

Abstract
The forkhead-box protein P3 (Foxp3) is a key transcription factor for the development and suppressive activity of regulatory T cells (Tregs), a T cell subset critically involved in the maintenance of self-tolerance and prevention of over-shooting immune responses. However, the transcriptional regulation of Foxp3 expression remains incompletely understood. We have previously shown that epigenetic modifications in the CpG-rich Treg-specific demethylated region (TSDR) in the Foxp3 locus are associated with stable Foxp3 expression. We now demonstrate that the methylation state of the CpG motifs within the TSDR controls its transcriptional activity rather than a Treg-specific transcription factor network. By systematically mutating every CpG motif within the TSDR, we could identify four CpG motifs, which are critically determining the transcriptional activity of the TSDR and which serve as binding sites for essential transcription factors, such as CREB/ATF and NF-κB, which have previously been shown to bind to this element. The transcription factor Ets-1 was here identified as an additional molecular player that specifically binds to the TSDR in a demethylation-dependent manner in vitro. Disruption of the Ets-1 binding sites within the TSDR drastically reduced its transcriptional enhancer activity. In addition, we found Ets-1 bound to the demethylated TSDR in ex vivo isolated Tregs, but not to the methylated TSDR in conventional CD4+ T cells. We therefore propose that Ets-1 is part of a larger protein complex, which binds to the TSDR only in its demethylated state, thereby restricting stable Foxp3 expression to the Treg lineage.
Electronic supplementary material
The online version of this article (doi:10.1007/s00109-010-0642-1) contains supplementary material, which is available to authorized users.
Keywords: Epigenetic control, DNA methylation, Gene expression, Transcription factors
Introduction
In recent years, the regulatory T cell (Treg) lineage has been recognized as a key player maintaining peripheral self-tolerance and modulating almost any type of immune responses. With this, Tregs are a promising target for therapeutic approaches in multiple clinical situations such as autoimmunity [1], organ transplantation and graft-versus-host disease [2] or cancer [3]. The transcription factor Foxp3 might represent an interesting target for therapeutic intervention as Foxp3 drives the suppressive activity of Tregs [4-7] and fixates their lineage identity [8]. Yet, despite its essential function in Treg physiology the molecular regulation of Foxp3 expression is only incompletely understood.
Common T cell signaling pathways derived from the T cell receptor (TCR), the costimulatory receptor CD28 and the interleukin-2 (IL-2) receptor have been shown to participate in the transcriptional regulation of the Foxp3 gene. Their downstream transcription factors (nuclear factor of activated T cells, activator protein-1, and signal transducer and activator of transcription-5, respectively) bind to the Foxp3 promoter upon activation and facilitate Foxp3 expression [9-12]. Additionally, other common transcription factors, such as the nuclear factor κB (NF-κB), the cAMP response element binding protein/activating transcription factor (CREB/ATF), and the runt-related transcription factor-1, have been described to be involved in Foxp3 regulation by binding to the Foxp3 locus [13-20]. Furthermore, transcription factors of the transforming growth factor-β (TGF-β)-signaling cascade (Sma/mothers against decapentaplegic (SMAD)-2/3 and TGF-β-inducible early gene-1) bind to a transcriptional enhancer element in the first intron of the Foxp3 gene or to the promoter, respectively, and facilitate TGF-β-mediated Foxp3 induction [21, 22].
We have recently demonstrated that Foxp3 expression is under epigenetic control. We could identify a highly conserved CpG-rich element in the Foxp3 gene, which was selectively demethylated in murine as well as human Tregs—the Treg-specific demethylated region (TSDR) [23-26]. Interestingly, only naturally occurring but not in vitro TGF-β-induced Foxp3+ Tregs displayed a demethylated TSDR, which correlated with stable Foxp3 expression. Further molecular characterization of the TSDR revealed that this element possesses transcriptional enhancer activity [23] and indeed determines the stability of Foxp3 expression [27]. Our finding that stable Foxp3 expression is under epigenetic control was supported by studies using histone deacetylase-inhibitors, which led to the induction of Foxp3 expression in vitro or to the expansion of the Treg population in vivo [28, 29]. Similar observations were made using the hypomethylating drug azacytidine [10, 13, 27, 30-32]. In mice harboring a T cell-restricted DNA methyltransferase-1 (DNMT-1) deficiency, Foxp3 expression could be rapidly induced in peripheral T cells by TCR-ligation in vitro even in the absence of TGF-β, a treatment, which does not lead to Foxp3 induction in murine wild-type T cells [33].
Taken together, these data strongly suggest that the epigenetic status of the Foxp3 locus is a critical determinant for the regulation of Foxp3 expression. The TSDR might serve as a molecular gatekeeper, which, by its methylation status, allows or prevents binding of widely expressed methylation-sensitive transcription factors, thereby restricting stable Foxp3 expression to a defined subset of cells. We here provide further molecular data to underpin this hypothesis. We found the TSDR enhancer activity to be strictly dependent on its demethylated status; in this state, transcriptional activity was even observed in Foxp3- conventional T cells. These results indicate that TSDR accessibility rather than a specific transcription factor repertoire mediates stable Foxp3 expression in Tregs. Furthermore, we show that the transcription factor Ets-1 binds to the demethylated TSDR in vitro as well as in vivo and might therefore participate in the transcriptional regulation of Foxp3 expression, probably as part of a larger protein complex also containing the transcription factors CREB/ATF and NF-κB.
Material and methods
Mice
Foxp3gfp-reporter mice [34], kindly provided by Alexander Rudensky (New York, USA), were back-crossed to the BALB/c background and bred at the Helmholtz Centre for Infection Research (Braunschweig, Germany). BALB/c wild-type mice were purchased from the Bundesinstitut fuer Risikobewertung (BfR) in Berlin (Germany). All animals were kept under specific pathogen-free conditions. Animal care and all procedures were performed in accordance with institutional, state and federal guidelines.
