

Abstract
Chronic morphine drives adaptations in synaptic transmission thought to underlie opiate dependence. Here we examine the role of μ-opioid receptor (MOR) trafficking in one of these adaptations, specifically, changes in GABA transmission in the ventral tegmental area (VTA). To address this question, we used a knock-in mouse, RMOR (for recycling MOR), in which genetic change in the MOR promotes morphine-induced receptor desensitization and endocytosis in GABA interneurons of the VTA. In wild-type mice (postnatal days 23–28) chronic morphine (10 mg/kg, s.c., twice daily for 5 d), induced a cAMP-dependent increase in the probability of GABA release onto VTA dopamine neurons. The increased GABA release frequency correlated with physical dependence on morphine measured by counting somatic signs of morphine withdrawal, such as, tremors, jumps, rears, wet-dog shakes, and grooming behavior precipitated by subcutaneous administration of naloxone (NLX) (2 mg/kg). This adaptation in GABA release was prevented in RMOR mice given the same morphine treatment, implicating MOR trafficking in this morphine-induced change in plasticity. Importantly, treatment with the cAMP activity inhibitor rp-cAMPS [(R)-adenosine, cyclic 3′,5′-(hydrogenphosphorothioate) triethylammonium] (50 ng/0.5 μl), directly to the VTA, attenuated somatic withdrawal signs to systemic morphine produced by intra-VTA NLX (500 ng/0.5 μl), directly tying enhanced cAMP-driven GABA release to naloxone-precipitated morphine withdrawal in the VTA.
Introduction
Opioid peptides, such as enkephalin, and small molecule drugs such as morphine, exert their anti-nociceptive effects by activating μ-opioid receptors (MORs). These compounds have varying propensities to promote tolerance and dependence. Indeed, when given at equal anti-nociceptive doses, morphine causes more elevated tolerance and physical dependence as measured by monitoring somatic signs of withdrawal than [d-Ala2, N-MePhe4, Gly5-ol]-enkephalin (DAMGO), methadone, or etorphine (Duttaroy and Yoburn, 1995; Walker and Young, 2001; Grecksch et al., 2006; Kim et al., 2008). Importantly, morphine-activated MORs fail to undergo substantial endocytosis both in vitro (Keith et al., 1996; Yu et al., 1997; Whistler et al., 1999; Koch et al., 2001) and in vivo (Keith et al., 1998; Trafton et al., 2000; Abbadie and Pasternak, 2001; He et al., 2002; He and Whistler, 2005), whereas endogenous opioid peptides and small molecule opioid drugs promote endocytosis and recycling (Keith et al., 1996; Yu et al., 1997; Whistler et al., 1999; Koch et al., 2001). These differences in trafficking also translate into differences in receptor desensitization. Whereas met-enkephalin causes rapid desensitization of the MOR as assessed by G-protein-coupled inward rectifying (GIRK) currents, morphine does not promote significant desensitization (Alvarez et al., 2002).
A mutant receptor, RMOR (for recycling MOR), containing a 28 aa substitution in the cytoplasmic tail of the MOR enables morphine-induced endocytosis and desensitization in vitro (Finn and Whistler, 2001). In addition, knock-in mice expressing the RMOR exhibit greatly reduced morphine tolerance and physical dependence measured as naloxone (NLX)-precipitated withdrawal (Kim et al., 2008).
Here, we used the RMOR and wild-type (WT) mice to examine synaptic plasticity in the ventral tegmental area (VTA) that occurs after chronic morphine treatment. The VTA is critically important for the motivational effects of opioids (Wise, 1989). Acute opioid activation of the MOR modulates dopamine neuron activity by inhibiting GABA release onto dopamine neurons (Johnson and North, 1992a,b), and MORs in the VTA mediate the rewarding effects of morphine (Phillips and LePiane, 1980; Stinus et al., 1990). In addition, intra-VTA infusion of an opioid antagonist in morphine-dependent animals precipitates behavioral signs of opioid withdrawal (Baumeister et al., 1989; Stinus et al., 1990) and the expression of conditioned place aversion (Stinus et al., 1990).
Chronic morphine exposure, both in vitro and in vivo, promotes a compensatory upregulation of adenylyl cyclase and a rebound increase in cAMP levels (Williams et al., 2001; Nestler, 2004). This cAMP superactivation, however, does not occur in cells expressing the RMOR (Finn and Whistler, 2001). Interestingly, withdrawal from chronic morphine promotes a cAMP-dependent increased probability of GABA release onto dopamine neurons in the VTA of guinea pig (Bonci and Williams, 1997). We hypothesized that cAMP superactivation in the VTA promotes changes in synaptic activity that manifest as dependence and that the RMOR mutation may reduce cAMP activation in the VTA as well. To test this, the effects of NLX-precipitated morphine withdrawal on GABA signaling in WT and RMOR mice were examined.
Materials and Methods
Animals.
