

Abstract
Choroidal neovascularization (CNV) is a common and severe complication in heterogeneous diseases affecting the posterior segment of the eye, the most frequent being represented by age-related macular degeneration. Although the term may suggest just a vascular pathological condition, CNV is more properly definable as an aberrant tissue invasion of endothelial and inflammatory cells, in which both angiogenesis and inflammation are involved. Experimental and clinical evidences show that vascular endothelial growth factor is a key signal in promoting angiogenesis. However, many other molecules, distinctive of the inflammatory response, act as neovascular activators in CNV. These include fibroblast growth factor, transforming growth factor, tumor necrosis factor, interleukins, and complement. This paper reviews the role of inflammatory mediators and angiogenic factors in the development of CNV, proposing pathogenetic assumptions of mutual interaction. As an extension of this concept, new therapeutic approaches geared to have an effect on both the vascular and the extravascular components of CNV are discussed.
1. Introduction
Choroidal neovascularization (CNV) represents the growth of new blood vessels from the choroid into the subretinal pigment epithelium which, in several patients, reaches the retina. CNV is a common pathological endpoint in a heterogeneous variety of chorioretinal diseases [1]. Virtually any pathologic process that involves the retinal pigment epithelium (RPE) and damages Bruch's membrane can be complicated by CNV. The most frequent cause of CNV is age-related macular degeneration (AMD) [2].
The clinical classification of AMD-related CNV is carried out according to the definitions of Treatment of Age-Related Macular Degeneration with Photodynamic Therapy (TAP) and Visudyne in Photodynamic Therapy (VIP) studies [3–6], distinguishing between four subtypes characterized by different patterns during the fluorescein angiography (FA):
classic CNV: a demarcated area of uniform hyperfluorescence with a hypofluorescent margin in FA early phase, and dye leakage obscuring the boundaries during the mid and late phases (Figures 1(a) and 1(b));
predominantly classic CNV: the classic component occupying 50% or more of the entire neovascular lesion (including occult CNV and all the fluorescence-blocking constituents) (Figures 2(a) and 2(b));
minimally classic CNV: the classic component occupying less than 50% of the neovascular complex (Figures 3(a) and 3(b));
occult CNV with no classic component: including two types: (a) fibrovascular RPE detachment appearing as stippled hyperfluorescence with irregular RPE elevation (Figures 4(a) and 4(b)); (b) an undefined area with a late-phase dye leakage from an undetermined source, not corresponding to classic CNV and/or fibrovascular RPE detachment in FA early or mid phases.
Figure 1.
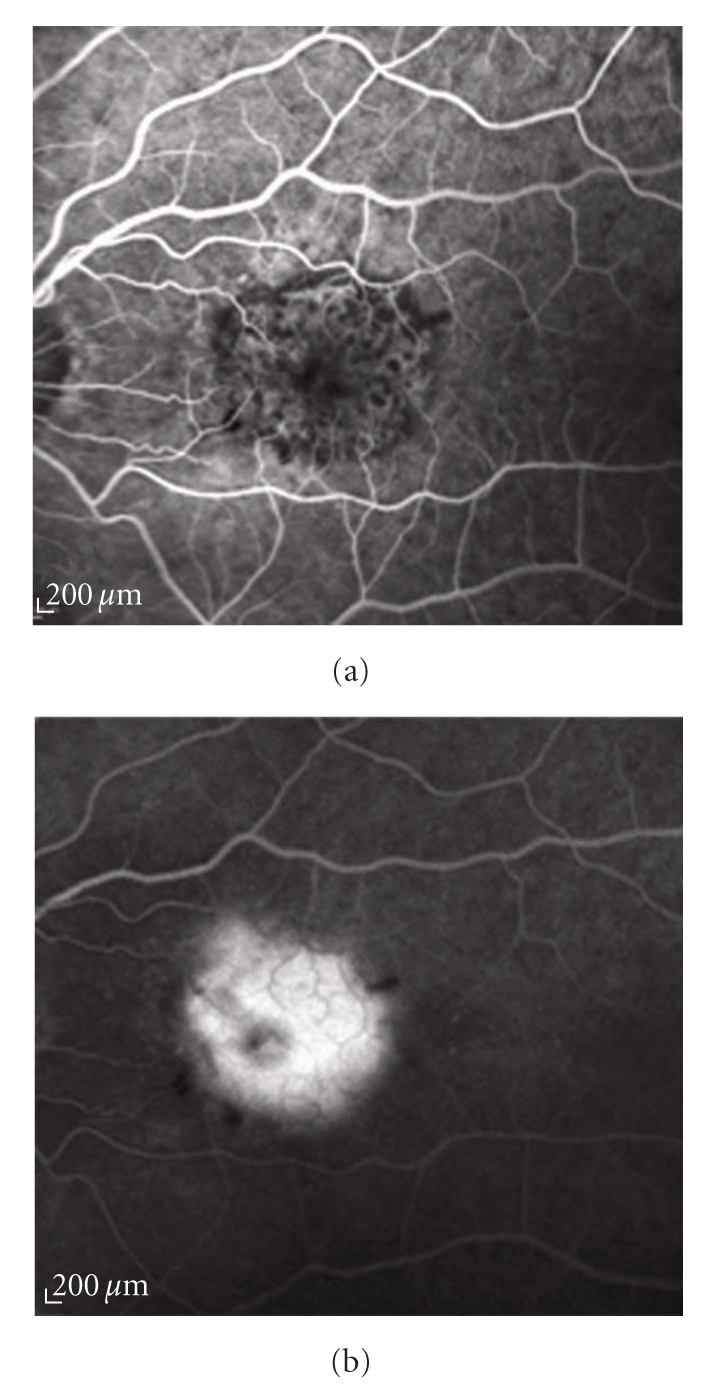
Fluorescein angiography of a classic choroidal neovascularization. (a) Early and (b) late angiograms: the lesion is characterized by a well demarcated area of early fluorescence with a progressive leakage of the dye to the subretinal space leading to blurring of the borders in the late phase of the exam.
Figure 2.
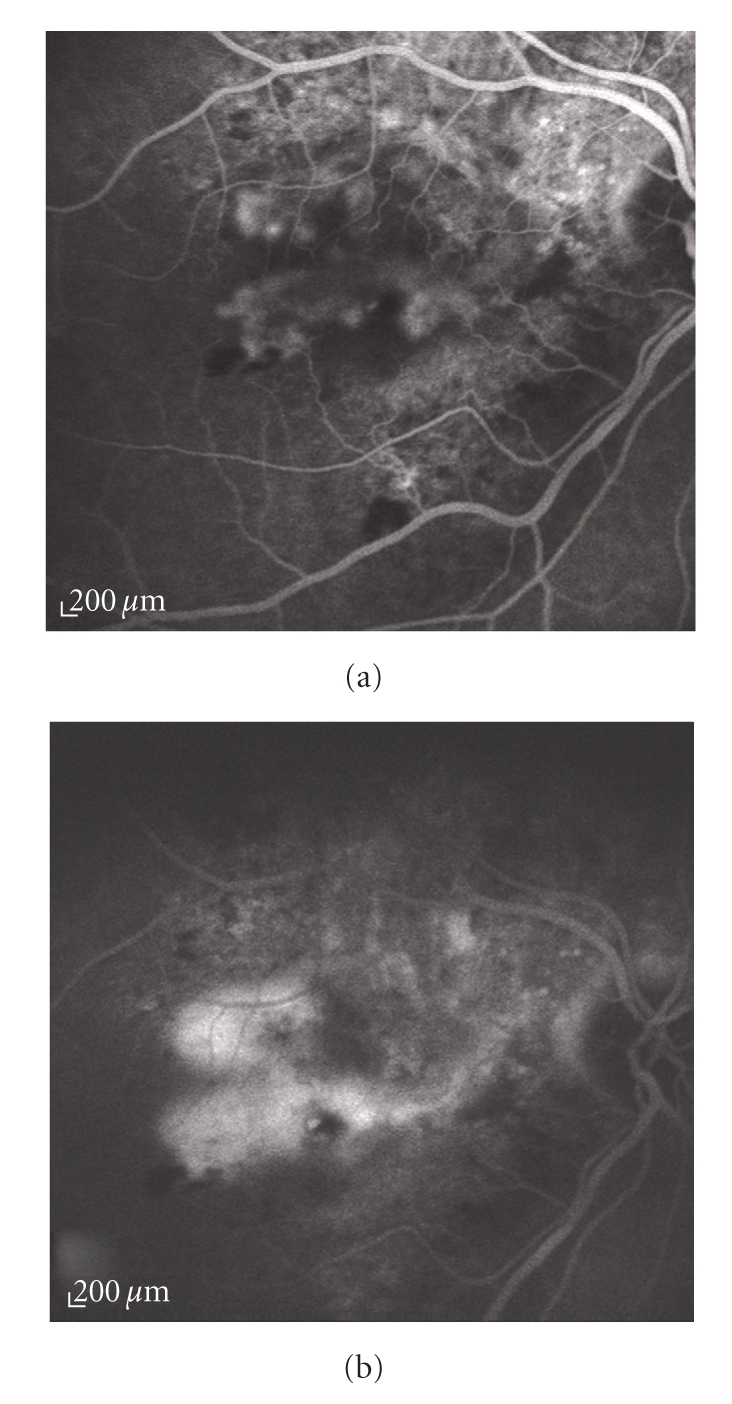
Fluorescein angiography of a predominantly classic choroidal neovascularization. (a) Early and (b) late angiograms: the lesion has a mixture of angiographic features of the classic and occult type, with the classic component making up more than 50% of the entire neovascular complex.
Figure 3.
