (Fig. 7B). NS was without effect on ACF abundance. Infectivity was nearly 100% at the maximal MOI as assessed by fluorescence microscopy visualizing GFP expression expressed off a CMV promoter 3′ to the shRNAi (data not shown). The abundance of secreted ApoB protein was evaluated by RIA in media harvested 48 hours following infection of primary hepatocyte cultures. H2 expression induced approximately 40% reduction of secreted ApoB protein compared to cells expressing NS (Fig. 7C). These findings are significant in light of the foregoing experiments as they underscore the possibility that hepatic ACF participates at an early stage in the regulation of apoB mRNA availability for translation in the cytoplasm.
Fig. 7.
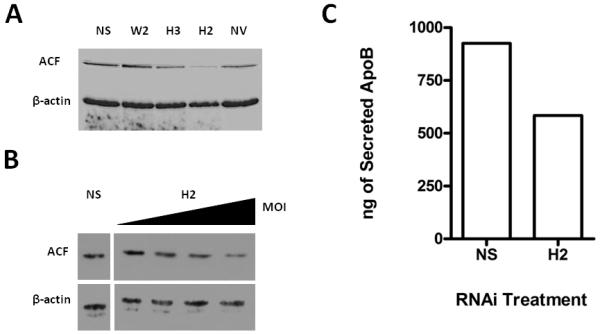
Reduction of ACF Abundance Results in Reduced ApoB Protein Secretion. A. Western blot analysis of McArdle whole cell lysates resolved by SDS-PAGE 48 hours after infection with virally expressed shRNAi against acf (W2, H2, H3) or scrambled sequence (NS). NV represents uninfected cells. Blots were probed sequentially for ACF, using antibody recognizing the C-terminus of ACF and β-actin, using monoclonal antibody (Sigma-Aldrich). Results are representative of duplicate experiments. B. Western blot analysis of whole cell protein from primary rat hepatocytes with increasing MOI (Multiplicity of Infection) of adenovirus shRNAi targeting acf, denoted H2. Blots were probed as described in A. The abundance of ACF with NS treatment is shown at the highest MOI. Results are representative of duplicate experiments. C. ApoB protein secreted from primary rat hepatocytes was assessed by radioimmunoassay as previously described [32]. Media samples were taken 48 hours following infection at the maximal MOI in panel B. Results are depicted graphically as the average nanograms of ApoB secreted for two trials.
4. Discussion
In this report we describe a novel role for ACF in the regulation of apoB mRNA export from the nucleus and the importance of this process in animal models for modulating the expression of ApoB protein. The function of ACF to date has been limited to its role as an auxiliary factor required for apoB mRNA editing in mammalian small intestine and in the liver of many species [49]. Our new findings provide an intriguing potential hypothesis for why ACF expression has been retained in human and non-human primate livers despite the loss of APOBEC-1 expression and lack of apoB mRNA editing activity [49].
An important implication of the findings from this study was that ACF may function early in the regulatory pathways that control lipoprotein synthesis. Prior to our study, research on ACF focused on its role in apoB mRNA editing [50] and studies of ApoB focused on lipoprotein assembly and secretion [51]. Early work from this lab showed that ACF trafficking was regulated by insulin-dependent serine phosphorylation [27; 28]. We reasoned that Ob/Ob mice might enable an evaluation of ACF trafficking and ApoB expression in the context of an insulin-resistant disease model. We observed that the levels of hepatic acf message and protein were markedly increased. This change stood out for the magnitude of change in acf mRNA levels relative to that of related RNA binding proteins.
The progression to obesity in Ob/Ob mice begins immediately after birth due to hyperphagia. This manifests in an increased abundance of adipose tissue, altered response to insulin stimulation and exposure of the liver to increased levels of serum insulin, adipokines (leptin [42] and resistin [43]), pro-inflammatory cytokines (Il-6 and TNFα [41]), and free fatty acid flux [44]. In testing several of these factors individually, under the defined conditions of primary hepatocyte culture, we found that only leptin had the ability to alter acf mRNA abundance, causing its suppression in a dose-dependent manner. Supporting a role for leptin in ACF expression was the finding that insulin resistance and obesity were not sufficient to change ACF expression in the Db/Db mice that retained leptin signaling in their livers.
