

Abstract
The structure-based design, synthesis and biological evaluation of a series of nonpeptidic macrocyclic HIV protease inhibitors are described. The inhibitors are designed to effectively fill in the hydrophobic pocket in the S1′ S2′ subsites and retain all major hydrogen bonding with the protein backbone similar to darunavir (1) or inhibitor 2. The ring size, the effect of methyl substitution and unsaturation within the macrocyclic ring structure were assessed. In general, cyclic inhibitors were significantly more potent than their acyclic homologues, saturated rings were less active than their unsaturated analogs and a preference for 10 and 13-membered macrocylic rings was revealed. The addition of methyl substituents resulted in the reduction of potency. Both inhibitors 14b and 14c exhibited marked enzyme inhibitory and antiviral activity and they exerted potent activity against multi-drug resistant HIV-1 variants. Protein-ligand X-ray structures of inhibitors 2 and 14c provided critical molecular insights into the ligand-binding site interactions.
Introduction
HIV/AIDS has become one of the major medical and humanitarian challenges in the 21st century.1 Highly active antiretroviral therapy (HAART), which combines protease inhibitors along with reverse-transcriptase inhibitors, is currently used to combat this pandemic. HAART therapy has resulted in a significant decline in the number of deaths due to HIV infection in the developed countries.2 One of the major challenges still faced is the emergence of drug resistant viral strains rendering the present drug regimen ineffective.3 There is an urgent need for development of antiretroviral agents with minimal side effects and broad-spectrum activity for current and future management of HIV/AIDS.
Recently, our structure-based design of inhibitors maximizing interactions within the active site protease backbone led to the development of nonpeptide inhibitors (1-2) that have shown picomolar enzyme affinity and exceptional antiviral activity against both wild-type and drug-resistant HIV-1 strains.4 6 The X-ray crystallographic studies revealed that the backbone conformation of mutant protease is minimally distorted compared to wild-type HIV-1 proteases. We, therefore, speculated that maximizing ‘backbone binding’ may be an important design strategy to combat drug-resistance.7 Inhibitor 1 (Darunavir, TMC 114) was recently approved by the FDA for the treatment of drug resistant HIV strains.8 More recently, it has been approved for all HIV/AIDS patients including pediatric AIDS patients.9 The protein-ligand X-ray structure of darunavir and its analog 2 (TMC 126) exhibited extensive hydrogen bonding interactions with the backbone atoms throughout the active site of the HIV-1 protease.10, 11
In our continuing efforts towards the conceptual design of novel PIs, we now plan to design PIs with functionalities that interact with the protein backbone as well as introduce flexible macrocycles involving P1′ -P2′ -ligands for effective repacking due to side chain mutations. The notion of such macrocyclic design evolved from the observation that certain mutations lead to decreased van der Waals interactions and increased the size of the S1 hydrophobic pocket. The X-ray structure and modeling studies of PI (2S,2′S) -N,N’ -((2S,3R,4R,5S)-3-hydroxy-4-methyl 1,6-diphenylhexane-2,5-diyl)bis(3-methyl-2-(3-methyl-3-(pyridin-2-ylmethyl)ureido)butanamide) (A-77003)12 indicated that the V82A mutant results in decreased van der Waals interaction with the phenyl rings in both the S1 and S1′ -subsites.13 There was also evidence of repacking of inhibitor side chain and enzyme atoms in the S1-subsite. Based upon this insight of enzyme flexibility in accommodating alternate packing, we planned to design flexible macrocycles between the P1′ -side chain and the P2′ -sulfonamide ring to fill in the S1′ and S2′ -subsites.13 In particular, we envisioned that 11-15-membered saturated and unsaturated macrocycles would effectively fill in the S1′ -S2′ hydrophobic pocket of the enzyme active site while retaining all major interactions of PIs 1 and 2 with the protein backbone. Conceivably, such inhibitors will maintain potency against both wild-type and mutant strains. Based on this presumption, we designed a series of PIs that incorporated various macrocycles that could effectively fill in the enzyme active site. Herein we report the structure-based design, synthesis, and preliminary biological results of these macrocylic inhibitors. Among these compounds, 14b and 14c were the most potent with excellent enzyme inhibitory and antiviral activity. Both inhibitors exerted potent activity against multi-drug resistant HIV-1 variants. Furthermore, protein-ligand X-ray structures of inhibitors 2 and 14c bound HIV-1 protease have revealed important insights regarding ligand-binding interactions.
Chemistry
The synthesis of sulfonyl chlorides 7a-d is shown in Scheme 1. A Mitsunobu-type reaction between 3-methoxyphenol and alcohols 4a-d in the presence of triphenylphosphine and diisopropyl azodicarboxylate (DIAD) afforded ethers 5a-d.14 Electrophilic aromatic substitution of ethers 5a-d with acetic anhydride and concentrated sulfuric acid in methanol furnished a mixture of sulfonic acid regioisomers in a 1:1 ratio that were separated by flash chromatography to give 6a-d. Structural confirmation of the isomers was determined by extensive 2D NMR experiments (NOESY and HMBC). Conversion to the sulfonyl chlorides 7a-d was achieved by reaction of the sulfonic acids 6a-d with thionyl chloride in the presence of pyridine.
Scheme 1.

Synthesis of sulfonyl chlorides 7a-d
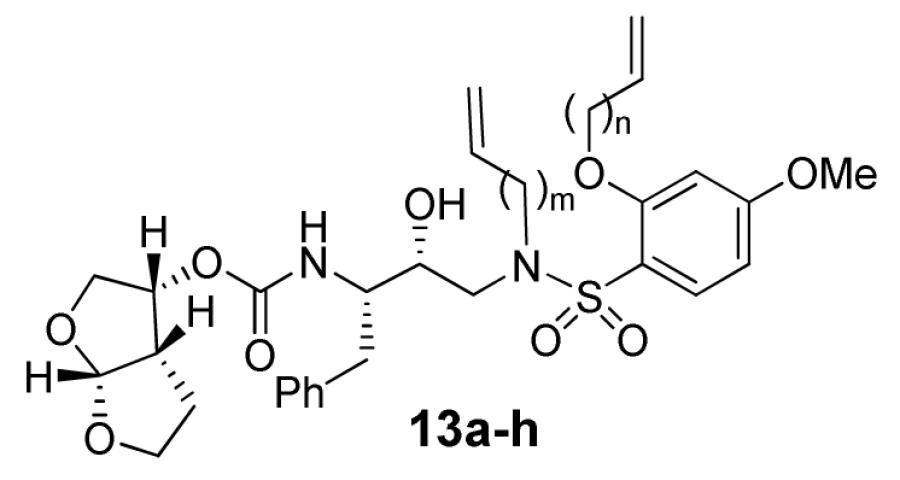
The synthesis of compounds 13a-h is outlined in Scheme 2. Nucleophilic attacks of amines 9a and 9b on commercially available epoxide 8 in the presence of isopropanol gave hydroxy amines 10a and 10b. The conversion of amines 10a and 10b to the sulfonamides 11a-h was realized by coupling with sulfonyl chlorides 7a-d in the presence of pyridine. Removal of the Boc protecting group from sulfonamides 11a-h using 30% trifluoroacetic acid in CH2Cl2 furnished the corresponding amines, which were then coupled with activated bis-THF15 (12) to give acyclic inhibitors 13a-h.
Scheme 2.

Synthesis of compounds 13a-h
The acyclic compounds 13a-h thus obtained were exposed to ring closing metathesis using Grubbs’ 1st or 2nd generation catalyst16(Scheme 3) to give the unsaturated macrocyclic inhibitors 14a-h. Interestingly, larger ring sizes (15-13) gave a mixture of E/Z isomers, while in the case of smaller rings (12-9) the Z isomer was obtained exclusively. The E and Z isomers were isolated using reversed-phase HPLC and the stereochemistry established by 2-D NMR (COSY and NOESY) experiments allowing their individual biological characterization. The unsaturated compounds were subsequently reduced using hydrogen and 10% Pd-C as a catalyst to yield inhibitors 15a-h.
Scheme 3.

Synthesis of macrocyclic inhibitors 14a-h and 15a-h
A series of selectively methylated inhibitors were prepared in a similar fashion. Nucleophilic attack of amines 16a-c on commercially available epoxide 8 gave hydroxy amines 17a-c (Scheme 4). The conversion of amines 17a-c to the sulfonamides 18a-c was realized by coupling the amines with sulfonyl chloride 7d in the presence of pyridine. Removal of the Boc protecting group from sulfonamides 18a-c using 30% trifluoroacetic acid in CH2Cl2 furnished the corresponding amines, which were then coupled with the mixed carbonate of activated bis THF15 (12) to give acyclic inhibitors 19a-c. A ring closing metathesis reaction using Grubbs’ 2nd generation catalyst16 (Scheme 5) provided E/Z mixture of unsaturated macrocyclic inhibitors 20a-c and 21a-c, which were separated by reversed phase HPLC and identified by 2-D NMR (COSY and NOESY). The unsaturated compounds 20a-c and 21a-c were subsequently hydrogenated over 10% Pd C as catalyst to yield inhibitors 22a-c.
Scheme 4.

Synthesis of compounds 19a-c
Scheme 5.

Synthesis of macrocyclic inhibitors 20a-c, 21a-c and 22a-c
Biological Evaluation and Discussion
The inhibitory potencies of acyclic and cyclic inhibitors were measured by the assay protocol of Toth and Marshall.17 Compounds that showed potent enzyme inhibition Ki values were further evaluated in an antiviral assay. Biological results for the acyclic compounds 13a-h are shown in Table 1. In general, elongation of the carbon chains resulted in lower enzyme inhibitory activity. Extension of the β hydroxyl amine chain by three methylene groups resulted in a 10 fold loss in activity (13a Ki = 16 nM versus 13e Ki = 1.7 nM). Similarly, extension of the ether carbon chain by three methylene groups resulted in a 17 fold loss in activity (13h Ki = 0.10 nM versus 13e Ki = 1.7 nM).
Table 1.
Enzyme inhibitory and antiviral activity of inhibitors 13a-h
 | |||||
|---|---|---|---|---|---|
| Compd | m | n | Ki(nM) | IC50(nM)a | Ring Size by RCM |
| 13a | 4 | 4 | 16.5 ± 0.5 | >1000 | 15 |
| 13b | 4 | 3 | 11.5 ± 0.4 | >1000 | 14 |
| 13c | 4 | 2 | 6.9 ± 0.1 | ND | 13 |
| 13d | 4 | 1 | 10 ± 2 | ND | 12 |
| 13e | 1 | 4 | 1.70 ± 0.07 | 270 | 12 |
| 13f | 1 | 3 | 1.02 ± 0.08 | 290 | 11 |
| 13g | 1 | 2 | 0.63 ± 0.01 | ND | 10 |
| 13h | 1 | 1 | 0.10 ± 0.01 | ND | 9 |
human T-lymphoid cells, MT-2 cells (2×103), were exposed to HIV-1LAI (100 TCID50), cultured in the presence of each PI, and IC50 values were determined by using the MTT assay. The IC50 values of saquinaivr (SQV) and amprenavir (APV) tested as reference agents were 16 nM and 27 nM, respectively. ND: not determined.
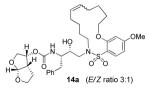
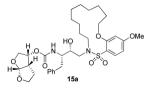
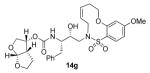
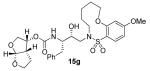
Interestingly, conversion of these acyclic inhibitors 13a-h to their cyclic analogs 14a-h and 15a-h resulted in significant improvements of enzyme inhibitory and antiviral activity as shown in Table 2. For example, the 14-membered macrocyclic inhibitors 14b and 15b have Ki values of less than 0.7 nM and IC50 values less than 49 nM whereas their corresponding acyclic inhibitor 13b had a Ki of 11 nM and IC50 value of >1000 nM (greater than 15-fold and 20-fold change, respectively). Another trend observed is a preference of the S2′ subsite for macrocylic rings of size 10 and 13. This preference is consistent with our energy-minimized model structure of a saturated 13-membered prototype inhibitor 15c where a macrocycle is incorporated in place of the P1′-isobutyl and attached at the ortho-position of the sulfonamide ring of inhibitor 2 as shown in Figure 2. The model of inhibitor 15c was created based upon the crystal structure of inhibitor 2-bound HIV-1 protease.
Table 2.
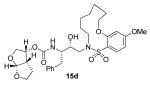
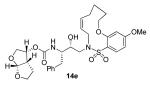
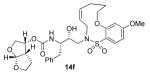
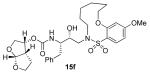
Enzyme inhibitory and antiviral activity of inhibitors 14a-h and 15a-h
| Ring size |
Inhibitor structure | Ki(nM) | IC50(nM)a |
|---|---|---|---|
| 15 |
|
0.38 ± 0.03 | 120 |
| 15 |
|
0.82 ± 0.03 | 318 |
| 14 |
|
0.082 ± 0.008 | 4 |
| 14 |
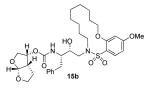
|
0.67 ± 0.02 | 49 |
| 13 |
|
0.0045 ± 0.005 | 2 |
| 13 |
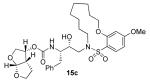
|
0.47 ± 0.002 | 22 |
| 12 |
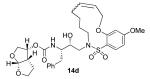
|
0.058 ± 0.005 | 14 |
| 12 |
|
0.017 ± 0.001 | 14 |
| 12 |
|
0.08 ± 0.02 | 23 |
| 11 |
|
0.077 ± 0.004 | 20 |
| 11 |
|
0.15 ± 0.01 | 95 |
| 10 |
|
0.051 ± 0.004 | 15 |
| 10 |
|
0.09 ± 0.01 | 5.5 |
| 9 |
|
2.4 ± 0.3 | n.t.b |
| 9 |
|
1.27 ± 0.03 | 77 |
MT-2 human T-lymphoid cells exposed to HIV-1LAI; Saquinavir and amprenavir exhibited IC50 values of 16 nM and 27 nM, respectively.
n.t. = not tested.
Figure 2.

An overlay of energy-minimized macrocyclic inhibitor 15c (magenta) with the X-ray structure of inhibitor 2 (green)-bound HIV-1 protease.
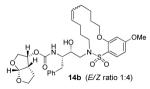
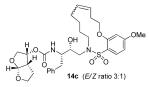
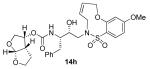
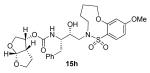
Indeed, the most potent compound of the series, inhibitor 14c incorporating a 13-membered ring, showed a Ki of 45 pM and IC50 of 2 nM. It would appear the 13-membered ring provides an optimally sized macrocyclic ring as increasing the ring size to 14 or 15 as well as decreasing the ring to 12 or 11 resulted in reduced enzymatic inhibitory and antiviral activity. Interestingly, inhibitors incorporating a 10-membered ring (14g and 15g) were also exceedingly potent displaying a similar activity profile as the 13-membered ring 14c (14g Ki = 51 pM; 15g Ki = 86 pM and IC50 = 5.5 nM).
In comparing the potency of 14c to its unsaturated analogue 15c we observed a dramatic improvement in the Ki and IC50 values for the unsaturated cyclic inhibitor 14c. Furthermore, effect appeared to remain consistent throughout the various sized unsaturated macrocycles 14a-h as compared to their saturated analogs 15a-h. For the 13 membered ring series, the presence of a double bond resulted in a 10-fold increase in both Ki and IC50 (14c Ki = 45 pM and IC50 = 2 nM as compared to 15c Ki = 470 pM and IC50 = 22 nM). Similar differences in potency can be seen for the 11, 14, and 15-membered macrocycles although for the smaller rings 9, 10, and 12 the effect is less pronounced. This effect may be explained by a restriction of conformation in the molecule that results from the presence of the double bond and leads to a better fit in the hydrophobic pocket of the S2′ subsite. These observations led us to take a closer look at the olefinic compounds and evaluate the importance of the stereochemistry of this group. As shown in Table 3, only minor variations in activity (less than 5-fold) were observed between the E and Z isomers of the 13, 14, and 15-membered macrocycles. For the 13-membered ring system, the Z isomer was favored over the E configuration.
Table 3.
Enzyme inhibitory and antiviral activity of E and Z isomers of 14a-c
| Ring size | Inhibitor structure | Ki (nM) | IC50 (nM)a |
|---|---|---|---|
| 15 |
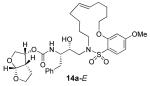
|
0.24 ± 0.07 | 360 |
| 15 |
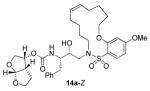
|
0.16 ± 0.04 | >1000 |
| 14 |
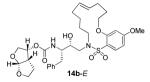
|
0.18 ± 0.01 | 6.6 |
| 14 |
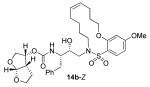
|
0.9 ± 0.3 | 2.9 |
| 13 |
|
0.06 ± 0.01 | 7 |
| 13 |
|
0.012 ± 0.004 | 4.6 |
MT-2 human T-lymphoid cells exposed to HIV-1LAI; Saquinavir and amprenavir exhibited IC50 values of 16 nM and 27 nM, respectively.
Based on these exciting results for this series of macrocyclic inhibitors, we began to envision possible substitutions that could be made across the macrocylic ring system in order to further enhance biological activity. The energy-minimized structure of 15b-bound HIV-1 protease with 2-bound HIV-1 protease (Figure 2) suggested that a single methyl substitution β to the macrocylic amine could fill an additional hydrophobic pocket previously filled by darunavir’s isobutyl methyl group. To this end, we designed and synthesized a series of mono and dimethylated 14-membered macrocylic ring systems and evaluated the impact of this substitution on the biological activity. Geminal dimethyl at this location (19a through 22a) significantly decreased (10-fold) the enzyme inhibition and greatly reduced antiviral activity. In most cases, the addition of a single methyl group to the ring also reduced biological activity as compared to 14b and 15b although results varied greatly depending upon the stereochemistry of the ring systems. Inhibitors 20c and 21b were the most potent compounds from this series with Ki = 0.31 and 2.8 nM and IC50 = 9.0 and 6.3 nM, respectively. In general, antiviral potency of cyclic inhibitors was superior to that of their acyclic homologues, unsaturated macrocyclic derivatives were more potent than the corresponding saturated derivatives and the addition of methyl substituents tended to decrease potency. The two most potent macrocyclic inhibitors identified were 14c and 14b having enzyme inhibitory Ki values of 45 pM and 82 pM and antiviral IC50 values of 2 nM and 4 nM, respectively.