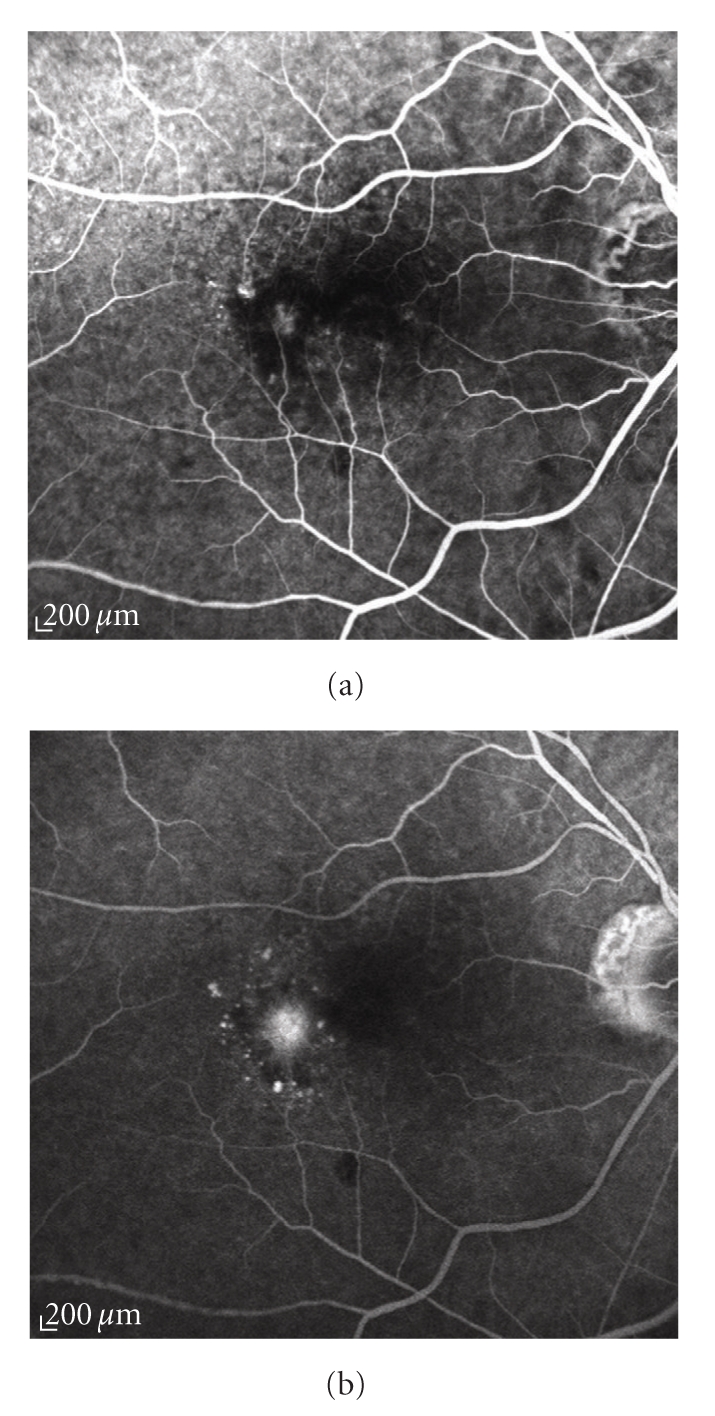
Fluorescein angiography of a minimally classic choroidal neovascularization. (a) Early and (b) late angiograms: the lesion has a mixture of the angiographic features of the classic and occult type, with the classic component making up less than 50% of the entire neovascular complex.
Figure 4.
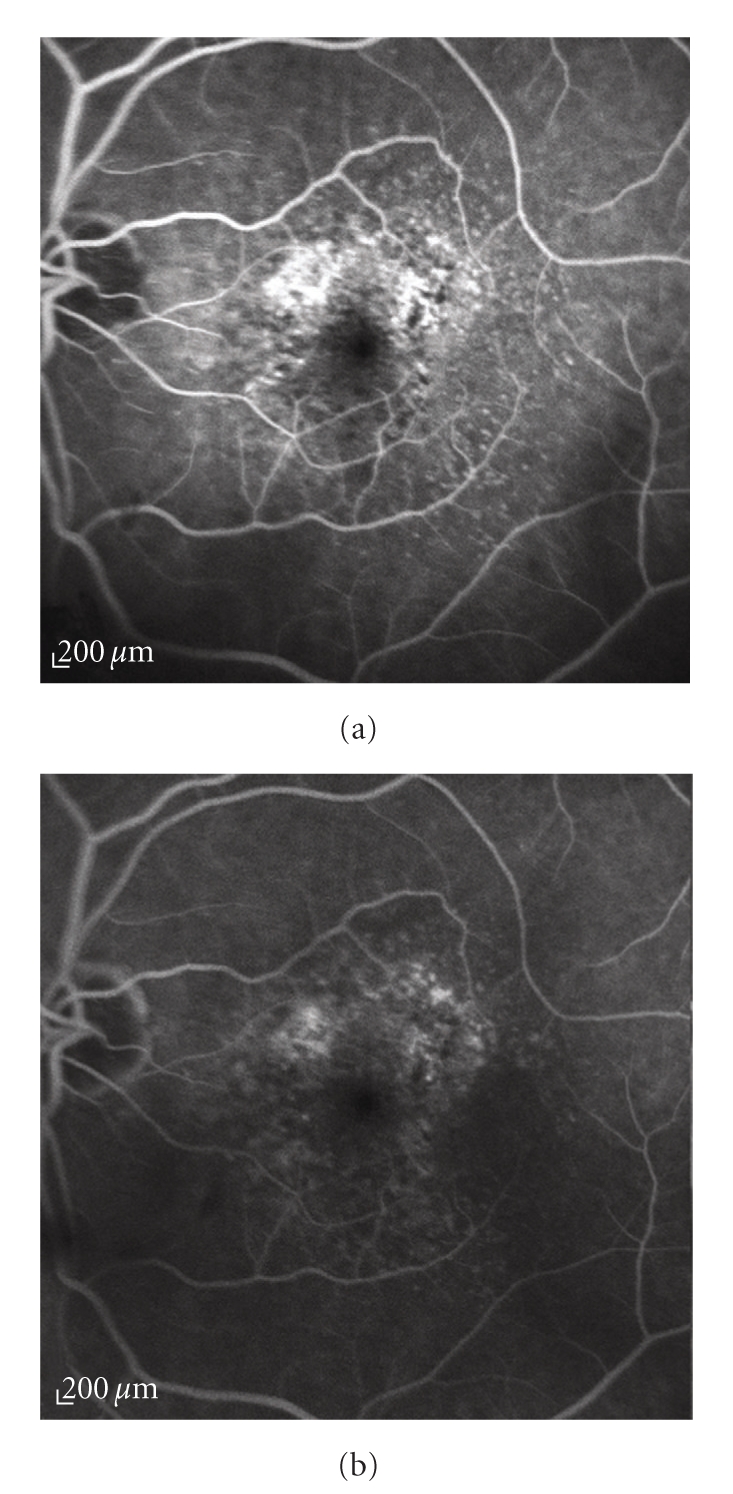
Fluorescein angiography of an occult choroidal neovascularization. (a) Early and (b) late angiograms: the lesion appears within 1-2 minutes from the start of the exams and persists during the late phase; it is characterized by areas of irregular elevation of the retinal pigment epithelium that present stippled hyperfluorescence.
Correspondingly, three basic patterns of CNV growth have been described [7]:
sub-RPE (type 1, clinically definable as occult CNV with no classic component);
subretinal (type 2, clinically definable as classic CNV);
sub-RPE and subretinal (combined, clinically definable as predominantly classic or minimally classic CNV).
2. Pathogenesis of Choroidal Neovascularization
In the surgically excised CNV of patients with AMD, the histopathologic examination indicates the presence of fragments of Bruch's membrane, RPE, photoreceptors, vascular endothelium, fibroblasts, macrophages, circulating progenitor/stem cells, and extracellular components including collagen, fibrin, and basal laminar deposits [8–14]. Hence, a two-component model for CNV has been proposed to describe CNV: (i) the vascular component of CNV is composed of vascular endothelial cells (ECs), pericytes and precursors of EC; (i i) the extravascular component is comprised of inflammatory cells (macrophages, lymphocytes, granulocytes, and foreign body giant cells), glial cells, RPE cells and fibroblasts [15]. Therefore, CNV is a process with a pathogenesis involving both inflammation and angiogenesis. The relevance of each component depends on both the underlying disease and dynamic stage of CNV development.
The natural history of CNV may be schematically divided in three stages: in the initiation stage, EC are derived from the chriocapillaris proliferate and migrate towards the retina through the Bruch's membrane; subsequently, during the active stage, the neovascular complex grows up to a certain size; finally, for reasons not yet well-known, the CNV becomes fibrotic and forms a disciform scar in the involutional stage [7].
2.1. Initiation Stage
2.1.1. Vascular Endothelial Growth Factor
In the initiation stage of the neovascular lesion, it is now well established that vascular endothelial growth factor (VEGF) plays a key role as inciting stimulus involved in CNV development. VEGF production has a number of potential sources, counting EC, pericytes, glial cells, Müller cells, ganglion cells, photoreceptors, and RPE [16–20]. Particularly, RPE secretes VEGF in a polarized manner, with higher basal secretion towards Bruch's membrane than apical secretion towards in situ photoreceptors [21]. VEGF-A, commonly named simply VEGF, is the prototype member of a gene family, also including placenta growth factor (PlGF), VEGF-B, VEGF-C, VEGF-D, and orf-virus-encoded VEGF-E [22, 23]. The increased VEGF production is mainly determined by hypoxic stimuli, and VEGF per se triggers the growth of vascular EC derived from arteries, veins, and lymphatics. VEGF is also a survival factor for endothelium; it enhances microvascular permeability and promotes monocyte chemotaxis [24].
Alternative exon splicing results in the generation of four main VEGF isoforms, named VEGF-121, VEGF-165, VEGF-189, and VEGF-206 basing on the number of amino acids after the signal sequence of slicing [24]. The smaller isoforms (VEGF-121 and VEGF-165) are freely diffusible whereas the larger ones (VEGF-189 and VEGF-206) are bound to heparin-containing proteoglycans on the cell surface or basement membrane [25]. These longer molecules can be released, after a plasmin-related proteolytic cleavage, as diffusible bioactive fragments [25, 26]. VEGF-165 can be proteolyzed by various matrix metalloproteinases (MMPs), especially the MMP-3, generating VEGF-113 which seems to be very similar to the plasmin-generated VEGF fragments [27].
Hitherto, three VEGF-receptors (VEGFRs) have been identified: (i) VEGFR-1 was firstly identified more than fifteen years ago, but its function is still debated [24, 28]; (i i) VEGFR-2 is considered the major mediator of the mitogenic, angiogenic and propermeability effects of VEGF [24]; (i i i) VEGFR-3 is identical to neuropilin-1, implicated in axon guidance as a receptor for collapsin/semaphorin molecules, and it appears to enhance the effectiveness of the signal transduction mediated by VEGFR-2 [24].
Experimental and clinical evidences indicate the central role of VEGF in CNV occurrence. In fact, VEGF is present in surgically excised CNV [16, 17, 29–31], and vitreous levels of VEGF were significantly higher in patients with neovascular AMD compared with healthy controls [32, 33]. The VEGF overexpression in mice's RPE is sufficient to cause the development of CNV [34, 35], and, in animal models, VEGF blocking treatments are able to cure the laser-induced CNV [36, 37]. Finally, intravitreal injections of VEGF elicit proliferation of choroidal EC in nonhuman primates [38]. In physiologic and pathophysiologic conditions, the upregulation of proangiogenic cytokine expression, such as VEGF, is involved in the response to tissue hypoxia that it is mediated by the family of transcription regulators, named hypoxia inducible factors (HIFs). It is therefore plausible that hypoxia represents a major stimulus for the submacular wound healing, and, within this context, CNV is other than one element of this process which, in turn, leads to HIF synthesis; HIF are the key signals for hyperproduction of VEGF with subsequent aberrant growth of the neovascular component of submacular tissue repair. In fact, the presence of HIF-1alpha and HIF-2alpha has been recently discovered in active CNV specimens [39]. In the ocular tissues, VEGF acts as both a specific EC mitogen and promoter of vascular permeability. However, in addition to angiogenesis, vasculogenesis appears to be implicated in CNV development, indeed: (i) VEGF is a chemoattractant for EC precursors, inducing their mobilization and promoting their differentiation [40]; (ii) chemokine stromal cell-derived factor 1-alpha and its receptor CXCR4, both involved in the recruitment of EC precursors, have been detected inside CNV [41]. VEGF induces CNV enlargement also by stimulating EC expression of MMPs, which degrade the extracellular matrix and facilitate neovascular tissue invasion [42]. Lastly, VEGF represents a potent chemotactic signal for macrophages [43, 44].