Experiments testing anti-nociceptive tolerance, naloxone-precipitated morphine withdrawal, immunohistochemistry, and electrophysiology were performed on male mice aged between postnatal days 23 and 28. Experiments testing the effects of cAMP inhibitor rp-cAMPS [(R)-adenosine, cyclic 3′,5′-(hydrogenphosphorothioate) triethylammonium] on naloxone-precipitated morphine withdrawal in the VTA were conducted in adult (postnatal days 50–70) male WT mice, which were surgically cannulated to receive drug infusions in the VTA. Generation of the RMOR knock-in mice was as described previously (Kim et al., 2008) and was based on the mutant receptor RMOR first described by Finn and Whistler (2001). Briefly, the MOR sequence EFCIPTSSTIEQQNSARIRQTREHPSTAN contained entirely within exon 3 was replaced with the corresponding sequence QLCRTPCGRQEPGSLRRPRQATTRERVTACTPS from the δ-opioid receptor. The RMOR mutation was knocked into TC-1 stem cells. A positive clone was injected into C57BL/6 blastocysts to create chimeric animals. F1 agouti progeny were genotyped for transmission of the mutant allele. The mice were backcrossed onto C57BL/6/H and intercrossed thereafter. Mice used as WT controls were littermates or cousins of RMOR mice. All mice were bred in-house, and procedures were performed in accordance with Institutional Animal Care and Use Committee guidelines at the Ernest Gallo Clinic and Research Center.
Assessment of anti-nociceptive tolerance and NLX-precipitated withdrawal.
Anti-nociceptive responses were measured using the hotplate assay. Mice (postnatal days 23–28) were placed on a hotplate maintained at 56°C. An observer recorded latency to lick the forepaws or hindpaws or flick the hindpaws. A cutoff time (either 2.5 × baseline latency or 30 s, whichever was lower) was used to prevent tissue damage. Morphine was dissolved in physiological saline and injected in a volume of 10 ml/kg. To assay tolerance development to chronic morphine, mice were injected subcutaneously twice daily (10:00 A.M. and 4:00 P.M.) with morphine (10 mg/kg) or saline for 5 consecutive days. The hotplate test was administered 30 min after the morning injection on days 1, 3, and 5. Data are reported as maximum possible effect (MPE), calculated as 100% × [(drug response latency − basal response latency)/(cutoff − basal response latency)]. The magnitude of tolerance used in the correlations (see Fig. 4) was calculated as 100 − MPE. Therefore, all morphine-treated mice used in this study were tested for the development of anti-nociceptive tolerance. On day 6, mice were divided into two groups. Group 1 was used for the patch-clamp electrophysiology experiments to test NLX-precipitated changes in GABA release probability. Mice in group 2 were injected with NLX (2 mg/kg, s.c.), 30 min after the final morphine injection, and somatic signs of withdrawal were scored for a 30 min period. Several withdrawal behaviors were used to score each mouse and included wet-dog shakes, rears, jumps, paw tremors, and paw licks/grooming behavior (Schulteis et al., 1994). The global withdrawal score was calculated by summing all observed behaviors over 30 min. This dose of NLX was chosen because it produces withdrawal signs in morphine-treated wild-type mice in our mixed background but does not produce significant withdrawal signs in naive mice. Withdrawal signs can be precipitated with lower doses of NLX in WT mice on a pure C57BL/6 background (data not shown).
Figure 4.
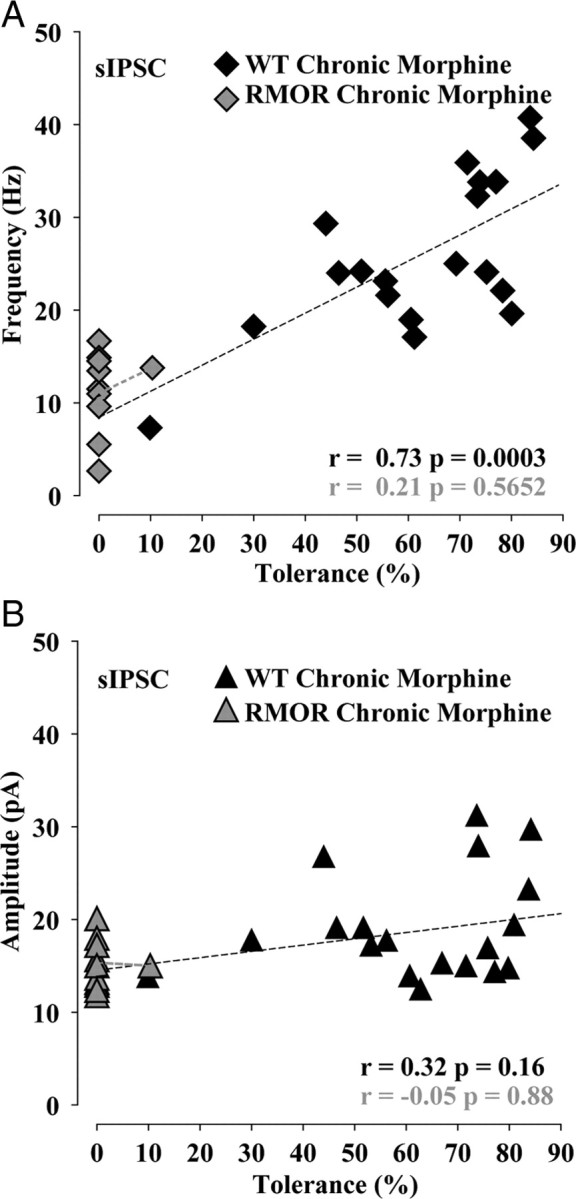
Tolerance and dependence covaried with frequency of GABA sIPSCs during NLX-precipitated morphine withdrawal. A, Magnitude of tolerance (100 − MPE) covaries with frequency of GABA sIPSC in chronic morphine-treated WT but not in chronic morphine-treated RMOR. B, The correlation between magnitude of tolerance in mice against sIPSC mean amplitude is not significant in chronic morphine-treated WT and chronic morphine-treated RMOR.