T cell isolation, culture, and flow cytometry
CD25+ and CD25-CD4+ T cell subsets were isolated from pooled spleen and lymph node cells of wild-type BALB/c mice using the MACS®-technology (Miltenyi Biotec) in a multi-step selection procedure. Firstly, CD4+ T cells were positively selected using anti-CD4-fluorescein isothiocyanate (FITC) antibody (clone RM4-5, eBioscience) and anti-FITC-multisort microbeads, followed by release of the beads using the Multisort release reagent (Miltenyi Biotec). Secondly, CD25− (=conventional T cells) and CD25+CD4+ T cells (natural Tregs) were separated by another positive selection using anti-CD25-APC antibody (clone PC61.5, eBioscience) and anti-APC-microbeads. CD4+ T cells from pooled spleen and lymph node cells of Foxp3gfp-reporter mice were pre-enriched using anti-CD4-microbeads and the MACS®-technology. The enriched population was then stained for CD25 and CD8 (clone 53–6.7, eBioscience) and sorted on a FACSAria instrument (BD Biosciences) according to their phenotype (conventional CD4+ T cells = Foxp3−CD25−CD4+CD8−; natural Tregs = Foxp3+CD25+CD4+CD8−). All isolated cell populations were >95% pure if not indicated otherwise. Cell culture was performed in RPMI 1640 (Gibco) supplemented with 10% fetal calf serum (Sigma), 25 mM Hepes, 1 mM sodium pyruvat, 50 μM β-mercaptoethanol, 100 U penicillin/streptomycin (all Gibco). For activation, cells were placed in cell culture dishes which had been coated with 1 μg/ml anti-CD3 (clone 145-2C11) and 1 μg/ml anti-CD28 (clone 37.51) antibodies (eBioscience). Fifty ng/ml (for natural Tregs) or 10 ng/ml recombinant mouse IL-2 (R&D Systems) was added to all cultures. For induction of Foxp3, 5 ng/ml recombinant porcine TGF-β (R&D Systems) or 1 μM 5-Aza-2′-deoxycytidine (Sigma-Aldrich) was added. On day 2 or 3 of culture, cells were removed from the stimulus and, if required, split at a ratio of 1:2 for further expansion or used directly for luciferase assays. The phenotype of the expanded cells was analyzed by flow cytometry using a FACS Calibur instrument (Becton Dickinson) and the FlowJo software (Tree Star). If indicated, cultured cells were FACS-sorted on day 5 for GFP+ cells to high purity (>95%).
Cloning of luciferase reporter plasmids
The murine Foxp3 promoter (FoxPro), the full-length TSDR as well as fragments or truncated versions thereof were amplified by PCR from the bacterial artificial chromosome RPCIB731D08143Q2 (German Science Centre for Genome Research), which contains the murine Foxp3 locus, and cloned into the pGL3-basic luciferase reporter vector (Promega). Point mutations for the disruption of CpG motifs or transcription factor binding sites were inserted by site-directed mutagenesis. For the generation of reporter vectors in a CpG-free background, the TSDR was inserted into the pCpGL-CMV/EF1 vector [35] in a way to replace the CMV-enhancer by the TSDR. Integrity of all cloned sequences was confirmed by sequencing. A complete listing of all primer sequences used in this study can be found in the Electronic supplementary material.
In vitro DNA methylation
Luciferase reporter vectors were methylated in vitro using the SssI methylase (New England Biolabs) according to the manufacturer's recommendations, followed by purification using the QIAquick PCR clean-up kit (Qiagen). In control samples, the methyl-group donor S-adenosylmethionine was omitted. Successful methylation was verified by excision of the TSDR and digestion with the methylation-sensitive and methylation-insensitive restriction enzymes HpaII and MspI (New England Biolabs), respectively.
Dual luciferase reporter assays
Luciferase assays in RLM-11-1 cells were performed as previously described [27] using 2 μg of the indicated firefly reporter vectors. For transfection of cultured primary T cells, the Amaxa® Mouse T cell Nucleofector Kit (Lonza) was used according to the manufacturer's recommendations using 3 μg of the firefly reporter vector and 0.5 μg of the Renilla control vector. Following transfection and a resting period of 3 h, cells were cultured for 12–20 h, with addition of the same cytokine cocktail, which was used during the culture before the transfection. If indicated, stimulation was carried out by addition of phorbol 12-myristate 13-acetate (PMA; 10 ng/ml, Sigma-Aldrich) and Ionomycin (0.5 μg/ml, Sigma-Aldrich) for additional 9 h. Luciferase activities were measured using the dual luciferase assay system on a GloMax Instrument (Promega). Mean values and standard deviations from the performed triplicates were calculated and expressed in relation to the indicated reference sample, which was set to 1.
Electromobility shift assays
Nuclear protein extracts of in vitro polarized Th1 cells [36] were generated using the NE-PER® nuclear and cytoplasmic extraction reagents (Pierce Biotechnology) according to the manufacturer's instructions and subsequently dialyzed against a protein storage buffer (20 mM Hepes pH 8.0; 20 mM KCl; 1 mM MgCl2; 0.5 mM DTT; 0.5 mM PMSF; 0.1 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0; 10% glycerol). Single-stranded DNA-oligonucleotides (sequences are provided in the Electronic supplementary material), which were labeled with the infrared dye IRD700 on the 5′ end, were purchased from Metabion and subsequently annealed to its reverse complementary sequence to generate double-strands. The electromobility shift assays (EMSA) binding reaction was performed in the EMSA reaction buffer (250 mM Tris-HCl pH 8.0; 62.5 mM MgCl2; 5 mM EDTA; 5 mM DTT) containing 65 μg/ml salmon sperm DNA and 5% glycerol for 30 min on ice. Optimal concentrations of DNA and protein were determined empirically. A 30-fold excess of unlabeled DNA-oligonucleotides was added for competition purposes. The binding reactions were separated by native gel-electrophoresis through a 5% TBE gel (Biorad) and visualized on the Fluorescent Image Analyzer FLA-9000 (Fuji).
Cell lysis and western blot analysis
Cells of interest were lysed in TPNE buffer (1 × PBS, 300 mM NaCl, 1 mM EDTA, 1% Triton-X100 and Halt Protease Inhibitors (Thermo Scientific)) for 15 min on ice and cell debris were removed by centrifugation. Protein samples were analyzed by western blot analysis using anti-Ets-1 (C-4; Santa Cruz Biotechnology) or anti-β-actin (AC-74; Sigma-Aldrich) as primary antibodies and horseradish peroxidase-conjugated secondary antibodies (Southern Biotech). As substrate, Super Signal West Dura (Thermo Scientific) was applied to the membranes and chemiluminescence was detected by the Fusion-FX7 imaging-system (PeqLab Biotechnology).