The inhibitors 14b and 14c were then examined for their activity against a clinical wild-type X4-HIV-1 isolate (HIV-1ERS104pre) along with various multidrug resistant clinical X4- and R5-HIV-1 isolates using PBMCs as target cells.6b The results are shown in Table 5. As can be seen, the potency of inhibitors 14b and 14c against HIV-1ERS104pre (IC50 = 7 and 5 nM, respectively) is superior to FDA approved inhibitors IDV, APV and LPV. It is comparable to saquinavir but nearly 2-fold less potent than darunavir (IC50 = 3 nM).6b In these studies, both indinavir and lopinavir were unable to suppress the replication of the multidrug-resistant clinical isolates examined that include HIV-1MDR/B, HIV-1MDR/C, HIV-1MDR/TM, HIV-1MRR/MM and HIV-1MDR/JSL. Of particular note, lopinavir which is widely used as a first-line agent in HAART treatment regimens, was not active against these multidrug-resistant clinical isolates. Amprenavir displayed a 10-fold or greater reduction in potency except against HIV-1MDR/MM where it showed a 7 fold reduction in potency. Inhibitor 14c, while less potent than darunavir, maintained 7-fold or better potency over amprenavir against HIV-1MDR/C, HIV-1MDR/G, HIV-1MDR/TM and HIV-1MDR/ISL. It maintained over 6-fold potency against HIV-1MDR/MM. Inhibitor 14b maintained superior potency against HIV-1MDR/C and HIV-1MDR/G (greater than 12- and 21-fold) compared to amprenavir. It maintained 3-fold or better potency compared to amprenavir against all other multidrug resistant clinical isolates tested. Both inhibitors 14b and 14c have shown low cytotoxicity (CC50 values 49 and 33 μM, respectively) in target CD4+ MT-2 cells. Furthermore, they prevented the replication of HIV-1NL4-3 variants selected against up to 5 μM of saquinavir, lopinavir and indinavir with IC50 values of 20 nM to 46 nM. More detailed virologic studies with inhibitors 14c and 14b will be published in due course. The reason why these macrocyclic inhibitors maintained impressive potency against multidrug-resistant clinical isolates is possibly due to their ability to make extensive hydrogen-bonds with protease backbone and effectively fill in the hydrophobic pockets in the S1′-S2′ subsites.
Table 5.
Antiviral activity of macrocyclic inhibitors against multi-drug resistant clinical isolates in PHA-PBMs
| Virus | SQV | IDV | APV | LPV | DRV | 14b | 14c |
|---|---|---|---|---|---|---|---|
| HIV-1ERS104pre (wild-type: X4) |
0.008 ± 0.005 | 0.043 ± 0.004 | 0.030 ± 0.005 | 0.034 ± 0.002 | 0.003 ± 0.0002 | 0.007 ± 0.002 | 0.005 ± 0.003 |
| HIV-1MDR/B (X4) | 0.27 ± 0.073 (34) |
>1 (>23) |
>1 (>33) |
>1 (>29) |
0.019 ± 0.012 (6) |
0.089 ± 0.016 (13) |
0.037 ± 0.016 (7) |
| HIV-1MDR/C (X4) | 0.032 ± 0.002 (11) |
>1 (>23) |
0.37 ± 0.011 (12) |
>1 (>29) |
0.008 ± 0.006 (3) |
0.029 ± 0.001 (4) |
0.044 ± 0.002 (9) |
| HIV-1MDR/G (X4) | 0.030 ± 0.002 (4) |
0.34 ± 0.14 (5) |
0.43 ± 0.004 (14) |
0.26 ± 0.04 (8) |
0.023 ± 0.006 (5) |
0.028 ± 0.004 (4) |
0.057 ± 0.012 (11) |
| HIV-1MDR/TM (X4) | 0.26 ± 0.04 (33) |
>1 (>23) |
0.32 ± 0.007 (11) |
>1 (>29) |
0.004 ± 0.001 (1) |
0.072 ± 0.014 (10) |
0.027 ± 0.001 (6) |
| HIV-1MDR/MM (R5) | 0.19 ± 0.05 (24) |
>1 (>23) |
0.21 ± 0.222 (7) |
>1 (>29) |
0.011 ± 0.002 (4) |
0.055 ± 0.025 (8) |
0.033 ± 0.010 (7) |
| HIV-1MDR/JSL (R5) | 0.30 ± 0.02 (37) |
>1 (>23) |
0.62 ± 0.02 (21) |
>1 (>29) |
0.027 ± 0.011 (9) |
0.21 ± 0.032 (30) |
0.073 ± 0.07 (15) |
Amino acid substitutions identified in the protease-encoding region compared to the consensus type B sequence cited from the Los Alamos database include L63P in HIV-1ERS104pre; L10I, K14R, L33I, M36I, M46I, F53I, K55R, I62V, L63P, A71V, G73S, V82A, L90M, and I93L in HIV-1 MDR/B; L10I, I15V, K20R, L24I, M36I, M46L, I54V, I62V, L63P, K70Q, V82A, and L89M in HIV-1 MDR/C; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, and L90M in HIV-1 MDR/G; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90M, I93L in HIV-1MDR/TM; L10I, K43T, M46L, I54V, L63P, A71V, V82A, L90M, and Q92K in HIV-1MDR/MM; L10I, L24I, I33F, E35D, M36I, N37S, M46L, I54V, R57K, I62V, L63P, A71V, G73S, and V82A in HIV-1 MDR/JSL.HIV-1ERS104pre served as a source of wild type HIV-1. IC50 values were determined by using PHA-PBMs as target cells, and inhibition of p24 Gag protein production by each drug was used as an end point. Numbers in parentheses represent n-fold changes of IC50 values for each isolate compared to IC50 values for wild type HIV-1ERS104pre. All assays were conducted in duplicate or triplicate, and data shown represent mean values (±1 standard deviation) derived from results of three independent experiments.
X-Ray Crystallography
To gain molecular insights into ligand-binding site interactions responsible for the potent antiviral activity of inhibitor 14c, we have determined an X-ray crystal structure of the inhibitor complexed with wild-type protease. The crystal structure was solved and refined at 1.17Å resolution with an R-factor and Rfree of 16.0% and 19.4%, respectively. In this high resolution structure, the inhibitor 14c was bound to the HIV-1 protease active site in the two orientations with a ratio of 1:1. A stereoview of the X-ray structure of 14c bound HIV-1 protease is shown in Figure 3. As can be seen, the inhibitor makes extensive interactions involving the P2 to P2′-ligands in the protease active site; most notably through favorable polar interactions including hydrogen bonds, and weaker C—H⋯O interactions in the active site. The transition-state hydroxyl group in 14c forms asymmetric hydrogen bonding interactions with all four carboxylate oxygen atoms of the Asp25 and Asp25′ with distances of 2.5-3.3Å. The conserved tetrahedral water molecule forms hydrogen bonds with one of the sulfonamide oxygens, the urethane carbonyl oxygen, and the backbone amide nitrogen of Ile50 and Ile50′ with distances of 2.6-3.1Å. These interactions have been observed in the majority of HIV-1 protease complexes with the inhibitors18 and substrate analogs.19 The flexible P1′-P2′ macrocyclic ligand nicely packs into the hydrophobic pocket in the S1′-subsite. It also makes weaker C—H⋯O interactions which play important roles as we have reported earlier.20-22 The macrocyclic ring zigzags into a crown shape and fits well in between the S1′ and S2′ pockets. The protein-ligand complex shows three major interactions with the carbonyl oxygen of backbone residues; one with Gly27′ and two with Gly48′, with distances ranging from 3.0-3.6Å. In comparison to the X-ray structures of the protease with 1 and 2, the P1-phenyl ring in 14c is rotated about 30° towards Asp 29′ along the backbone. The macrocycle acts more or less like a spring that pushes against the P1 phenyl ring causing this rotation.
Figure 3.

A stereoview of the X-ray structure of macrocyclic inhibitor 14c (light gray)-bound HIV-1 protease. All strong hydrogen bonding interactions are shown as dotted lines.
Both P2-and P2′-ligands form five strong N—H⋯O hydrogen bonds with protease backbone. Of these, three hydrogen bonds are formed between the P2-bis-THF ring oxygens and the backbone amide nitrogens of Asp29 and Asp30 with distances of 3.1, 3.0 and 3.2Å. The fourth interaction is between the P2-urethane NH and the carbonyl oxygen of Gly27 with distance of 3.0Å. The fifth backbone interaction is between the p-methoxy group of the P2′-sulfonamide and the amide nitrogen of Asp30′ with a distance of 3.0Å. All of these ligand-backbone interactions are present in the X-ray structure of 2-bound HIV-1 protease as well. This backbone binding with the main chain atoms of the protease may be responsible for both inhibitors’ (2 and 14c) abilities to maintain robust potency against multi-drug resistant HIV-1 variants.
Conclusions
In summary, we have designed novel macrocyclic protease inhibitors modifying P1′-P2′ ligands of darunavir-like PIs and investigated their biological activity. The inhibitors were designed to maintain key hydrogen bonding interactions with protease backbone similar to darunavir. The design of macrocycles involving the P1′-P2′ ligands is based upon the premise that a flexible macrocycle would effectively repack the hydrophobic pocket in the S1′ to S2′ subsites when it is altered by mutations. We have investigated inhibitors containing 9-15-membered macrocycles containing both E/Z olefins and the corresponding saturated derivatives. Grubbs’ metathesis reaction was the key step in building these inhibitors in very good yields. Most remarkably, all macrocyclic inhibitors are significantly more potent than their acyclic counterpart. The saturated inhibitors are in general less active than the corresponding unsaturated derivatives. Our investigation resulted in the identification of inhibitors 14b and 14c, which have displayed significantly better antiviral activity than many of the currently FDA-approved inhibitors. Inhibitor 14b contains a 14-membered ring with a Z-olefin and 14c contains a 13-membered ring with an E-olefin. Both inhibitors exerted potent activity against HIV-1LAI with IC50 values of 4 nM and 2 nM, respectively. They have maintained excellent potency against multidrug-resistant HIV-1 variants. These inhibitors have shown low cytotoxicity (CC50 values 49 and 33 μM, respectively) in target CD4+ MT-2 cells. Furthermore, both inhibitors 14b and 14c blocked the replication of HIV-1NL4-3 variants, selected after exposure of up to 5 μM of saquinavir, lopinavir and indinavir, with IC50 values of 20 nM to 46 nM. Protein-ligand X-ray structure of 14c showed critical ligand-binding site interactions in the protease active site. Particularly, it maintained all key backbone hydrogen bonding interactions similar to darunavir and inhibitor 2. Also, the conformational flexibility of the P1′-P2′-macrocycle most likely contributed to its impressive activity against multidrug-resistant clinical variants. Further design and optimization of P1′-P2′macrocyclic ligands are in progress.
Experimental Section
General Experimental Methods
Chemicals and reagents were purchased from commercial suppliers and used without further purification. Anhydrous solvents were obtained as follows: pyridine and dichloromethane were distilled from calcium hydride; tetrahydrofuran and diethyl ether were distilled from sodium wire with benzophenone as an indicator. All other solvents were reagent grade. All moisture sensitive reactions were carried out in oven dried glassware under argon. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance ARX-400, Bruker DRX-500, or Bruker Avance-III-800 spectrometer. Chemical shifts are given in ppm and are referenced against the diluting solvent. For chloroform-d: 13C triplet = 77.00 CDCl3 and 1H singlet = 7.26 ppm. For methanol-d4: 13C septuplet = 49.05 and 1H quintuplet = 3.31 ppm. Characteristic splitting patterns due to spin spin coupling are expressed as follows: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, sept = septuplet. All coupling constants are measured in hertz. FTIR spectra were recorded on a Mattson Genesis II FT-IR spectrometer or a Perkin Elmer spectrometer #L1185247 using a NaCl plate or KBr pellet. Optical rotations were recorded on a Perkin Elmer 341 or Rudolph Research Autopol III polarimeter. Low resolution mass spectra were recorded on a FinniganMAT LCQ or Hewlett-Packard Engine mass spectrometer. High resolution mass spectra were recorded on a FinniganMAT XL95 mass spectrometer calibrated against PPG. Column chromatography was performed with Whatman 240-400 mesh silica gel under low pressure of 3-5 psi. TLC was carried out with E. Merck silica gel 60-F-254 plates. HPLC data was collected using a system composed of an Agilent 1100 series degasser, quaternary pump, thermostatable column compartment, variable wavelength detector, and Agilent 1200 series autosampler and fraction collector controlled by Chemstation software. All chromatographic reagents used were HPLC grade. The reported inhibitors were found to be >95% pure by reversed phase gradient HPLC (see supporting information for specific method conditions).
1-(hex-5-enyloxy)-3-methoxybenzene (5a)
To a stirred solution of 3-methoxy phenol (1.24 g, 10 mmol), 5-hexen-1-ol, 4a (1.4 mL, 12 mmol) and Ph3P (3.14g, 12 mmol) in THF (20 mL) at 0 °C was added DIAD (2.3 mL, 12 mmol) dropwise. After stirring the solution for 30 min at 0 °C the reaction mixture was warmed to 23 °C and stirred for 3 h. The reaction mixture was concentrated in vacuo and the residue was subjected to column chromatography (98:2 hexanes:EtOAc) to yield 5a (1.98g, 96% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.58-1.66 (m, 2H), 1.80-1.88 (m, 2H), 2.15-2.20 (m, 2H), 3.82 (s, 3H), 3.98 (t, J = 6.4 Hz, 2H), 5.02-5.12 (m, 2H), 5.83-5.92 (m, 1H), 6.52-6.56 (m, 3H), 7.19-7.24 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 25.3, 28.7, 33.4, 55.1, 67.6, 100.9, 106.0, 106.6, 114.7, 129.8, 138.5, 160.3, 160.8; FT-IR (film, NaCl) νmax = 3075, 2939, 1599, 1493, 1287, 1200, 1152, 1046 cm−1; CI LRMS (m/z): 207.25 [M + H]+.
1-methoxy-3-(pent-4-enyloxy)benzene (5b)
Title compound was obtained from 4-penten-1-ol 4b, as described for 5a in 95% yield after flash chromatography (98:2 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.88-1.94 (m, 2H), 2.25-2.30 (m, 2H), 3.81 (s, 3H), 3.98 (t, J = 6.4 Hz, 2H), 5.03-5.13 (m, 2H), 5.85-5.95 (m, 1H), 6.51-6.56 (m, 3H), 7.20 (t, J = 8.1 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 28.3, 30.0, 55.1, 67.0, 100.9, 106.0, 106.6, 115.1, 129.7, 137.7, 160.2, 160.7; FT-IR (film, NaCl) νmax = 3076, 2940, 1599, 1492, 1287, 1200, 1152, 1048 cm−1; CI LRMS (m/z): 193.25 [M + H]+.
1-(but-3-enyloxy)-3-methoxybenzene (5c)
Title compound was obtained from 3-buten-1-ol 4c, as described for 5a in 96% yield after flash-chromatography (98:2 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 2.54-2.60 (m, 2H), 3.81 (s, 3H), 4.02 (t, J = 6.7 Hz, 2H), 5.13-5.23 (m, 2H), 5.89-5.97 (m, 1H), 6.51-6.56 (m, 3H), 7.20 (t, J = 8.1 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 33.6, 55.1, 67.1, 100.9, 106.2, 106.6, 116.9, 129.8, 134.4, 160.1, 160.8; FT-IR (film, NaCl) νmax = 3136, 2378, 1644, 1509 cm−1; CI LRMS (m/z): 179.20 [M + H]+.
1-(allyloxy)-3-methoxybenzene (5d)
Title compound was obtained from allyl alcohol 4d, as described for 5a in 96% yield after flash chromatography (98:2 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 3.81 (s, 3H), 4.02 (d, J = 6.7 Hz, 2H), 5.13-5.23 (m, 2H), 5.89-5.97 (m, 1H), 6.51-6.56 (m, 3H), 7.20 (t, J = 8.1 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 55.1, 67.1, 100.9, 106.2, 106.6, 116.9, 129.8, 134.4, 160.1, 160.8.
2-(hex-5-enyloxy)-4-methoxybenzenesulfonic acid (6a)
To 6a (2 g, 9.7 mmol) was added acetic anhydride (1.4 mL, 14.5 mmol) and the resulting mixture was stirred at 0 °C. To this was then added concentrated H2SO4 (1.1 g) followed by methanol (20 mL). The resulting solution was warmed to 23 °C and stirred for 12 h. After this time the reaction mixture was concentrated in vacuo and the resulting red oil was subjected to column chromatography (88:12 CH2Cl2:MeOH) to give 6a (1.06 g, 38%) as a red waxy solid. 1H NMR (400 MHz, D2O): δ 1.44 (quintet, J = 7.4 Hz, 2H), 1.66-1.73 (m, 2H), 1.99 (q, J = 7.1 Hz, 2H), 3.70 (s, 3H), 3.98 (t, J = 6.5 Hz, 2H), 4.85-4.97 (m, 2H), 5.74-5.92 (m, 1H), 6.52-6.56 (m, 2H), 7.19-7.24 (m, 1H); 13C NMR (100 MHz, D2O): δ 25.3, 28.7, 33.4, 55.1, 67.6, 100.9, 106.0, 106.6, 114.7, 129.8, 138.5, 160.3, 160.8; ESI (m/z): 285.09 [M − H]−.
4-methoxy-2-(pent-4-enyloxy)benzenesulfonic acid (6b)
Title compound was obtained from ether 5b as described for 6a in 36% yield after flash-chromatography (88:12 CH2Cl2:MeOH) as a white solid. 1H NMR (400 MHz, D2O): δ 1.75-1.82 (m, 2H), 2.10-2.16 (m, 2H), 3.69 (s, 3H), 3.98 (t, J = 6.4 Hz, 2H), 4.88-5.00 (m, 2H), 5.77-5.87 (m, 1H), 6.45 (dd, J = 8.6, 2.2 Hz, 1H), 6.51 (d, J = 2.1 Hz, 1H), 7.58 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, D2O): δ 28.0, 29.9, 56.1, 68.7, 100.5, 104.9, 115.5, 123.7, 130.2, 139.4, 157.8, 163.5; ESI (m/z): 271.07 [M − H]−.
2-(but-3-enyloxy)-4-methoxybenzenesulfonic acid (6c)
Title compound was obtained from ether 5c as described for 6a in 30% yield after flash-chromatography (88:12 CH2Cl2:MeOH) as a white solid. 1H NMR (400 MHz, D2O): δ 2.38-2.43 (m, 2H), 3.62 (s, 3H), 3.98 (t, J = 6.8 Hz, 2H), 4.93-5.05 (m, 2H), 5.77-5.87 (m, 1H), 6.37 (dd, J = 8.6, 2.2 Hz, 1H), 6.43 (d, J = 2.4 Hz, 1H), 7.58 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, D2O): δ 33.3, 56.1, 68.8, 100.6, 105.0, 117.5, 123.7, 130.2, 135.4, 157.5, 163.4; ESI (m/z): 257.10 [M − H]−.