VTA cannulation surgeries.
Male B6–129 mice, postnatal days 50–70, were anesthetized with a mixture of ketamine and xylazine, their heads were shaved and disinfected, and a 1 cm incision was cut in the scalp to reveal the skull. Mice were placed in a digital stereotaxic alignment instrument (model 1900; David Kopf Instruments). The skull was cleaned and leveled in the coronal and sagittal planes using bregma and lambda as reference points. Holes were drilled bilaterally in the skull at the anteroposterior (AP) (in reference to bregma) and mediolateral (ML) coordinates corresponding to VTA (−3.2 mm AP; ±0.5 mm ML), based on the mouse brain atlas of Paxinos and Franklin (2001), using a 0.3 mm carbide drill bit. Bilateral cannulae (Plastics One) measuring −4.7 mm dorsoventral and ±0.5 mm ML were inserted to 4.5 mm below the skull level and secured to the skull using dental acrylic (Geristore kit; Denmat). Bilateral stainless steel dummy cannula and caps were inserted into the guide cannula and remained in place when the guide cannula was not in use. Animals were allowed at least a 1 week recovery period before behavioral testing.
Intra-VTA infusions and NLX-precipitated morphine withdrawal testing.
For infusions, an injector cannula designed to reach 4.7 mm below the skull was used. Mice (postnatal days 50–70) mice were injected subcutaneously twice daily (10:00 A.M. and 4:00 P.M.) with morphine (10 mg/kg) or left untreated for 5 consecutive days. On day 6, mice were (1) administered morphine (10 mg/kg) and 30 min later were injected with NLX (500 ng/0.5 μl dissolved in physiological saline) or (2) administered morphine (10 mg/kg), followed by rp-cAMPS (50 ng/0.5 μl dissolved in physiological saline), followed 30 min later by NLX (500 ng/0.5 μl), at the rate of 0.25 μl/min using an infusion pump (Harvard Apparatus). Withdrawal signs were monitored beginning 1 min after infusion of naloxone. Somatic signs of withdrawal, including wet-dog shakes, rears, jumps, paw tremors, and paw licks/grooming behavior (Schulteis et al., 1994) were scored for each mouse. The global withdrawal score was calculated by summing all observed behaviors over 30 min.
After monitoring withdrawal signs, mice were infused with India ink dye and anesthetized with 5% isoflurane, and their brains were removed and submerged in 4% paraformaldehyde dissolved in 0.1 m PBS overnight. Brains were sectioned coronally at 50 μm, mounted, and stained with cresyl violet. Only animals with injection sites within the VTA, confirmed using The Mouse Brain in Stereotaxic Coordinates by Paxinos and Franklin (2001), were included in the data presented in Figure 9.
Figure 9.
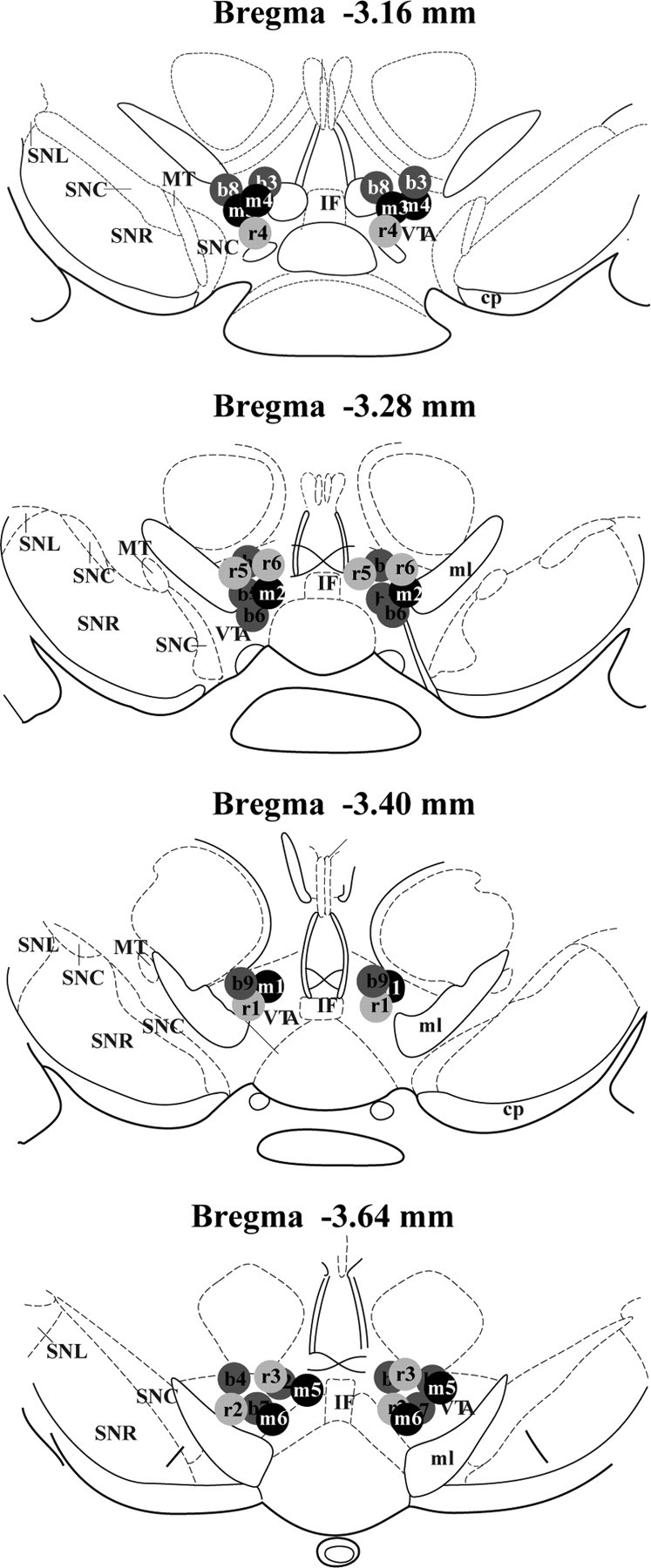
Schematic representation of cannula placements in the VTA. Coordinates of slides are in relation to bregma. b1–b9, NLX mice; m1–m6, chronic morphine + NLX-treated mice; r1–r6, chronic morphine + rp-cAMPS + NLX-treated mice; SNC, substantia nigra compacta; SNR, substantia nigra reticulata; MT, medial terminal nucleus of the accessory optic tract; IF, interfascicular nucleus; ml, medial lemniscus.