Chromatin immunoprecipitation
ChIP assays were performed essentially as described in the protocol of the chromatin immunoprecipitation (ChIP) assay kit by Miltenyi Biotec. Briefly, cells were fixed with 1% formaldehyde and DNA was sheared using the Sonopuls Ultrasonic homogenizer (Bandelin Electronic). Chromatin extracts were incubated with 2 μg anti-Ets1 antibody (C-20; Santa Cruz Biotechnology) or normal rabbit IgG (Cell Signaling) at 4°C overnight. ChIP was performed by incubation with magnetic labeled protein-A beads (Miltenyi Biotec) and subsequent loading onto a μ column (Miltenyi Biotec). Immunoprecipitated chromatin was reverse-crosslinked by incubation with 200 mM NaCl at 65°C for 4 h and DNA was isolated with the DNA extract II kit (Machery and Nagel). The amount of immunoprecipitated DNA was determined by real time PCR with the Mx3000 system (Stratagene) using QuantiTect SYBR Green RT-PCR Kit (Qiagen) and the following primer pair: 5′-CCGGCTACAGGATAGACTAGCC-3′ (forward); 5′-CTGGGCCCTGTTGTCACAAC-3′ (reverse). ChIP PCR products were set in relation to input DNA.
Bioinformatics and statistics
Transcription factor binding sites were identified using the TRANSFAC® database [37] and the search tool MATCH™ [38]. The inter-species conservation plots of the TSDR fragments were generated using the mVISTA-tool comparing the sequences of the murine TSDR fragments to the human Foxp3 locus (http://genome.lbl.gov/vista/index.shtml). For the evaluation of statistical significance, the Wilcoxon signed-rank test was used.
Results
The TSDR acts as a demethylation-dependent transcriptional enhancer in T cells
We have shown previously that the TSDR displays transcriptional enhancer activity in combination with a minimal SV40 promoter [23, 27]. However, it is more physiological to assess its enhancer capacity in conjunction with the promoter of Foxp3 itself. As depicted in Fig. 1, the TSDR drastically enhanced the Foxp3 promoter activity in a classical dual luciferase assay in the murine CD4+ T cell hybridoma RLM-11-1 (>30-fold increase after stimulation with PMA and ionomycin). The TSDR alone did not show any transcriptional activity. Interestingly, also the Foxp3 promoter alone was not sufficient to drive luciferase expression, indicating that the promoter is dependent on an enhancer element to initiate transcription.
Fig. 1.
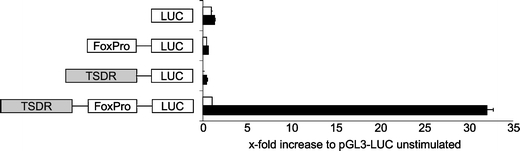
The TSDR acts as a transcriptional enhancer. The TSDR and the Foxp3 promoter (FoxPro) were cloned into the pGL3-luciferase-reporter vector, and their transcriptional activities were assessed in a dual luciferase assay. The indicated constructs were transfected into RLM-11-1 cells and cells rested for 3 h after transfection. The luciferase activity was measured 20 h after stimulation with PMA and ionomycin (black bars). Unstimulated samples are shown in white bars. All values are depicted in relation to the indicated reference sample, which was set to 1. The graph shows mean values of performed triplicates with standard deviation. LUC coding sequence for the firefly luciferase
Next we addressed the question, whether the TSDR displays a cell-type restricted enhancer activity, as would be expected when Foxp3+ Tregs, but not Foxp3− conventional T cells were equipped with a selective transcription factor repertoire addressing the TSDR. Alternatively, the methylation status of the TSDR alone could determine its functionality. In this case, activity of the demethylated, but not the methylated TSDR would be found even in Foxp3− conventional T cells.
To test these hypotheses, we took advantage of a CpG-free luciferase reporter plasmid [35]. It contains a CpG-free human EF1 promoter and allowed us to selectively methylate the inserted CpG-rich TSDR by incubation of the whole reporter plasmid with the DNA methylase SssI. Methylated and unmethylated plasmids were subsequently tested for their enhancer activities in primary murine T cells. For this, we isolated murine Foxp3+CD25+CD4+ natural Tregs and Foxp3-CD25-CD4+ conventional T cells from Foxp3gfp reporter mice and cultured them in the presence of IL-2 and TCR/CD28-stimulation for 5 days. Half of the conventional T cells were cultured in the presence of TGF-β, leading to the upregulation of Foxp3 (Fig. 2a). As shown previously, induced Tregs express Foxp3 only transiently and lose it as soon as TGF-β is withdrawn from the culture medium, a finding correlating with a fully methylated TSDR [23, 27]. The expanded CD4+ T cells (Fig. 2a) were transfected with methylated or unmethylated reporter vectors. Following a resting period, the cells were restimulated with PMA and ionomycin for 9 h and luciferase activity was subsequently measured. As shown in Fig. 2b, the unmethylated TSDR showed transcriptional enhancer activity in all three cell types, albeit with varying degrees and weaker activity in conventional T cells. Importantly, the transcriptional activity in all samples was abrogated upon methylation of the TSDR, resulting in a luciferase signal equal to or below that of the hEF1 promoter alone.
Fig. 2.
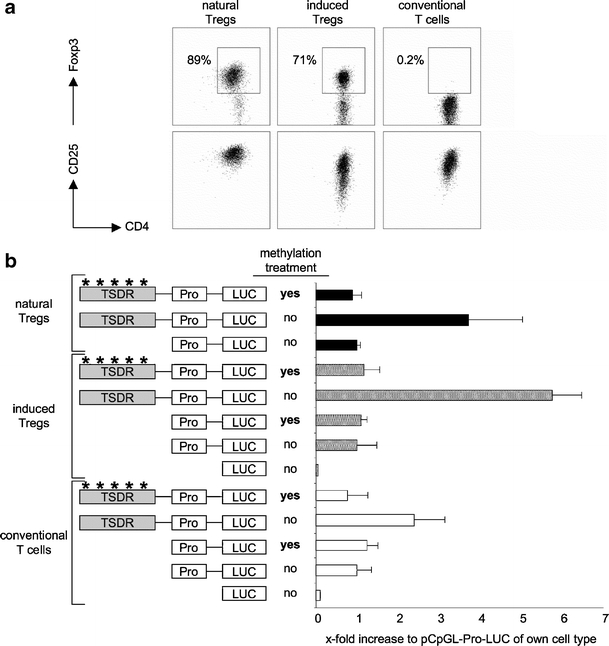
Functionality of the TSDR is determined by its methylation status. a Primary T cells from Foxp3gfp-reporter mice were sorted and expanded for 5 days in the presence of IL-2 (natural Tregs and conventional T cells) and IL-2 plus TGF-β (induced Tregs). The phenotypes of the expanded cells are shown. b The TSDR was cloned into the CpG-free pCpGL-luciferase reporter plasmid carrying the hEF-promoter (Pro). Subsequently, the plasmids were subjected to in vitro methylation or not. Primary expanded T cell subsets (from a) were transfected with the indicated plasmids and rested over night. On the next day, cells were stimulated for 9 h with PMA and ionomycin and luciferase activity was measured. Asterisks indicate methylated regions of the plasmid. Values are depicted in relation to the indicated reference sample, which was set to 1. The graph shows mean values of performed triplicates with standard deviation. One representative out of two to three independent experiments is depicted. LUC coding sequence for the firefly luciferase
The observed differences in the luciferase activity in Foxp3- conventional T cells and Foxp3+ cells might be either attributed to different potentials of the tested populations to activate TSDR function or, alternatively, to address the hEF1 promoter of the CpG-free luciferase reporter plasmid. We therefore tested the TSDR in conjunction with the murine Foxp3 promoter in the same primary murine T cell populations. Here, in all three cell types tested, the TSDR was equally able to enhance luciferase expression under the control of the original Foxp3 promoter (Fig. 3), indicating that the TSDR in its demethylated form possesses enhancer activity even in Foxp3− conventional T cells.