2.1.2. Macrophages and Other Cytokines
Macrophages are other important players in the process of CNV development. In experimental model of CNV, macrophages depletion diminishes both size and severity of the neovascular lesion [45]. Leukocytes are recruited in CNV not only by VEGF but also by vascular adhesion protein-1, an EC adhesion molecule [46]. It is unknown whether either macrophages actively cause breaks in Bruch's membrane (via production of collagenase/elastase), or they are introduced into the ill area after the CNV breaks of Bruch's membrane [8]. The arriving of macrophages elicits the production of tumor necrosis factor-α (TNF-α) which, in particular, stimulates the synthesis of type-8 interleukin (IL), monocyte colonization protein, and RPE-secreted VEGF [11]. This autocrine/paracrine loop is completed with the recruitment of further macrophages by type-1 monocyte colonization protein [47]. At the level of RPE cells, integrins α3 and α5 are expressed, mediating the migration of vascular ECs or macrophages in the early stage of CNV development [48]. As well, IL-2, IL-6, and IL-10 might participate to CNV expansion, but their exact roles have not been deeply investigated yet [49–51].
2.1.3. Angiogenic and Antiangiogenic Agents
Several angiogenic/antiangiogenic molecules, different from VEGFs, are implicated in CNV development [52].
Insulin-like growth factor is a mediator of anabolic and mitogenic actions of the growth hormone; inside AMD-related CNV present are mRNAs of this factor and of its receptor [53]; in vivo, both VEGF expression and CNV activity are downregulated by a specific receptorial inhibitor (picropodophyllin) [54].
Nitric oxide is a signaling molecule with pleiotropic effects, and it is a well-known mediator of vascular dilatation and permeability [55]; experimental findings indicate that nitric oxide is an important CNV stimulator, and that its reduction, obtained in pharmacologic or genetic manner, represents a potential therapeutic strategy for CNV [56].
Angiostatin is a plasminogen fragment firstly recognized as endogenous angiogenic inhibitor [57]; subretinal injection of recombinant adenoassociated virus vector expressing mouse angiostatin is able to suppress experimental CNV in a rat model [58].
Endostatin, another endogenous inhibitor of angiogenesis, is generated by the cleavage of a collagen XVIII fragment [59]; in mice, intravenous administration of adenoviral vectors containing an expression construct for endostatin results in prevention of laser-induced CNV [60].
Pigment epithelium-derived factor (PEDF) is a potent antiangiogenic and neuroprotective protein, normally produced by RPE [61]; in pigs and mice, the periocular injection of an adenoviral vector encoding PEDF inhibits CNV development [62, 63].
CCR3 (also known as CD193) is a chemokine receptor best known for its role in promoting eosinophil and mast cell trafficking; it is specifically expressed in CNV endothelial cells in humans with AMD [64]. CCR3 targeting reduces CNV in a mouse model through a direct antivascular effect which does not appear to involve modulation of cellular inflammation [64].
2.2. Active Stage
In the course of the natural history of CNV, active stage is characterized by the progressive enlargement of neovascular complex. This neovascular enlargement is mainly related to the presence of many inflammatory cells, synergistically acting with aberrant cytokines produced via autocrine/paracrine mechanisms. Vascular endothelium and macrophages produce MMPs which, in turn, degrade extracellular matrix allowing CNV infiltration through Bruch's membrane [8]. Several MMPs were detected in the vitreous of AMD patients and in surgically excised subfoveal CNV [65, 66]. Particularly, peculiar MMPs seem to play a key role in CNV growth; in fact: (i) MMP-2/MMP-9 deficient mice have more difficulty in developing laser-induced CNV [67, 68]; (i i) overexpression of tissue inhibitor for MMP-3 (TIMP-3) in RPE cells reduces the activity of experimental CNV [69]. At the same time, macrophages express tissue factor, a protein involved in fibrinogenesis, which is able to generate a fibrin scaffold for the growing of CNV [8]. During active stage, also angiopoietins and their receptors (Tie-1, Tie-2) play a role in angiogenic process. Histological examination of AMD-related CNV showed that angiopoietin-1 and -2 and Tie-2 were present [70]. Angiopoietins are essential for maturity and integrity of vessels: angiopoietin-1 appears crucial for this structural stabilization in normal vascular system whereas the dramatic upregulation of angiopoietin-2, in presence of VEGF, is a prerequisite for vascular remodeling and/or normal angiogenesis [71]. Moreover, at this stage, RPE produces a basic fibroblast growth factor (FGF-2), a heparin-binding peptide, that stimulates pathologic angiogenesis. However, FGF-2 expression alone is neither necessary nor sufficient for CNV development [71, 72]. Transforming growth factor-beta (TGF-β) is another important factor secreted by RPE during active CNV phase [73]; since it is a potent inducer of extracellular matrix synthesis, reliably it limits the extent of neovascular complex, starting that process then resulting in progressive CNV fibrosis [8]. Recent experimental evidences suggest a role also for hepatocyte growth factor (HGF), a mitogen for hepatic and renal cells that may be involved in CNV progression [74]; whether this is a result of its dedifferentiating effect on RPE cells is currently unclear.
Activation of complement, specifically the formation of membrane attack complex (MAC), is essential for the development of laser-induced CNV in mice [75] underlining, once again, the importance of inflammation in the pathogenesis of the neovascular complex. MAC can mediate the release of several factors from various nucleated cells, such as VEGF, FGF-2, and platelet-derived growth factor (PDGF), with consequent amplification of the angiogenic processes [75]. Gene variants of the complement components have been evaluated as risk factors for AMD among different ethnic clusters [76–101]. Hitherto, one of the most investigated single nucleotide polymorphism (SNP) has been the rs1061170, which causes a Tyr402His amino acid substitution in complement factor H (CFH). Independent studies have documented that the allele 402His is related to increased incidence and/or severity of AMD in several populations [76–100, 102]. The frequency of 402His allele varies greatly between populations and, notionally, it may contribute to the observed variability in the incidence of AMD among different ethnic groups [103]. Finally, also elevated plasma levels of C3a complement compound in patients with AMD-related CNV have recently suggested the presence of an association between systemic complement activation and this pathologic occurrence [104].
In the course of initiation stage, the neovascularization is highly dependent on VEGF but, afterward, it undergoes a process of maturation which makes it less dependent on this growth factor. In fact, once the new vessels are formed, ECs start to secrete other factors to recruit mural cells (pericytes) that promote vessel stabilization, endothelium differentiation, and growth arrest. The most important of these factors is PDGF-B which works through its receptors expressed by pericytes [105–107]. In animal models of CNV, inhibition of both VEGF-A and PDGF-B signaling appeared more effective for vessel regression than blocking VEGF-A alone [108].
2.3. Involutional Stage
Whilst there is considerable overlapping between the phases of CNV progress, at certain time the balance of these pathologic events shifts towards antiangiogenic and antiproteolytic activities, resulting in the involutional stage of CNV [8]. Indeed, it is quite frequent to find different developmental stages within the same neovascular complex, mostly characterized as either cellular or fibrotic regions [14]. At this stage, the most important players are TGF-β and TIMP-3, produced by RPE, which are able to markedly influence both the secretion of extracellular matrix and the tissue remodeling. Concurrently, angiogenesis continues until a state of normoxia or hyperoxia exists, thereby switching off VEGF synthesis. The outcomes of these processes are the maturation of established vessels and the occurrence of scar tissue. The origin of vascular elements contributing to the subretinal fibrosis is not yet clear, but it is known that RPE cells themselves, directed by TNF-α, TGF-β, and other growth factors, dedifferentiate and proliferate showing, together with choroidal fibroblasts, a wound repair pattern [109].
3. Therapeutic Approaches for Choroidal Neovascularization
The wider knowledge of the mechanisms involved in the pathogenesis and development of CNV has led, in the last few years, to a remarkable increase of the possible pharmacological strategies toward this severe macular lesion (Table 1).
Table 1.
Therapeutic approaches for choroidal neovascularization.
| Therapies directed against the vascular component of choroidal neovascularization. | ||
|---|---|---|
| Agent | Class | Molecular target |
|
| ||
| Pegaptanib sodium | aptamer | VEGF-165 |
|
| ||
| Ranibizumab | monoclonal antibody fragment | all VEGF isoforms |
|
| ||
| Bevacizumab | full-length monoclonal antibody | all VEGF isoforms |
|
| ||
| VEGF-trap | decoy receptor | all VEGF isoforms and PlGF |
|
| ||
| Pazopanib | tyrosine kinase inhibitors | VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-a/β, and c-kit |
| TG100801 | ||
| TG101095 | ||
| Vatalanib | ||
| AL39324 | ||
|
| ||
| Bevasiranib | siRNA | VEGF mRNA |
|
| ||
| E10030 | aptamer | PDGF |
|
| ||
| Combretastat in A4 phosphate | vascular | VE-cadherin, beta-catenin/AKT |
|
| ||
| Therapies directed against the extra-vascular component of choroidal neovascularization | ||
|
| ||
| Agent | Class | Molecular target |
|
| ||
| Anecortave acetate | corticosteroid | uPA, stromelysin (MMP-3) |
|
| ||
| JSM6427 | integrins antagonist | α5β1 integrin |
|
| ||
| sirolimus | immunosuppressant | mTORC1 |
|
| ||
| infliximab | Monoclonal antibody | TNF α |
VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; PlGF, placenta growth factor; PDGFR, platelet-derived growth factor receptor; PDGF, platelet-derived growth factor; uPA, urokinase plasminogen activator; MMP, matrix metalloproteinase; mTORC1, mammalian target of rapamycin complex 1.
3.1. Therapies Directed against the Vascular Component of Choroidal Neovascularization
In view of the fact that VEGF plays a key role in the pathogenesis of CNV, targeting VEGF has soon appeared as an effective strategy to treat CNV [110]. Antivascular endothelial growth factor (anti-VEGF) drugs not only can arrest choroidal angiogenesis, but they also reduce vascular hyperpermeability which, in patients with CNV, is often the main cause of visual acuity (VA) deterioration.