Electrophysiology.
The mice were anesthetized with 5% isoflurane and immediately decapitated using a guillotine. Horizontal brain slices 190 μm thick were cut in ice-cold modified artificial CSF (aCSF) solution. All solutions were saturated with 95% O2–5% CO2 (carbogen). The composition of the solution included the following (in mm): 85 choline Cl, 40 NaCl, 4 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 10 dextrose, 1 ascorbate, 3 Na pyruvate, and 3 myo inositol (310–320 osmolarity). Slices recovered first for ∼10–15 min at 32°C in the cutting solution and were later transferred to recording aCSF of the following composition (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, 25 dextrose, and 25 NaHCO3 (295–300 osmolarity). GABA currents were recorded in the presence of DNQX (10 μm), strychnine (10 μm), [S-(R*,R*)]-[3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-hydroxypropyl(cyclohexylmethyl) phosphinic acid (CGP 54626 hydrochloride) (10 μm), and eticlopride (100 nm) to block glutamate, glycine, GABAB, and dopamine D2 receptors, respectively. AP-5 (10 μm) was used in some experiments to block NMDA receptors, and the results did not differ in the presence or absence of AP-5. In experiments in which the effect of NLX-precipitated morphine withdrawal was being studied, naloxone (1 μm) was used in solution to precipitate withdrawal. NLX was not used in experiments in which the acute effects of DAMGO and forskolin were studied (see Figs. 6, 7).
Figure 6.

MOR and RMOR show equivalent acute responses to opioid agonist but different degrees of desensitization to morphine. A, DAMGO (DG) dose–response curves of peak inhibition (black bars, 3 min window, inset) of GABA IPSCs plotted against log10 (agonist in nanomolar). Sigmoidal dose–response curves fitted for WT and RMOR were not significantly different for DAMGO (WT EC50 of 89.22 ± 0.28 nm, Emax of 90.57 ± 7.6; RMOR EC50 of 112.6 ± 0.32 nm, Emax of 86.96 ± 10.6; n = 17 each) or morphine (WT EC50 of 106.2 ± 0.78 nm, Emax of 52.82 ± 9.8; RMOR EC50 of 125.7 ± 0.51 nm, Emax of 48.66 ± 6.52; n = 8 each). B, Cumulative interevent interval distribution showing equivalent presynaptic inhibition of GABA frequency by 300 nm DAMGO (DG) in WT and RMOR mice. B, Inset, DAMGO significantly inhibits GABA mIPSC frequency in WT (n = 9) and RMOR (n = 9). C, Cumulative amplitude distribution of GABA mIPSCs shows no effect of DAMGO in either WT or RMOR. C, Inset, DAMGO does not inhibit GABA mIPSC amplitude in WT (n = 9) or RMOR (n = 9). The distributions of frequencies were not significantly different comparing WT and RMOR in either the absence (n = 9) or presence of DAMGO (n = 9). D, Morphine (30 μm) induces a sustained membrane hyperpolarization in WT Ih-negative GABA neurons (n = 8; black trace) that lasts throughout the time period of morphine application and a membrane hyperpolarization in RMOR Ih-negative GABA neurons (n = 6; gray trace) that returns to baseline during morphine application. E, Normalized changes in membrane potential after morphine application in Ih-negative GABA neurons of WT and RMOR [(% desensitization = 100 × (final response − peak response)/(peak response − baseline)] (gray bar in E is final response). Percentage desensitization was as follows: WT (n = 8), 16.37 ± 12.81; RMOR (n = 6), 78.06 ± 27.01. **p < 0.01, ***p < 0.001.
Figure 7.

Elevated cAMP signaling in chronic morphine-treated WT mice during NLX-precipitated morphine withdrawal. A, Effect of forskolin (3 μm) on GABA IPSCs in WT, WT treated with 10 mg/kg morphine for 5 d (WT Chronic Morphine), RMOR, and RMOR treated with 10 mg/kg morphine for 5 d (RMOR Chronic Morphine). B, Effect of forskolin on paired pulse ratio of GABA currents in all four conditions. C, Cumulative effects of forskolin showing greater enhancement of GABA IPSCs in WT morphine-treated slices (WT Chronic Morphine, n = 6) than in WT controls (WT, n = 6) and no difference between similar morphine-treated RMOR slices (RMOR Chronic Morphine, n = 6) and RMOR controls (RMOR, n = 6). D, The percentage change (peak change minus baseline; black bar in Fig. 4C) in GABA IPSCs after forskolin application in all four conditions. ***p < 0.0001.