2-(allyloxy)-4-methoxybenzenesulfonic acid (6d)
Title compound was obtained from ether 5d as described for 6a in 35% yield after flash chromatography (88:12 CH2Cl2:MeOH) as a white solid. 1H NMR (400 MHz, D2O): δ 3.71 (s, 3H), 4.58-4.75 (m, 2H), 5.16-5.38 (m, 2H), 5.92-5.99 (m, 1H), 6.47-6.57 (m, 2H), 7.58 (d, J = 8.8. Hz, 1H); 13C NMR (100 MHz, D2O): δ 56.1, 69.7, 101.1, 105.3, 118.0, 123.7, 130.6, 133.2, 157.1, 163.5; ESI (m/z): 243.13 [M − H]−.
2-(hex-5-enyloxy)-4-methoxybenzene-1-sulfonyl chloride (7a)
To a stirring solution of sulfonic acid 6a (266 mg, 0.9 mmol) in pyridine (2 mL) was added thionyl chloride (0.2 mL, 2.8 mmol) dropwise. The resulting solution was allowed to stir for 4 h and then the reaction mixture concentrated in vacuo. The resulting residue was purified using column chromatography (5:1 hexanes:EtOAc) to give 7a (140 mg, 50%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.63-1.70 (m, 2H), 1.87-1.93 (m, 2H), 2.10-2.16 (m, 2H), 3.88 (s, 3H), 4.14 (t, J = 6.2 Hz, 2H), 4.95-5.05 (m, 2H), 5.76-5.86 (m, 1H), 6.51-6.54 (m, 2H), 7.84 (d, J = 9.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 24.9, 28.1, 33.1, 55.9, 69.2, 99.9, 104.6, 114.7, 124.3, 131.7, 138.3, 158.7, 166.8.
4-methoxy-2-(pent-4-enyloxy)benzene-1-sulfonyl chloride (7b)
Title compound was obtained from ether 6b as described for 7a in 48% yield after flash chromatography (6:1 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.96-1.2.03 (m, 2H), 2.31-2.37 (m, 2H), 3.88 (s, 3H), 4.15 (t, J = 6.2 Hz, 2H), 4.99-5.09 (m, 2H), 5.79-5.90 (m, 1H), 6.51-6.54 (m, 2H), 7.84 (d, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 27.8,29.7, 55.9, 68.4. 99.9, 104.6, 115.6, 124.3, 131.7, 137.3, 158.6, 166.8.
2-(but-3-enyloxy)-4-methoxybenzene-1-sulfonyl chloride (7c)
Title compound was obtained from ether 6c as described for 7a in 52% yield after flash-chromatography (6:1 hexanes:EtOAc) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 2.62-2.70 (m, 2H), 3.88 (s, 3H), 4.19 (t, J = 6.2 Hz, 2H), 5.12-5.25 (m, 2H), 5.91-6.05 (m, 1H), 6.52-6.56 (m, 2H), 7.86 (d, J = 9.2 Hz, 1H).
2-(allyloxy)-4-methoxybenzene-1-sulfonyl chloride (7d)
Title compound was obtained from ether 6d as described for 7a in 58% yield after flash chromatography (6:1 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 3.88 (s, 3H), 4.72 (d, J = 4.4 Hz, 2H), 5.33 (d, J = 10.6 Hz, 1H), 5.57 (d, J = 17.3 Hz, 1H), 5.99-6.07 (m, 1H), 6.52-6.55 (m, 2H), 7.83 (d, J = 8.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 55.9, 69.7, 100.5, 105.0, 118.1, 124.4, 131.0, 131.7, 158.0, 166.7.
tert-butyl (2S,3R)-4-(hex-5-enylamino)-3-hydroxy-1-phenylbutan-2-ylcarbamate (10a)
A solution of hex-5-en-1-amine 9a (297 mg, 3 mmol) and epoxide 8 (263 mg, 1 mmol) was heated to 60 °C in isopropanol (4 mL) for 4 h. The solvent was then evaporated under reduced pressure and the resulting residue was purified by silica chromatography (5:95 MeOH:CHCl3) to give 10a (350 mg, 97%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 1.34 (s, 9H), 1.39-1.50 (m,4H), 2.05 (q, J = 7 Hz, 2H), 2.55-2.68 (m, 6H), 2.81-2.86 (m, 1H), 2.96 (dd, J = 4.5, 14 Hz, 1H), 3.44-3.49 (m, 1H), 3.79 (br s, 1H), 4.72 (d, J = 8.7 Hz, 1H), 4.92-5.02 (m, 2H), 5.74-5.84 (m, 1H), 7.17-7.28 (m, 5H); 13C NMR (100 MHz, CDCl3): δ 26.4, 28.2, 29.4, 33.4, 36.5, 49.6, 51.3, 54.1, 70.7, 79.3, 114.5, 126.2, 128.3, 129.4, 137.8, 138.6,155.9; CI LRMS (m/z): 363.55 [M + H]+.
tert-butyl (2S,3R)-4-(allylamino)-3-hydroxy-1-phenylbutan-2-ylcarbamate (10b)
Title compound was obtained from allylamine 9b and epoxide 8 as described for 10a in 99% yield after flash-chromatography (5:95 MeOH:CHCl3) as a white solid. 1H NMR (400 MHz, CDCl3): δ 1.35 (s, 9H), 2.60-2.86 (m, 5H), 2.96 (dd, J = 4.5, 14.1 Hz, 1H), 3.16-3.29 (m, 2H), 3.50-3.53 (m, 1H), 3.81 (br s, 1H), 4.73 (d, J = 9.1 Hz, 1H), 5.10 (d, J = 10.3 Hz, 1H), 5.17 (d, J = 17 Hz, 1H), 5.81-5.91 (m, 1H), 7.13-7.32 (m, 5H); 13C NMR (100 MHz, CDCl3): δ 28.2, 36.5, 50.7, 52.1, 54.1, 70.9, 79.3, 116.2, 126.2, 128.3, 129.4, 136.3, 137.8, 155.9; CI LRMS (m/z) 321.50 [M + H]+.
tert-butyl-(2S,3R)-4-(N-(hex-5-enyl)-2-(hex-5-enyloxy)-4-ethoxyphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (11a)
To a stirring solution of 10a (50 mg, 0.14 mmol) in pyridine (2 mL) was added 7a (64 mg, 0.20 mmol) and the resulting solution was allowed to stir for 2 h at 23 °C. The reaction mixture was concentrated under reduced pressure and the resulting residue was purified by flash column chromatography (3:1 hexanes:EtOAc) to yield 11a (74 mg 84% yield) as a colorless oil. [α]D20 −1.4 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.25-1.34 (m, 11 H), 1.47-1.50 (m, 2H), 1.57-1.60 (m, 2H), 1.82-1.90 (m, 2H), 1.97 (q, J = 7.1 Hz, 2H), 2.11 (q, J = 7.0 Hz, 2H), 2.92-2.95 (m, 2H), 3.10-3.17 (m, 1H), 3.30 (br s, 3H), 3.76 (br s, 2H), 3.84 (s, 3H), 3.90-3.92 (m, 1H) 4.03 (t, J = 6.7 Hz, 2H), 4.65 (d, J = 7.1 Hz, 1H), 4.89-5.04 (m, 4H), 5.66-5.82 (m, 2H), 6.46 (d, J = 2 Hz, 1H), 6.49 (dd, J = 8.8, 2.3 Hz, 1H), 7.17-7.29 (m, 5H), 7.83 (d, J = 7.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 24.9, 25.7, 27.9, 28.2, 28.3, 33.1, 33.2, 35.1, 49.9, 52.3, 54.5, 55.6, 69.1, 72.2, 79.4, 100.2, 104.1, 114.7, 115.0, 119.4, 126.2, 128.3, 129.5, 133.5, 137.8, 138.1, 138.2, 156.0, 157.6, 164.7; FT-IR (film, NaCl) νmax = 3395, 2935, 1705, 1597, 1325 cm−1; ESI (+) LRMS (m/z): 653.13 [M + Na]+.
tert-butyl-(2S,3R)-4-(N-(hex-5-enyl)-4-methoxy-2-(pent-4enyloxy)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (11b)
Title compound was obtained from 10a and 7b, as described for 11a in 79% yield after flash chromatography (3:1 hexanes:EtOAc) as a colorless oil. [α]D20 −1.6 (c 1.20, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.25-1.37 (m, 12H), 1.43-1.51 (m, 2H), 1.88-1.98 (m, 4H), 2.27 (q, J = 7.1 Hz, 2H), 2.86-2.97 (m, 2H), 3.10-3.17 (m, 1H), 3.25-3.31 (m, 3H), 3.76 (br s, 2H), 3.84 (s, 3H), 4.04 (t, J = 6.6 Hz, 2H), 4.66 (d, J = 7.1 Hz, 1H), 4.88-5.10 (m, 4H), 5.65-5.72 (m, 1H), 5.78-5.85 (m, 1H), 6.46 (d, J = 2 Hz, 1H), 6.49 (dd, J = 8.8, 2.0 Hz, 1H), 7.17-7.29 (m, 5H), 7.83 (d, J = 8.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 25.7, 27.9, 28.2, 29.7, 33.1, 35.2, 49.8, 52.2, 54.6, 55.6, 68.4, 72.2, 79.4, 100.2, 104.1, 114.7, 115.7, 119.4, 126.2, 128.3, 129.5, 133.5, 137.0, 137.8, 138.2, 156.0, 157.6, 164.7; FT-IR (film, NaCl) νmax = 3398, 2931, 1706, 1596, 1325 cm−1; ESI (+) LRMS (m/z): 639.06 [M + Na]+.
tert-butyl-(2S,3R)-4-(2-(but-3-enyloxy)-N-(hex-5-enyl)-4-methoxyphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (11c)
Title compound was obtained from 10a and 7c, as described for 11a in 50% yield after flash-chromatography (3:1 hexanes:EtOAc) as a colorless oil. [α]D20 +0.6 (c 2.00, CHCl3); 1H NMR (500 MHz, CDCl3): δ 1.28-1.38 (m, 12H), 1.48-1.52 (m, 2H), 2.00 (q, J = 6.7 Hz, 2H), 2.65 (q, J = 6.7, 2H), 2.96 (br s, 2H), 3.14-3.18 (m, 1H), 3.30-3.39 (m, 3H), 3.79 (br s, 2H), 3.88 (s, 3H), 4.13 (t, J = 7.1 Hz, 2H), 4.68 (d, J = 5.1 Hz, 1H), 4.92-4.98 (m, 2H), 5.17-5.24 (m, 2H), 5.68-5.76 (m, 1H), 5.91-5.99 (m, 1H), 6.51 (d, J = 2 Hz, 1H), 6.54 (dd, J = 8.8, 2.0 Hz, 1H), 7.21-7.32 (m, 5H), 7.88 (d, J = 8.8 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.9, 28.2, 28.4, 29.9, 33.4, 35.4, 50.1, 52.5, 54.8, 55.9, 68.7, 72.5, 79.7, 100.6, 104.5, 114.9, 118.0, 119.8, 126.5, 128.6, 129.8, 133.7, 133.9-138.1, 138.5, 156.3, 157.7, 165.0; FT-IR (film, NaCl) νmax = 3394, 2931, 1704, 1596, 1325 cm−1; ESI (+) (m/z): 625.05 [M + Na]+.
tert-butyl(2S,3R)-4-(2-(allyloxy)-N-(hex-5-enyl)-4-methoxyphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (11d)
Title compound was obtained from 10a and 7d, as described for 11a in 64% yield after flash-chromatography (3:1 hexanes:EtOAc) as a colorless oil. [α]D20 +1.1 (c 2.80, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.25-1.34 (m, 11H), 1.39-1.46 (m, 2H), 1.96 (q, J = 7.0 Hz, 2H), 2.89-2.96 (m, 2H), 3.08-3.15 (m, 1H), 3.24-3.30 (m, 3H), 3.77 (br s, 2H), 3.84 (s, 3H), 4.60 (d, J = 5.4 Hz, 2H), 4.66 (d, J = 7.2 Hz, 1H), 4.88-4.94 (m, 2H), 5.33 (d, J = 10.4, 2H), 5.43 (d, J = 17.4 Hz, 1H), 5.63-5.73 (m, 1H), 6.00-6.10 (m, 1H), 6.47 (d, J = 1.8 Hz, 1H), 6.54 (dd, J = 8.9, 1.9 Hz, 1H), 7.18-7.29 (m, 5H), 7.85 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 25.7, 27.8, 28.2, 33.1, 35.2, 49.9, 52.5, 54.5, 55.6, 69.9, 72.2, 79.4, 100.6, 104.4, 114.7, 119.4, 119.7-126.2, 128.3, 129.5, 131.9, 133.5, 137.9, 138.2, 155.9, 157.0, 164.6; FT-IR (film, NaCl) νmax = 3400, 2929, 1704, 1596, 1324 cm−1; ESI (+) LRMS (m/z): 611.03 [M + Na]+.
tert-butyl-(2S,3R)-4-(N-allyl-2-(hex-5-enyloxy)-4-methoxyphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (11e)
Title compound was obtained from 10b and 7a, as described for 11a in 77% yield after flash-chromatography (3:1 hexanes:EtOAc) as a colorless oil. [α]D20 −6.6 (c 1.96, CHCl3); 1H NMR (500 MHz, CDCl3): δ 1.36 (s, 9H), 1.60-1.66 (m, 2H), 1.87-1.97 (m, 2H), 2.13-2.17 (m, 2H), 2.91-2.98 (m, 2H), 3.27-3.39 (m, 2H), 3.79 (br s, 2H), 3.87 (s, 3H), 3.91-3.99 (m, 2H), 4.08 (t, J = 6.7 Hz, 2H), 4.65 (d, J = 7.1 Hz, 1H), 4.98 (d, J = 10.1, 1H), 5.04 (d, J = 17.1, 1H), 5.13-5.19 (m, 2H), 5.63-5.73 (m, 1H), 5.78-5.86 (m, 1H), 6.50-6.53 (m, 2H), 7.21-7.31 (m, 5H), 7.88 (d, J = 8.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 25.0, 28.3, 33.3, 35.3, 51.2, 52.2, 54.6, 55.7, 69.2, 71.8, 79.5, 100.3, 104.2, 115.1, 119.0, 119.6, 126.3, 128.4, 129.6, 133.6, 137.9, 138.2, 156.1, 157.7, 164.8; FT-IR (film, NaCl) νmax = 3392, 2932, 1702, 1595, 1324 cm−1; ESI (+) LRMS(m/z): 611.04 [M + Na]+.
tert-butyl-(2S,3R)-4-(N-allyl-4-methoxy-2-(pent-4-enyloxy)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (11f)
Title compound was obtained from 10b and 7b, as described for 11a in 86% yield after flash-chromatography (3:1 hexanes:EtOAc) as a colorless oil. [α]D20 −5.6 (c 1.10, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.32 (s, 9H), 1.94 (quintet, J = 7 Hz, 2H), 2.65 (q, J = 7 Hz, 2H), 2.86-2.96 (m, 2H), 3.27 (dd, J = 7.4, 14.8 Hz, 1H), 3.34-3.38 (m, 1H), 3.76 (br s, 2H), 3.82 (s, 3H), 3.87-3.92 (m, 2H), 4.04 (t, J = 6.5 Hz, 2H), 4.65 (d, J = 8.2 Hz, 1H), 4.98-5.15 (m, 4H), 5.57-5.63 (m, 1H), 5.75-5.86 (m, 1H), 6.46-6.49 (m, 2H), 7.12-7.27 (m, 5H), 7.83 (d, J = 8.5 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 27.8, 28.1, 29.7, 35.3, 51.0, 51.9, 54.5, 55.6, 68.4, 71.8, 79.3, 100.2, 104.2, 115.7, 119.0, 119.5, 126.1, 128.2, 129.5, 133.4, 137.2, 137.9, 155.9, 157.6, 164.7; FT-IR (film, NaCl) νmax = 3390, 2976, 1710, 1597, 1325 cm−1; ESI (+) LRMS (m/z): 597.13 [M + Na]+.
tert-butyl-(2S,3R)-4-(N-allyl-2-(but-3-enyloxy)-4-methoxyphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (11g)
Title compound was obtained from 10b and 7c, as described for 11a in 78% yield after flash chromatography (3:1 hexanes:EtOAc) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.34 (s, 9H), 2.62 (q, J = 7 Hz, 2H), 2.88-2.96 (m, 2H), 3.27 (dd, J = 7.8, 15.1 Hz, 1H), 3.34-3.38 (m, 1H), 3.76 (br s, 2H), 3.85 (s, 3H), 3.87-3.99 (m, 2H), 4.10 (t, J = 6.5 Hz, 2H), 4.65 (br s, 1H), 5.10-5.22 (m, 4H), 5.57-5.63 (m, 1H), 5.87-5.95 (m, 1H), 6.49 (d, J = 2.2 Hz, 1H), 6.51 (dd, J = 2.3, 8.7 Hz 1 H) 7.18-7.37 (m, 5H), 7.85 (d, J = 8.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): 27.8, 28.1, 35.3, 51.0, 51.9, 54.5, 55.6, 68.4, 71.8, 79.3, 100.2, 104.2, 115.7, 119.0, 119.5, 126.2, 128.2, 129.5, 133.4, 137.2, 137.9, 155.9, 157.6, 164.7; FT-IR (film, NaCl) νmax = 3390, 2976, 1710, 1597, 1325 cm−1; ESI LRMS (m/z): 582.95 [M + Na]+.
tert-butyl-(2S,3R)-4-(N-allyl-2-(allyloxy)-4-methoxyphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (11h)
Title compound was obtained from 10b and 7d, as described for 11a in 80% yield after flash chromatography (3:1 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.33 (s, 9H), 2.83-2.96 (m, 2H), 3.25-3.36 (m, 2H), 3.77 (br s, 2H), 3.83 (s, 3H), 3.87-3.94 (m, 2H), 4.56-4.67 (m, 3H), 5.07-5.14 (m, 2H), 5.33 (d, J = 10.5 Hz, 1H), 5.44 (d, J = 17.2 Hz, 1H), 5.57-5.64 (m, 1H), 6.00-6.10 (m, 1H), 6.47 (d, J = 1.9 Hz, 1H), 6.51 (dd, J = 1.9, 8.9 Hz, 1 H) 7.16-7.28 (m, 5H), 7.85 (d, J = 8.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 28.2, 35.3, 51.3, 52.1, 54.5, 55.6, 69.9, 71.8, 79.3, 100.6, 104.5, 119.0, 119.4, 119.8, 126.2, 128.2, 129.5, 131.8, 133.4, 137.9, 155.9, 157.0, 164.7; FT-IR (film, NaCl) νmax = 3390, 2976, 1710, 1597, 1325 cm−1; ESI (+) LRMS (m/z): 569.06 [M + Na]+.