Fig. 3.
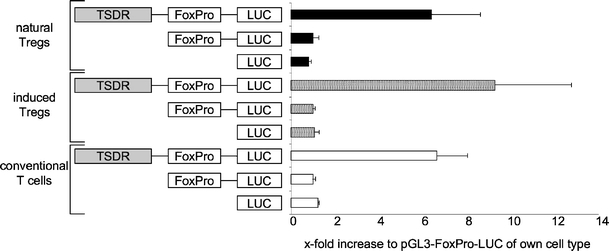
The TSDR acts as a transcriptional enhancer even in Foxp3− conventional T cells. Primary murine T cells from BALB/c mice, which had been cultured for 3 days in the presence of IL-2 (natural Tregs and conventional T cells) and IL-2 plus TGF-β (induced Tregs), were transfected with the indicated constructs. Luciferase activity was measured 20 h post-transfection. All values are depicted in relation to the indicated reference sample, which was set to 1. Both graphs show mean values of performed triplicates with standard deviation. One out of three independent experiments is depicted. FoxPro murine Foxp3 promoter; LUC coding sequence for the firefly luciferase
Taken together, these results indicate that both, Foxp3+ and Foxp3− cells, are equipped with a transcription factor repertoire that can address the TSDR and that solely the methylation status of this element determines its transcriptional enhancer activity.
Certain CpG motifs in a highly conserved fragment are essential for the TSDR enhancer activity
To identify the essential elements of the TSDR for its transcriptional enhancer activity, we generated truncated versions that were cloned in Foxp3 promoter containing luciferase reporter vectors as depicted in Fig. 4a, and applied these constructs in luciferase reporter assays in RLM-11-1 cells. The results showed a sequential reduction of the transcriptional activity with each truncation step (Fig. 4a). The D-segment alone did not show any activity. To test which of the four TSDR fragments displays enhancer function on its own, we performed luciferase assays with the cloned single fragments (Fig. 4b). Of the four fragments, only fragment C was capable of significantly enhancing transcriptional activity to about 55% of the full TSDR. All other fragments did not display any enhancer activity.
Fig. 4.
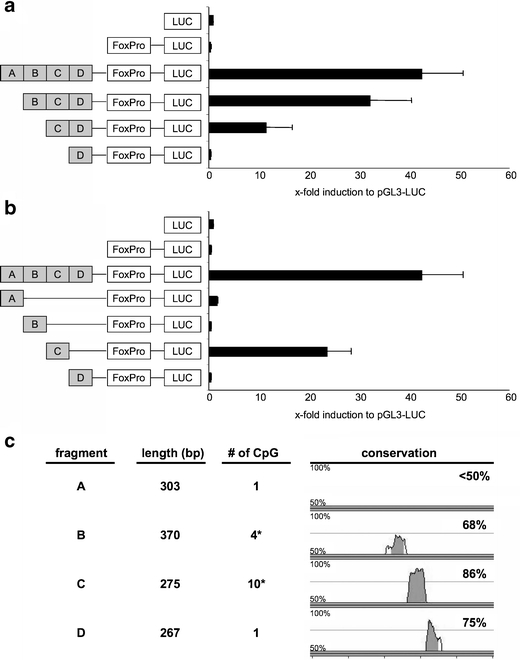
The CpG-rich, highly conserved fragment C of the TSDR is of special importance for the transcriptional activity. a and b Dual luciferase reporter assays were performed in RLM-11-1 cells using the indicated constructs. Luciferase activity was measured 20 h after stimulation with PMA and ionomycin. All values are depicted in relation to the indicated reference sample, which was set to 1. Both graphs show mean values of performed triplicates with standard deviation. One out of three independent experiments is depicted. c Characteristics of the tested TSDR fragments. The inter-species conservation was assessed using the mVISTA-tool comparing the murine and the human Foxp3 locus. Due to the primer-design, CpG #5 is present in fragments B and C (indicated by the asterisks)
These data clearly demonstrate that for full enhancer activity the entire TSDR is required. However, fragment C is able to enhance transcription on its own, suggesting that it contains some of the most important transcription factor binding sites. This idea is supported by the notion that fragment C is the one showing the highest inter-species conservation (86%) and contains the majority of the CpG motifs (Fig. 4c).
As CpG motifs play an important role in the regulation of the TSDR function, we aimed at identifying which of the 15 motifs were essential for the TSDR transcriptional activity. For this, each CpG motif was disrupted by site-specific mutagenesis resulting in 15 TSDR mutants with one point mutation each. These were all tested in luciferase reporter assays, using RLM-11-1 cells as described before. Most mutations did not influence the TSDR activity significantly (Fig. 5). Surprisingly, three mutations (CpG #8; #10 and #15) even led to an enhanced luciferase signal, probably due to accidental introduction of novel transcription factor binding sites or due to disruption of binding sites for repressive transcription factors. Most importantly, mutations of CpG motifs #6, #11 and #13, all of which are located in fragment C (Fig. 4c), led to a reduction of more than 60% compared with the unmutated TSDR. A moderate reduction level of approximately 30% was caused by mutation of CpG motif #7. Although there was a rather high variation between experiments, the reduction for these four sites was highly significant (p values ≤ 0.002 by Wilcoxon signed-rank test). Together, these results clearly show that some selective CpG motifs, which cluster in a highly conserved fragment of the TSDR, are essential for its transcriptional activity.