The regulation of ACF abundance by leptin is only the second example of hormonal regulation of acf message. Previously, thyroid hormone was shown to increase the abundance of ACF in neonatal Pax8−/− mice [12]. Treatment with thyroid hormone resulted in a mobilization of hepatic triglycerides, which the authors hypothesized was due to increases in the proportion of ApoB48 availability [12]. It is of interest that the PPARα agonist, ciprofibrate, also reduced ACF abundance in Low Density Lipoprotein Receptor (LDLR) knockout animals [52]. In these studies, the amount of edited apoB mRNA varied in direct proportion with ACF abundance. In contrast, we observed no significant changes in the basal level of apoB mRNA editing in Ob/Ob mice relative to that of the lean control. These findings together with the data showing that Ob/Ob hepatocytes failed to up-regulate ACF retention in the nucleus underscore the importance of insulin for controlling the function of ACF in the lean physiological state.
Our studies employed two animal models for obesity. Ob/Ob mice express leptin receptors, but do not express leptin and consequently become hyperphagic and obese. Db/Db mice express leptin but lack the leptin receptor in their CNS and become hyperphagic and obese. Obesity led to insulin resistance in both animal models. We found that hepatic ACF nuclear retention and apoB mRNA editing activity were insulin-resistant in both animal models. Db/Db hepatic acf mRNA and ACF protein abundances were not elevated above control whereas Ob/Ob acf mRNA and ACF protein levels were markedly elevated. We attribute this difference to the expression of leptin and leptin signal in Db/Db liver but not in the Ob/Ob liver [45]. There are several splice variants of the leptin receptor that differ in the length of their cytoplasmic domain required for docking of Janus kinase and STAT proteins, through which leptin signals [53; 54]. Signaling through the long form of leptin receptor (Ob-Rb) is unlikely to have contributed to acf mRNA suppression as only the truncated leptin receptor (Ob-Ra) is expressed [46]. The Ob-Ra receptor lacks the consensus box 1 and 2 motifs utilized for docking of Janus kinase and therefore its signaling does not go through a STAT3 dependent pathway [55]. Signaling through the Ob-Ra receptor has been described through MAPK activation [55]. Functional Ob-Ra receptor signaling has been shown through induced expression of c-fos, c-jun, and jun-B [56]. Leptin signaling in the Db/Db liver may have remained responsive and therefore continued to the suppression of acf mRNA abundance. The regulatory factors that normally modulate ACF expression will be an important area for future research.
We can not rule out that over expression of ACF may have compounded the defect in regulating ACF and apoB mRNA nuclear export and ApoB translation in the Ob/Ob mouse but our data suggested that insulin resistance was the primary reason for why ACF nuclear retention and apoB mRNA editing activity were not elevated above the lean control values as would have been anticipated given the hyperinsulinemic condition in these animals. Considering that ACF was hyper-phosphorylated on serine residues in Ob/Ob liver and phosphoACF was restricted in the nucleus, PKC activity on nuclear ACF and factors determining nuclear retention must have retained a basal level of function. However, the possibility remains that insulin resistance involved reduced activation of PKC and/or heightened protein phosphatase I activity. This might explain why the radioactivity per μg of ACF protein in Ob/Ob nuclei was only 2-fold higher than that observed in the lean control when an earlier study showed that nuclear ACF specific radioactivity increased approximately 5-fold in lean mice following a 10 nm insulin injection [27].
An equally plausible explanation for our findings is that nuclear protein kinase C activity may have become insufficient for the level of ACF phosphorylate necessary to affect nuclear restriction because this pathway was less responsive to insulin. Given that apoB mRNA editing efficiency was suppressed in Ob/Ob and Db/Db mouse liver compared to lean control liver, our study suggested that the insulin-dependent processes that otherwise regulate ACF assembly into editosomes may have been compromised by the inability to restrict adequate amounts of ACF to the nucleus.