Whole-cell patch-clamp recordings with 3–5 MΩ electrodes were made with a Multiclamp 700B amplifier using Clampex 10.0 (Molecular Devices) and Igor Pro 6.0 (Wavemetrics) for data acquisition. The series resistance (Rs), input resistance (Ri), and holding current (Ihold) of all recordings were continuously monitored. Recordings with large deviations in any of these properties were discarded from analysis. VTA neurons included in this study were located medial to the medial terminal nucleus. The neurons were confirmed to be dopamine neurons by postrecording immunohistochemistry for tyrosine hydroxylase (TH). Only TH-positive neurons were considered in the analysis. To record GABA events and IPSCs, we used an internal solution of the following composition: 125 mm KCl, 10 mm NaCl, 1 mm MgCl2, 10 mm HEPES, 1 mm EGTA, 2 mm Na-ATP, 0.6 mm Na-GTP, 5 mm creatine phosphate, and 0.1 mg/ml Alexa Fluor 488, pH 7.2–7.4 (275–285 osmolarity). To record the effect of morphine on cationic currents in GABA neurons, the potassium methanesulfonate-based internal solution consisted of 0.95% (v/v) KOH, 0.38% (v/v) methanesulfonic acid, 20 mm HEPES, 0.2 mm EGTA, 2.8 mm NaCl, 2.5 mg/ml MgATP, and 0.25 mg/ml GTP, pH 7.2–7.4 (275–285 osmolarity). Neurons were visualized with an upright microscope equipped with infrared differential interference contrast using Axiovision camera and software (Carl Zeiss). aCSF at 30–32°C was continuously perfused at 2–3 ml/min over brain slices. Whole-cell voltage-clamp recording at −70 mV holding potential in all recordings of GABA IPSCs and whole-cell current clamp at I = 0 pA in recordings of GABA neuron membrane potential was performed. Using this technique, all cells recorded were first tested for the presence of Ih currents, a cellular marker of dopamine neurons in the VTA (Grace and Onn, 1989). Miniature events were recorded in the presence of tetrodotoxin (TTX) (0.5 μm), a sodium channel blocker. Spontaneous action-potential driven events (sIPSCs) and evoked GABA IPSCs (acute effects and forskolin experiments) were recorded in the absence of TTX.
Immunohistochemistry.
Horizontal VTA slices were treated with morphine (30 μm, 25 min) or left untreated. The slices were fixed with 4% paraformaldehyde in 0.1 m PBS, resectioned into 30 μm slices using a cryostat at −20°C. The sections were preincubated in PBT solution (0.1 m phosphate buffer plus 0.2% BSA and 0.2% Triton X-100) for 30 min, blocked in 5% normal goat serum in PBT solution for another 30 min, and then incubated with rabbit anti-MOR receptor antibody (rabbit Ab1580, 1:1000; Millipore Bioscience Research Reagents) overnight at 4°C. Sections were then washed with PBT and incubated in Alexa Fluor 594-conjugated goat anti-rabbit antibody (1:1000; Invitrogen) for 2 h at room temperature. The sections were then washed, mounted on slides, and visualized using an LSM 510 Meta confocal imaging system. For identifying dopamine neurons after electrophysiology, slices were incubated with 1:2000 mouse anti-tyrosine hydroxylase (Sigma) and 1:3000 anti-mouse Alexa Fluor 594 (Invitrogen). In the experiments requiring recording of GABA IPSCs from dopamine neurons, 92% of the Ih-positive cells (n = 161 of 175 neurons) recorded in the VTA were identified to be dopamine neurons, and neurons that stained negative for the TH antibody were not considered for additional analysis.
Data analysis.
Spontaneous and miniature events were analyzed using Minianalysis software (Synaptosoft). In each event recording, at least 500 events were analyzed. The threshold for minimum amplitude was set at 8 pA. We did not see any variability in waveform in each recording, suggesting different populations of events. Data analysis was performed using GraphPad Software Prizm or Microsoft Excel. For the correlation graphs, individual cells in each animal were pooled together, and the mean event properties were plotted against the value of tolerance for that mouse. Data are represented as mean ± SEM, unless specified. t tests [unpaired and paired (see Fig. 6B,C)] or one-way ANOVA was used to compare data for significance. All data in which four groups were used in a 2 (morphine exposure: no, yes) × 2 (genotype: WT, RMOR) are analyzed with a two-way ANOVA, and statistics of the interaction and the morphine effect are reported. Degrees of freedom (DF) for F and t statistics are marked as t(DF), F(1,DF) or for two-way ANOVA as F(DFn,DFd). Pearson's χ2 test (p < 0.05) was used to compare the correlations. *p < 0.05, **p < 0.01, ***p < 0.001; not significant is p > 0.05.