Fig. 5.
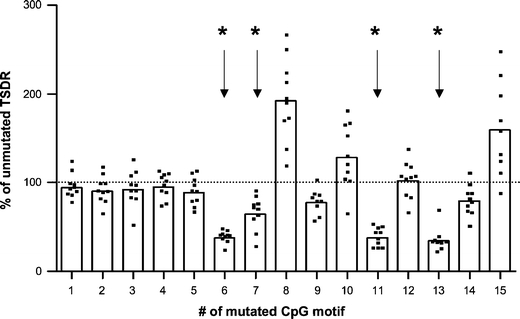
Selective CpG motifs are essential for the transcriptional activity of the TSDR. Point mutations were introduced to destroy single CpG motifs in the TSDR-FoxPro-LUC reporter vector. The resulting 15 mutant-TSDR-plasmids were used in dual luciferase reporter assays in RLM-11-1 cells. After transfection, the cells were rested for 3 h and subsequently stimulated with PMA and ionomycin. The luciferase activity was measured 20 h later. The luciferase activity of the FoxPro-control-plasmid (carrying no TSDR) was subtracted from each value to yield the unmutated TSDR value, which was set to 100%. All other values are depicted in relation to that. Bars represent the mean value of 9–10 replicates (shown as individual dots) taken from three independent experiments. Data sets marked with an asterisk show a statistically significant reduction to 100% according to the Wilcoxon signed-rank test (p ≤ 0.002)
The transcription factor Ets-1 binds to the TSDR and contributes to its transcriptional enhancer activity
In silico analysis for potential transcription factor binding sites spanning one of the critical CpG motifs identified in Fig. 5 revealed two Ets-1 binding sites (EBSs) spanning CpG #6 (EBS-1) and CpG #11 (EBS-2; Fig. 6a), a NF-κB binding site spanning CpG #7 and a binding site for CREB/ATF (CBS-2) spanning CpG #13. An additional CREB/ATF binding site (CBS-1) was postulated spanning CpG #5.
Fig. 6.
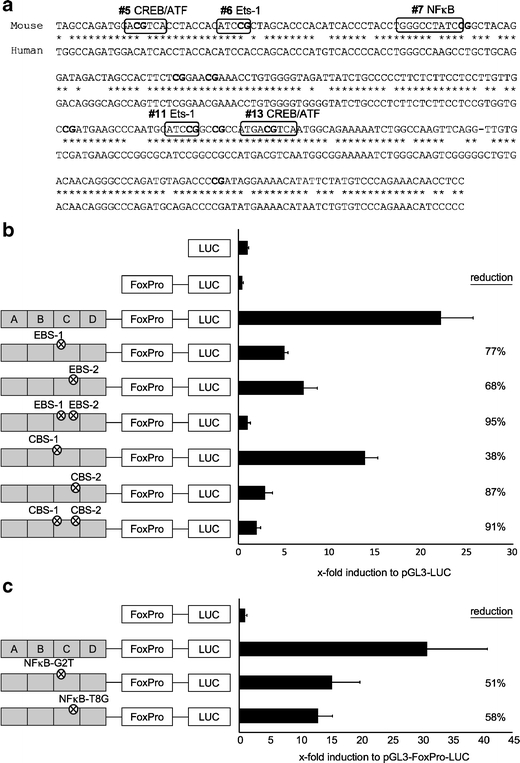
Mutation of the Ets-1, the CREB/ATF or the NF-κB binding sites in the TSDR leads to reduction of the transcriptional activity. a Sequence alignment of the murine (upper) and the human (lower) sequence of the TSDR fragment C. Postulated transcription factor binding sites, which span CpG motifs (in bold), are indicated with boxes. b Dual luciferase reporter assays were performed in RLM-11-1 cells using the indicated constructs. Circled multiplication symbol indicates inserted point mutations which destroy the indicated transcription factor binding sites. Luciferase activity was measured 20 h after stimulation with PMA and ionomycin. c Two-point mutations at distinct positions of the NF-κB-binding site were introduced and the resulting TSDR mutants were tested for transcriptional activity as in b. All values are depicted in relation to the indicated reference sample, which was set to 1. Both graphs show mean values of performed triplicates with standard deviation. One out of at least two independent experiments is depicted
To test the potential involvement of these binding sites in the TSDR function, we created additional TSDR mutants carrying point mutations in the core binding sequences for these transcription factors. These TSDR mutants were tested in luciferase assays, using RLM11-1 cells as described above. Interestingly, disruption of either one of the EBSs strongly reduced the TSDR functionality by about 70% (Fig. 6b). The combined disruption of both EBSs even completely abolished the enhancer activity. Disruption of the CBS-2 strongly affected the functionality of the TSDR, confirming previously reported observations [13]. The mutation of the CBS-1 only showed a moderate reduction of about 38% (Fig. 6b), indicating that this site might only weakly (if at all) contribute to transcriptional regulation, taking into account that the mutation of the corresponding CpG #5 did not result in a significant reduction of the TSDR activity (Fig. 5). Disrupting the NF-κB binding site, however, reduced the luciferase signal intensity to about half (Fig. 6c). Although this effect was not as pronounced as for the EBS-1 and -2 or CBS-2, it was consistently observed in two independent mutants, underlining the significance of this observation.
Taken together, these data support the hypothesis that the CpG motifs #6, #7, #11 and #13 are crucial for the functionality of the TSDR and allow speculations that in addition to CREB/ATF and NF-κB, the transcription factor Ets-1 might bind to the TSDR and contribute to the regulation of Foxp3 expression. To further evaluate this hypothesis, we tested whether the putative EBSs indeed could be bound by Ets-1 protein. Thereto, we performed EMSAs using fluorescently labeled oligonucleotides containing EBS-1 or EBS-2 of the TSDR and nuclear protein extract from in vitro differentiated Th1 cells, which express high levels of Ets-1 protein [39]. Oligonucleotides containing EBS-1 or EBS-2 both were shifted, resulting in two distinct bands (bands A and B; Fig. 7), the lower one of which (band B) could be outcompeted by adding excess of two different unlabeled oligonucleotides containing published Ets-1 binding sequences (Ets-1 ctrl.1 and 2, [40, 41], respectively). In contrast, the unlabeled oligonucleotide of a control sequence containing CBS-2 did not outcompete band B.
Fig. 7.