In published studies and in this study, the bulk of total cellular ACF was recovered with cytoplasmic extracts from whole liver or hepatocytes [17; 25]. Immuno electron microscopy showed that cytoplasmic ACF was predominantly localized along the exterior of the endoplasmic reticulum [25]. This is also the intracellular site of apoB mRNA translation where microsomal triglyceride transfer protein facilitates co-translational lipid loading onto nascent ApoB [57; 58]. Differential centrifugation of liver cytoplasmic extracts yielded microsomal fractions in which the rough endoplasmic reticulum (high density microsomal fraction, HDM), smooth endoplasmic reticulum and Golgi stacks (low density microsomal fraction, LDM) and residual cytoplasm were separated [59]. The bulk of nascent ApoB protein co-fractionated with the LDM fraction [31]. Using this protocol, we found that most of the cytoplasmic ACF was not recovered as a soluble protein with the residual cytoplasm fraction but rather, was enriched in the LDM and HDM factions. This characteristic of ACF was conserved in mice and rats. In fact, despite the high level of ACF expression in Ob/Ob mouse liver, the association of ACF with microsomes was maintained. The relative recovery of ACF with the Ob/Ob HDM fraction compared with the LDM fraction, suggested that there was more ACF in the HDM fraction of obese mice compared with HDM of lean control mice. This association is consistent with the hypothesis that ACF maintained it association with apoB mRNA following nuclear export to the sites of ApoB protein synthesis in the cytoplasm. The importance of ACF abundance and trafficking for ApoB production was underscored by the finding that ApoB secretion was inhibited through RNAi knockdown of ACF. An additional correlation between ACF trafficking and ApoB synthesis and secretion was that nuclear retention of ACF through PP1 inhibition was associated with reduced cytoplasmic apoB mRNA and increased nuclear apoB mRNA in primary hepatocytes. Consistent with our hypothesis, intracellular and secreted ApoB protein was reduced in primary hepatocytes treated with Cantharidin. We propose therefore that the inability of the liver in obese animals to appropriately regulate nuclear retention of ACF is what led to dysregulation of ApoB.
More detailed analysis will be necessary to de-convolute the regulation of ACF trafficking in the insulin-resistant state. Future studies will focus on developing genetic models to determine the role of ACF nuclear and cytoplasmic trafficking in mRNA export from the nucleus and identify the signaling pathways that regulate ACF expression and post-translational modification. Specifically, this study has demonstrated using animal models a role for leptin signaling in the regulation of ACF expression and shown that deficiencies in nuclear retention of ACF was part of the obesity-induced, insulin-resistant phenotype. Importantly our studies suggested that independent of the function ACF has in apoB mRNA editing activity (resulting from its interaction with APOBEC-1), ACF may have a primary function as an apoB mRNA recognition factor and chaperone. This function suggested a new hypothesis that may explain why ACF continues to be expressed in nonhuman primate and human livers even though apoB mRNA editing activity is no longer possible. It is also possible that ACF serves to chaperone other mRNAs that are important for development and it is the loss of this function that accounts for why ACF knockouts were not viable. It will be important to develop a conditional ACF knockout animal model to address this possibility.
Supplementary Material
{kind=link}
Acknowledgements
The authors are grateful to Dr. Alicia Clementi and Allison Gaudy for assistance in the preparation of mouse primary hepatocytes. This work was supported in part by Public Health Services Grants [DK43739, DK78131 and DK38138 awarded to HCS, JDS and RAM].