Results
Endocytosis of RMOR and WT MOR in VTA
Morphine-induced endocytosis of the WT MOR and the RMOR was examined in VTA neurons. VTA slices from WT and RMOR mice were treated with a saturating concentration of morphine (30 μm) for 25 min or left untreated, and receptor distribution was examined by immunostaining. A saturating dose of morphine was used because of limitations in assessing the ex vivo responses of morphine on the MOR in brain slices (Alvarez et al., 2002). Both WT MOR and RMOR receptors were distributed primarily on the cell surface in the absence of drug (Fig. 1, MOR NT and RMOR NT). Treatment of slices from WT MOR mice with morphine did not change receptor distribution (Fig. 1, WT Morphine), consistent with the poor ability of morphine to induce MOR endocytosis in previous studies (Keith et al., 1998; Trafton et al., 2000; Abbadie and Pasternak, 2001; He et al., 2002; He and Whistler, 2005). In contrast, treatment of slices from RMOR mice with morphine caused a redistribution of receptors to the cytoplasm (Fig. 1, RMOR Morphine). These data indicate that, in response to morphine, the RMOR receptor undergoes substantial endocytosis in the VTA but the WT MOR does not.
Figure 1.
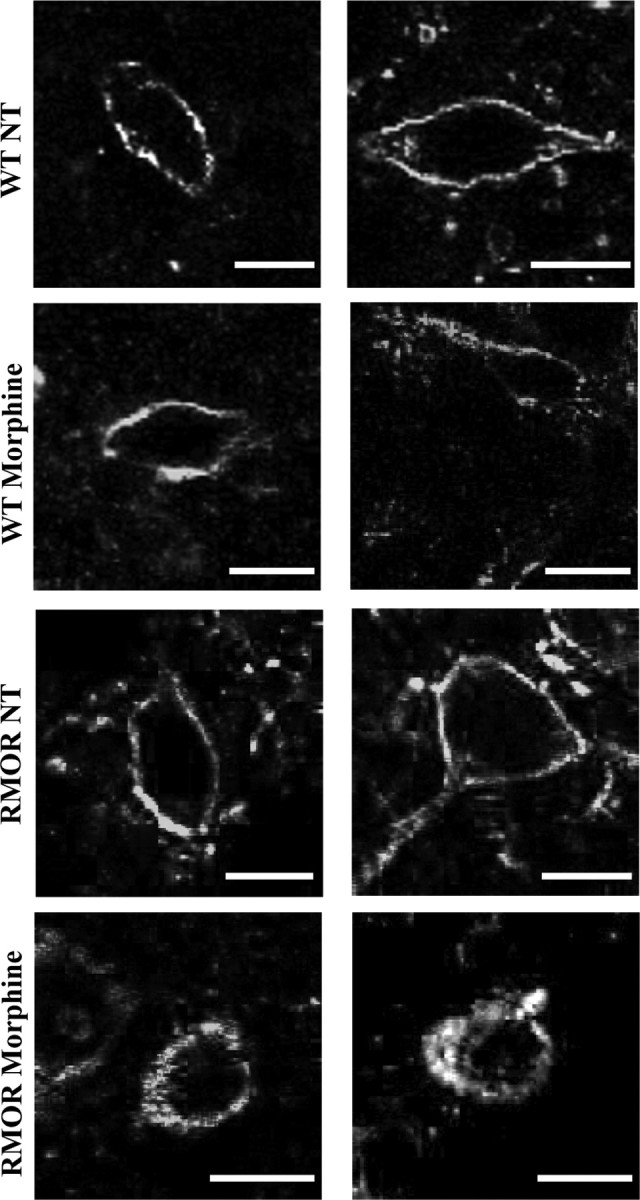
Morphine induces RMOR endocytosis in the VTA. WT and RMOR VTA slices were treated with 30 μm morphine or left untreated (NT) for 25 min. Slices were immunostained for MOR. Morphine induces internalization of the receptor in RMOR but not MOR VTA (both right and left columns). Scale bars, 10 μm.
Behavioral tolerance and dependence in RMOR and WT MOR mice
The effect of the RMOR mutation on the development of anti-nociceptive tolerance and physical dependence was examined in WT and RMOR mice (postnatal days 23–28). Mice were treated with morphine (10 mg/kg) twice per day for 5 d. Morphine-induced anti-nociception (expressed as MPE) was measured 30 min after the morphine injection on days 1, 3, and 5. Chronic morphine treatment caused a significant reduction in the MPE over time in the WT mice (Fig. 2A; n = 20, t(38) = 11.4, p < 0.0001, day 1 vs day 5), indicative of anti-nociceptive tolerance to the drug. However, knock-in mice expressing RMOR did not develop significant anti-nociceptive tolerance (Fig. 2A, n = 20, t(38) = 1.4, p = 0.16, day 1 vs day 5). Physical dependence on morphine was examined by counting somatic withdrawal signs including, tremors, jumps, wet-dog shakes, rears, and grooming behavior, induced by NLX (2 mg/kg, s.c.) injection 30 min after the final morphine dose on day 6, and the global withdrawal score was computed by adding all signs (Fig. 2B). WT mice treated chronically with morphine exhibited robust signs of NLX-precipitated morphine withdrawal compared with saline-treated WT mice, RMOR mice, and chronic morphine-treated RMOR mice [Fig. 2B; WT (n = 4), WT Chronic Morphine (n = 15), RMOR (n = 5), RMOR Chronic Morphine (n = 16); for interaction, F(1,36) = 4.33, two-way ANOVA, p = 0.04; for morphine effect, F(1,36) = 12.65, two-way ANOVA, p = 0.001] to morphine after the same treatment. Therefore, in line with our previous results in adult WT and RMOR mice (Kim et al., 2008), the morphine treatment regimen used here was sufficient to induce significantly higher analgesic tolerance and physical dependence in adolescent postnatal days 23–28 WT mice than in RMORs.
Figure 2.