Pegaptanib sodium and ranibizumab were the first intraocular anti-VEGF treatments evaluated in large, randomized, controlled clinical trials for the treatment of neovascular AMD. Both have been administered locally by repeated intravitreal injections.
Pegaptanib sodium is a pegylated oligonucleotide aptamer that selectively binds to and inactivates just the VEGF-165, the most abundant isoform of VEGF. In the VISION trial—a phase III double-masked, sham-controlled, dose-ranging study including all lesion subtypes and dimensional categories—pegaptanib sodium prevented moderate vision loss (the primary endpoint, which was defined as loss of <15 letters of vision) in 70% of the treated patients compared with 55% for the control group at one year. On average, patients in the pegaptanib sodium group lost 8 letters at one year, compared with a loss of 15 letters in the sham injection group. The proportion of patients who experienced a moderate gain in vision (defined as a change of ≥15 letters at one year from baseline) was small in both groups −6% in the pegaptanib sodium group versus 2% in the sham-injection group [111]. In two years, 59% of eyes treated with pegaptanib lost less than 15 letters compared with 45% of sham-treated eyes [112].
Ranibizumab is a recombinant, humanized antibody fragment that binds to and potently neutralizes the biological activities of all known human VEGF isoforms, as well as the proteolytic cleavage product VEGF-110. In a large randomized, multicentre, sham-controlled phase III study (MARINA trial) that included only patients with minimally classic or occult CNVs, at the 12-month visit, 95% of ranibizumab-treated eyes maintained stable vision (within 15 letters) compared with 62% of sham-treated eyes. An improvement of VA by ≥15 letters was found in 34% of eyes treated as compared with 5% of the sham-injection group. After 24 months, 90% of eyes in the ranibizumab group versus 53% in the control group demonstrated stable vision. A mean improvement of 7 letters was documented at 12 and 24 months of follow up in the ranibizumab arms. Improved vision by ≥15 letters occurred in 33% of eyes treated with ranibizumab, and 42% of patients in this group obtained a final VA of 20/40 or better [113]. The ANCHOR study was designed as a prospective, randomized phase III trial including patients with predominantly classic CNV secondary to AMD. Repeated injections of ranibizumab were compared with an arbitrary treatment with photodynamic therapy. In one year, 96% of ranibizumab-treated eyes lost less than 15 letters versus 64% of photodynamic-treated eyes. An improvement ≥15 letters was found in 40% of eyes injected with ranibizumab compared with 6% in the other treatment group. A vision gain of ≥30 letters was achieved in 12% of ranibizumab-treated patients. Mean VA demonstrated an improvement of 11 letters in one year and a final VA of 20/40 or better was found in 39% of ranibizumab-treated patients [114]. Consistent with results obtained at the 12-month check, at the 24th month of follow up the VA benefit due to ranibizumab was statistically significant and clinically meaningful [115].
Bevacizumab is a full-length recombinant, humanized antibody binding to all VEGF isoforms. The drug was originally developed to target pathologic angiogenesis in tumors and was approved by the FDA for the treatment of metastatic colorectal cancer. It is widely used as off-label drug for the treatment of various ocular neovascular diseases, including CNV, because numerous clinical studies have documented the ability of this compound to reduce angiogenesis and vascular hyperpermeability rapidly following its intravitreal repeated administrations [116–126]. Both favorable efficacy and safety profiles and lower costs are the major arguments to consider for an off-label use of this drug [116–131].
Several other molecules targeting VEGF and its signaling pathway are in various stages of clinical development for the treatment of CNV [132].
VEGF-Trap is a fusion protein that combines ligand-binding elements taken from the extracellular domains of VEGFR-1 and VEGFR- 2 fused to the Fc portion of IgG [132]. Unlike pegaptanib, ranibizumab, and bevacizumab, which all act through inhibition of VEGF-A, VEGF-Trap is designed to inhibit all members of the VEGF family: VEGF-A, -B, -C, -D, PlGF-1 and -2. The CLEAR-IT 2 study (phase II, randomized, controlled, clinical trial) has demonstrated that VEGF-Trap is able to significantly decrease retinal edema and CNV size in patients with neovascular AMD [133].
Another method of blocking the effects of VEGF is through inhibition of the downstream tyrosine kinase cascade activated by the VEGF binding to its receptors. Tyrosine kinase inhibitors, interacting with VEGF receptors currently in early stages of clinical development, include the two topical pazopanib formulations (TG100801 and TG101095), the oral formulation vatalanib (formerly PTK/ZK), and the intravitreal formulation AL39324 [132].
The levels of VEGF in the ocular tissues may be reduced also acting on its gene expression via small interfering RNA (siRNA). Therapeutic approach with siRNA works by downregulating the production of certain proteins as a result of degradation of specific mRNA. Bevasiranib (formerly Cand5) is an siRNA targeting mRNA of VEGF: it is currently tested in a phase III clinical trial (COBALT study) for the treatment of neovascular AMD. Since bevasiranib just blocks the VEGF production, the amount of VEGF already present in the eye at the time of administration precludes an immediate impact of the drug on CNV. For this reason, bevasiranib is evaluated as a combination therapy together with an agent that binds VEGF (ranibizumab) with the aim of reducing the dosage of the latter [134].
A major limitation of the anti-VEGF therapy is the difficulty to maintain the neovessels regression. This phenomenon is ascribed to the progressive maturation of CNV which then becomes less dependent on VEGF. As mentioned above, PDGF plays a key role in this process of maturation through the recruitment of pericytes [105, 106]. E10030 is an anti-PDGF aptamer that strips the pericytes from the neovascular tissue rendering it highly sensitive to an anti-VEGF attack. In a phase I clinical study evaluating E10030 in association with an anti-VEGF drug for the treatment of AMD-related CNV, 85% of patients exhibited neovascular regression [135]. This effect is further supported by the fact that, in ocular angiogenesis models, the pharmacologic inhibition of PDGF binding to its receptor (PDGFR-β) plus an anti-VEGF agent result in the regression of the neovascularization [136].
Beyond the inhibition of angiogenesis there is a new approach in the vascular targeting strategies: the so-called vascular disabling treatment. Vascular disabling agents (VDAs) target endothelial cells of the already established neovascular tissue leaving other blood vessels relatively unscathed. Zybrestat (combretastatin A4 phosphate, CA-4-P) is poised to become the first therapeutic product in this novel class of drug candidates. Zybrestat has a dual mode of action, targeting both VE-cadherin, a junction protein that is important for endothelial cell survival, and the associated beta-catenin/AKT signaling pathway [137]. In experimental models, CA-4-P was able to suppress the development of VEGF-induced retinal neovascularization and to promote CNV regression, showing potential for both prevention and treatment of ocular neovessels [138].
3.2. Therapies Directed against the Extravascular Component of Choroidal Neovascularization
The development of CNV is a complex process in which both angiogenesis and inflammation take part. As previously covered, VEGF apart, many cytokines and inflammatory cells are involved in CNV pathogenesis and, thus, they represent a potential target for therapy.
Anecortave acetate is a modified steroid derivative without glucocorticoid activity. It inhibits protease synthesis, which is necessary for cell migration through nonvascularized extracellular matrix in response to angiogenic stimulation. Juxtascleral depot of anecortave acetate at 6-month intervals was statistically superior to vehicle in a monotherapy trial at both 12 and 24 months for maintenance of vision and inhibition of CNV growth in patients with AMD; moreover, in a comparative trial, it was comparable to photodynamic therapy with verteporfin for maintaining vision over a 24-month period [139]. However, additional data about efficacy/safety profile have been requested for the clinical approval of this drug.
Integrins are cell adhesion receptors involved in the linkage to extracellular matrix and to adjacent cells. Integrins antagonists have effectively inhibited CNV progression in animal models, suggesting that these molecules may be beneficial in the treatment of CNV [140, 141]. Single intravitreal injections of JSM6427, a highly potent and specific small antagonist of α5β1 integrin, were well tolerated in patients with neovascular AMD and showed evidences of biological activity in phase I clinical study [142].
Whilst it has been demonstrated that MMPs are the key enzymes involved in the degradation of the extracellular matrix and have been shown to contribute to the growth of CNV, the strategies developed to inhibit CNV by overexpression of tissue inhibitors of MMP-3 have been somewhat disappointing [143]. The glycoprotein thrombospondin-1 suppresses angiogenesis by acting as both an activator of transforming growth factor-β, and a negative regulator of MMP-9 activation, and an activator of apoptotic pathways [52]. Recently a peptide derived from type 1 thrombospondin repeat-containing protein WISP-1 (wispostatin-1) has been shown to have inhibitory effect in vitro as well as in vivo in ocular neovascularization [144].
Several other drugs, already in use for several inflammatory disorders, are currently under investigation as potential treatments for CNV. They include cyclooxygenase inhibitors, sirolimus, and infliximab [145–149]; in particular, for these latter two, the preliminary data seem to be more interesting. Sirolimus, also known as rapamycin, acts on the protein kinase mammalian target of rapamycin, which regulates cell growth and metabolism. In addition to its anti-inflammatory, antifibrotic, and antiproliferative activities, sirolimus inhibits also angiogenesis by decreasing VEGF and transforming growth factor-β1 and by downregulating hypoxia-inducible factor-1α [132]. Systemic rapamycin is able to inhibit retinal and choroidal neovascularizations in mice [146]. A phase II clinical study is ongoing to assess the safety and efficacy of intravitreal ranibizumab plus subconjunctival sirolimus versus intravitreal ranibizumab plus placebo in patients with treatment-naive subfoveal CNV secondary to AMD [147]. Infliximab is a chimeric human IgG1 with a mouse Fv variable fragment of high TNF-α affinity and neutralizing capacity. In vivo, intravenous infliximab has been indicated in the treatment of rheumatologic, gastrointestinal, and dermatologic diseases, and recent studies have described its efficacy in the treatment of chronic ocular inflammation. Preclinical trials have demonstrated a reduction in CNV size in mice intravitreally treated with infliximab. However, there seems to be a dose-response relationship in which low doses of anti-TNF-α decrease angiogenesis while high doses increase it [148, 149].