Abbreviations
- ACF
APOBEC-1 Complementation Factor
- ApoB
Apolipoprotein B
- APOBEC-1
Apolipoprotein editing enzyme catalytic subunit 1
- ARE
AU rich element
- BW
body weight
- CNS
Central Nervous System
- ELAV
embryonic lethal abnormal vision
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- HDM
high density microsome
- Il-6
interleukin 6
- LDLR
low density lipoprotein receptor
- LDM
low density microsome
- NLS
nuclear localization sequence
- NMD
nonsense mediated decay
- PKC
protein kinase C
- PP1
protein phosphatase 1
- PPAR
peroxisome proliferation-activated receptor
- RIA
radioimmunoassay
- RNAi
RNA interference
- RRM
RNA recognition motif
- STAT
signal transducer and activators of transcription
- TNFα
tumor necrosis factor α
- VLDL
very low density lipoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- [2].Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58:266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mehta A, Driscoll DM. Identification of Domains in APOBEC-1 Complementation Factor Required for RNA Binding and Apolipoprotein B mRNA editing. RNA. 2002;8:69–82. doi: 10.1017/s1355838202015649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Backus JW, Smith HC. Three distinct RNA sequence elements are required for efficient apolipoprotein B (apoB) RNA editing in vitro. Nucleic Acids Res. 1992;20:6007–6014. doi: 10.1093/nar/20.22.6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Backus JW, Smith HC. Specific 3′ sequences flanking a minimal apolipoprotein B (apoB) mRNA editing ‘cassette’ are critical for efficient editing in vitro. Biochim Biophys Acta. 1994;1217:65–73. [PubMed] [Google Scholar]
- [6].Mehta A, Banerjee S, Driscoll DM. Apobec-1 interacts with a 65-kDa complementing protein to edit apolipoprotein-B mRNA in vitro. J Biol Chem. 1996;271:28294–28299. doi: 10.1074/jbc.271.45.28294. [DOI] [PubMed] [Google Scholar]
- [7].Lellek H, Kirsten R, Diehl I, Apostel F, Buck F, Greeve J. Purification and molecular cloning of a novel essential component of the apolipoprotein B mRNA editing enzyme-complex. J Biol Chem. 2000;275:19848–19856. doi: 10.1074/jbc.M001786200. [DOI] [PubMed] [Google Scholar]
- [8].Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- [9].Chen SH, Habib G, Yang CY, Gu ZW, Lee BR, Weng SA, Silberman SR, Cai SJ, Deslypere JP, Rosseneu M, et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science. 1987;238:363–366. doi: 10.1126/science.3659919. [DOI] [PubMed] [Google Scholar]
- [10].Powell LM, Wallis SC, Pease RJ, Edwards YH, Knott TJ, Scott J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell. 1987;50:831–840. doi: 10.1016/0092-8674(87)90510-1. [DOI] [PubMed] [Google Scholar]
- [11].Funahashi TF, Giannoni, DePaoli AM, Skarosi SF, Davidson NO. Tissue-specific, developmental and nutritional regulation of the gene encoding the catalytic subunit of the rat apoB mRNA editing enzyme: functional role in the modulation of apoB mRNA editing. J. Lipid Res. 1995;36:414–428. [PubMed] [Google Scholar]
- [12].Mukhopadhyay D, Plateroti M, Anant S, Nassir F, Samarut J, Davidson NO. Thyroid hormone regulates hepatic triglyceride mobilization and apolipoprotein B messenger ribonucleic Acid editing in a murine model of congenital hypothyroidism. Endocrinology. 2003;144:711–719. doi: 10.1210/en.2002-220741. [DOI] [PubMed] [Google Scholar]
- [13].Chen Z, Eggerman TL, Potosky D, Arborati M, Patterson AP. Calcium increases apolipoprotein B mRNA editing. Biochem Biophys Res Commun. 2000;277:221–227. doi: 10.1006/bbrc.2000.3668. [DOI] [PubMed] [Google Scholar]