Compound 13a
To a stirring solution of 11a (63 mg, 0.1 mmol) in CH2Cl2 (2 mL) was added a solution of 30% trifluoroacetic acid in CH2Cl2 and the resulting mixture was stirred for 30 min. The solvent was evaporated under reduced pressure and the residue was dissolved in CH3CN (2 mL). To this solution was added 12 (31 mg, 0.11 mmol), followed by i-Pr2NEt. After stirring for 24 h the reaction mixture was concentrated in vacuo and the resulting residue was subjected to flash-chromatography (1:1 hexanes:EtOAc) to give 13a (36 mg, 53% yield) as a colorless oil. [α]D20 −13.1 (c 1.50, CHCl3); 1H NMR (500 MHz, CDCl3): δ 1.28-1.31 (m, 6H), 1.47-1.51 (m, 3H), 1.58-1.61 (m, 3H), 1.84-1.88 (m, 2H), 1.96-1.99 (m, 2H), 2.01-2.13 (m, 2H), 2.79-2.84 (m, 1H), 2.88-2.92 (m, 1H), 3.09-3.15 (m, 1H), 3.24-3.40 (m, 3H), 3.61 (br s, 1H), 3.64-3.72 (m, 2H), 3.83-3.92 (m, 4H), 3.93-3.96 (m, 1H), 4.05 (t, J = 6.7 Hz, 2H), 4.90-5.03 (m, 4H), 5.64 (d, J = 5 Hz, 1H), 5.66-5.82 (m, 2H), 6.46 (s, 1H), 6.51 (d, J = 8.8 Hz, 1H), 7.17-7.26 (m, 5H), 7.83 (d, J = 8.8 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 27.9, 28.2, 28.1, 29.4, 31.7, 32.9, 33.1, 35.1, 45.1, 49.8, 52.1, 54.8, 55.5, 68.9, 69.3, 70.5, 72.0, 73.1, 100.1, 103.9, 109.0, 114.6, 114.9, 118.9, 126.3, 128.2, 129.1, 133.4, 137.4, 137.8, 138.0, 155.2, 157.4, 164.6; FT-IR (film, NaCl) νmax = 3343, 2928, 1721, 1595, 1325 cm−1; ESI (+) HRMS (m/z): [M + Na]+ calcd for C36H50N2O9S, 709.3135; found, 709.3136.
Compound 13b
Title compound was obtained from 11b and 12 as described for 13a in 55% yield after flash-chromatography (1:1 hexanes:EtOAc) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.27-1.32 (m, 3H), 1.41-1.53 (m, 3H), 1.57-1.62 (m, 1H), 1.92-1.99 (m, 4H), 2.27 (q, J = 7.05, 2H), 2.80 (dd, J = 10, 14 Hz, 1H), 2.86-2.91 (m, 1H), 3.01 (dd, J = 4, 14 Hz, 1H), 3.10-3.15 (m, 1H), 3.27-3.33 (m, 2H), 3.38 (dd, J = 8.6, 15.2 Hz, 1H), 3.61 (br s, 1H), 3.64-3.70 (m, 2H), 3.81-3.84 (m, 5H), 3.88-3.95 (m, 2H), 4.05 (t, J = 6.6 Hz, 2H), 4.89-5.09 (m, 5H), 5.63 (d, J = 5.2 Hz, 1H), 5.65-5.73 (m, 1H), 5.77-5.85 (m, 1H), 6.46 (d, J = 2 Hz, 1H), 6.51 (dd, J = 2.1, 8.8 Hz, 1H), 7.17-7.26 (m, 5H), 7.83 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 25.7, 27.9, 29.7, 33.1, 35.4, 45.1, 49.8, 52.1, 55.1, 55.7, 68.5, 69.5, 70.7, 72.2-73.2, 100.3, 104.2, 109.2, 114.8, 115.7, 119.2, 126.4, 128.4, 129.3, 133.6, 137.1, 137.6, 138.2, 155.4, 157.5, 164.8; ESI (+) HRMS (m/z): [M + H]+ calcd for C35H48N2O9S, 673.3159; found, 673.3153.
Compound 13c
Title compound was obtained from 11c and 12 as described for 13a in 81% yield after flash-chromatography (1:1 hexanes:EtOAc) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.31-1.35 (m, 3H), 1.50-1.54 (m, 3H), 1.62-1.67 (m, 1H), 2.00 (q, J = 7 Hz, 2H), 2.65 (q, J = 6.7 Hz, 2H), 2.82-2.87 (m, 1H), 2.91-2.94 (m, 1H), 3.06 (dd, J = 4.1, 14.2 Hz, 1H), 3.13-3.19 (m, 1H), 3.30-3.43 (m, 3H), 3.62 (br s, 1H), 3.68-3.74 (m, 2H), 3.85-3.88 (m, 4H), 3.92-3.99 (m, 2H), 4.13 (t, J = 6.9 Hz, 2H), 4.93-5.05 (m, 2H), 5.07-5.09 (m, 2H), 5.17-5.24 (m, 2H), 5.66 (d, J = 5.1 Hz, 1H), 5.69-5.77 (m, 1H), 5.90-5.99 (m, 1H), 6.52 (s, 1H), 6.55 (dd, J = 2.05, 8.8 Hz, 1H), 7.22-7.30 (m, 5H), 7.87 (d, J = 8.8 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.8, 25.9, 28.1, 29.8, 33.3, 35.6, 45.4, 50.0, 52.4, 55.2, 55.8, 68.7, 69.7, 70.8, 72.4, 73.4, 100.6, 104.5, 109.4, 114.9, 118.0, 119.5, 126.6, 128.6, 129.5, 133.5, 137.8, 138.3, 155.9, 157.6, 164.9; FT-IR (film, NaCl) νmax = 3339, 2929, 1719, 1596, 1324 cm−1; ESI (+) HRMS (m/z): [M + Na]+ calcd for C34H46N2O9S, 681.2822; found, 681.2812.
Compound 13d
Title compound was obtained from 11d and 12 as described for 13a in 55% yield after flash-chromatography (1:1 hexanes:EtOAc) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 1.24-1.30 (m, 2H), 1.43-1.51 (m, 3H), 1.57-1.63 (m, 1H), 1.96 (q, J = 7 Hz, 2H), 2.80 (dd, J = 10, 14 Hz, 1H), 2.86-2.91 (m, 1H), 3.01 (dd, J = 4.5, 14.5 Hz, 1H), 3.08-3.14 (m, 1H), 3.25-3.30 (m, 2H), 3.40 (dd, J = 8.5, 15 Hz, 1H) 3.58 (br s, 1H), 3.65-3.72 (m, 2H), 3.80-3.82 (m, 2H), 3.87 (s, 3H), 3.88-3.96 (m, 2H), 4.60 (d, J = 5.5 Hz, 2H), 4.90-4.95 (m, 2H), 4.98-5.06 (m, 2H), 5.33 (d, J = 10 Hz, 1H), 5.45 (d, J = 18 Hz, 1H), 5.64 (d, J = 5.5 Hz, 1H), 5.65-5.72 (m, 1H), 6.02-6.10 (m, 1H), 6.47 (d, J = 2 Hz, 1H), 6.53 (dd, J = 2.5, 9 Hz, 1H), 7.17-7.27 (m, 5H), 7.85 (d, J = 9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.7, 27.8, 33.0, 35.3, 45.2, 49.9, 52.4, 55.0, 55.6, 69.5, 69.9, 70.6, 72.2, 73.3, 100.6, 104.5, 109.2, 114.7, 119.4, 126.4, 128.4, 129.3, 131.9, 133.5, 137.6, 138.1, 155.4, 157.0, 164.7; FT-IR (film, NaCl) νmax = 3339, 1718, 1594, 1324 cm−1; ESI (+) HRMS (m/z): [M + Na]+ calcd for C33H44N2O9S, 667.2665; found, 667.2661.
Compound 13e
Title compound was obtained from 11e and 12 as described for 13a in 68% yield after flash chromatography (1:1 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.52-1.61 (m, 3H), 1.81-1.93 (m, 2H), 2.09 (q, J = 7 Hz, 2H), 2.76 (dd, J = 10, 13.7 Hz, 1H), 2.83-2.88 (m, 1H), 3.01 (dd, J = 3.6, 14.2 Hz, 1H), 3.28-3.32 (m, 2H), 3.61-3.69 (m, 2H), 3.78-3.85 (m, 6H), 3.87-3.93 (m, 4H), 4.04 (t, J = 6.5 Hz, 2H), 4.92-5.01 (m, 3H), 5.09-5.16 (m, 3H), 5.60-5.71 (m, 2H), 5.72-5.81 (m, 1H), 6.47-6.51 (m, 2H), 7.14-7.26 (m, 5H), 7.82 (d, J = 8.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 24.9, 25.7, 28.2, 33.2, 35.4, 45.3, 51.0, 52.1, 54.9, 55.7, 69.1, 69.5, 70.7, 71.8, 73.1, 100.3, 104.2, 109.2, 115.0, 119.0, 119.2, 126.3, 128.3, 129.3, 133.4, 133.6, 137.5, 138.0, 155.4, 157.6, 164.8; FT-IR (film, NaCl) νmax = 3368, 1720, 1596, 1325 cm−1; ESI (+) HRMS (m/z): [M + Na]+ calcd for C33H44N2O9S, 667.2665; found, 667.2668.
Compound 13f
Title compound was obtained from 11f and 12 as described for 13a in 64% yield after flash chromatography (1:1 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.37-1.43 (m, 1H), 1.54-1.62 (m, 1H), 1.91-1.97 (m, 2H), 2.27 (q, J = 7 Hz, 2H), 2.76 (dd, J = 10.1, 13.9 Hz, 1H), 2.84-2.89 (m, 1H), 3.02 (dd, J = 4, 14.1 Hz, 1H), 3.30-3.37 (m, 2H), 3.54 (br s, 1H), 3.62-3.69 (m, 2H), 3.79-3.87 (m, 5H), 3.88-3.95 (m, 4H), 4.06 (t, J = 6.7 Hz, 2H), 4.96-5.00 (m, 2H), 5.04-5.16 (m, 4H), 5.59-5.67 (m, 2H), 5.76-5.85 (m, 1H), 6.47 (d, J = 1.9 Hz, 1H), 6.50 (dd, J = 2.1, 8.9 Hz, 1H), 7.16-7.26 (m, 5H), 7.82 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 25.7, 27.9, 29.7, 35.5, 45.3, 51.0, 52.1, 54.9, 55.7, 68.5, 69.5, 70.7, 71.8, 73.2, 100.3, 104.2, 109.2, 115.7, 119.1, 119.3, 126.4, 128.3, 129.3, 133.5, 137.1, 137.7, 155.4, 157.6, 164.8; FT-IR (film, NaCl) νmax = 3350, 1720, 1596, 1325 cm−1; ESI (+) HRMS (m/z): [M + Na]+ calcd for C32H42N2O9S, 653.2509; found, 653.2509.
Compound 13g
Title compound was obtained from 11g and 12 as described for 13a in 68% yield after flash-chromatography (1:1 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.39-1.45 (m, 1H), 1.56-1.63 (m, 1H), 1.81 (br s, 1H), 2.61 (q, J = 6.6 Hz, 2H), 2.75-2.80 (m, 1H), 2.86-2.91 (m, 1H), 3.02 (dd, J = 4.1, 14.1 Hz, 1H), 3.32 (d, J = 5.8 Hz, 2H), 3.53 (br s, 1H), 3.64-3.70 (m, 2H), 3.81-3.95 (m, 8H), 4.10 (t, J = 6.7 Hz, 2H), 4.97-5.01 (m, 2H), 5.12-5.20 (m, 4H), 5.62-5.66 (m, 2H), 5.86-5.94 (m, 1H), 6.49 (s, 1H), 6.51 (d, J = 9.1 Hz, 1H), 7.18-7.26 (m, 5H), 7.85 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): 25.8, 33.2, 35.6, 45.4, 51.2, 52.3, 55.0, 55.8, 68.7, 69.6, 70.8, 71.9, 73.3, 100.5, 104.5, 109.3, 117.9, 119.2, 119.4, 126.5, 128.5, 129.4, 133.5, 133.7, 137.8, 155.4, 157.5, 164.9; FT-IR (film, NaCl) νmax = 3350, 1722, 1596, 1325 cm−1; ESI (+) HRMS (m/z): [M + H]+ calcd for C31H40N2O9S, 617.2533; found, 617.2540.
Compound 13h
Title compound was obtained from 11h and 12 as described for 13a in 52% yield after flash chromatography (1:1 hexanes:EtOAc) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.37-1.43 (m, 1H), 1.54-1.65 (m, 1H), 2.73-2.80 (m, 1H), 2.85-2.90 (m, 1H), 3.02 (dd, J = 2.9, 13.7 Hz, 1H), 3.28-3.39 (m, 2H), 3.51 (br s, 1H), 3.63-3.70 (m, 2H), 3.77-3.95 (m, 10H), 4.62 (d, J = 5.2 Hz, 2H), 4.96-5.06 (m, 2H), 5.10-5.16 (m, 2H), 5.33 (d, J = 10.4 Hz, 1H), 5.45 (d, J = 17.2 Hz, 1H), 5.62-5.66 (m, 2H), 6.01-6.10 (m, 1H), 6.49 (s, 1H), 6.52 (d, J = 8.9 Hz, 1H), 7.18-7.33 (m, 5H), 7.85 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 25.7, 35.4, 45.3, 51.4, 52.2, 54.9, 55.7, 69.6, 70.0, 70.7, 71.8, 73.1, 100.7, 104.5, 109.2, 119.1, 119.4, 119.5, 126.4, 128.4, 129.3, 131.8, 133.5, 137.7, 155.3, 157.0, 164.8; FT-IR (film, NaCl) νmax = 3351, 1717, 1596, 1324 cm−1; ESI (+) HRMS (m/z): [M + H]+ calcd for C30H38N2O9S, 603.2376; found, 603.2375.
Inhibitor 14a
To stirring solution of 13a (32 mg, 0.047 mmol) in CH2Cl2 (15 mL) was added Grubbs’ 1st generation catalyst (4 mg, 0.0046 mmol). After stirring at 23 °C for 16 h, the solvent was evaporated under reduced pressure and the residue was subjected to flash column chromatography to yield 14a (27 mg, 88% yield) as a white solid and E/Z mixture (3:1, determined by HPLC). The isomers were isolated by reversed phase HPLC using the following conditions: YMC Pack ODS A column (250 × 100 mm, 5 μm); Flow rate = 2.75 mL/min; Isocratic 60:40 CH3CN:H2O; T = 35 °C; λ = 215 nm; E isomer Rt = 16.5 min; Z isomer Rt = 14.5 min.
14aE
1H NMR (800 MHz, CDCl3): δ 1.39-1.44 (m, 2H), 1.48-1.53 (m, 1H), 1.57-1.64 (m, 2H), 1.68-1.74 (m, 3H), 1.77-1.82 (m, 1H), 1.83-.188 (m, 1H), 1.94-1.98 (m, 1H), 2.10-2,14 (m, 3H), 2.71 (dd, J = 9.6, 14.1 Hz, 1H), 2.86-2.89 (m, 1H), 2.90 (dd, J = 4.3, 14.1 Hz, 1H), 2.91-2.99 (m, 2H), 3.31-3.33 (m, 1H), 3.61-3.64 (m, 1H), 3.65-3.70 (m, 2H), 3.73 (br s, 1H), 3.76-3.78 (m, 1H), 3.79-3.85 (m, 5H), 3.93 (dd, J = 6.6, 9.4 Hz, 1H), 3.96-3.98 (m, 1H), 4.02-4.05 (m,1H), 4.87 (d, J = 9.1 Hz, 1H), 4.97-5.00 (m, 1H), 5.44-5.48 (m, 1H), 5.54-5.56 (m, 1H), 5.63 (d, J = 5.1 Hz, 1H), 6.44 (d, J = 2 Hz, 1H), 6.49 (dd, J = 2.2, 8.8 Hz, 1H), 7.13-7.24 (m, 5H), 7.84 (d, J = 8.8 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.1, 25.3, 25.7, 26.8, 29.8, 30.4, 32.5, 35.5, 45.2, 50.6, 51.6, 54.7, 55.6, 68.8, 69.5, 70.7, 71.6, 73.3, 99.9, 103.9, 109.2, 118.1, 126.4, 128.4, 129.3, 130.5, 132.1, 134.0, 137.4, 155.2, 157.9, 164.9; ESI (+) HRMS (m/z): [M + Na]+ calcd for C34H46N2O9S, 681.2822; found, 681.2815.
14aZ
1H NMR (800 MHz, CDCl3): δ 1.28-1.33 (m, 2H), 1.38-1.47 (m, 2H), 1.52-1.64 (m, 5H), 1.84-1.90 (m, 2H), 2.04-2.08 (m, 2H), 2.16-2.22 (m, 2H), 2.74 (dd, J = 9.5, 14.1 Hz, 1H), 2.88-2.90 (m, 1H), 3.00 (dd, J = 4.5, 14.1 Hz, 1H), 3.05-3.14 (m, 2H), 3.15 (dd, J = 9.4, 15.1 Hz, 1H), 3.44-3.48 (m, 1H), 3.61 (br s, 1H), 3.66-3.71 (m, 2H), 3.76-3.89 (m, 6H), 3.95 (dd, J = 6.2, 9.6 Hz, 1H), 4.10-4.15 (m, 1H), 4.87 (d, J = 9.1 Hz, 1H), 5.00-5.03 (m, 1H), 5.35-5.39 (m, 1H), 5.49-5.52 (m, 1H), 5.63 (d, J = 5.2 Hz, 1H), 6.50 (s, 1H), 6.49 (dd, J = 2.3, 8.8 Hz, 1H), 7.17-7.26 (m, 5H), 7.84 (d, J = 8.8 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.0, 25.1, 25.7, 26.0, 26.6, 27.8, 29.6, 35.5, 45.2, 48.5, 52.0, 54.7, 55.6, 68.8, 69.5, 70.6, 71.9, 73.3, 100.7, 104.2, 109.2, 117.8, 126.4, 128.4, 129.2, 129.8, 130.0, 134.1, 137.5, 155.2, 158.0, 165.0; ESI (+) HRMS (m/z): [M + Na]+ calcd for C34H46N2O9S, 681.2822; found, 681.2819.