4. Conclusions and Perspectives
The pathogenesis of CNV represents a highly complex process where not only angiogenesis but also inflammation plays an important role. Nowadays, the most frequent utilized treatment for the different typologies of subfoveal CNV is based on the pharmacological block of VEGF, which can be combined with the selective laser photothrombosis of the lesion (photodynamic therapy with verteporfin) [6]. However, neither therapy is ideal; in fact, verteporfin protocol is not usually associated with a functional improvement, and intravitreal drugs acting against VEGF are estimated to substantially improve vision in less than a third of patients, with one-sixth of treated subjects still progressing to legal blindness. Furthermore, in an elderly population, often already at risk for cerebrovascular accidents, there are concerns about possible systemic thromboembolic complications with repeated high dosages of anti-VEGF compounds [150]. Numerous intravitreal injections over many years may be also relatively contraindicated in some patients, such as diabetics, in who the underlying disease may favor infections and slow down the healing of the wound. In the last few years, following extensive immuno-histochemical and molecular biologic characterization of CNV, several innovative pharmacological treatments have come to notice. Although many of them are still in the early phase of development, it is likely that in the next future they will break new therapeutic ground in the treatment of CNV. Similar to cancer therapy, where a combination of agents have been found to be more effective than monotherapy, many retina specialists are starting to believe that a combination of two or more curative approaches will result in a better visual outcome than that of a single therapy for CNV. By targeting different mechanisms with individual agents, it should be possible to not only enhance efficacy, but also minimize unwanted collateral effects by using lower concentrations of each drug than those which would be used in monotherapy. In view of this new scenario, the relationships among pharmacogenetic predictors and diverse treatments towards CNV have been recently investigated considering the different responsiveness of subfoveal CNV to either photodynamic therapy with verteporfin [151–158] or intravitreal anti-VEGF agents [159, 160]. At present, it is very difficult to draw any unequivocal conclusion regarding the therapeutic influence towards CNV of those common immunologic gene polymorphisms, such as CFH Y402H and LOC387715/ARMS2 A69S, described as determinants of both phenotype and/or severity of AMD [76–101] and of the efficacy of very dissimilar CNV treatments [156–160], even if not in all published studies [161]. Unfortunately, there is no study in which has been verified the possible correlation between the allelic variants in complement cascade genes and the curative impact on CNV of immunotherapies, employed in single or combined modality, against the extravascular component of the lesion. Future investigations are warranted to outline these and other pharmacogenetic aspects about the possible treatments of subfoveal CNVs, stratifying the enrolled patients also on the basis of their different genotypic backgrounds for a remarkable optimization of each anti-CNV therapeutic strategy.
Acknowledgment
The authors are indebted to Ms. Graziella Ferraresi for the logistic support.
References
- 1.Green WR, Wilson DJ. Choroidal neovascularization. Ophthalmology. 1986;93(9):1169–1176. doi: 10.1016/s0161-6420(86)33609-1. [DOI] [PubMed] [Google Scholar]
- 2.Klein R, Peto T, Bird A, Vannewkirk MR. The epidemiology of age-related macular degeneration. American Journal of Ophthalmology. 2004;137(3):486–495. doi: 10.1016/j.ajo.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 3.Bressler NM. Photodynamic therapy of subfoveal choroidal neovascularization in age- related macular degeneration with verteporfin: one-year results of 2 randomized clinical trials—TAP report 1. Archives of Ophthalmology. 1999;117(10):1329–1345. [PubMed] [Google Scholar]
- 4.Arnold J, Barbezetto I, Birngruber R, et al. Verteporfin therapy of subfoveal choroidal neovascularization in age-related macular degeneration: two-year results of a randomized clinical trial including lesions with occult with no classic choroidal neovascularization—verteporfin in photodynamic therapy report 2. American Journal of Ophthalmology. 2001;131(5):541–560. doi: 10.1016/s0002-9394(01)00967-9. [DOI] [PubMed] [Google Scholar]
- 5.Bressler NM. Guidelines for using verteporfin (Visudyne) in photodynamic therapy for choroidal neovascularization due to age-related macular degeneration and other causes: update. Retina. 2005;25(2):119–134. doi: 10.1097/00006982-200502000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Chakravarthy U, Soubrane G, Bandello F, et al. Evolving European guidance on the medical management of neovascular age related macular degeneration. British Journal of Ophthalmology. 2006;90(9):1188–1196. doi: 10.1136/bjo.2005.082255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gass JDM. Stereoscopic Atlas of Macular Diseases: Diagrams and Treatment. Vol. 1. St. Louis, Mo, USA: Mosby; 1997. [Google Scholar]
- 8.Grossniklaus HE, Green WR. Choroidal neovascularization. American Journal of Ophthalmology. 2004;137(3):496–503. doi: 10.1016/j.ajo.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 9.Killingsworth MC. Angiogenesis in early choroidal neovascularization secondary to age-related macular degeneration. Graefe’s Archive for Clinical and Experimental Ophthalmology. 1995;233(6):313–323. doi: 10.1007/BF00200479. [DOI] [PubMed] [Google Scholar]
- 10.Grossniklaus HE, Green WR. Histopathologic and ultrastructural findings of surgically excised choroidal neovascularization. Archives of Ophthalmology. 1998;116(6):745–749. doi: 10.1001/archopht.116.6.745. [DOI] [PubMed] [Google Scholar]
- 11.Grossniklaus HE, Ling JX, Wallace TM, et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Molecular Vision. 2002;21(8):119–126. [PubMed] [Google Scholar]
- 12.Tsutsumi-Miyahara C, Sonoda KH, Egashira K, et al. The relative contributions of each subset of ocular infiltrated cells in experimental choroidal neovascularisation. British Journal of Ophthalmology. 2004;88(9):1217–1222. doi: 10.1136/bjo.2003.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Espinosa-Heidmann DG, Reinoso MA, Pina Y, Csaky KG, Caicedo A, Cousins SW. Quantitative enumeration of vascular smooth muscle cells and endothelial cells derived from bone marrow precursors in experimental choroidal neovascularization. Experimental Eye Research. 2005;80(3):369–378. doi: 10.1016/j.exer.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Sheridan CM, Rice D, Hiscott PS, Wong D, Kent DL. The presence of AC133-positive cells suggests a possible role of endothelial progenitor cells in the formation of choroidal neovascularization. Investigative Ophthalmology & Visual Science. 2006;47(4):1642–1645. doi: 10.1167/iovs.05-0779. [DOI] [PubMed] [Google Scholar]
- 15.Spaide RF. Rationale for combination therapies for choroidal neovascularization. American Journal of Ophthalmology. 2006;141(1):149–156. doi: 10.1016/j.ajo.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 16.Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Investigative Ophthalmology & Visual Science. 1996;37(5):855–868. [PubMed] [Google Scholar]
- 17.Kvanta A, Algvere PV, Berglin L, Seregard S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Investigative Ophthalmology & Visual Science. 1996;37(9):1929–1934. [PubMed] [Google Scholar]
- 18.Ishibashi T, Hata Y, Yoshikawa H, Nakagawa K, Sueishi K, Inomata H. Expression of vascular endothelial growth factor in experimental choroidal neovascularization. Graefe’s Archive for Clinical and Experimental Ophthalmology. 1997;235(3):159–167. doi: 10.1007/BF00941723. [DOI] [PubMed] [Google Scholar]
- 19.Wada M, Ogata N, Otsuji T, Uyama M. Expression of vascular endothelial growth factor and its receptor (KDR/flk-1) mRNA in experimental choroidal neovascularization. Current Eye Research. 1999;18(3):203–213. doi: 10.1076/ceyr.18.3.203.5368. [DOI] [PubMed] [Google Scholar]
- 20.Otani A, Takagi H, Oh H, et al. Vascular endothelial growth factor family and receptor expression in human choroidal neovascular membranes. Microvascular Research. 2002;64(1):162–169. doi: 10.1006/mvre.2002.2407. [DOI] [PubMed] [Google Scholar]
- 21.Blaauwgeers HGT, Holtkamp GM, Rutten H, et al. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris: evidence for a trophic paracrine relation. American Journal of Pathology. 1999;155(2):421–428. doi: 10.1016/S0002-9440(10)65138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrara N, Gerber HP, Le Couter J. The biology of VEGF and its receptors. Nature Medicine. 2003;9(6):669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 23.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438(7070):946–953. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 24.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocrine Reviews. 2004;25(4):581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 25.Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor. The Journal of Biological Chemistry. 1992;267(36):26031–26037. [PubMed] [Google Scholar]
- 26.Keyt BA, Berleau LT, Nguyen HV, et al. The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. The Journal of Biological Chemistry. 1996;271(13):7788–7795. doi: 10.1074/jbc.271.13.7788. [DOI] [PubMed] [Google Scholar]
- 27.Lee S, Jilani SM, Nikolova GV, Carpizo D, Luisa Iruela-Arispe M. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. Journal of Cell Biology. 2005;169(4):681–691. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255(5047):989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 29.Grossniklaus HE, Martinez JA, Brown VB, et al. Immunohistochemical and histochemical properties of surgically excised subretinal neovascular membranes in age-related macular degeneration. American Journal of Ophthalmology. 1992;114(4):464–472. doi: 10.1016/s0002-9394(14)71859-8. [DOI] [PubMed] [Google Scholar]
- 30.Frank RN, Amin RH, Eliott D, Puklin JE, Abrams GW. Basic fibroblast growth factor and vascular endothelial growth factor are present in epiretinal and choroidal neovascular membranes. American Journal of Ophthalmology. 1996;122(3):393–403. doi: 10.1016/s0002-9394(14)72066-5. [DOI] [PubMed] [Google Scholar]
- 31.Hera R, Keramidas M, Peoc’h M, Mouillon M, Romanet JP, Feige JJ. Expression of VEGF and angiopoietins in subfoveal membranes from patients with age-related macular degeneration. American Journal of Ophthalmology. 2005;139(4):589–596. doi: 10.1016/j.ajo.2004.11.064. [DOI] [PubMed] [Google Scholar]
- 32.Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. The New England Journal of Medicine. 1994;331(22):1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 33.Wells JA, Murthy R, Chibber R, et al. Levels of vascular endothelial growth factor are elevated in the vitreous of patients with subretinal neovascularisation. British Journal of Ophthalmology. 1996;80(4):363–366. doi: 10.1136/bjo.80.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spilsbury K, Garrett KL, Shen WY, Constable IJ, Rakoczy PE. Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization. American Journal of Pathology. 2000;157(1):135–144. doi: 10.1016/S0002-9440(10)64525-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Csaky KG, Baffi JZ, Byrnes GA, et al. Recruitment of marrow-derived endothelial cells to experimental choroidal neovascularization by local expression of vascular endothelial growth factor. Experimental Eye Research. 2004;78(6):1107–1116. doi: 10.1016/j.exer.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Krzystolik MG, Afshari MA, Adamis AP, et al. Prevention of experimental choroidal neovascularization with intravitreal anti-vascular endothelial growth factor antibody fragment. Archives of Ophthalmology. 2002;120(3):338–346. doi: 10.1001/archopht.120.3.338. [DOI] [PubMed] [Google Scholar]