- [14].Van Mater D, Sowden MP, Cianci J, Sparks JD, Sparks CE, Ballatori N, Smith HC. Ethanol increases apolipoprotein B mRNA editing in rat primary hepatocytes and McArdle cells. Biochem Biophys Res Commun. 1998;252:334–339. doi: 10.1006/bbrc.1998.9647. [DOI] [PubMed] [Google Scholar]
- [15].von Wronski MA, Hirano KI, Cagen LM, Wilcox HG, Raghow R, Thorngate FE, Heimberg M, Davidson NO, Elam MB. Insulin increases expression of apobec-1, the catalytic subunit of the apolipoprotein B mRNA editing complex in rat hepatocytes. Metabolism. 1998;47:869–873. doi: 10.1016/s0026-0495(98)90128-7. [DOI] [PubMed] [Google Scholar]
- [16].Lau PP, Cahill DJ, Zhu HJ, Chan L. Ethanol modulates apolipoprotein B mRNA editing in the rat. J Lipid Res. 1995;36:2069–2078. [PubMed] [Google Scholar]
- [17].Sowden MP, Lehmann DM, Lin X, Smith CO, Smith HC. Identification of Novel Alternative Splice Variants of APOBEC-1 Complementation Factor with Different Capacities to Support ApoB mRNA Editing. J. Biol. Chem. 2004;278:197–206. doi: 10.1074/jbc.M307920200. [DOI] [PubMed] [Google Scholar]
- [18].Baum CL, Teng BB, Davidson NO. Apolipoprotein B messenger RNA editing in the rat liver. Modulation by fasting and refeeding a high carbohydrate diet. J Biol Chem. 1990;265:19263–19270. [PubMed] [Google Scholar]
- [19].Nakamuta M, Chang BHJ, Zsigmond E, Kobayashi K, Lei H, Ishida BY, Oka K, Li E, Chan L. Complete phenotypic characterization of the apobec-1 knockout mice with a wild-type genetic background and a human apolipoprotein B transgenic background, and restoration of apolipoprotein B mRNA editing by somatic gene transfer of Apobec-1. J. Biol. Chem. 1996;271:25981–25988. doi: 10.1074/jbc.271.42.25981. [DOI] [PubMed] [Google Scholar]
- [20].Blanc V, Henderson JO, Newberry EP, Kennedy S, Luo J, Davidson NO. Targeted deletion of the murine apobec-1 complementation factor (acf) gene results in embryonic lethality. Mol Cell Biol. 2005;25:7260–7269. doi: 10.1128/MCB.25.16.7260-7269.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Farese RV, Jr., Ruland SL, Flynn LM, Stokowski RP, Young SG. Knockout of the mouse apolipoprotein B gene results in embryonic lethality in homozygotes and protection against diet-induced hypercholesterolemia in heterozygotes. Proc Natl Acad Sci U S A. 1995;92:1774–1778. doi: 10.1073/pnas.92.5.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Grosjean H, editor. Fine-Tuning of RNA Functions by Modification and Editing. Springer-Verlag; Heidelberg: 2005. [Google Scholar]
- [23].Lau PP, Xiong W, Zhu H-J, Chen S-H, Chan L. Apo B mRNA editing is an intranuclear event that occurs posttranscriptionally coincident with splicing and polyadenylation. J. Biol. Chem. 1991;266:20550–20554. [PubMed] [Google Scholar]
- [24].Sowden M, Hamm JK, Spinelli S, Smith HC. Determinants involved in regulating the proportion of edited apolipoprotein B RNAs. Rna. 1996;2:274–288. [PMC free article] [PubMed] [Google Scholar]
- [25].Sowden MP, Ballatori N, de Mesy Jensen KL, Hamilton Reed L, Smith HC. The editosome for cytidine to uridine mRNA editing has a native complexity of 27S: identification of intracellular domains containing active and inactive editing factors. J. Cell Science. 2002;115:1027–1039. doi: 10.1242/jcs.115.5.1027. [DOI] [PubMed] [Google Scholar]
- [26].Blanc V, Kennedy S, Davidson NO. A novel nuclear localization signal in the auxiliary domain of apobec-1 complementation factor regulates nucleocytoplasmic import and shuttling. J Biol Chem. 2003;278:41198–41204. doi: 10.1074/jbc.M302951200. [DOI] [PubMed] [Google Scholar]