Inhibitor 14b
The title compound was obtained from a ring closing metathesis reaction of 13b using Grubbs’ 1st generation catalyst as described for 14a. The crude material was purified by silica chromatography (60:40 EtOAc:hexanes) to give the desired product (50% yield) as a mix of E/Z isomers (27:73 by HPLC). The isomers were isolated by chiral HPLC using the following conditions: Chiralpak IA column (250 × 4.6mm, 5 μm); Flow rate = 0.75 mL/min; Isocratic 60:40 IPA:hexanes; T = 25 °C; λ = 215 nm; E isomer Rt = 7.56 min; Z isomer Rt = 8.89 min.
14bE
1H NMR (800 MHz, CDCl3): δ 1.60-1.20 (m, 4H), 2.30-1.90 (m, 7H), 3.10-2.80 (m, 5H), 3.32 (m, 1H), 4.00-3.50 (m, 12 H), 4.15 (m, 2H), 4.98 (m, 2H), 5.45 (m, 1H), 5.63 (m, 2H), 6.51 (m, 2H), 7.25-7.15 (m, 5H), 7.76 (d, J = 19.2 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 23.1, 24.8, 25.0, 25.7, 26.2, 26.6, 29.0, 35.5, 45.2, 48.2, 52.0, 54.8, 55.6, 67.1, 69.5, 70.7, 71.6, 73.3, 100.1, 104.0, 109.2, 114.6, 118.4, 126.4, 128.4, 128.9, 129.3, 130.6, 133.8, 137.4, 155.3, 157.7, 164.9; ESI (+) HRMS (m/z): [M + Na]+ calcd for C33H44N2O9S, 667.2665; found, 667.2660.
14bZ
1H NMR (800 MHz, CDCl3): δ 1.60-1.0 (m, 7H); 1.72 (m, 2H), 1.90 (m, 3H), 2.21 (m, 2H), 2.43 (m, 1H), 2.55 (m, 1H), 2.67 (m, 1H), 2.76 (m, 1H), 2.84 (m, 1H), 3.01 (m, 1H), 3.26 (m, 1H), 3.41 (m, 3H), 3.60 (m, 4H), 3.69 (m, 1H), 3.86 (m, 2H), 4.43 (d, J = 15.2 Hz, 1H), 4.83 (m, 1H), 5.24 (m, 1H), 5.33 (m, 1H), 5.51 (d, J = 8.8 Hz, 1H), 6.45 (m, 2H), 7.17 (m, 3H), 7.24 (m, 2H), 7.87 (d, J = 13.6 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 24.7, 25.7, 27.3, 28.6, 30.9, 32.1, 35.5, 45.2, 49.3, 51.8, 54.8, 55.6, 69.5, 70.7, 71.0, 71.9, 73.3, 101.2, 104.3, 109.2, 119.3, 126.4, 128.4, 128.9, 129.3, 130.8, 130.9, 133.4, 137.5, 155.3, 158.2, 164.5; ESI (+) HRMS (m/z): [M + Na]+ calcd for C33H44N2O9S, 667.2665; found, 667.2667.
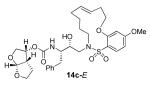
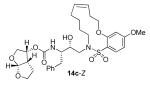
Inhibitor 14c
Title compound was obtained from 13c and Grubbs’ 1st generation catalyst as described for 14a in 89% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid and E/Z mixture (3:1, determined by HPLC). The isomers were isolated using reversed-phase HPLC under the following conditions: YMC Pack ODS-A column (250 × 100mm, 5 μm); Flow rate = 2.75 mL/min; Isocratic 60:40 CH3CN:H2O; T = 35 °C; λ = 215 nm; E isomer Rt = 13.43 min; Z isomer Rt = 11.76 min.
14cE
1H NMR (800 MHz, CDCl3): δ 1.45-1.54 (m, 3H), 1.56-1.64 (m, 2H), 1.68-1.72 (m, 1H), 2.11-2.20 (m, 2H), 2.52-2.62 (m, 2H), 2.79 (dd, J = 9.6, 14 Hz, 1H), 2.87-2.90 (m, 1H), 2.96-3.00 (m, 2H), 3.04 (dd, J = 4.2, 14.2 Hz, 1H), 3.40-3.44 (m, 1H), 3.53-3.56 (m, 1H), 3.66-3.70 (m, 3H), 3.82-3.89 (m, 6H), 3.94 (dd, J = 6.3, 9.5 Hz, 1H), 4.07-4.14 (m, 2H), 4.96 (d, J = 9.4 Hz, 1H), 4.98-5.01 (m, 1H), 5.53-5.56 (m, 1H), 5.64-5.68 (m, 2H), 6.52-6.53 (m, 2H), 7.18-7.27 (m, 5H), 7.76 (d, J = 9.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 24.4, 25.7, 28.3, 32.3, 32.4, 35.6, 45.2, 49.6, 51.9, 54.9, 55.6, 69.5, 69.9, 70.7, 71.9, 73.2, 101.9, 104.9, 109.2, 119.3, 126.4, 128.0, 128.4, 129.3, 133.4, 133.9, 137.6, 155.3, 158.0, 164.5; ESI (+) HRMS (m/z): [M + H]+ calcd for C32H42N2O9S, 631.2689; found, 631.2698.
14cZ
1H NMR (500 MHz, CDCl3): δ 1.48-1.52 (m, 3H), 1.57-1.72 (m, 3H), 2.10-2.14 (m, 1H), 2.28-2.32 (m, 1H), 2.47-2.51 (m, 1H), 2.78 (dd, J = 9, 14 Hz, 2H), 2.87-2.91 (m, 1H), 2.98-3.08 (m, 3H), 3.45-3.60 (m, 2H), 3.65-3.71 (m, 3H), 3.80-3.90 (m, 6H), 3.95 (dd, J = 6, 9.5 Hz, 1H), 4.07-4.10 (m, 1H), 4.18-4.21 (m, 1H), 4.95 (d, J = 9.5 Hz, 1H), 4.98-5.02 (m, 1H), 5.44-5.49 (m, 2H), 5.63 (d, J = 5.5 Hz, 1H), 6.51 (dd, J = 2.5, 9.8 Hz, 1H), 6.53 (d, J = 2 Hz, 1H), 7.18-7.27 (m, 5H), 7.81 (d, J = 9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 24.7, 24.8, 25.7, 26.3, 27.7, 35.5, 45.2, 46.9, 50.2, 54.9, 55.6, 69.5, 69.9, 70.6, 70.7, 73.2, 101.9, 104.8, 109.1, 120.5, 126.4, 127.6, 128.4, 129.3, 129.6, 132.5, 133.2, 137.6, 155.3, 158.1, 164.6; ESI (+) HRMS (m/z): [M + H]+ calcd for C32H42N2O9S, 631.2689; found, 631.2706.
Inhibitor 14d
Title compound was obtained from 13d and Grubbs’ 1st generation catalyst as described for 14a in 71% yield after flash-chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.40-1.47 (m, 1H), 1.58-1.62 (m, 2H), 1.92-1.95 (m, 1H), 2.11-2.15 (m, 1H), 2.28-2.39 (m, 2H), 2.73-2.78 (m, 1H), 2.80-3.05 (m, 6H), 3.64-3.70 (m, 3H), 3.80-3.89 (m, 6H), 3.93-3.96 (m, 2H), 4.12-4.15 (m, 1H), 4.62 (br s, 1H), 4.97-4.99 (m, 1H), 5.11 (d, J = 9.2 Hz, 1H), 5.52-5.54 (m, 2H), 5.61 (d, J = 5.1 Hz, 1H), 6.46-6.49 (m, 2H), 7.18-7.26 (m, 5H), 7.80 (d, J = 9.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 22.6, 22.9, 23.4, 25.7, 25.9, 29.6, 35.4, 45.1, 45.2, 47.9, 55.0, 55.6, 62.0, 69.5, 70.7, 73.2, 100.2, 103.6, 109.2, 119.9, 123.5, 126.4, 128.4, 129.3, 132.6, 137.6, 140.4, 155.4, 156.4, 164.3; ESI (+) HRMS (m/z): [M + H]+ calcd for C31H40N2O9S, 617.2533; found, 617.2534.
Inhibitor 14e
Title compound was obtained from 13e and Grubbs’ 2nd generation catalyst as described for 14a in 52% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.46-1.51 (m, 1H), 1.60-1.76 (m, 4H), 1.88-1.92 (m, 2H), 2.23-2.37 (m, 2H), 2.79 (dd, J = 9, 14 Hz, 1H), 2.88-3.01 (m, 2H), 3.10-3.13 (m, 2H), 3.57 (br s, 1H), 3.66-3.72 (m, 2H), 3.72-3.89 (m, 5H), 3.92-3.99 (m, 2H), 4.07-4.15 (m, 2H), 4.94 (d, J = 8.5 Hz, 1H), 5.00-5.04 (m, 1H), 5.44-5.54 (m, 3H), 5.64 (d, J = 5.1 Hz, 1H), 6.43 (d, J = 2 Hz, 1H), 6.49-6.51 (m, 1H), 7.16-7.28 (m, 5H), 7.81 (d, J = 9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.7, 26.3, 26.5, 27.4, 29.6, 35.5, 43.9, 45.2, 50.7, 54.7, 55.6, 69.5, 69.7, 70.7, 73.3, 100.0, 103.9, 109.2, 117.7, 122.9, 126.4, 128.4, 129.3, 133.7, 135.1, 137.4, 155.3, 157.7, 164.9; ESI (+) HRMS (m/z): [M + Na]+ calcd for C31H40N2O9S, 639.2352; found, 639.2345.
Inhibitor 14f
Title compound was obtained from 13f and Grubbs’ 2nd generation catalyst as described for 14a in 81% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.40-1.47 (m, 1H), 1.58-1.62 (m, 2H), 1.92-1.95 (m, 1H), 2.11-2.15 (m, 1H), 2.28-2.39 (m, 2H), 2.73-2.78 (m, 1H), 2.80-3.05 (m, 5H), 3.64-3.70 (m, 2H), 3.80-3.89 (m, 6H), 3.93-3.96 (m, 2H), 4.12-4.15 (m, 1H), 4.62 (br s, 1H), 4.97-4.99 (m, 1H), 5.11 (d, J = 9.2 Hz, 1H), 5.52-5.54 (m, 2H), 5.61 (d, J = 5.1 Hz, 1H), 6.46-6.49 (m, 2H), 7.18-7.26 (m, 5H), 7.80 (d, J = 9.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 22.7, 25.1, 25.7, 29.6, 35.5, 41.2, 45.3, 47.8, 54.8, 55.6, 65.9, 69.5, 70.7, 73.2, 101.0, 104.3, 109.2, 118.7, 123.3, 126.4, 128.4, 129.3, 133.4, 133.6, 137.5, 155.4, 157.8, 164.7; ESI (+) HRMS (m/z): [M + H]+ calcd for C30H38N2O9S, 603.2376; found, 603.2369.
Inhibitor 14g
Title compound was obtained from 13g and Grubbs’ 2nd generation catalyst as described for 14a in 81% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.47-1.55 (m, 1H), 1.64-1.70 (m, 2H), 2.51-2.54 (m, 1H), 2.73-2.78 (m, 1H), 2.84-3.94 (m, 2H), 3.04-3.11 (m, 2H), 3.19 (d, J = 13.6 Hz, 1H), 3.70-3.75 (m, 2H), 3.86 (s, 4H), 3.94-3.99 (m, 4H), 4.07-4.08 (m, 1H), 4.18 (br s, 1H), 4.58 (s, 1H), 5.02-5.06 (m, 1H), 5.12 (s, 1H), 5.61 (d, J = 5 Hz, 1H), 5.76-5.82 (m, 1H), 5.88-5.93 (m, 1H), 6.35 (d, J = 1.1 Hz, 1H), 6.51 (dd, J = 1.5, 8.7 Hz, 1H), 7.20-7.31 (m, 5H), 7.79 (d, J = 8.8 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.8, 26.7, 35.4, 43.6, 45.3, 47.1, 55.1, 55.7, 67.4, 69.6, 70.5, 70.8, 73.3, 99.4, 104.1, 109.2, 120.5, 126.5, 126.9, 128.5, 129.4, 131.2, 132.0, 137.6, 155.6, 157.4, 164.7; ESI (+) HRMS (m/z): [M + Na]+ calcd for C29H36N2O9S, 611.2039; found, 611.2040.
Inhibitor 14h
Title compound was obtained from 13h and Grubbs’ 2nd generation catalyst as described for 14a in 79% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (400 MHz, CDCl3): δ 1.45-1.51 (m, 1H), 1.60-1.68 (m, 2H), 2.80-2.90 (m, 2H), 3.00-3.12 (m, 3H), 3.53 (br s, 1H), 3.66-3.71 (m, 2H), 3.83 (s, 4H), 3.92-3.95 (m, 3H), 4.25 (br s, 1H), 4.88-5.01 (m, 3H), 5.13 (d, J = 8.3 Hz, 1H), 5.63 (d, J = 5.1 Hz, 1H), 5.72-5.76 (m, 2H), 6.59-6.64 (m, 2H), 7.19-7.26 (m, 5H), 7.74 (d, J = 8.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 25.7, 29.6, 35.3, 43.9, 45.2, 47.8, 55.1, 55.7, 69.5, 70.7, 71.1, 73.3, 104.5, 107.2, 109.2, 123.4, 126.1, 126.4, 128.4, 129.3, 131.3, 131.5, 137.6, 155.6, 157.7, 164.5; ESI (+) HRMS (m/z): [M + Na]+ calcd for C28H34N2O9S, 597.1883; found, 597.1887.
Inhibitor 15a
To a stirring solution of 14a (10 mg, 0.015 mmol) in EtOAc (2 mL) was added 10% Pd on carbon and the reaction was stirred under H2 atmosphere for 12 h. After this time the reaction was filtered through a pad of celite and solvent was evaporated under reduced pressure. The residue was then purified by flash chromatography to give 15a (9 mg, 93% yield) as a white solid. 1H NMR (500 MHz, CDCl3): δ 0.87-0.92 (m, 2H), 1.25-1.28 (m, 8H), 1.38-1.49 (m, 5H), 1.60-1.64 (m, 3H), 1.71-1.83 (m, 4H), 2.32-2.38 (m, 1H), 2.75 (dd, J = 9.5, 13.7 Hz, 1H), 2.86-2.91 (m, 1H), 3.01-3.10 (m, 3H), 3.64-3.71 (m, 2H), 3.85 (s, 4H), 3.94 (dd, J = 6.4, 9.5 Hz, 1H), 4.01-4.04 (m, 1H), 4.10-4.16 (m,1H), 4.94 (d, J = 9.2 Hz, 1H), 5.01-5.15 (m, 1H), 5.67 (d, J = 5.1 Hz, 1H), 6.52-6.54 (m, 2H), 7.19-7.29 (m, 5H), 7.85 (d, J = 9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 23.8, 24.2, 25.5, 26.3, 26.4, 28.2, 29.1, 29.4, 35.3, 45.1, 51.3, 52.8, 54.6, 55.4, 69.2, 69.3, 69.5, 70.5, 71.9, 73.1, 100.2, 103.9, 109.0, 118.7, 126.3, 128.2, 129.1, 133.5, 137.3, 155.1, 157.9, 164.6; ESI (+) HRMS (m/z): [M + Na]+ calcd for C34H48N2O9S, 683.2978; found, 683.2984.
Inhibitor 15b
Title compound was obtained from 14b as described for 15a in 90% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.30-1.47 (m, 8H), 1.57-1.70 (m, 5H), 1.72-1.79 (m, 1H), 1.82-1.87 (m, 2), 2.77 (dd, J = 10, 14 Hz, 1H), 2.86-2.91 (m, 1H),3.00 (dd, J = 4.5, 14 Hz, 1H), 3.05 (d, J = 2.5, 15 Hz, 1H), 3.14-3.23 (m, 2H), 3.56-3.62 (m, 2H), 3.65-3.72 (m, 2H), 3.81-3.90 (m, 6H), 3.94 (dd, J = 6.5, 10 Hz, 1H), 4.05-4.15 (m, 2H), 4.96 (d, J = 9.5 Hz, 1H), 4.99-5.02 (m, 1H), 5.64 (d, J = 5.5 Hz, 1H), 6.49-6.52 (m, 2H), 7.17-7.27 (m, 5H), 7.82 (d, J = 9.5 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 23.2, 24.2, 24.9, 25.2, 25.7, 26.1, 27.6, 28.9, 35.4, 45.2, 49.2, 52.7, 54.8, 55.6, 68.9, 69.5, 70.7, 71.7, 73.2, 100.5, 104.2, 109.2, 118.5, 126.4, 128.4, 129.3, 133.8, 137.5, 155.3, 158.1, 164.8; ESI (+) HRMS (m/z): [M + Na]+ calcd for C33H46N2O9S, 669.2822; found, 669.2828.
Inhibitor 15c
Title compound was obtained from 14c as described for 15a in 90% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.35-1.39 (m, 3H), 1.40-1.54 (m, 4H), 1.60-1.66 (m, 6H), 1.85-1.98 (m, 2H), 2.78 (dd, J = 9.1, 14 Hz, 1H), 2.88-2.91 (m, 1H),2.97-3.03 (m, 1H), 3.06-3.14 (m, 1H), 3.39-3.43 (m, 1H), 3.50-3.56 (m, 1H), 3.66-3.72 (m, 3H), 3.82-3.85 (m, 6H), 3.95 (dd, J = 6.3, 9.6 Hz, 1H), 4.07-4.10 (m, 2H), 4.95-5.03 (m, 2H), 5.64 (d, J = 5 Hz, 1H), 6.50-6.53 (m, 2H), 7.17-7.27 (m, 5H), 7.82 (d, J = 9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 22.6, 23.5, 24.0, 24.6, 25.3, 25.7, 26.1, 35.5, 44.8, 45.2, 46.5, 51.0, 54.8, 55.6, 69.5, 70.1, 70.7, 70.8, 73.2, 100.9, 104.3, 109.2, 118.5, 126.4, 128.4, 129.4, 133.9, 137.5, 155.2, 158.2, 164.8; ESI (+) HRMS (m/z): [M + Na]+ calcd for C32H44N2O9S, 655.2668; found, 655.2667.