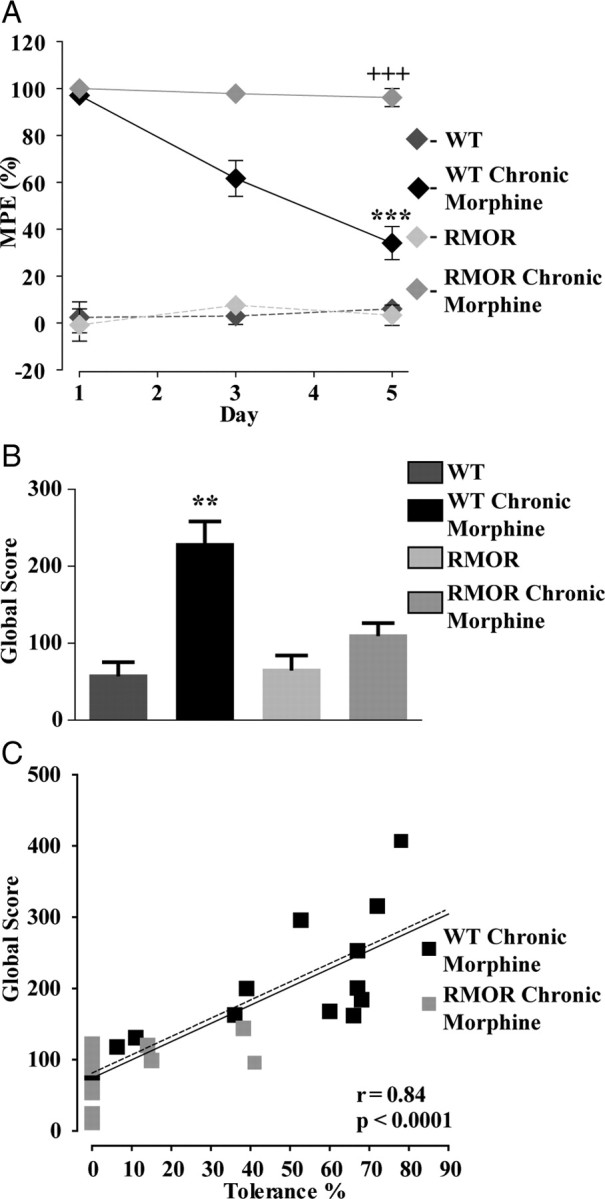
Anti-nociceptive tolerance and physical dependence in WT and RMORs. A, MPE to morphine or saline (control) was assessed in WT and RMOR mice using the hotplate test. WT mice (n = 20), but not RMOR knock-in mice (n = 20), show pronounced and progressive tolerance to morphine over 5 d. MPE values were significantly different between morphine-treated WT and RMOR mice. B, Global withdrawal scores after NLX treatment (2 mg/kg) were assessed for all four conditions: WT, WT treated with 10 mg/kg morphine for 5 d (WT Chronic Morphine), RMOR knock-in (RMOR), and RMOR treated with 10 mg/kg morphine for 5 d (RMOR Chronic Morphine). WT mice chronically treated with morphine (n = 15) show significantly elevated levels of NLX-precipitated somatic withdrawal signs than similarly treated RMOR mice (n = 16) or untreated WT (n = 4) and RMOR mice (n = 5). C, Scatter plot of global withdrawal score versus the magnitude of tolerance (100 − MPE) for morphine-treated WTs (WT Chronic Morphine) and RMORs (RMOR Chronic Morphine). Greater tolerance to morphine predicts increased somatic signs of withdrawal for WT Chronic Morphine and RMOR Chronic Morphine (n = 15) and WT Chronic Morphine alone (n = 15). **p < 0.01; ***,+++p < 0.001.
Mice within a genotype showed varying degrees of tolerance and NLX-precipitated withdrawal, even with identical morphine treatment. Importantly, the magnitude of tolerance was a faithful predictor of withdrawal for both WT MORs and RMORs [Fig. 2C; WT Chronic Morphine (n = 15), RMOR Chronic Morphine (n = 16); r = 0.84, p < 0.0001] and for WT MORs alone (n = 15, r = 0.81, p < 0.0001; dashed line). Mice exhibiting higher levels of tolerance were also more dependent on morphine. Importantly, RMOR mice as a group showed a significant reduction in development of morphine tolerance and withdrawal compared with WT mice (Fig. 2C).