- [27].Lehmann DM, Galloway CA, Sowden MP, Smith HC. Metabolic regulation of apoB mRNA editing is associated with phosphorylation of APOBEC-1 complementation factor. Nucleic Acids Res. 2006;34:3299–3308. doi: 10.1093/nar/gkl417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lehmann DM, Galloway CA, Macelrevey C, Sowden MP, Wedekind JE, Smith HC. Functional characterization of APOBEC-1 complementation factor phosphorylation sites. Biochim Biophys Acta. 2007;1773:408–418. doi: 10.1016/j.bbamcr.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chester A, Somasekaram A, Tzimina M, Jarmuz A, Gisbourne J, O’Keefe R, Scott J, Navaratnam N. The apolipoprotein B mRNA editing complex performs a multifunctional cycle and suppresses nonsense-mediated decay. Embo J. 2003;22:3971–3982. doi: 10.1093/emboj/cdg369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Smith HC. Analysis of protein complexes assembled on apolipoprotein B mRNA for mooring sequence-dependent RNA editing. Methods. 1998;15:27–39. doi: 10.1006/meth.1998.0603. [DOI] [PubMed] [Google Scholar]
- [31].Phung TL, Roncone A, Jensen KL, Sparks CE, Sparks JD. Phosphoinositide 3-kinase activity is necessary for insulin-dependent inhibition of apolipoprotein B secretion by rat hepatocytes and localizes to the endoplasmic reticulum. J Biol Chem. 1997;272:30693–30702. doi: 10.1074/jbc.272.49.30693. [DOI] [PubMed] [Google Scholar]
- [32].Sparks JD, Bolognino M, Trax PA, Sparks CE. The production and utility of monoclonal antibodies to rat apolipoprotein B lipoproteins. Atherosclerosis. 1986;61:205–211. doi: 10.1016/0021-9150(86)90139-5. [DOI] [PubMed] [Google Scholar]
- [33].Backus JW, Eagleton MJ, Harris SG, Sparks CE, Sparks JD, Smith HC. Quantitation of endogenous liver apolipoprotein B mRNA editing. Biochem Biophys Res Commun. 1990;170:513–518. doi: 10.1016/0006-291x(90)92121-f. [DOI] [PubMed] [Google Scholar]
- [34].Gomez-Lechon MJ, Donato MT, Martinez-Romero A, Jimenez N, Castell JV, O’Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact. 2007;165:106–116. doi: 10.1016/j.cbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- [35].Paddison PJ, Caudy AA, Sachidanandam R, Hannon GJ. Short hairpin activated gene silencing in mammalian cells. Methods Mol Biol. 2004;265:85–100. doi: 10.1385/1-59259-775-0:085. [DOI] [PubMed] [Google Scholar]
- [36].Ro S, Hwang SJ, Ordog T, Sanders KM. Adenovirus-based short hairpin RNA vectors containing an EGFP marker and mouse U6, human H1, or human U6 promoter. Biotechniques. 2005;38:625–627. doi: 10.2144/05384RN01. [DOI] [PubMed] [Google Scholar]
- [37].Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- [38].Anant S, Henderson JO, Mukhopadhyay D, Navaratnam N, Kennedy S, Min J, Davidson NO. Novel role for RNA-binding protein CUGBP2 in mammalian RNA editing. J. Biol. Chem. 2001;276:47338–47351. doi: 10.1074/jbc.M104911200. [DOI] [PubMed] [Google Scholar]
- [39].Lau PP, Chang BH, Chan L. Two-hybrid cloning identifies an RNA-binding protein, GRY-RBP, as a component of apobec-1 editosome. Biochem Biophys Res Commun. 2001;282:977–983. doi: 10.1006/bbrc.2001.4679. [DOI] [PubMed] [Google Scholar]
- [40].Blanc V, Navaratnam N, Henderson JO, Anant S, Kennedy S, Jarmuz A, Scott J, Davidson NO. Identification of GRY-RBP as an apolipoprotein B RNA-binding protein that interacts with both apobec-1 and apobec-1 complementation factor to modulate C to U editing. J Biol Chem. 2001;276:10272–10283. doi: 10.1074/jbc.M006435200. [DOI] [PubMed] [Google Scholar]
- [41].Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- [42].Uygun A, Kadayifci A, Yesilova Z, Erdil A, Yaman H, Saka M, Deveci MS, Bagci S, Gulsen M, Karaeren N, Dagalp K. Serum leptin levels in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2000;95:3584–3589. doi: 10.1111/j.1572-0241.2000.03297.x. [DOI] [PubMed] [Google Scholar]