Inhibitor 15d
Title compound was obtained from 14d or 14e as described for 15a in 94% yield after flash-chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.33-1.54 (m, 6H), 1.57-1.74 (m, 3H), 1.86-1.94 (m, 2H), 2.77 (dd, J = 9.5, 14 Hz, 1H), 2.86-2.90 (m, 1H), 2.96-3.08 (m, 3H), 3.24-3.29 (m, 1H), 3.64-3.72 (m, 2H), 3.74-3.86 (m, 7H), 3.94-4.00 (m, 2H), 4.15-4.22 (m, 2H), 4.92 (d, J = 9.2 Hz, 1H), 4.97-5.02 (m, 1H), 5.62 (d, J = 5.2 Hz, 1H), 6.48 (d, J = 2.1Hz, 1H), 6.52 (dd, J = 2, 8.9 Hz, 1H), 7.17-7.27 (m, 5H), 7.87 (d, J = 8.6 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 23.2, 25.0, 25.6, 26.1, 26.3, 35.4, 42.9, 45.2, 48.5, 54.7, 55.6, 69.3, 69.5, 70.7, 73.3, 99.9, 103.8, 109.1, 117.1, 126.4, 128.4, 129.3, 134.6, 137.4, 155.1, 157.7, 165.2; ESI (+) HRMS (m/z): [M + Na]+ calcd for C31H42N2O9S, 641.2509; found, 641.2512.
Inhibitor 15f
Title compound was obtained from 14f as described for 15a in 93% yield after flash-chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.33-1.48 (m, 4H), 1.56-1.66 (m, 3H), 1.67-1.71 (m, 2H), 1.83-1.86 (m, 1H), 2.04-2.12 (m, 2H), 2.74 (dd, J = 9.7, 14 Hz, 1H), 2.83-2.99 (m, 4H), 3.18 (br s, 1H), 3.64-3.71 (m, 2H), 3.76-3.85 (m, 6H), 3.89-3.99 (m, 3H), 4.08 (br s, 1H), 4.19-4.23 (m, 1H), 4.96-5.00 (m, 2H), 5.63 (d, J = 5 Hz, 1H), 6.45 (d, J = 2Hz, 1H), 6.50 (dd, J = 2.5, 9 Hz, 1H), 7.15-7.26 (m, 5H), 7.88 (d, J = 9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 22.2, 24.4, 24.6, 25.7, 25.8, 35.4, 45.2, 46.8, 51.8, 54.7, 55.7, 69.2, 69.6, 70.5, 70.8, 73.3, 99.8, 103.9, 109.2, 117.5, 126.5, 128.4, 129.3, 134.3, 137.4, 155.2, 157.8, 165.3; ESI (+) HRMS (m/z): [M + Na]+ calcd for C30H40N2O9S, 605.2533; found, 605.2526.
Inhibitor 15g
Title compound was obtained from 14g as described for 15a in 90% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.46-1.50 (m, 3H), 1.59-1.67 (m, 4H), 1.96-2.04 (m, 2H), 2.81-2.91 (m, 2H), 3.03 (dd, J = 3, 14 Hz, 1H), 3.07-3.12 (m, 2H), 3.56 (br s, 1H), 3.57-3.71 (m, 2H), 3.83-3.91 (m, 6H), 3.93-4.02 (m, 3H), 4.25 (br s, 1H), 4.98-5.02 (m, 2H), 5.63 (d, J = 5 Hz, 1H), 6.40 (d, J = 2 Hz, 1H), 6.49 (dd, J = 2, 9 Hz, 1H), 7.18-7.28 (m, 5H), 7.76 (d, J = 9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 22.4, 24.4, 25.3, 25.7, 35.4, 43.1, 45.2, 47.3, 54.9, 55.6, 69.5, 70.1, 70.6, 70.7, 73.2, 100.5, 104.2, 109.2, 121.4, 126.4, 128.4, 129.3, 131.5, 137.5, 155.4, 157.7, 164.5; ESI (+) HRMS (m/z): [M + H]+ calcd for C29H38N2O9S, 591.2376; found, 591.2381.
Inhibitor 15h
Title compound was obtained from 14h as described for 15a in 92% yield after flash chromatography (2:3 hexanes:EtOAc) as a white solid. 1H NMR (500 MHz, CDCl3): δ 1.39-1.51 (m, 3H), 1.60-1.68 (m, 3H), 1.72-1.81 (m, 2H), 2.82-2.92 (m, 2H), 3.00-3.06 (m, 3H), 3.66-3.71 (m, 3H), 3.80-3.87 (m, 6H), 3.95 (dd, J = 6, 9.5 Hz, 1H), 3.97-4.02 (m, 1H), 4.25 (br s, 1H), 4.98-5.02 (m, 2H), 5.63 (d, J = 5 Hz, 1H), 6.62-6.64 (m, 2 H), 7.18-7.28 (m, 5H), 7.77 (d, J = 10.5 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 24.3, 25.0, 25.7, 35.3, 44.5, 45.2, 46.5, 55.0, 55.6, 69.5, 70.6, 73.2, 73.9, 105.6, 107.3, 109.1, 126.4, 128.4, 129.3, 130.9, 137.3, 155.5, 157.4, 164.4; ESI (+) HRMS (m/z): [M + H]+ calcd for C28H36N2O9S, 577.2220; found, 577.2222.
tert-butyl (2S,3R)-4-(2,2-dimethylpent-4-enylamino)-3-hydroxy-1-phenylbutan-2-ylcarbamate (17a)
A stirring solution of amine 16a (1.58 g, 4.33 mmol) and epoxide 8 (304 mg, 1.15 mmol) was heated to 60 °C for 4 h and allowed to stir at 23 °C overnight. The reaction mixture was concentrated and purified by silica chromatography (3:97 MeOH:CH2Cl2) to give 370 mg (98% yield) of product as a clear oil that solidified upon refrigeration. [α]D20 +0.9 (c 0.14, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 0.90 (s, 6H), 1.36 (s, 9H), 2.01 (d, J = 7.6 Hz, 2H), 2.35 (s, 2H), 2.69 (d, J = 4.8 Hz, 2H), 2.87 (dd, J = 8, 14.8 Hz, 1H), 2.98 (dd, J = 4.8, 9.2 Hz, 1H), 3.45 (m, 1H), 3.82 (m, 1H), 4.79 (d, J = 9.2 Hz, 1H), 5.03 (m, 2H), 5.81 (m, 1H), 7.26 (m, 5H); 13C NMR (CDCl3, 100 MHz): δ 25.4, 25.4, 29.6, 34.3, 36.8, 44.5, 52.1, 54.3, 60.1, 70.3, 79.1, 116.9, 126.2, 128.3, 129.4, 135.2, 137.9, 155.8; FTIR (NaCl) νmax = 3349, 3064, 2928, 1693, 1391, 1366, cm−1; ESI LRMS (m/z): 376.06 [M+H]+.
tert-butyl (2S,3R)-4-(N-(2,2-dimethylpent-4-enyl)-2-(hex-5-enyloxy)-4-methoxyphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (18a)
To a mixture of amine 17a (188 mg, 0.5 mmol) and sulfonyl chloride 7d (183 mg, 0.6 mmol) at 0 °C under argon atmosphere was added pyridine (8 mL, freshly distilled over KOH) and reaction was allowed to warm to 23 °C while stirring. The reaction turned orange and was allowed to stir overnight. The reaction was condensed under reduced pressure, washed with sat. CuSO4, and the product extracted with dichloromethane. The organic layer was washed with H2O, brine, dried over sodium sulfate and purified by silica chromatography (20:80 EtOAc:hexanes) to give 18a (180 mg, 56% yield) as a clear oil. [α]D20 +18.3 (c 0.12, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 0.90 (s, 3H), 0.92 (s, 3H), 1.25 (m, 1H), 1.31 (s, 9H), 1.59 (m, 2H), 1.86 (quintet, J = 8 Hz, 2H), 1.99 (d, J = 7.2 Hz, 2H), 2.12 (q, J = 6.8 Hz, 2H), 2.77 (dd, J = 8, 13.2 Hz, 1H), 2.86 (dd, J = 4.8, 14.4 Hz, 1H), 3.20 (m, 4H), 3.66 (m, 1H), 3.80 (m, 1H), 3.83 (s, 3H), 4.00 (m, 2H), 4.40 (d, J = 8.8 Hz, 1H), 5.00 (m, 4H), 5.79 (m, 2H), 6.47 (m, 2H), 7.20 (m, 5H),7.78 (d, J = 8.4 Hz, 1H); 13C NMR (CDCl3, 100 MHz): δ 25.0, 25.5, 25.6, 28.0, 28,2, 28.4, 33.2, 35.8, 45.2, 54.2, 55.5, 55.6, 62.4, 69.1, 72.0, 79.3, 100.1, 104.2, 115.1, 117.5, 118.5, 126.2, 128.3, 129.4, 134.0, 134.7, 137.6, 138.0, 155.5, 157.5, 164.8; FTIR (NaCl) νmax = 3412, 2852, 1701, 1596, 1496, 1456, 1391, 1367 cm−1; ESI (+) HRMS (m/z): [M+Na+] calcd for C35H52N2O7S 667.3393; found, 667.3399.
(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl (2S,3R)-4-(N-(2,2-dimethylpent-4-enyl)-2-(hex-5-enyloxy)-4-methoxyphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (19a)
To a stirring solution of 18a (180 mg, 0.28 mmol) in CH2Cl2 (5 mL) at 0 °C was added trifluoroacetic acid (1.5 mL). The reaction was allowed to warm to 23 °C and stirred overnight. Solvents were removed under reduced pressure and saturated aqueous NaHCO3 and 1N NaOH (1 mL) was added and extracted with ether. Solvents were removed under reduced pressure to afford the crude amine product.
To a solution of above amine (0.28 mmol) in MeCN (20 mL) was added carbonate 12 (92 mg, 0.31 mmol) under argon followed by dropwise addition of i-Pr2NEt (1 mL) and pyridine (1 mL) and the reaction was allowed to stir overnight at 23 °C. After stirring for 2 days, the solvent was removed under reduced pressure and the crude material purified by silica gel column chromatography (50:50 EtOAc:hexanes) to give 19a (144 mg, 73% yield over two steps) as a white solid. m.p. = 49-52 °C; [α]D20 +3.9 (c 1.03, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 0.90 (s, 3H), 0.91 (s, 3H), 1.35 (m, 1H), 1.58 (m, 3H), 1.86 (m, 2H), 2.02 (m, 3H), 2.11 (m, 2H), 2.69 (dd, J = 9.6, 14 Hz, 1H), 2.84 (m, 1H), 2.94 (dd, J = 4, 14.4 Hz, 1H), 3.08 (m, 2H), 3.29 (m, 2H), 3.63 (m, 2H), 3.78 (m, 2H), 3.83 (s, 3H), 3.91 (m, 3H), 4.03 (m, 2H), 4.87 (d, J = 9.6 Hz, 1H), 5.02 (m, 4H), 5.60 (d, J = 5.2 Hz, 1H), 5.77 (m, 2H), 6.48 (m, 2H), 7.18 (m, 5H), 7.79 (d, J = 8.4 Hz, 1H),; 13C NMR (CDCl3, 100 MHz) δ 24.9, 25.4, 25.5, 25.7, 28.3, 33.2,35.5, 35.7, 45.1, 45.2, 54.8, 55.6, 55.6, 62.6, 69.2, 69.5, 70.6, 72.3, 73.1, 100.1, 104.3, 109.2, 115.0, 117.6, 118.0, 126.3, 128.3, 129.2, 134.0, 134.5, 137.5, 137.9, 155.0, 157.6, 164.9; FTIR (NaCl) νmax = 1323, 1595, 1722, 2922, 3487 cm−1; ESI (+) HRMS (m/z): [M+H]+ calcd for C37H52N2O9S 701.3472; found, 701.3473.
Inhibitors 20a and 21a
19a (100 mg, 0.14 mmol) was dissolved in CH2Cl2 (90 mL). Grubbs’ 2nd generation catalyst (12 mg, 0.014 mmol) was added and the reaction was allowed to stir overnight at 23 °C. Solvent was removed under reduced pressure and the material purified by silica gel column chromatography (50:50 → 75:25 EtOAc:hexanes) to give 92 mg (96% yield) of product as a mixture of stereoisomers (31:69 Z:E by HPLC) as a white solid. The individual stereoisomers were isolated by reversed-phase HPLC YMC-Pack ODSA (250×10 mm, 5 μm); flow rate = 1.5 mL/min; isocratic 80:20 MeOH:H2O; T=25 °C; λ = 210 nm, Rt Z = 17 min, Rt E = 18 min).
21a
[α]D20 −0.8 (c 2.36, CHCl3); 1H NMR (CDCl3, 800 MHz) δ 1.05 (s, 3H), 1.12 (s, 3H), 1.24 (t, J = 6.4 Hz, 1H), 1.25 (s, 1H), 1.39 (m, 1H), 1.58 (m, 1H), 1.90-1.72 (m, 4H), 1.94-2.10 (m, 4H), 2.68 (dd, J = 9.6, 14.4 Hz, 1H), 2.86 (q, J = 7.2 Hz, 1H), 2.92 (dd, J = 4, 14.4 Hz, 1H), 2.97 (dd, J = 2.4, 15.2 Hz, 1H), 3.11 (dd, J = 9.6, 15.2 Hz, 1H), 3.64 (m, 1H), 3.68 (m, 1H), 3.71 (dd, J = 6.4, 13.6 Hz, 1H), 3.77 (m, 1H), 3.82 (m, 1H), 3.84 (s, 3H), 4.04-3.88 (m, 4H), 4.71 (d, J = 9.6 Hz, 1H), 4.98 (q, J = 7.2 Hz, 1H), 5.60 (d, J = 5.6 Hz, 1H), 5.62 (d, J = 5.6 Hz, 1H), 5.68 (m, 1H), 6.47 (m, 1H), 6.50 (dd, J = 1.6, 8.8 Hz, 1H), 7.12 (d, J = 7.2 Hz, 2H), 7.18 (t, J = 7.2 Hz, 1H), 7.24 (t, J = 8 Hz, 2H), 7.79 (d, J = 8.8 Hz, 1H),; 13C NMR (CDCl3, 125 MHz) δ 25.7, 26.1, 27.6, 29.2, 29.6, 29.7, 35.6, 35.8, 45.3, 46.1, 54.7, 55.7, 56.1, 63.0, 69.6, 69.7, 70.7, 72.2, 73.3, 100.6, 104.4, 109.3, 117.8, 126.5, 128.4, 128.5, 129.3, 133.6, 134.5, 137.5, 155.1, 158.1, 164.9; FTIR (film, NaCl) νmax = 3445, 2926, 1720, 1596, 1575, 1469, 1369 cm−1; ESI (+) HRMS (m/z): [M+Na]+ calcd for C35H48N2O9S 695.2978; found, 695.2989.
20a
[α]D20 −0.5 (c 0.99, CHCl3); 1H NMR (CDCl3, 800 MHz) δ 1.07 (s, 3H), 1.13 (s, 3H), 1.25 (s, 3H), 1.38 (m, 1H), 1.57 (m, 1H), 1.62-1.76 (m, 2H), 1.90 (br s, 2H), 1.98-2.20 (m, 4H), 2.70 (dd, J = 9.6, 14.4 Hz, 1H), 2.86 (m, 1H), 2.91 (dd, J = 0.8, 15.2 Hz, 1H), 2.93 (dd, J = 4, 14.4 Hz, 1H), 3.08 (dd, J = 8.8, 15.2 Hz, 1H), 3.65 (m, 1H), 3.68 (dd, J = 6.4, 9.6 Hz, 1H), 3.79 (m, 1H), 3.82 (dt, J = 2.4, 8.8 Hz, 1H), 3.85 (s, 3H), 3.93 (dd, J = 6.4, 9.6 Hz, 1H), 3.96 (m, 1H), 4.08 (t, J = 4.8 Hz, 2H), 4.74 (d, J = 9.6 Hz, 1H), 4.98 (q, J = 5.6 Hz, 1H), 5.53 (q, J = 8.8 Hz, 1H), 5.62 (d, J = 5.6 Hz, 1H), 5.65 (q, J = 8.8 Hz, 1H), 6.47 (d, J = 2.4 Hz, 1H), 6.51 (dd, J = 2.4, 8.8 Hz, 1H), 7.13 (d, J = 7.2 Hz, 2H), 7.18 (d, J = 7.2 Hz, 1H), 7.23 (t, J = 8 Hz, 2H), 7.81 (d, J = 8.8 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 164.9, 157.9, 155.1, 137.5, 134.8, 131.0, 129.3, 128.4, 126.7, 126.5, 117.3, 109.3, 104.1, 100.2, 73.3, 72.6, 70.7, 69.6, 68.8, 64.1, 56.1, 55.7, 54.7, 45.3, 40.5, 36.1, 35.6, 29.7, 27.4, 26.7, 26.1, 25.8, 25.3; FTIR (film, NaCl) νmax = 3344, 2925, 1718, 1595, 1575, 1388 cm−1; [M+Na]+ calcd for C35H48N2O9S 695.2978; found, 695.2970.