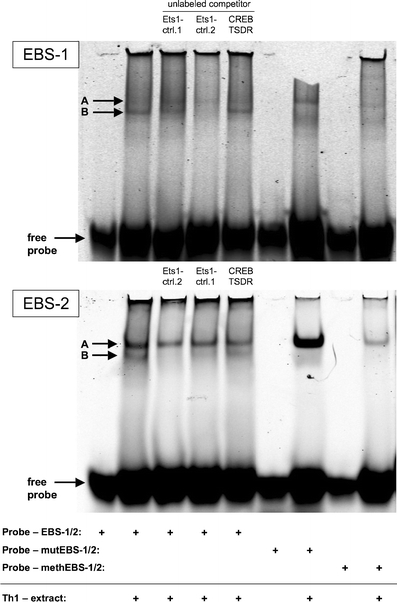
Two protein complexes bind to the Ets-1 binding sites (EBSs) of the TSDR in vitro. Electromobility shift assays were performed using nuclear protein extracts of in vitro differentiated Th1 cells and oligo-deoxynucleotides (probes) containing the EBS-1 or EBS-2 of the TSDR or their mutated or methylated versions. To compete for Ets-1 binding, unlabeled competitor oligonucleotides containing other known Ets-1 binding sites (Ets-1 contr.1 and 2) were used in 30-fold excess. The CREB/ATF binding site-2 of the TSDR (CREB-TSDR) served as a competition negative control. The gels present one out of two independent experiments
We further aimed to investigate whether introduction of the same mutation in EBS-1 and -2, which led to a strong reduction of the TSDR transcriptional enhancer activity (Fig. 6b), would also lead to the disappearance of band B in the EMSA. Indeed, when we used a mutated oligonucleotide as a labeled probe, we could not detect band B (Fig. 7). Most interestingly, band B was also absent, when we used the unmutated EBSs, in which the CpG motif was methylated (Fig. 7), indicating that Ets-1 only binds to the demethylated form of the TSDR. In none of these conditions, band A was outcompeted. Thus, the molecular identity of this complex remains to be determined.
The data from the luciferase assays and EMSAs demonstrated that the Ets-1 protein is able to interact with the demethylated TSDR in vitro and that mutation of the Ets-1 binding sequence abrogates the TSDR function. To confirm these in vitro observations under physiological conditions in vivo, we analyzed Ets-1 binding to the TSDR in ex vivo isolated Tregs and Foxp3- conventional CD4+ T cells by ChIP. Although Ets-1 protein was present in both cell types at similar amounts (Fig. 8a), binding to the TSDR could only be detected in the Treg population (Fig. 8b), which harbors a demethylated TSDR [23]. To ensure that the selective binding of Ets-1 in ex vivo Tregs was facilitated by the TSDR methylation status only, we generated induced Foxp3+ Treg populations, which would display a methylated or demethylated TSDR. This can be achieved by cultivation of conventional CD4+ T cells in the presence of TGF-β or the demethylating drug 5-Aza-2′-deoxycytidine (Aza), respectively, as we have shown earlier [27]. Indeed, Ets-1-binding to the (demethylated) TSDR could only be found in Aza-induced Foxp3+ Tregs, but not in TGF-β-induced Foxp3+ Tregs (Fig. 8c), which harbor a fully methylated TSDR [27]. These findings proof that Ets-1 binds to the TSDR in vivo only in its demethylated state and strongly suggest that Ets-1 is critically involved in the transcriptional regulation of Foxp3 expression in Tregs.
Fig. 8.
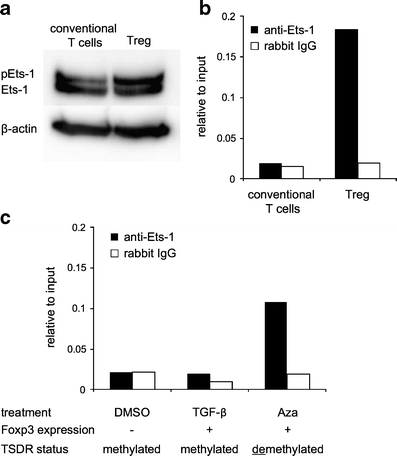
In vivo binding of Ets-1 to the demethylated TSDR in stable Foxp3+ Tregs. CD4+CD25+Foxp3gfp+ Tregs and CD4+CD25-Foxp3gfp- conventional T cells were isolated from BALB/c Foxp3gfp reporter mice and analyzed a for Ets-1 protein expression by Western Blotting and b for Ets-1 binding to the TSDR by ChIP. c Isolated conventional T cells were cultured with TCR-stimulation and IL-2 for 5 days. TGF-β (5 ng/ml) or 5-Aza-2′-deoxycytidine (Aza, 1 μM) was added to the culture to induce Foxp3 expression. Control cultures contained DMSO. On day 5, cultures were harvested and sorted for Foxp3gfp+ cells by FACS. Control cultures not containing Foxp3gfp+ cells were used unsorted. All populations were subjected to ChIP using anti-Ets-1 or control antibody. Both graphs show one representative out of three independent experiments
Discussion
Despite its critical role in Tregs, the transcriptional regulation of Foxp3 expression is only incompletely understood and molecular data elucidating its selective and sustained expression in Tregs are lacking so far. In previous studies, we have defined the TSDR, which acts as an expression stabilizer for the Foxp3 gene [12, 23, 24, 27]. We now present molecular data proving that the methylation state of this element is the crucial determinant, allowing or preventing the binding of the methylation-sensitive transcription factor Ets-1 and thereby controlling stability of Foxp3 expression in CD4+ T cells.
The cloned demethylated version of the TSDR, but not the methylated one, showed transcriptional enhancer activity not only in Foxp3+ Tregs, but also in Foxp3− conventional T cells, suggesting that also conventional T cells are equipped with all relevant transcription factors needed to address the TSDR. Apparently, only the constitutive methylation of the TSDR in conventional T cells prevents transcription factor binding and hence stable Foxp3 expression. This scenario would explain why sustained Foxp3 expression is confined to the natural Treg lineage only although also other T cell types can respond to the signals which have been implicated in Foxp3 induction so far: TCR-ligation, CD28-stimulation and IL-2 receptor triggering. Indeed, signaling via these common pathways is able to induce Foxp3 expression in situations where the DNA-methylation level is reduced either genetically (deficiency in the maintenance DNA methyltransferase DNMT-1) or by drug-treatment using the DNA methyltransferase inhibitor azacytidine [10, 13, 27, 30-33], which further substantiates our hypothesis.