- [43].Yagmur E, Trautwein C, Gressner AM, Tacke F. Resistin serum levels are associated with insulin resistance, disease severity, clinical complications, and prognosis in patients with chronic liver diseases. Am J Gastroenterol. 2006;101:1244–1252. doi: 10.1111/j.1572-0241.2006.00543.x. [DOI] [PubMed] [Google Scholar]
- [44].Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- [45].Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- [46].Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM, Whitington PF. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1035–1043. doi: 10.1152/ajpgi.00199.2004. [DOI] [PubMed] [Google Scholar]
- [47].Thorngate FE, Raghow R, Wilcox HG, Werner CS, Heimberg M, Elam MB. Insulin promotes the biosynthesis and secretion of apolipoprotein B-48 by altering apolipoprotein B mRNA editing. Proc Natl Acad Sci U S A. 1994;91:5392–5396. doi: 10.1073/pnas.91.12.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Baetens D, Stefan Y, Ravazzola M, Malaisse-Lagae F, Coleman DL, Orci L. Alteration of islet cell populations in spontaneously diabetic mice. Diabetes. 1978;27:1–7. doi: 10.2337/diab.27.1.1. [DOI] [PubMed] [Google Scholar]
- [49].Greeve J, Altkemper I, Dieterich JH, Greten H, Windler E. Apolipoprotein B mRNA editing in 12 different mammalian species: hepatic expression is reflected in low concentrations of apoB-containing plasma lipoproteins. J Lipid Res. 1993;34:1367–1383. [PubMed] [Google Scholar]
- [50].Smith HC. Measuring editing activity and identifying cytidine-to-uridine mRNA editing factors in cells and biochemical isolates. Methods Enzymol. 2007;424:389–416. doi: 10.1016/S0076-6879(07)24018-2. [DOI] [PubMed] [Google Scholar]
- [51].Gibbons GF, Wiggins D, Brown AM, Hebbachi AM. Synthesis and function of hepatic very-low-density lipoprotein. Biochem Soc Trans. 2004;32:59–64. doi: 10.1042/bst0320059. [DOI] [PubMed] [Google Scholar]
- [52].Fu T, Mukhopadhyay D, Davidson NO, Borensztajn J. The peroxisome proliferator-activated receptor alpha (PPARalpha) agonist ciprofibrate inhibits apolipoprotein B mRNA editing in low density lipoprotein receptor-deficient mice: effects on plasma lipoproteins and the development of atherosclerotic lesions. J Biol Chem. 2004;279:28662–28669. doi: 10.1074/jbc.M403271200. [DOI] [PubMed] [Google Scholar]
- [53].Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- [54].Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- [55].Yamashita T, Murakami T, Otani S, Kuwajima M, Shima K. Leptin receptor signal transduction: OBRa and OBRb of fa type. Biochem Biophys Res Commun. 1998;246:752–759. doi: 10.1006/bbrc.1998.8689. [DOI] [PubMed] [Google Scholar]
- [56].Murakami T, Yamashita T, Iida M, Kuwajima M, Shima K. A short form of leptin receptor performs signal transduction. Biochem Biophys Res Commun. 1997;231:26–29. doi: 10.1006/bbrc.1996.6030. [DOI] [PubMed] [Google Scholar]
- [57].Allister EM, Borradaile NM, Edwards JY, Huff MW. Inhibition of microsomal triglyceride transfer protein expression and apolipoprotein b100 secretion by the citrus flavonoid naringenin and by insulin involves activation of the mitogen-activated protein kinase pathway in hepatocytes. Diabetes. 2005;54:1676–1683. doi: 10.2337/diabetes.54.6.1676. [DOI] [PubMed] [Google Scholar]
- [58].Liao W, Hui TY, Young SG, Davis RA. Blocking microsomal triglyceride transfer protein interferes with apoB secretion without causing retention or stress in the ER. J Lipid Res. 2003;44:978–985. doi: 10.1194/jlr.M300020-JLR200. [DOI] [PubMed] [Google Scholar]
- [59].Graham JM. Fractionation of a microsomal fraction from rat liver or cultured cells. Methods Mol Biol. 1993;19:51–57. doi: 10.1385/0-89603-236-1:51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.