22a
The corresponding olefin 20a or 21a (22 mg, 0.032 mmol) was dissolved in ethyl acetate (5 mL). Pd/C (10%) was added and the reaction flask flushed with H2 gas. After stirring overnight, the reaction was filtered over celite and purified by silica chromatography (50:50 → 75:25 EtOAc:hexanes) to give 19 mg (90% yield) of the desired product. [α]D20 −2.9 (c 0.70, CHCl3); 1H NMR (CDCl3, 800 MHz) δ 0.97 (s, 3H), 1.03 (s, 3H), 1.18 (m, 1H), 1.27 (m, 2H), 1.38 (m, 3H), 1.45 (m, 4H), 1.54-1.64 (m, 3H), 1.69 (m, 1H), 1.81 (m, 2H), 2.71 (dd, J = 9.6, 14.4 Hz, 1H), 2.87 (m, 1H), 2.98 (dd, J = 4, 14.4 Hz, 1H), 3.01 (dd, J = 1.6, 15.2 Hz, 1H), 3.19 (dd, J = 9.6, 15.2 Hz, 1H), 3.63-3.70 (m, 2H), 3.80 (m, 1H), 3.82 (dt, J = 1.6, 8 Hz, 1H), 3.86 (s, 3H), 3.93 (dd, J = 6.4, 9.6 Hz, 1H), 3.94 (m, 1H), 4.07 (m, 1H), 4.10 (m, 1H), 4.75 (d, J = 9.6 Hz, 1H), 4.98 (q, J = 5.6 Hz, 1H), 5.63 (d, J = 4.8 Hz, 1H), 6.51 (m, 2H), 7.15 (d, J = 7.2 Hz, 2H), 7.18 (t, J = 7.2 Hz, 1H), 7.24 (t, J = 8 Hz, 2H), 7.82 (d, J = 8.8 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 19.6, 22.3, 25.0, 25.4, 25.8, 26.2, 27.4, 28.3, 35.7, 35.8, 39.8, 45.3, 54.8, 55.7, 56.1, 61.8, 68.9, 69.6, 70.7, 72.5, 73.3, 100.2, 104.0, 109.3, 117.7, 126.5, 128.4, 129.3, 135.6, 137.5, 155.1, 158.1, 164.9; FTIR (NaCl) νmax = 3449, 2930, 1718, 1596, 1534 cm−1; ESI (+) HRMS (m/z): [M+H]+ calcd for C35H50N2O9S 675.3315; found, 675.3318.
tert-butyl (2S,3R)-3-hydroxy-4-(2-methylpent-4-enylamino)-1-phenylbutan-2-ylcarbamate (17b and 17c)
A mixture of 2-methylpent-4-en-1 amine (460 mg, 4.6 mmol) and epoxide 8 (361.5 mg, 1.3 mmol) was allowed to stir at 60 °C overnight under argon atmosphere. The crude material was purified by silica gel column chromatography (3:97 MeOH:CH2Cl2) to give 17b and 17c (470 mg, 95%) as a white solid as a mix of diastereomers (50:50 by NMR). mp 89-90 °C; [α]D20 +5.4 (c 2.58, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 0.91 (d, J = 2 Hz, 3H), 0.92 (d, J = 2.4 Hz, 3H), 1.35 (s, 18H), 1.67 (m, 2H), 1.92 (m, 2H), 2.13 (m, 2H), 2.40 (dd, J = 6.8, 11.6 Hz, 2H), 2.52 (dd, J = 6, 12 Hz, 2H), 2.60-3.01 (m, 10H), 3.45 (m, 2H), 3.59-3.45 (m, 4H), 4.55-4.77 (m, 2H), 5.04 (m, 4H), 5.77 (m, 2H), 7.22 (m, 6H), 7.29 (m, 4H); 13C NMR (CDCl3, 100 MHz): δ 17.8, 28.3, 33.2, 36.7, 39.2, 39.2, 51.4, 51.4, 54.1, 55.7, 70.6, 79.3, 116.0, 126.3, 128.4, 129.5, 137.0, 137.9, 155.9; FTIR (NaCl) νmax = 3365, 1683, 1520, 1455, 1391, cm−1; ESI (+) LRMS m/z (relative intensity): 363.03 [M+H]+. Pure 17c was prepared in a similar fashion by using enantiopure (S) 2-methylpent-4-en-1 amine. 1H NMR (CDCl3, 400 MHz): δ 0.99 (d, J = 6 Hz, 3H, 1.32 (s, 9H), 1.99 (m, 2H), 2.12 (m, 1H), 2.70-2.95 (m, 4H), 3.08 (m, 2H), 3.80 (m, 2H), 5.04 (s, 1H), 5.07 (m, 1H), 5.71 (m, 1H), 6.41 (br s, 3H),7.15-7.34 (m, 5H).
tert-butyl (2S,3R)-4-(2-(hex-5-enyloxy)-4-methoxy-N-(2-methylpent-4-enyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate(18b and 18c)
A mixture of amines 17b and 17c (166 mg, 0.46 mmol) and sulfonyl chloride 7d (167 mg, 0.55 mmol) were cooled to 0 °C. Pyridine (5 mL) was added and the solution stirred overnight. Solvents were removed by reduced pressure and the crude material purified by silica gel column chromatography (20:80 EtOAc:hexanes) to give a mixture of 18b and 18c (148 mg, 51% yield) as a colorless oil. [α]D20 −1.8 (c 0.67, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 0.80 (d, J = 6 Hz, 3H), 0.88 (d, J = 6 Hz, 3H), 1.33 (s, 18H), 1.52-1.65 (m, 4H), 1.70-1.92 (m, 9H), 2.06-2.26 (m, 6H), 3.36-2.80 (m, 12H), 3.73 (br, 4H), 3.84 (s, 6H), 4.06-3.94 (m, 5H), 4.59 (m, 2H), 4.90-5.06 (m, 8H), 5.58-5.87 (m, 4H), 6.44-6.54 (m, 4H), 7.16-7.30 (m, 10H), 7.83 (d, J = 8.7 Hz, 2H); 13C NMR (CDCl3, 75 MHz): δ 17.1, 25.0, 28.2, 28.3, 31.7, 31.9, 33.2, 35.3, 38.7, 53.2, 53.3, 54.4, 54.6, 55.6, 56.8, 57.1, 69.1, 2.3, 72.7, 79.4, 100.2, 104.1, 115.1, 116.3, 119.0, 126.3, 128.3, 129.5, 133.7, 136.3, 137.7, 137.9, 138.1, 155.9, 157.6, 164.7; FTIR (NaCl) νmax = 3392, 2929, 1702, 1640, 1445, 1391, 1366, cm−1; ESI (+) HRMS (m/z): [M+H]+ calcd for C34H50N2O7S 631.3417; found, 631.3408.
Pure 18c was prepared in a similar fashion using pure 17c. 1H NMR (CDCl3, 400 MHz): δ 0.80 (d, J = 6 Hz, 3H), 1.34 (s, 9H), 1.59 (m, 2H), 1.74 (m, 2H), 1.87 (m, 2H), 2.12 (q, J = 7.2 Hz, 2H), 2.19 (m, 1H), 2.94 (m, 3H), 3.25 (m, 3H), 3.74 (m, 2H), 3.85 (s, 3H), 3.96 (m, 1H), 4.03 (t, J = 6.8 Hz, 2H), 4.57 (d, J = 7.2 Hz, 1H), 4.93-5.05 (m, 4H), 5.66 (m, 1H), 5.80 (m, 1H), 6.46 (d, J = 2.4 Hz, 1H), 6.50 (dd, J = 2.4, 8.8 Hz, 1H), 7.20 (m, 3H), 7.27 (m, 2H), 7.84 (d, J = 8.4 Hz, 1H),; 13C NMR (CDCl3, 100 MHz): δ 17.1, 25.0, 28.2, 28.4, 31.8, 33.3, 35.2, 38.1, 53.2, 54.5, 55.7, 56.9, 9.1, 72.3, 79.5, 100.2, 104.1, 115.1, 116.3, 119.1, 126.3, 128.4, 129.6, 133.7, 136.4, 137.7, 138.1, 155.9, 157.6, 164.7.
(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl (2S,3R)-4-(2-(hex-5-enyloxy)-4-methoxy-N-(2-methylpent-4-enyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (19b and 19c)
To a stirred solution of 18b and 18c (140 mg, 0.22 mmol) in CH2Cl2 (4.5 mL) was added TFA (1.5 mL). The resulting mixture was stirred for 4 hr and then quenched with saturated aqueous NaHCO3 (1.5 mL). 2N NaOH was added until the solution turned basic. The aqueous layer was extracted with ether, washed with brine, and dried over NasSO4. Solvents were removed under reduced pressure to give crude N-((2R,3S)-3-amino-2-hydroxy-4-phenylbutyl)-2-(hex-5-enyloxy)-4 methoxy-N-(2-methylpent-4-enyl)benzensulfonamide.
To the above crude amine in MeCN (5 mL) were added (3R,3aS,6aR) hexahydrofuro[2,3-b]furan-3-yl 4-nitrophenyl carbonate 12 (71 mg, 0.24 mmoL) and i-Pr2NEt (0.75 mL) under argon atmosphere and the reaction stirred overnight. Solvents were removed under reduced pressure and the crude material purified by silica gel column chromatography (50:50 EtOAc:hexanes) to give 68 mg (45% yield) of product as a clear oil. [α]D20 −5.7 (c 0.17, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 0.80 (d, J = 6.4 Hz, 3H) 0.90 (d, J = 6.4 Hz, 3H), 1.40-2.24 (m, 22H), 2.70-3.40 (m, 14H), 3.62-4.06 (m, 24H), 4.92-5.05 (m, 12H), 5.60-5.85 (m, 6H), 6.50 (m, 4H), 7.18 (m, 6H), 7.24 (m, 4H), 7.83 (d, J = 8.8 Hz, 2H),; 13C NMR (CDCl3, 100 MHz): δ 17.1, 25.0, 25.7, 28.4, 31.9, 32.1, 33.2, 35.4, 38.1, 38.7, 45.3, 53.2, 53.4, 54.9, 55.0, 55.7, 56.9, 57.3, 69.2, 69.6, 70.7, 72.3, 72.8, 73.3, 100.3, 104.2, 109.2, 115.1, 116.5, 118.7, 118.8, 126.5, 128.4, 129.3, 133.8, 136.2, 137.5, 137.6, 138.0, 155.3, 157.6, 164.8; FTIR (film, NaCl) νmax = 3436, 2970, 2927, 1718, 1600, 1458, 1374 cm−1; ESI (+) HRMS (m/z): [M+Na]+ calcd for C36H50N2O9S 709.3135; found, 709.3131.
Pure 19c was prepared in a similar fashion using pure 18c. 1H NMR (CDCl3, 400 MHz) δ 0.81 (d, J = 6 Hz, 3H), 1.45-1.90 (m, 8H), 2.12 (m, 2H), 2.23 (m, 1H), 2.80 (dd, J = 9.2, 14 Hz, 1H), 2.93 (m, 3H), 3.15 (dd, J = 2.4, 15.2 Hz, 1H), 3.24 (dd, J = 8, 14 Hz, 1H), 3.38 (dd, J = 9.2, 15.6 Hz, 1H), 3.65-3.75 (m, 3H), 3.77-3.89 (m, 6H), 3.95 (dd, J = 6.4, 9.6 Hz, 1H), 4.05 (t, J = 6.8 Hz, 2H), 4.85-5.05 (m, 6H), 5.62-5.85 (m, 3H), 6.48 (d, J = 2.4 Hz, 1H), 6.52 (dd, J = 2, 8.8 Hz, 1H), 7.20 (m, 3H), 7.26 (m, 2H), 7.84 (d, J = 8.8 Hz, 1H). ESI (+) HRMS (m/z): [M+Na]+ calcd for C36H50N2O9S 709.3135; found, 709.3139.
Inhibitors 20b, 20c, 21b, and 21c
To a solution of carbamates 19b and 19c (119 mg, 0.17 mmol) in CH2Cl2 (125 mL) was added Grubb’s 2nd generation catalyst (15 mg, 0.02 mmol). The resulting solution was stirred overnight. The solvent was removed under reduced pressure and the crude material purified by silica gel chromatography (50:50 EtOAc:hexanes) to give 110 mg (97% yield) of product as an oil. Reversed-phase HPLC (Waters Sunfire C18 50 × 4.6 mm, 5 μm coupled to Agilent Eclipse XDB C18 150 × 4.6 mm, 5 μm and YMC Pack C8 250 × 4.6 mm, 5 μm, Flow rate = 0.95 mL/min, λ = 215 nm, T = 30 °C, isocratic 60:40 MeCN:H2O) was used to isolate the individual isomers: Rt (RZ) = 22 min, Rt (S Z) = 24 min, Rt (R E) = 25.3 min, Rt (S E) = 26.8 min.
20b
[α]D20 −29 (c 0.60, CHCl3); 1H NMR (CDCl3, 800 MHz): δ 1.12 (d, J = 6.4 Hz, 3H) 1.50-1.62(m, 2H), 1.73 (m, 2H), 1.86 (m, 1H), 1.95 (m, 1H), 2.01-2.11 (m, 3H), 2.18 (m, 1H), 2.74 (dd, J = 9.6, 14.4 Hz, 1H), 2.81 (d, J = 15.2 Hz, 1H), 2.87 (m, 1H), 2.99 (dt, J = 4, 13.6 Hz, 2H), 3.16 (dd, J = 9.6, 15.2 Hz, 1H), 3.65-3.75 (m, 5H), 3.79-3.85 (m, 2H), 3.85 (s, 3H), 3.93 (dd, J = 6.4, 9.6 Hz, 1H), 4.11 (m, 2H), 4.25 (m, 1H), 4.84 (d, J = 9.6 Hz, 1H), 4.99 (q, J = 6.4 Hz, 1H), 5.44 (q, J = 9.6 Hz, 1H), 5.57 (q, J = 7.2 Hz, 1H), 5.63 (d, J = 4.8 Hz, 1H), 6.48 (d, J = 1.6 Hz, 1H), 6.50 (dd, J = 2.4, 8.8 Hz, 1H), 7.17 (m, 3H), 7.24 (m, 2H), 7.86 (d, J = 8.8 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δ 17.4, 25.4, 25.8, 26.1, 27.5, 32.2, 32.8, 35.7, 45.3, 53.9, 54.7, 55.7, 59.1, 68.4, 69.6, 70.7, 73.0, 73.3, 100.3, 103.9, 109.3, 118.1, 126.5, 127.2, 128.4, 129.4, 131.1, 134.4, 137.5, 155.2, 157.8, 164.9; FTIR (film, NaCl) νmax = 3467, 2923, 1717, 1595, 1444, 1384, 1256 cm−1; ESI (+) HRMS (m/z): [M+Na]+ calcd for C34H46N2O9S 681.2822; found, 681.2829.
20c
[α]D20 −30.6 (c 0.83, CHCl3); 1H NMR (CDCl3, 800 MHz): δ 1.13 (d, J = 7.2 Hz, 3H) 1.61 (m, 1H), 1.69 (m, 1H), 1.78-1.90 (m, 4H), 1.94-2.09 (m, 5H), 2.18 (m, 1H), 2.74 (dd, J = 8.8, 13.6 Hz, 1H), 2.89 (m, 2H), 2.96 (dd, J = 3.2, 13.6 Hz, 1H), 3.11 (dd, J = 8, 14.4 Hz, 1H), 3.65-3.74 (m, 3H), 3.78-3.86 (m, 6H), 3.88-4.02 (m, 4H), 4.84 (d, J = 8.8 Hz, 1H), 5.00 (q, J = 5.6 Hz, 1H), 5.52 (m, 1H), 5.64 (d, J = 4.8 Hz, 1H), 5.66 (m, 1H), 6.78 (d, J = 2.4 Hz, 1H), 6.50 (dd, J = 1.6, 8.8 Hz, 1H), 7.17 (m, 3H), 7.25 (m, 2H), 7.83 (d, J = 8.8 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δ 18.4, 25.8, 26.2, 28.6, 29.7, 30.0, 32.3, 35.4, 37.9, 45.3, 54.4, 54.8, 55.7, 58.1, 58.5, 69.6, 70.8, 72.6, 73.4, 100.7, 104.3, 109.3, 118.8, 126.5, 128.1, 128.5, 129.3, 133.3, 134.0, 137.6, 155.3, 158.1, 164.8; FTIR (NaCl) νmax = 3442, 3339, 2924, 1717, 1595, 1495, 1444, 1325, 1256, 1206 cm−1; ESI (+) HRMS (m/z): [M+Na]+ calcd for C34H46N2O9S 681.2822; found, 681.2825.
21b
[α]D20 +27.6 (c 0.36, CHCl3); 1H NMR (CDCl3, 800 MHz): δ 1.09 (d, J = 6.4 Hz, 3H) 1.50-1.60 (m, 2H), 1.72 (m, 1H), 1.86 (m, 2H), 1.94-2.12 (m, 4H), 2.18 (m, 1H), 2.78 (dd, J = 10.4, 14.4 Hz, 1H), 2.90 (m, 2H), 3.02 (dd, J = 4.8, 14.4 Hz, 1H), 3.12 (dd, J = 4, 14.4 Hz, 1H), 3.17 (m, 2H), 3.46 (m, 1H), 3.64-3.71 (m, 3H), 3.84-3.90 (m, 5H), 3.96 (m, 2H), 4.12 (m, 1H), 4.15 (m, 1H), 4.88 (d, J = 8.8 Hz, 1H), 5.00 (q, J = 8 Hz, 1H), 5.47 (q, J = 8.8 Hz, 1H), 5.57 (q, J = 7.2 Hz, 1H), 5.64 (d, J = 5.6 Hz, 1H), 6.49 (m, 2H), 7.20 (m, 3H), 7.27 (m, 2H), 7.83 (d, J = 8.8 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δ 18.0, 25.3, 25.8, 26.0, 27.3, 32.2, 32.7, 35.4, 45.3, 53.3, 55.1, 55.7, 58.0, 68.4, 69.6, 70.9, 72.3, 73.4, 100.2, 104.0, 109.3, 118.3, 126.5, 127.6, 128.5, 129.3, 130.8, 134.3, 137.7, 155.6, 157.8, 164.8; FTIR (film, NaCl) νmax = 3467, 2927, 1717, 1595, 1456, 1325, cm−1; ESI (+) HRMS (m/z): [M+Na]+ calcd for C34H46N2O9S 681.2822; found, 681.2819.
21c
[α]D20 +20.5 (c 1.17, CHCl3); 1H NMR (CDCl3, 800 MHz): δ 1.06 (d, J = 6.4 Hz, 3H), 1.47 (m, 1H), 1.63 (m, 1H), 1.70-1.82 (m, 4H), 1.87 (m, 1H), 1.93 (m, 1H), 2.17 (m, 2H), 2.26 (m, 1H), 2.47 (br, 1H), 2.76 (dd, J = 8.8, 14.4 Hz, 1H), 2.91 (m, 2H), 2.98 (m, 2H), 3.55 (m, 1H), 3.69 (m, 2H), 3.75 (m, 1H), 3.80 (m, 1H), 3.85 (m, 4H), 3.88 (t, J = 8.8 Hz, 1H), 3.94 (dd, J = 6.4, 9.6 Hz, 1H), 4.11 (m, 1H), 4.17 (br, 1H), 4.76 (d, J = 9.6 Hz, 1H), 5.00 (q, J = 8 Hz, 1H), 5.53 (m, 1H), 5.64 (d, J = 4.8 Hz, 1H), 5.67 (m, 1H), 6.47 (d, J = 2.4 Hz, 1H), 6.50 (dd, J = 2.4, 8.8 Hz, 1H), 7.14 (d, J = 7.2 Hz, 2H), 7.18 (m, 1H), 7.24 (t, J = 7.2 Hz, 2H),7.83 (d, J = 8.8 Hz, 1H); 13C NMR (CDCl3, 125 MHz): δ 19.0, 25.8, 26.2, 28.1, 29.8, 30.3, 33.6, 35.7, 38.2, 45.3, 54.6, 54.9, 55.7, 58.6, 69.6, 70.8, 71.5, 73.4, 100.5, 104.2, 109.3, 119.0, 126.5, 128.5, 129.4, 129.6, 132.5, 134.2, 137.4, 155.2, 158.1, 164.8; FTIR (film, NaCl) νmax = 3463, 3339, 1717, 1595, 1495, 1387, 1326 cm−1; ESI(+) HRMS (m/z): [M+Na]+ calcd for C34H46N2O9S 681.2822; found, 681.2812.