As an exception to this, TGF-β is able to induce Foxp3 expression, even in the absence of enhanced DNA (and TSDR) demethylation. However, TGF-β-induced Foxp3 expression is unstable and rapidly lost as soon as the inducing cytokine is withdrawn [12, 23, 27]. This transient Foxp3 induction has been shown to be mediated by a distinct transcriptional enhancer element in the Foxp3 locus, which can be addressed by TGF-β-activated SMAD-transcription factors [21].
Since the murine Foxp3 promoter alone seems not to be sufficient to initiate transcription in murine T cells (Figs. 1 and 3), we propose a model, in which at least one of the two enhancer elements needs to be activated: either the TSDR in its demethylated state (present in natural Tregs), resulting in sustained Foxp3 expression, or the TGF-β-sensitive enhancer (in TGF-β-induced Tregs), leading to transient Foxp3 expression.
For the question of how TSDR mediates its transcriptional enhancer capacity, two explanations are possible: (1) Methylation of certain CpG motifs blocks the binding of essential demethylation-dependent transcription factors to these sites, or (2) the whole methylated TSDR restricts (methylation-independent) transcription factor binding by condensation of the chromatin structure. Both mechanisms are not mutually exclusive and might complement each other. Our present results, including the selective effects of mutations of four CpG motifs of the TSDR in reporter assays as well as the distinct demethylation-dependent interactions of transcription factors with crucial CpG-containing binding sites favor the first scenario, even though a possible contribution of the chromatin structure cannot be excluded.
Using in silico analyses, site-specific mutagenesis and EMSAs we here provide comprehensive evidence that the transcription factors CREB/ATF, NF-κB and Ets-1 are critical for the molecular regulation of Foxp3 expression. The binding of CREB/ATF to the TSDR was previously published [13]. Interestingly, this binding vitally depends on the demethylated state of the TSDR.
Additionally, binding of the NF-κB-subunit c-Rel to the TSDR has been shown recently [19], which complements our present data and strongly suggests a role for this transcription factor in Foxp3 regulation. However, in that study the authors did not address whether the transcriptional activity of the TSDR is hampered by disrupting the NF-κB binding site. Furthermore, it is still unclear whether or not the NF-κB-subunit c-Rel binds to the TSDR only in its demethylated state.
In our present study, such a demethylation-dependent binding was observed for the transcription factor Ets-1, which has not been reported in the context of Foxp3 regulation so far. Methylation-sensitive binding of Ets proteins has first been reported for the promoters of the housekeeping genes Surf-1 and Surf-2 [42]. For Ets-1 in particular, binding to and activation of the early B-cell-specific mb-1 gene in conjunction with the transcription factor Pax-5 was shown to be strictly dependent on the demethylated state of the CpG motif, which is part of the target sequence in the mb-1 promoter [43]. Our finding that Ets-1 binds to the TSDR in a demethylation-dependent manner in vitro as well as in stable Foxp3+ Tregs in vivo and that the disruption of its binding sites dramatically reduced the TSDR transcriptional enhancer activity, clearly states that Ets-1 is controlling the stability of Foxp3 expression.
Since the binding sites for the transcription factors Ets-1, CREB/ATF and NF-κB are all in close proximity within the TSDR, these proteins might physically interact for the transcriptional regulation of the Foxp3 gene. A c-Rel-containing multi-component protein complex, which might function as an “enhanceosome” on the Foxp3 locus, has recently been postulated by others, too, although in this study, in contrast to our present results, c-Rel seems to bind to the Foxp3 promoter but not to the TSDR [20]. The reason for this discrepancy remains to be investigated and could involve different NF-κB subunits.
It has previously been suggested that Foxp3 is involved in regulating its own expression in a positive feedback loop [44] similar to what has been observed for GATA-3 in Th2 cells [45]. However, our observation that even Foxp3- conventional T cells can address the TSDR to a similar extent as Foxp3+ Tregs makes a direct effect of Foxp3 on the transcriptional activity of the Foxp3 promoter and the TSDR unlikely. Nevertheless, a contribution of the Foxp3 protein to the opening of the Foxp3 locus might still be a possible scenario.
In conclusion, we propose that a large protein complex including Ets-1, CREB/ATF, and NF-κB assembles on the demethylated version of the TSDR and stabilizes Foxp3 expression in natural Tregs. In conventional T cells, binding of this complex to the TSDR is blocked by DNA methylation and the weak Foxp3 promoter alone is insufficient to drive Foxp3 expression unless the second transcriptional enhancer element in the Foxp3 gene [21] is targeted by TGF-β-mediated signals. However, under these circumstances Foxp3 expression remains transient and depends on the continuous presence of TGF-β. A central question still remains to be solved: Which signals and pathways lead to the demethylation of the TSDR during differentiation into stable Tregs? An answer to this issue might be crucial for the generation of stable Treg populations to be used for therapeutic interventions in the future.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 55.8 kb)
Acknowledgements and disclosure statement
We thank Sieglinde Keilholz-Gast and Kerstin Schlawe for expert technical assistance, Michael Rehli for provision of the CpG-free luciferase reporter vector, Alexander Rudensky and Ludger Klein for providing Foxp3 reporter mice, Lothar Gröbe for cell sorting, and Frank Klawonn for help with statistics. In addition, we are grateful to all members of the Huehn and Hamann labs for fruitful discussions. This work has been supported by the German Research Foundation (DFG, SFB738-C7 and SFB650-TP1). The authors disclose any financial interest in connection with the submitted material.
Contributions
J.K.P., L.S., C.T., L.L, M.K. and S.C. performed experiments. J.K.P., L.S., C.T. and L.L planned experiments. J.K.P., S.F., A.H. and J.H. designed and directed the project, R.B., A.H., and J.H. took responsibility for funding, J.K.P., L.S., and J.H. wrote the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Julia K. Polansky and Lisa Schreiber contributed equally to this study.