- 37.Saishin Y, Saishin Y, Takahashi K, et al. VEGF-TRAPR1R2 suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. Journal of Cellular Physiology. 2003;195(2):241–248. doi: 10.1002/jcp.10246. [DOI] [PubMed] [Google Scholar]
- 38.Tolentino MJ, Miller JW, Gragoudas ES, Chatzistefanou K, Ferrara N, Adamis AP. Vascular endothelial growth factor is sufficient to produce iris neovascularization and neovascular glaucoma in a nonhuman primate. Archives of Ophthalmology. 1996;114(8):964–970. doi: 10.1001/archopht.1996.01100140172010. [DOI] [PubMed] [Google Scholar]
- 39.Sheridan CM, Pate S, Hiscott P, Wong D, Pattwell DM, Kent D. Expression of hypoxia-inducible factor-1α and -2α in human choroidal neovascular membranes. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2009;247(10):1361–1367. doi: 10.1007/s00417-009-1133-3. [DOI] [PubMed] [Google Scholar]
- 40.Asahara T, Takahashi T, Masuda H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. The EMBO Journal. 1999;18(14):3964–3972. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guerin E, Sheridan CM, Assheton D, et al. SDF1-alpha is associated with VEGFR-2 in human choroidal neovascularisation. Microvascular Research. 2008;75(3):302–307. doi: 10.1016/j.mvr.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 42.Lamoreaux WJ, Fitzgerald MEC, Reiner A, Hasty KA, Charles ST. Vascular endothelial growth factor increases release of gelatinase A and decreases release of tissue inhibitor of metalloproteinases by microvascular endothelial cells in vitro. Microvascular Research. 1998;55(1):29–42. doi: 10.1006/mvre.1997.2056. [DOI] [PubMed] [Google Scholar]
- 43.Clauss M, Gerlach M, Gerlach H, et al. Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. Journal of Experimental Medicine. 1990;172(6):1535–1545. doi: 10.1084/jem.172.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marmé D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87(8):3336–3343. [PubMed] [Google Scholar]
- 45.Espinosa-Heidmann DG, Suner IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Investigative Ophthalmology & Visual Science. 2003;44(8):3586–3592. doi: 10.1167/iovs.03-0038. [DOI] [PubMed] [Google Scholar]
- 46.Noda K, She H, Nakazawa T, Hafezi-Moghadam A, et al. Vascular adhesion protein-1 blockade suppresses choroidal neovascularization. The FASEB Journal. 2008;22(8):2928–2935. doi: 10.1096/fj.07-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bian ZM, Elner SG, Strieter RM, Kunkel SL, Lukacs NW, Elner VM. IL-4 potentiates IL-1β- and TNF-α-stimulated IL-8 and MCP-1 protein production in human retinal pigment epithelial cells. Current Eye Research. 1999;18(5):349–357. doi: 10.1076/ceyr.18.5.349.5353. [DOI] [PubMed] [Google Scholar]
- 48.Mousa SA, Lorelli W, Campochiaro PA. Role of hypoxia and extracellular matrix-integrin binding in the modulation of angiogenic growth factors secretion by retinal pigmented epithelial cells. Journal of Cellular Biochemistry. 1999;74(1):135–143. [PubMed] [Google Scholar]
- 49.Izumi-Nagai K, Nagai N, Ozawa Y, et al. Interleukin-6 receptor-mediated activation of signal transducer and activator of transcription-3 (STAT3) promotes choroidal neovascularization. American Journal of Pathology. 2007;170(6):2149–2158. doi: 10.2353/ajpath.2007.061018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roh MI, Kim HS, Song JH, Lim JB, Koh HJ, Kwon OW. Concentration of cytokines in the aqueous humor of patients with naive, recurrent and regressed cnv associated with amd after bevacizumab treatment. Retina. 2009;29(4):523–529. doi: 10.1097/IAE.0b013e318195cb15. [DOI] [PubMed] [Google Scholar]
- 51.Apte RS, Richter J, Herndon J, Ferguson TA. Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PLoS Medicine. 2006;3(8, article e310) doi: 10.1371/journal.pmed.0030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barouch FC, Miller JW. Potential future targets for treating ocular neovascularization. Ophthalmology Clinics of North America. 2006;19(3):401–409. doi: 10.1016/j.ohc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Lambooij AC, van Wely KHM, Lindenbergh-Kortleve DJ, Kuijpers RWAM, Kliffen M, Mooy CM. Insulin-like growth factor-I and its receptor in neovascular age-related macular degeneration. Investigative Ophthalmology & Visual Science. 2003;44(5):2192–2198. doi: 10.1167/iovs.02-0410. [DOI] [PubMed] [Google Scholar]
- 54.Economou MA, Wu J, Vasilcanu D, et al. Inhibition of VEGF secretion and experimental choroidal neovascularization by picropodophyllin (PPP), an inhibitor of the insulin-like growth factor-1 receptor. Investigative Ophthalmology & Visual Science. 2008;49(6):2620–2626. doi: 10.1167/iovs.07-0742. [DOI] [PubMed] [Google Scholar]
- 55.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43(2):109–142. [PubMed] [Google Scholar]
- 56.Ando A, Yang A, Nambu H, Campochiaro PA. Blockade of nitric-oxide synthase reduces choroidal neovascularization. Molecular Pharmacology. 2002;62(3):539–544. doi: 10.1124/mol.62.3.539. [DOI] [PubMed] [Google Scholar]
- 57.O’Reilly MS, Holmgren L, Shing Y, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79(2):315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 58.Lai CC, Wu WC, Chen SL, et al. Suppression of choroidal neovascularization by adeno-associated virus vector expressing angiostatin. Investigative Ophthalmology & Visual Science. 2001;42(10):2401–2407. [PubMed] [Google Scholar]
- 59.O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 60.Mori K, Ando A, Gehlbach P, et al. Inhibition of choroidal neovascularization by intravenous injection of adenoviral vectors expressing secretable endostatin. American Journal of Pathology. 2001;159(1):313–320. doi: 10.1016/S0002-9440(10)61697-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dawson DW, Volpert OV, Gillis P, et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285(5425):245–248. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- 62.Saishin Y, Silva RL, Saishin Y, et al. Periocular gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization in a human-sized eye. Human Gene Therapy. 2005;16(4):473–478. doi: 10.1089/hum.2005.16.473. [DOI] [PubMed] [Google Scholar]
- 63.Gehlbach P, Demetriades AM, Yamamoto S, et al. Periocular injection of an adenoviral vector encoding pigment epithelium-derived factor inhibits choroidal neovascularization. Gene Therapy. 2003;10(8):637–646. doi: 10.1038/sj.gt.3301931. [DOI] [PubMed] [Google Scholar]
- 64.Takeda A, Baffi JZ, Kleinman ME, et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460(7252):225–230. doi: 10.1038/nature08151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De La Paz MA, Itoh Y, Toth CA, Nagase H. Matrix metalloproteinases and their inhibitors in human vitreous. Investigative Ophthalmology & Visual Science. 1998;39(7):1256–1260. [PubMed] [Google Scholar]
- 66.Steen B, Sejersen S, Berglin L, Seregard S, Kvanta A. Matrix metalloproteinases and metalloproteinase inhibitors in choroidal neovascular membranes. Investigative Ophthalmology & Visual Science. 1998;39(11):2194–2200. [PubMed] [Google Scholar]
- 67.Lambert V, Munaut C, Jost M, et al. Matrix metalloproteinase-9 contributes to choroidal neovascularization. American Journal of Pathology. 2002;161(4):1247–1253. doi: 10.1016/S0002-9440(10)64401-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lambert V, Wielockx B, Munaut C, et al. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. The FASEB Journal. 2003;17(15):2290–2292. doi: 10.1096/fj.03-0113fje. [DOI] [PubMed] [Google Scholar]
- 69.Takahashi T, Nakamura T, Hayashi A, et al. Inhibition of experimental choroidal neovascularization by overexpression of tissue inhibitor of metalloproteinases-3 in retinal pigment epithelium cells. American Journal of Ophthalmology. 2000;130(6):774–781. doi: 10.1016/s0002-9394(00)00772-8. [DOI] [PubMed] [Google Scholar]
- 70.Otani A, Takagi H, Oh H, Koyama S, Matsumura M, Honda Y. Expressions of angiopoietins and Tie2 in human choroidal neovascular membranes. Investigative Ophthalmology & Visual Science. 1999;40(9):1912–1920. [PubMed] [Google Scholar]
- 71.Lu M, Adamis AP. Molecular biology of choroidal neovascularization. Ophthalmology Clinics of North America. 2006;19(3):323–334. doi: 10.1016/j.ohc.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 72.Amin R, Puklin JE, Frank RN. Growth factor localization in choroidal neovascular membranes of age- related macular degeneration. Investigative Ophthalmology & Visual Science. 1994;35(8):3178–3188. [PubMed] [Google Scholar]
- 73.Ogata N, Yamamoto C, Miyashiro M, Yamada H, Matsushima M, Uyama M. Expression of transforming growth factor-β mRNA in experimental choroidal neovascularization. Current Eye Research. 1997;16(1):9–18. doi: 10.1076/ceyr.16.1.9.5121. [DOI] [PubMed] [Google Scholar]
- 74.Hu W, Criswell MH, Fong SL, et al. Differences in the temporal expression of regulatory growth factors during choroidal neovascular development. Experimental Eye Research. 2009;88(1):79–91. doi: 10.1016/j.exer.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 75.Bora PS, Sohn JH, Cruz JMC, et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. The Journal of Immunology. 2005;174(1):491–497. doi: 10.4049/jimmunol.174.1.491. [DOI] [PubMed] [Google Scholar]
- 76.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(20):7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 78.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 79.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wegscheider BJ, Weger M, Renner W, et al. Association of complement factor H Y402H gene polymorphism with different subtypes of exudative age-related macular degeneration. Ophthalmology. 2007;114(4):738–742. doi: 10.1016/j.ophtha.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 81.Seddon JM, Francis PJ, George S, Schultz DW, Rosner B, Klein ML. Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. Journal of the American Medical Association. 2007;297(16):1793–1800. doi: 10.1001/jama.297.16.1793. [DOI] [PubMed] [Google Scholar]
- 82.Brantley MA, Jr., Edelstein SL, King JM, Apte RS, Kymes SM, Shiels A. Clinical phenotypes associated with the complement factor H Y402H variant in age-related macular degeneration. American Journal of Ophthalmology. 2007;144(3):404–408. doi: 10.1016/j.ajo.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seitsonen SP, Onkamo P, Peng G, et al. Multifactor effects and evidence of potential interaction between complement factor HY402H and LOC387715 A69S in age-related macular degeneration. PLoS ONE. 2008;3(12, article e3833) doi: 10.1371/journal.pone.0003833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shuler RK, Jr., Schmidt S, Gallins P, et al. Phenotype analysis of patients with the risk variant LOC387715 (A69S) in age-related macular degeneration. American Journal of Ophthalmology. 2008;145(2):303–307. doi: 10.1016/j.ajo.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 85.Droz I, Mantel I, Ambresin A, Faouzi M, Schorderet DF, Munier FL. Genotype-phenotype correlation of age-related macular degeneration: influence of complement factor H polymorphism. British Journal of Ophthalmology. 2008;92(4):513–517. doi: 10.1136/bjo.2007.127811. [DOI] [PubMed] [Google Scholar]
- 86.Kim NR, Kang JH, Kwon OW, Lee SJ, Oh JH, Chin HS. Association between complement factor H gene polymorphisms and neovascular age-related macular degeneration in koreans. Investigative Ophthalmology & Visual Science. 2008;49(5):2071–2076. doi: 10.1167/iovs.07-1195. [DOI] [PubMed] [Google Scholar]
- 87.Kaur I, Katta S, Hussain A, et al. Variants in the 10q26 gene cluster (LOC387715 and HTRA1) exhibit enhanced risk of age-related macular degeneration along with CFH in Indian patients. Investigative Ophthalmology & Visual Science. 2008;49(5):1771–1776. doi: 10.1167/iovs.07-0560. [DOI] [PubMed] [Google Scholar]