22b
To a stirred solution of the corresponding R E olefin (8 mg, 0.013 mmol) was dissolved into EtOAc (5 mL) a spatula tip of Pd on carbon (10 wt %) was added and the mixture was stirred under H2 atmosphere. The reaction stirred overnight. Solvents were removed under reduced pressure and the crude material was purified by silica gel column chromatograph (70:30 Et2O:hexanes → 100% Et2O) to give 22b (8 mg, quantitative yield) as a white solid. TLC 100% Et2O. [α]D23 −7.8 (c 0.84, CHCl3). 1H NMR (CDCl3, 500 MHz) δ 0.92 (d, J = 7 Hz, 3H) 1.16-1.84 (m, 15H), 2.74 (dd, J = 9.5, 14 Hz, 1H), 2.89 (m, 1H), 3.05 (m, 3H), 3.28 (m, 2H), 3.65-3.87 (m, 9H), 3.94 (m, 1H), 4.10 (m, 1H), 4.21 (m, 1H), 4.84 (d, J = 9.5 Hz, 1H), 5.01 (q, J = 7 Hz, 1H), 5.64 (d, J = 5.5 Hz, 1H), 6.51 (m, 2H), 7.19 (m, 3H), 7.26 (m, 2H),7.88 (d, J = 9.5 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ 17.8, 23.4, 23.5, 25.5, 25.8, 26.1, 27.5, 31.8, 31.9, 35.7, 45.3, 52.5, 54.8, 55.2, 55., 68.8, 69.6, 70.7, 71.3, 73.3, 100.4, 104.0, 109.3, 118.0, 126.5, 128.5, 129.4, 134.4, 137.6, 155.2, 158.0, 165.0. FTIR (NaCl) νmax = 3463, 2924, 2853, 1721, 1596, 1323, 1256 cm−1. ESI(+) HRMS (m/z): [M+Na]+ calcd for C34H48N2O9S 683.2978; found, 683.2987.
22c
To a solution of the corresponding S E olefin (12 mg, 0.018 mmol) in EtOAc (5 mL) a spatula tip of Pd on carbon (10 wt-%) was added and the mixture was stirred under H2 atmosphere. The reaction stirred overnight. Solvents were removed under reduced pressure and the crude material purified by silica chromatograph (70:30 Et2O:hexanes → 100% Et2O) to give 22c (9 mg, 78% yield) as a white solid. [α]D23 −6.6 (c 0.91, CHCl3). 1H NMR (CDCl3, 800 MHz) δ 0.90 (d, J = 6.4 Hz, 3H) 1.20-1.90 (m, 15H), 2.73 (m, 1H), 2.79 (dd, J = 8.8, 13.6 Hz, 1H), 2.90 (m, 1H), 2.96 (m, 1H), 3.08 (m 1H), 3.33 (dd, J = 9.6, 15.2 Hz, 1H), 3.47 (m, 1H), 3.61 (m, 1H), 3.70 (m, 2H), 3.85 (m, 6H), 3.95 (m, 1H), 4.10 (m, 1H), 4.25 (m, 1H), 4.86 (d, J = 8.8 Hz, 1H), 5.03 (q, J = 7.2 Hz, 1H), 5.65 (d, J = 4.8 Hz, 1H), 6.52 (m, 2H), 7.18 (m, 3H), 7.25 (m, 2H), 7.87 (d, J = 9.6 Hz, 1H). 13C NMR (CDCl3, 200 MHz) δ 18.4, 23.4, 24.3, 25.0, 25.8, 26.5, 27.2, 32.8, 33.1, 35.4, 45.3, 53.5, 54.8, 55.7, 57.0, 69.3, 69.6, 70.8, 72.2, 73.3, 100.6, 104.2, 109.3, 118.0, 126.5, 128.5, 129.4, 134.5, 137.5, 155.3, 158.1, 165.1. FTIR (NaCl) νmax = 3463, 2854, 1717, 1596, 1444, 1323, 1256 cm−1. ESI (+) HRMS (m/z): [M+Na]+ calcd for C34H48N2O9S 683.2978; found, 683.2976.
Determination of X-ray structures of HIV-1 protease (wt)-inhibitor complexes for inhibitors 14c and 2
X-ray structure for 14c
The HIV-1 protease construct with the substitutions Q7K, L33I, L63I, C67A and C95A to optimize protein stability without altering protease structure and activity was expressed and purified as described.23, 24 Crystals were grown by the hanging drop vapor diffusion method using 1:15 molar ratio of protease at 2.0 mg/mL and inhibitor 14c dissolved in dimethylsulfoxide. The reservoir contained 5% glycerol, 0.5 M NaI in 0.2 M MES buffer with pH 6.0. Subsequently, crystals were mounted on nylon loops in liquid nitrogen with additional 28% glycerol (v/v) as cyro-protectant. X-ray diffraction data were collected on SER-CAT 22-ID beam line of the Advanced Photon Source, Argonne National Laboratory, with wavelength 0.8 Å under the stream of liquid nitrogen at 90 K. Data were processed by HKL200025 resulting in Rmerge of 10.8% for 14c, the structure was solved by molecular replacement with our previous structure [PDB code 3B7V] using PHASER26 in CCP4i Suite,27,28 refined by SHELX-97,29,30 and refitted manually with the graphic programs O10,31 and COOT.32 The inhibitor structure was modeled by PRODRG-2,33 which also provided restraints for refinement. The crystal structure of HIV-1 protease with inhibitor 14c was determined at 1.17 Å resolution. The RMSD for Cα atoms in comparison with our wild type protease with DRV [PDB code: 2IEN,19] is only 0.33 Å . Alternate conformations were modeled for the protease residues when obvious in the electron density maps. Anisotropic atomic displacement parameters (B-factors) were applied for all atoms including solvent molecules. The occupancies of iodine atoms were refined and vary from 0.24 to 0.60. Hydrogen atoms were added at the final stages of the refinement. The final structure includes 2 Na+ ions, 19 I ions, 2 glycerols and 181 waters in PR-14c complex. The R-factor and Rfree are 16.0%, 19.4%. The crystallographic statistics are listed in Table 1 (supporting information). The crystal structure and structure factors have been deposited in the RCSB Protein Data Bank,34 with PDB code: 3I6O for inhibitor 14c.
X-ray structure for 2
The wild type HIV-1 protease was expressed and purified as described previously.35 After purification, the protease was concentrated to 4.0 mg/ml. Inhibitor 2, at a 10-fold molar excess, was mixed with the protease. The mixture was incubated at room temperature for 2 hours and then clarified with a 0.2 mm spin filter. The crystallization trials employed the hanging drop method using equal volumes of enzyme-inhibitor and well solution. The reservoir contained 20% (NH4)2SO4, 200 mM NaPO4/citric acid buffer, pH 5.5. The well solutions also included 10% dimethyl sulfoxide, 30 mM β-mercaptoethanol, and 4% isopropanol.
Diffraction data were collected at room temperature with an Raxis-IV image plate, integrated and reduced with the HKL program package.25 The diffraction data set has a resolution of 1.7 Å with an Rmerge value of 0.052. Data completeness was 95.5%. The space group was determined to be P212121 with unit cell dimensions of a = 51.9 Å, b = 59.0 Å, c = 62.6 Å with one dimer in the asymmetric unit. The initial phase was solved by molecular replacement using the program AMoRe.36 A protease dimer (PDB code 2AID)37 from a previously determined HIV-1 protease crystal structure was used as the search model. Crystallographic refinement was carried out using X-PLOR 3.1. Molecular graphics program O,31 was used for map display and model building. Water molecules were added to the structure as identified in the ∣Fo∣-∣Fc∣ map contoured at the 3σ level. Data collection and refinement statistics are listed in Table 2 (supporting information). The final Rwork was 21.6 % and Rfree was 28.9% for data between the resolution of 20 and 1.7 Å (Table 1). The RMSD values from ideal bond distances and angles were 0.009 Å and 1.5°, respectively. The average B-factor was 28.3 and 19.6 Å2 for protein and inhibitor atoms, respectively, and 41.0 Å2 for solvent atoms. The crystal structures and structure factors have been deposited in the RCSB Protein Data Bank,34 with PDB code: 3I7E for inhibitor 2.
Supplementary Material
Figure 1.

Structure of inhibitors 1, 2, 14c and 15c
Table 4.
Enzyme inhibitory activity of 19a-c, 20a-c, 21a-c
| Inhibitor structure | Ki (nM) | IC50 (nM)a |
|---|---|---|
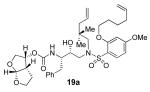
|
82 ± 2 | >1000 |
|
16 ± 1 | >1000 |
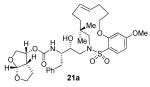
|
6.9 ± 0.2 | >1000 |
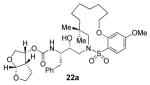
|
6.8 ± 0.8 | >1000 |
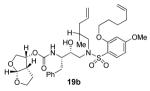
|
15 ± 3 | 380 |
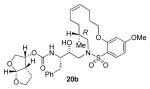
|
6 ± 1 | 200 |
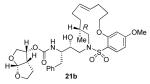
|
2.8 ± 0.3 | 6.3 |
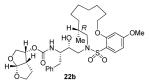
|
3.7 ± 0.3 | 650 |
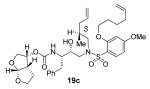
|
58 ± 6 | >1000 |
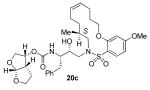
|
0.31 ± 0.03 | 9 |
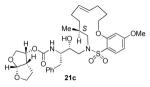
|
0.15 ± 0.01 | 52 |
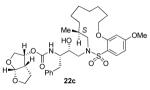
|
0.07 ± 0.02 | 170 |
MT-2 human T-lymphoid cells exposed to HIV-1LAI; Saquinavir and amprenavir exhibited IC50 values of 16 nM and 27 nM, respectively.
Acknowledgements
The research was supported by grants from the National Institutes of Health (GM53386, AKG and GM062920, ITW). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health and in part by a Grant-in-aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan (Kosei Rohdosho: H15-AIDS-001), and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Re emerging Infectious Diseases (Renkei Jigyo: No. 78, Kumamoto University) of Monbu-Kagakusho. We thank the staff at the SER-CAT beamline at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The authors’ thank Dr. John Harwood, Purdue University, for many helpful discussions surrounding NMR spectroscopy.
a Abbreviations
- HIV
human immunodeficiency virus
- bis-THF
bis-tetrahydrofuran
- PI
protease inhibitor
- HAART
highly active antiretroviral therapy
- APV
amprenavir
- DRV
darunavir
- SQV
saquinavir
- IDV
indinavir
- LPV
lopinavir
- DIAD
diisopropyl azodicarboxylate
Footnotes
Supporting Information Available. HPLC and HRMS data of all inhibitors; crystallographic data collection and refinement statistics. This information is available free of charge via the internet at http://pubs.acs.org.
The PDB accession code for 2-bound HIV-1 protease X-ray structure is 3I7E and 14c-bound HIV-1 protease X-ray structure is 3I6O.
References
- 1.UNAIDS, WHO 2007 AIDS epidemic update. 2007 December;
- 2.Spekowitz KA. AIDS-The First 20 years. Engl. J. Med. 2001;344:1764–1772. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- 3.Hertogs K, Bloor S, Kemp SD, Van den Enyde C, Alcorn TM, Pauwels R, Van Houtte M, Staszewski S, Miller V, Larder B. A Phenotypic and genotypic analysis of clinical HIV-1 isolated reveals extensive protease inhibitor cross-resistance: a survey of over 6000 samples. AIDS. 2000;14:1203–1210. doi: 10.1097/00002030-200006160-00018. [DOI] [PubMed] [Google Scholar]
- 4 (a).Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Holland J, Buthod J. Potent HIV-1 inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino)sulofonamide isostere. Bioorg. Med. Chem. Lett. 1998;8:687–690. doi: 10.1016/s0960-894x(98)00098-5. [DOI] [PubMed] [Google Scholar]; (b) Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. A novel bis tetrahydrofuranylurethane-containing non-peptide protease inhibitor (PI) UIC-94017 (TMC-114) potent against multi-PI-resistant HIV in vitro. Antimicrob. Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh AK, Pretzer E, Cho H, Hussain KA, Duzgunes N. Antiviral activity of UIC-PI, a novel inhibitor of human immunodeficiency virus type 1 protease. Antiviral Res. 2002;54:29–36. doi: 10.1016/s0166-3542(01)00209-1. [DOI] [PubMed] [Google Scholar]
- 5.Surleraux DLNG, Tahri A, Verchueren WG, Pille GME, de Kock HA, Jonckers THM, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune M-P, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. Discovery and selection of TMC-114, a next generation of HIV-1 protease inhibitor. J. Med. Chem. 2005;48:1813–1822. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- 6 (a).Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Husssin KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya HA. A potent human immunodeficiency virus type 1 protease inhibitor, UIC-94003 (TMC-126), and selection of a novel (A28S) mutation in the protease active site. J. Virol. 2002;76:1349–1358. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. A novel bis-tetrahydrofuranylurethane-containing non-peptide protease inhibitor (PI) UIC-94017 (TMC-114) potent against multi-PI-resistant HIV in vitro. Antimicrob. Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 8. [On June 23, 2006];FDA approved new HIV treatment for patients who do not respond to existing drugs. Please see: http://www.fda.gov/bbs/topics/NEWS/2006/NEW01395.html.
- 9.On October 21, 2008, FDA granted traditional approval to Prezista (darunavir), co-administered with ritonavir and with other antiretroviral agents, for the treatment of HIV-1 infection in treatment-experienced adult patients. In addition to the traditional approval, a new dosing regimen for treatment nal̈ve patients was approved.
- 10.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High resolution crystal structures of HIV-1 protease with a potent non peptide inhibitor (UIC-94017) active against multidrug-resistant clinical strains. J. Mol. Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 11.Kovalevsky AY, Tie Y, Liu F, Boross PI, Wang YF, Leshchenko S, Ghosh AK, Harrison RW, Weber IT. Effectiveness of nonpeptide clinical inhibitor TMC-114 on HIV-1 protease with highly drug resistant mutations D30N, I50V, and L90M. J. Med. Chem. 2006;49:1379–1387. doi: 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kempf DJ, Marsh KC, Paul DA, Knige MF, Norbeck DW, Kohlbrenner WE, Codacovi L, Vasavanonda S, Bryant P, Wang XC, Wideburg NE, Clement JJ, Plattner JJ, Erickson J. Antiviral and Pharmacokinetic Properties of C2-Symmetric Inhibitors of the Human Immunodeficiency Virus Type 1 Protease. Antimicrob. Agents Chemother. 1991;35:2209–2214. doi: 10.1128/aac.35.11.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldwin ET, Bhat TN, Liu B, Pattabriaman N, Erickson JW. Structural Basis of Drug Resistance for V82A mutant of HIV-1 Proteinase. Struct. Biol. 1995;2:244–49. doi: 10.1038/nsb0395-244. [DOI] [PubMed] [Google Scholar]
- 14.Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis. 1981:1–28. [Google Scholar]
- 15.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. Bis-Tetrahydrofuran: a Privileged Ligand for Darunavir and a New Generation of HIV Protease Inhibitors That Combat Drug Resistance. ChemMedChem. 2006;1:937–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
-
16(a).Schwab P, France MB, Ziller JW, Grubbs RH. A Series of Well-Defined Metathesis Catalysts-Synthesis of [RuCl2(
 CHR’)(PR3)2] and Its Reactions. Angew.Chem., Int. Ed. Engl. 1995;34:2039–2041. [Google Scholar]; (b) Scholl M, Ding S, Lee C, Grubbs RH. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3 Dimesityl-4,5 dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
CHR’)(PR3)2] and Its Reactions. Angew.Chem., Int. Ed. Engl. 1995;34:2039–2041. [Google Scholar]; (b) Scholl M, Ding S, Lee C, Grubbs RH. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3 Dimesityl-4,5 dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar] - 17.Toth MV, Marshall GR. A simple, continuous fluorometric assay for HIV protease. Int. J. Pep. Protien Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 18.Gustchina A, Sansom C, Prevost M, Richelle J, Wodak S, Wlodawer A, Weber I. Energy calculations and analysis of HIV-1 protease-inhibitor crystal structures. Protein Eng. 1994;7:309–317. doi: 10.1093/protein/7.3.309. [DOI] [PubMed] [Google Scholar]
- 19.Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 Å resolution crystal structures of HIV-1 protease mutants with substrate analogs. FEBS J. 2005;272:5265–5277. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Panigrahi S, Desiraju G. Strong and weak hydrogen bonds in the protein-ligand interface. Proteins. 2007;67:128–141. doi: 10.1002/prot.21253. [DOI] [PubMed] [Google Scholar]
- 21.Steiner T. Hydrogen bonds from water molecules to aromatic acceptors in very high-resolution protein crystal structures. Biophys. Chem. 2002;95:195–201. doi: 10.1016/s0301-4622(01)00256-3. [DOI] [PubMed] [Google Scholar]
- 22.Wang YF, Tie Y, Boross PI, Tozser J, Ghosh AK, Harrison RW, Weber IT. Potent new antiviral compound shows similar inhibition and structural interactions with drug resistant mutants and wild type HIV-1 protease. J. Med. Chem. 2007;50:4509–4515. doi: 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 24.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug resistant mutants of HIV-1 protease: High resolution crystal structures of the mutant protease/substrate analog complexes. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Carter CW Jr., Sweet RM, editors. Processing of X ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276 : Macromolecular Crystallography(part A):307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project Number 4 The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Cryst. 2003;D59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 29.Sheldrick GM. A short history of SHELX. Acta Cryst. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 30.Sheldrick GM, Schneider TR. SHELXL: High resolution refinement. Methods in Enzymology. 1997;277:319–343. [PubMed] [Google Scholar]
- 31.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: Model Building Tools for Molecular Graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Schuettelkopf AW, van Aalten DMF. PRODRG a tool for high throughput crystallography of protein ligand complexes. Acta Cryst. 2004;D60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 34.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong L, Treharne A, Hartsuck JA, Foundling S. Steve, Tang J. Crystal Structures of Complexes of a Peptidic Inhibitor with Wild Type and Two Mutant HIV-1 Protease. Biochemistry. 1996;35:10627–10633. doi: 10.1021/bi960481s. [DOI] [PubMed] [Google Scholar]
- 36.Navaja JA. MoRe: an Automated Package for Molecular Replacement. Acta Crystallogr. 1994;A 50:157–163. [Google Scholar]
- 37.Rutenber E, Fauman EB, Keenan RJ, Fong S, Furth PS, de Montellano P. R. Ortiz, Meng E, Kuntz ID, DeCamp DL, Salto R. Structure of a non-peptide inhibitor complexed with HIV-1 protease. Developing a cycle of structure-based drug design. J. Biol. Chem. 1993;268:15343–15346. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.