References
- 1.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008;223:371–390. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 2.Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity. 2009;30:656–665. doi: 10.1016/j.immuni.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curiel TJ. Regulatory T cells and treatment of cancer. Curr Opin Immunol. 2008;20:241–246. doi: 10.1016/j.coi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4 + CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 5.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 6.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 7.Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- 8.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 9.Mantel PY, Ouaked N, Ruckert B, Karagiannidis C, Welz R, Blaser K, Schmidt-Weber CB. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. 2006;176:3593–3602. doi: 10.4049/jimmunol.176.6.3593. [DOI] [PubMed] [Google Scholar]
- 10.Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, Ritz J. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–1579. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 12.Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. 2009;9:83–89. doi: 10.1038/nri2474. [DOI] [PubMed] [Google Scholar]
- 13.Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007;204:1543–1551. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klunker S, Chong MM, Mantel PY, Palomares O, Bassin C, Ziegler M, Ruckert B, Meiler F, Akdis M, Littman DR, Akdis CA. Transcription factors RUNX1 and RUNX3 in the induction and suppressive function of Foxp3+ inducible regulatory T cells. J Exp Med. 2009;206:2701–2715. doi: 10.1084/jem.20090596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruno L, Mazzarella L, Hoogenkamp M, Hertweck A, Cobb BS, Sauer S, Hadjur S, Leleu M, Naoe Y, Telfer JC, Bonifer C, Taniuchi I, Fisher AG, Merkenschlager M. Runx proteins regulate Foxp3 expression. J Exp Med. 2009;206:2329–2337. doi: 10.1084/jem.20090226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitoh A, Ono M, Naoe Y, Ohkura N, Yamaguchi T, Yaguchi H, Kitabayashi I, Tsukada T, Nomura T, Miyachi Y, Taniuchi I, Sakaguchi S. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity. 2009;31:609–620. doi: 10.1016/j.immuni.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Rudra D, Egawa T, Chong MM, Treuting P, Littman DR, Rudensky AY. Runx-CBFbeta complexes control expression of the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2009;10:1170–1177. doi: 10.1038/ni.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, Banerjee A, Proietto A, Gugasyan R, Li W, McNally A, Steptoe RJ, Thomas R, Shannon MF, Gerondakis S. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. 2009;206:3001–3014. doi: 10.1084/jem.20091411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kB modulates regulatory T cell development by directly regulating expression of the Foxp3 transcription factor. Immunity. 2009;31:921–931. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 20.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Green MI, Tone M, Chen YH. Development of Foxp3+ regulatory T cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 22.Venuprasad K, Huang H, Harada Y, Elly C, Subramaniam M, Spelsberg T, Su J, Liu YC. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol. 2008;9:245–253. doi: 10.1038/ni1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, Dong J, Thiel A, Boeld TJ, Hoffmann P, Edinger M, Turbachova I, Hamann A, Olek S, Huehn J. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur J Immunol. 2007;37:2378–2389. doi: 10.1002/eji.200737594. [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, Olek S, Dietmaier W, Andreesen R, Edinger M. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol. 2009 doi: 10.1002/eji.200838904. [DOI] [PubMed] [Google Scholar]
- 26.Wieczorek G, Asemissen A, Model F, Turbachova I, Floess S, Liebenberg V, Baron U, Stauch D, Kotsch K, Pratschke J, Hamann A, Loddenkemper C, Stein H, Volk HD, Hoffmuller U, Grutzkau A, Mustea A, Huehn J, Scheibenbogen C, Olek S. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res. 2009;69:599–608. doi: 10.1158/0008-5472.CAN-08-2361. [DOI] [PubMed] [Google Scholar]
- 27.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–1663. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 28.Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD, Hancock WW. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 29.Johnson J, Pahuja A, Graham M, Hering B, Hancock WW, Bansal-Pakala P. Effects of histone deacetylase inhibitor SAHA on effector and FOXP3+ regulatory T cells in rhesus macaques. Transplant Proc. 2008;40:459–461. doi: 10.1016/j.transproceed.2008.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagar M, Vernitsky H, Cohen Y, Dominissini D, Berkun Y, Rechavi G, Amariglio N, Goldstein I. Epigenetic inheritance of DNA methylation limits activation-induced expression of FOXP3 in conventional human CD25-CD4+ T cells. Int Immunol. 2008;20:1041–1055. doi: 10.1093/intimm/dxn062. [DOI] [PubMed] [Google Scholar]
- 31.Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, Reid SP, Levy DE, Bromberg JS. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng Q, Xu Y, Liu Y, Zhang B, Li X, Guo F, Zhao Y. Induction of Foxp3 demethylation increases regulatory CD4+CD25+ T cells and prevents the occurrence of diabetes in mice. J Mol Med. 2009;87(12):1191–1205. doi: 10.1007/s00109-009-0530-8. [DOI] [PubMed] [Google Scholar]
- 33.Josefowicz SZ, Wilson CB, Rudensky AY. Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J Immunol. 2009;182:6648–6652. doi: 10.4049/jimmunol.0803320. [DOI] [PubMed] [Google Scholar]
- 34.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 35.Klug M, Rehli M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics. 2006;1:127–130. doi: 10.4161/epi.1.3.3327. [DOI] [PubMed] [Google Scholar]
- 36.Feuerer M, Eulenburg K, Loddenkemper C, Hamann A, Huehn J. Self-limitation of Th1-mediated inflammation by IFN-gamma. J Immunol. 2006;176:2857–2863. doi: 10.4049/jimmunol.176.5.2857. [DOI] [PubMed] [Google Scholar]
- 37.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moisan J, Grenningloh R, Bettelli E, Oukka M, Ho IC. Ets-1 is a negative regulator of Th17 differentiation. J Exp Med. 2007;204:2825–2835. doi: 10.1084/jem.20070994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wai PY, Mi Z, Gao C, Guo H, Marroquin C, Kuo PC. Ets-1 and runx2 regulate transcription of a metastatic gene, osteopontin, in murine colorectal cancer cells. J Biol Chem. 2006;281:18973–18982. doi: 10.1074/jbc.M511962200. [DOI] [PubMed] [Google Scholar]
- 41.Pei H, Yordy JS, Leng Q, Zhao Q, Watson DK, Li R. EAPII interacts with ETS1 and modulates its transcriptional function. Oncogene. 2003;22:2699–2709. doi: 10.1038/sj.onc.1206374. [DOI] [PubMed] [Google Scholar]
- 42.Gaston K, Fried M. CpG methylation has differential effects on the binding of YY1 and ETS proteins to the bi-directional promoter of the Surf-1 and Surf-2 genes. Nucleic Acids Res. 1995;23:901–909. doi: 10.1093/nar/23.6.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maier H, Colbert J, Fitzsimmons D, Clark DR, Hagman J. Activation of the early B-cell-specific mb-1 (Ig-alpha) gene by Pax-5 is dependent on an unmethylated Ets binding site. Mol Cell Biol. 2003;23:1946–1960. doi: 10.1128/MCB.23.6.1946-1960.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 45.Ouyang W, Lohning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A, Murphy KM. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/S1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 55.8 kb)