- 88.Deangelis MM, Ji F, Adams S, et al. Alleles in the HtrA serine peptidase 1 gene alter the risk of neovascular age-related macular degeneration. Ophthalmology. 2008;115(7):1209–1215. doi: 10.1016/j.ophtha.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leveziel N, Zerbib J, Richard F, et al. Genotype-phenotype correlations for exudative age-related macular degeneration associated with homozygous HTRA1 and CFH genotypes. Investigative Ophthalmology & Visual Science. 2008;49(7):3090–3094. doi: 10.1167/iovs.07-1540. [DOI] [PubMed] [Google Scholar]
- 90.Gotoh N, Yamada R, Nakanishi H, et al. Correlation between CFH Y402H and HTRA1 rs11200638 genotype to typical exudative age-related macular degeneration and polypoidal choroidal vasculopathy phenotype in the Japanese population. Clinical and Experimental Ophthalmology. 2008;36(5):437–442. [PubMed] [Google Scholar]
- 91.Xu Y, Guan N, Xu J, et al. Association of CFH, LOC387715, and HTRA1 polymorphisms with exudative age-related macular degeneration in a northern Chinese population. Molecular Vision. 2008;14:1373–1381. [PMC free article] [PubMed] [Google Scholar]
- 92.Ricci F, Zampatti S, D’Abbruzzi F, et al. Typing of ARMS2 and CFH in age-related macular degeneration: case-control study and assessment of frequency in the Italian population. Archives of Ophthalmology. 2009;127(10):1368–1372. doi: 10.1001/archophthalmol.2009.237. [DOI] [PubMed] [Google Scholar]
- 93.Francis PJ, Zhang H, DeWan A, Hoh J, Klein ML. Joint effects of polymorphisms in the HTRA1, LOC387715/ARMS2, and CFH genes on AMD in a Caucasian population. Molecular Vision. 2008;14:1395–1400. [PMC free article] [PubMed] [Google Scholar]
- 94.Kim IK, Ji F, Morrison MA, et al. Comprehensive analysis of CRP, CFH Y402H and environmental risk factors on risk of neovascular age-related macular degeneration. Molecular Vision. 2008;14:1487–1495. [PMC free article] [PubMed] [Google Scholar]
- 95.Klein R, Knudtson MD, Klein BE, et al. Inflammation, complement factor H, and age-related macular degeneration: the multi-ethnic study of atherosclerosis. Ophthalmology. 2008;115(10):1742–1749. doi: 10.1016/j.ophtha.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marioli DI, Pharmakakis N, Deli A, Havvas I, Zarkadis IK. Complement factor H and LOC 387715 gene polymorphisms in a Greek population with age-related macular degeneration. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2009;247(11):1547–1553. doi: 10.1007/s00417-009-1129-z. [DOI] [PubMed] [Google Scholar]
- 97.Jiang H, Qu Y, Dang G, et al. Analyses of single nucleotide polymorphisms and haplotype linkage of LOC387715 and the HTRA1 gene in exudative age-related macular degeneration in a Chinese cohort. Retina. 2009;29(7):974–979. doi: 10.1097/IAE.0b013e3181a3b90e. [DOI] [PubMed] [Google Scholar]
- 98.Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Investigative Ophthalmology & Visual Science. 2009;50(12):5818–5827. doi: 10.1167/iovs.09-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lederman M, Weiss A, Chowers I. Association of neovascular age-related macular degeneration with specific gene expression patterns in peripheral white blood cells. Investigative Ophthalmology & Visual Science. 2010;51(1):53–58. doi: 10.1167/iovs.08-3019. [DOI] [PubMed] [Google Scholar]
- 100.Kaur I, Katta S, Reddy RK, et al. The involvement of complement factor B and complement component C2 in an Indian cohort with age-related macular degeneration. Investigative Ophthalmology & Visual Science. 2010;51(1):59–63. doi: 10.1167/iovs.09-4135. [DOI] [PubMed] [Google Scholar]
- 101.Francis PJ, Hamon SC, Ott J, Weleber RG, Klein ML. Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss. Journal of Medical Genetics. 2009;46(5):300–307. doi: 10.1136/jmg.2008.062737. [DOI] [PubMed] [Google Scholar]
- 102.de Córdoba SR, de Jorge EG. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clinical and Experimental Immunology. 2008;151(1):1–13. doi: 10.1111/j.1365-2249.2007.03552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hageman GS, Hancox LS, Taiber AJ, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Annals of Medicine. 2006;38(8):592–604. [PMC free article] [PubMed] [Google Scholar]
- 104.Machalińska A, Dziedziejko V, Mozolewska-Piotrowska K, Karczewicz D, Wiszniewska B, MacHaliński B. Elevated plasma levels of C3a complement compound in the exudative form of age-related macular degeneration. Ophthalmic Research. 2009;42(1):54–59. doi: 10.1159/000219686. [DOI] [PubMed] [Google Scholar]
- 105.Jain RK. Molecular regulation of vessel maturation. Nature Medicine. 2003;9(6):685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 106.Gaengel K, Genové G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(5):630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 107.Thomas M, Augustin HG. The role of the angiopoietins in vascular morphogenesis. Angiogenesis. 2009;12(2):125–137. doi: 10.1007/s10456-009-9147-3. [DOI] [PubMed] [Google Scholar]
- 108.Jo N, Mailhos C, Ju M, et al. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. American Journal of Pathology. 2006;168(6):2036–2053. doi: 10.2353/ajpath.2006.050588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kent D, Sheridan C. Choroidal neovascularization: a wound healing perspective. Molecular Vision. 2003;9:747–755. [PubMed] [Google Scholar]
- 110.Ferrara N, Mass RD, Campa C, Kim R. Targeting VEGF-A to treat cancer and age-related macular degeneration. Annual Review of Medicine. 2007;58:491–504. doi: 10.1146/annurev.med.58.061705.145635. [DOI] [PubMed] [Google Scholar]
- 111.Gragoudas ES, Adamis AP, Cunningham ET, Jr., Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. The New England Journal of Medicine. 2004;351(27):2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- 112.Chakravarthy U, Adamis AP, Cunningham ET, Jr., et al. Year 2 efficacy results of 2 randomized controlled clinical trials of pegaptanib for neovascular age-related macular degeneration. Ophthalmology. 2006;113(9):1508–1521. doi: 10.1016/j.ophtha.2006.02.064. [DOI] [PubMed] [Google Scholar]
- 113.Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. The New England Journal of Medicine. 2006;355(14):1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 114.Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. The New England Journal of Medicine. 2006;355(14):1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- 115.Brown DM, Michels M, Kaiser PK, Heier JS, Sy JP, Ianchulev T. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study. Ophthalmology. 2009;116(1):57–65. doi: 10.1016/j.ophtha.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 116.Avery RL, Pieramici DJ, Rabena MD, Castellarin AA, Nasir MA, Giust MJ. Intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Ophthalmology. 2006;113(3):363–372. doi: 10.1016/j.ophtha.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 117.Bashshur ZF, Bazarbachi A, Schakal A, Haddad ZA, El Haibi CP, Noureddin BN. Intravitreal bevacizumab for the management of choroidal neovascularization in age-related macular degeneration. American Journal of Ophthalmology. 2006;142(1):1–9. doi: 10.1016/j.ajo.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 118.Spaide RF, Laud K, Fine HF, et al. Intravitreal bevacizumab treatment of choroidal neovascularization secondary to age-related macular degeneration. Retina. 2006;26(4):383–390. doi: 10.1097/01.iae.0000238561.99283.0e. [DOI] [PubMed] [Google Scholar]
- 119.Lazic R, Gabric N. Intravitreally administered bevacizumab (Avastin) in minimally classic and occult choroidal neovascularization secondary to age-related macular degeneration. Graefe’s Archives for Clinical and Experimental Ophthalmology. 2007;245(1):68–73. doi: 10.1007/s00417-006-0466-4. [DOI] [PubMed] [Google Scholar]
- 120.Algvere PV, Steén B, Seregard S, Kvanta A. A prospective study on intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration of different durations. Acta Ophthalmologica. 2008;86(5):482–489. doi: 10.1111/j.1600-0420.2007.01113.x. [DOI] [PubMed] [Google Scholar]
- 121.Ehrlich R, Weinberger D, Priel E, Axer-Siegel R. Outcome of bevacizumab (Avastin) injection in patients with age-related macular degeneration and low visual acuity. Retina. 2008;28(9):1302–1307. doi: 10.1097/IAE.0b013e3181803c2a. [DOI] [PubMed] [Google Scholar]
- 122.Costagliola C, Romano M, Corte MD, et al. Intravitreal bevacizumab for treatment-naive patients with subfoveal occult choroidal neovascularization secondary to age-related macular degeneration: a 12-month follow-up study. Retina. 2009;29(9):1227–1234. doi: 10.1097/IAE.0b013e3181b773e1. [DOI] [PubMed] [Google Scholar]
- 123.Landa G, Amde W, Doshi V, et al. Comparative study of intravitreal bevacizumab (Avastin) versus ranibizumab (Lucentis) in the treatment of neovascular age-related macular degeneration. Ophthalmologica. 2009;223(6):370–375. doi: 10.1159/000227783. [DOI] [PubMed] [Google Scholar]
- 124.Stepien KE, Rosenfeld PJ, Puliafito CA, et al. Comparison of intravitreal bevacizumab followed by ranibizumab for the treatment of neovascular age-related macular degeneration. Retina. 2009;29(8):1067–1073. doi: 10.1097/IAE.0b013e3181b1bb06. [DOI] [PubMed] [Google Scholar]
- 125.Subramanian ML, Ness S, Abedi G, et al. Bevacizumab vs ranibizumab for age-related macular degeneration: early results of a prospective double-masked, randomized clinical trial. American Journal of Ophthalmology. 2009;148(6):875–882. doi: 10.1016/j.ajo.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 126.Fong DS, Custis P, Howes J, Hsu JW. Intravitreal bevacizumab and ranibizumab for age-related macular degeneration. A multicenter, retrospective study. Ophthalmology. 2010;117(2):298–302. doi: 10.1016/j.ophtha.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 127.Wu L, Martínez-Castellanos MA, Quiroz-Mercado H, et al. Twelve-month safety of intravitreal injections of bevacizumab (Avastin®): results of the pan-american collaborative retina study group (PACORES) Graefe’s Archive for Clinical and Experimental Ophthalmology. 2008;246(1):81–87. doi: 10.1007/s00417-007-0660-z. [DOI] [PubMed] [Google Scholar]
- 128.Wong LJ, Desai RU, Jain A, et al. Surveillance for potential adverse events associated with the use of intravitreal bevacizumab for retinal and choroidal vascular disease. Retina. 2008;28(8):1151–1158. doi: 10.1097/IAE.0b013e31817e100f. [DOI] [PubMed] [Google Scholar]
- 129.Raftery J, Clegg A, Jones J, Seng CT, Lotery A. Ranibizumab (Lucentis) versus bevacizumab (Avastin): modelling cost effectiveness. British Journal of Ophthalmology. 2007;91(9):1244–1246. doi: 10.1136/bjo.2007.116616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cruess AF, Zlateva G, Xu X, et al. Economic burden of bilateral neovascular age-related macular degeneration: multi-country observational study. PharmacoEconomics. 2008;26(1):57–73. doi: 10.2165/00019053-200826010-00006. [DOI] [PubMed] [Google Scholar]
- 131.Smiddy WE. Economic implications of current age-related macular degeneration treatments. Ophthalmology. 2009;116(3):481–487. doi: 10.1016/j.ophtha.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 132.Chappelow AV, Kaiser PK. Neovascular age-related macular degeneration: potential therapies. Drugs. 2008;68(8):1029–1036. doi: 10.2165/00003495-200868080-00002. [DOI] [PubMed] [Google Scholar]
- 133.Slakter S, CLEAR-IT 2 Investigators A phase 2, randomized, controlled dose-and interval-ranging study of intravitreal VEGF trap-eye in patients with neovascular age-related macular degeneration: optical coherence tomography (OCT) and fluorescein angiography (FA) outcomes at 1 Year. Investigative Ophthalmology & Visual Science. 2009;50 abstract no. 1890. [Google Scholar]
- 134. Safety & Efficacy Study Evaluating the Combination of Bevasiranib & Lucentis Therapy in Wet AMD (COBALT). [ClinicalTrials.gov identifier: NCT00499590], http://clinicaltrials.gov/ct2/show/ NCT00499590.
- 135.Boyer DS, Ophthotech Anti-PDGF in AMD Study Group Combined inhibition of platelet derived (PDGF) and vascular endothelial (VEGF) growth factors for the treatment of neovascular age-related macular degeneration (NV-AMD)—results of a phase 1 study. Investigative Ophthalmology & Visual Science. 2009;50 abstract no. 1260. [Google Scholar]
- 136.Jo N, Mailhos C, Ju M, et al. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. American Journal of Pathology. 2006;168(6):2036–2053. doi: 10.2353/ajpath.2006.050588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Siemann DW, Chaplin DJ, Walicke PA. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P) Expert Opinion on Investigational Drugs. 2009;18(2):189–197. doi: 10.1517/13543780802691068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nambu H, Nambu R, Melia M, Campochiaro PA. Combretastatin A-4 phosphate suppresses development and induces regression of choroidal neovascularization. Investigative Ophthalmology & Visual Science. 2003;44(8):3650–3655. doi: 10.1167/iovs.02-0985. [DOI] [PubMed] [Google Scholar]
- 139.Russell SR, Hudson HL, Jerdan JA. Anecortave acetate for the treatment of exudative age-related macular degeneration—a review of clinical outcomes. Survey of Ophthalmology. 2007;52(supplement 1):S79–S90. doi: 10.1016/j.survophthal.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 140.Yasukawa T, Hoffmann S, Eichler W, Friedrichs U, Wang YS, Wiedemann P. Inhibition of experimental choroidal neovascularization in rats by an α(v)-integrin antagonist. Current Eye Research. 2004;28(5):359–366. doi: 10.1076/ceyr.28.5.359.28678. [DOI] [PubMed] [Google Scholar]
- 141.Fu Y, Ponce ML, Thill M, Yuan P, Nam SW, Csaky KG. Angiogenesis inhibition and choroidal neovascularization suppression by sustained delivery of an integrin antagonist, EMD478761. Investigative Ophthalmology & Visual Science. 2007;48(11):5184–5190. doi: 10.1167/iovs.07-0469. [DOI] [PubMed] [Google Scholar]
- 142.Capone A, Jr., Gonzalez VH, Heier JS. A phase I open-label study of single and repeated doses of intravitreal JSM6427, a small molecule integrin 5β1 antagonist, in neovascular age-related macular degeneration (AMD) Investigative Ophthalmology & Visual Science. 2009;50 abstract no. 1259. [Google Scholar]
- 143.Takahashi T, Nakamura T, Hayashi A, et al. Inhibition of experimental choroidal neovascularization by overexpression of tissue inhibitor of metalloproteinases-3 in retinal pigment epithelium cells. American Journal of Ophthalmology. 2000;130(6):774–781. doi: 10.1016/s0002-9394(00)00772-8. [DOI] [PubMed] [Google Scholar]
- 144.Cano MV, Karagiannis ED, Soliman M, et al. A peptide derived from type 1 thrombospondin repeat-containing protein WISP-1 inhibits corneal and choroidal neovascularization. Investigative Ophthalmology & Visual Science. 2009;50(8):3840–3845. doi: 10.1167/iovs.08-2607. [DOI] [PubMed] [Google Scholar]
- 145.Takahashi K, Saishin Y, Saishin Y, et al. Topical nepafenac inhibits ocular neovascularization. Investigative Ophthalmology & Visual Science. 2003;44(1):409–415. doi: 10.1167/iovs.02-0346. [DOI] [PubMed] [Google Scholar]
- 146.Dejneka NS, Kuroki AM, Fosnot J, Tang W, Tolentino MJ, Bennett J. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Molecular Vision. 2004;10:964–972. [PubMed] [Google Scholar]
- 147. Phase 2 Study of an Ocular Sirolimus (Rapamycin) Formulation in Combination With Lucentis® in Patients With Age-Related Macular Degeneration (EMERALD) [ClinicalTrials.gov identifier: NCT00766337], http://clinicaltrials.gov/ct2/show/NCT00766337.
- 148.Regatieri CV, Dreyfuss JL, Melo GB, et al. Dual role of intravitreous infliximab in experimental choroidal neovascularization. Effect on the expression of sulfated glycosaminoglycans. Investigative Ophthalmology & Visual Science. 2009;50(11):5487–5494. doi: 10.1167/iovs.08-3171. [DOI] [PubMed] [Google Scholar]
- 149.Olson JL, Courtney RJ, Mandava N. Intravitreal infliximab and choroidal neovascularization in an animal model. Archives of Ophthalmology. 2007;125(9):1221–1224. doi: 10.1001/archopht.125.9.1221. [DOI] [PubMed] [Google Scholar]
- 150.Ueta T, Yanagi Y, Tamaki Y, Yamaguchi T. Cerebrovascular accidents in ranibizumab. Ophthalmology. 2009;116(2):p. 362. doi: 10.1016/j.ophtha.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 151.Parmeggiani F, Costagliola C, Gemmati D, et al. Predictive role of coagulation-balance gene polymorphisms in the efficacy of photodynamic therapy with verteporfin for classic choroidal neovascularization secondary to age-related macular degeneration. Pharmacogenetics and Genomics. 2007;17(12):1039–1046. doi: 10.1097/FPC.0b013e3282f12a4e. [DOI] [PubMed] [Google Scholar]
- 152.Parmeggiani F, Costagliola C, Gemmati D, et al. Coagulation-balance genetic predictors for efficacy of photodynamic therapy in occult choroidal neovascularization secondary to age-related macular degeneration. Investigative Ophthalmology & Visual Science. 2008;49(7):3100–3106. doi: 10.1167/iovs.07-1654. [DOI] [PubMed] [Google Scholar]
- 153.Parmeggiani F, Gemmati D, Costagliola C, Sebastiani A, Incorvaia C. Predictive role of C677T MTHFR polymorphism in variable efficacy of photodynamic therapy for neovascular age-related macular degeneration. Pharmacogenomics. 2009;10(1):81–95. doi: 10.2217/14622416.10.1.81. [DOI] [PubMed] [Google Scholar]
- 154.Parmeggiani F, Gemmati D, Costagliola C, Sebastiani A, Incorvaia C. Predictive role of gene polymorphisms affecting thrombin-generation pathway in variable efficacy of photodynamic therapy for neovascular age-related macular degeneration. Recent Patents on DNA and Gene Sequences. 2009;3(2):114–122. doi: 10.2174/187221509788654151. [DOI] [PubMed] [Google Scholar]
- 155.Parmeggiani F, Gemmati D, Costagliola C, et al. Impact of coagulation-balance gene predictors on efficacy of photodynamic therapy for choroidal neovascularization in pathologic myopia. Ophthalmology. 2010;117(3):517–523. doi: 10.1016/j.ophtha.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 156.Goverdhan SV, Hannan S, Newsom RB, Luff AJ, Griffiths H, Lotery AJ. An analysis of the CFH Y402H genotype in AMD patients and controls from the UK, and response to PDT treatment. Eye. 2008;22(6):849–854. doi: 10.1038/sj.eye.6702830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chowers I, Meir T, Lederman M, et al. Sequence variants in HTRA1 and LOC387715/ARMS2 and phenotype and response to photodynamic therapy in neovascular age-related macular degeneration in populations from Israel. Molecular Vision. 2008;14:2263–2271. [PMC free article] [PubMed] [Google Scholar]
- 158.Brantley MA, Jr., Edelstein SL, King JM, et al. Association of complement factor H and LOC387715 genotypes with response of exudative age-related macular degeneration to photodynamic therapy. Eye. 2009;23(3):626–631. doi: 10.1038/eye.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Brantley MA, Jr., Fang AM, King JM, Tewari A, Kymes SM, Shiels A. Association of complement factor H and LOC387715 genotypes with response of exudative age-related macular degeneration to intravitreal bevacizumab. Ophthalmology. 2007;114(12):2168–2173. doi: 10.1016/j.ophtha.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 160.Lee AY, Raya AK, Kymes SM, Shiels A, Brantley MA., Jr. Pharmacogenetics of complement factor H (Y402H) and treatment of exudative age-related macular degeneration with ranibizumab. British Journal of Ophthalmology. 2009;93(5):610–613. doi: 10.1136/bjo.2008.150995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Feng X, Xiao J, Longville B, et al. Complement factor H Y402H and C-reactive protein polymorphism and photodynamic therapy response in age-related macular degeneration. Ophthalmology. 2009;116(10):1908–1912. doi: 10.1016/j.ophtha.2009.03.011. [DOI] [PubMed] [Google Scholar]