Modulation of GABA signaling in morphine-treated WT MORs and RMORs
Opioid agonists acting at MORs present in GABA cells in the VTA directly control postsynaptic dopamine neuron activity by inhibiting GABA release (Johnson and North, 1992b) (see Fig. 6). To determine whether NLX-precipitated withdrawal from chronic morphine treatment modulates this GABA signaling, the frequency and amplitude of GABA sIPSCs were recorded from VTA dopamine neurons ex vivo from saline- or morphine-treated (10 mg/kg, s.c., twice daily for 5 d) mice. Changes in the frequency of sIPSCs are typically attributed to alterations in presynaptic release probability, whereas changes in amplitude are attributed to modifications in GABA receptor function, number, or both (Bonci and Williams, 1997). GABA sIPSCs were recorded from TH-positive dopamine neurons from WT and RMOR mice treated with morphine (10 mg/kg twice per day for 5 d) or left untreated (Fig. 3A). Withdrawal from morphine was precipitated in the slice with the addition of NLX (1 μm). There was a higher frequency of GABA sIPSCs from morphine-treated WT mice compared with untreated control mice, but, interestingly, morphine treatment and withdrawal did not change the frequency of GABA sIPSCs in morphine-treated RMOR mice compared with control RMOR mice (Fig. 3B,D; WT, 13.75 ± 1.67 Hz, n = 32 neurons, n = 19 mice; WT Chronic Morphine, 26.11 ± 1.68 Hz, n = 28 neurons, n = 20 mice; RMOR, 12.52 ± 1.82 Hz, n = 15 neurons, n = 5 mice; RMOR Chronic Morphine, 12.74 ± 1.82 Hz, n = 31 neurons, n = 11 mice; for interaction, F(1,102) = 10.41, two-way ANOVA, p = 0.002; for morphine effect, F(1,102) = 11.20, two-way ANOVA, p = 0.001). A small, but significant, increase in mean amplitude of sIPSCs was also observed during NLX-precipitated morphine withdrawal in cells from morphine-treated WT mice compared with WT control and with control or morphine-treated RMOR mice (Fig. 3C,E; WT, 15.55 ± 0.44 pA, n = 32; WT Chronic Morphine, 19.12 ± 1.17 pA, n = 28; RMOR, 15.27 ± 0.93 pA, n = 15; RMOR Chronic Morphine, 16.48 ± 0.82 pA, n = 31; for interaction, F(1,102) = 1.68, two-way ANOVA, p = 0.2; for morphine effect, F(1,102) = 6.92, two-way ANOVA, p = 0.009).
Figure 3.

NLX-precipitated withdrawal from chronic morphine treatment enhances frequency of GABA sIPSCs in VTA dopamine neurons from WT but not RMOR mice. A, GABA sIPSCs recorded in WT, WT treated with 10 mg/kg morphine for 5 d (WT Chronic Morphine), RMOR knock-in (RMOR), and RMOR treated with 10 mg/kg morphine for 5 d (RMOR Chronic Morphine). Calibration: 13.3 pA, 153.6 ms. B, Cumulative interevent distribution of sIPSCs shows that events in morphine-withdrawing WTs are more frequent than in other conditions. C, Cumulative amplitude distribution of sIPSCs shows that larger-amplitude events are more frequent in WT morphine-withdrawing slices. D, Mean frequency of GABA sIPSCs is significantly higher after NLX-precipitated withdrawal in wild-type mice (WT Chronic Morphine, n = 32) compared with WT controls (WT, n = 28). Mean frequency of GABA sIPSCs is not significantly different in RMOR controls (RMOR, n = 15) and morphine-treated RMOR mice (RMOR Chronic Morphine, n = 31) or when compared with WT controls (WT). E, Mean amplitude of GABA sIPSCs is higher during NLX-precipitated withdrawal in morphine-treated WT (WT Chronic Morphine) than in the other three conditions. F, Paired pulse ratio of GABA IPSCs is significantly reduced during NLX-precipitated withdrawal in morphine-treated WT mice (WT Chronic Morphine; n = 30) than in control conditions (WT, n = 29; RMOR, n = 34; RMOR Chronic Morphine, n = 37). *p < 0.05; **p < 0.01.
An increase or decrease in the paired pulse ratio (PPR) of evoked current amplitudes also indicates reduction or enhancement of transmitter release, respectively. The paired pulse ratio (PPR = IPSC2/IPSC1) of GABA IPSC amplitudes resulting from two current pulses each 160 μs in duration, separated by 100 ms, was determined from cells taken from morphine-treated and control WT and RMOR mice. The PPR of GABA IPSCs was significantly enhanced exclusively in dopamine neurons from morphine-treated WT mice (Fig. 3F; WT, 0.75 ± 0.04, n = 30; WT Chronic Morphine, 0.60 ± 0.04, n = 34; RMOR, 0.72 ± 0.04, n = 29; RMOR Chronic Morphine, 0.73 ± 0.02, n = 37; for interaction, F(1,126) = 5.38, two-way ANOVA, p = 0.02; for morphine effect, F(1,126) = 4.52, two-way ANOVA, p = 0.03).
We next determined whether the magnitude of tolerance correlated with the probability of GABA release onto VTA dopamine neurons. The frequency (Fig. 4A) and amplitude (Fig. 4B) of spontaneous GABA release events in WTs during NLX-precipitated morphine withdrawal was plotted against the magnitude of tolerance of the mouse from which the recordings were made (Fig. 4A,B). The frequency of sIPSCs covaried with values of behavioral tolerance (n = 20 mice, n = 28 neurons; r = 0.73; p = 0.0003) (Fig. 4A). The correlation between tolerance and amplitude of sIPSCs (n = 20 mice, n = 28 neurons; r = 0.33; p = 0.16) (Fig. 4B) was weaker and not statistically significant. In the case of RMORs with the same morphine treatment, there was no correlation between tolerance (rarely seen and only to a moderate degree in RMOR mice) and either GABA sIPSC frequency (n = 11 mice, n = 31 neurons; r = 0.21; p = 0.5652) (Fig. 4A) or amplitude (n = 11 mice, n = 31 neurons; r = −0.05; p = 0.88) (Fig. 4B).
The increase in GABA release could be modulated by changes in the excitability of the presynaptic cell as evidenced by changes in the action potential-driven sIPSCs. Furthermore, the increased release could also be attributable to changes in release probability independent of action potentials. To clarify the role of presynaptic action potentials in driving changes in GABA release during withdrawal, we assessed the effects of morphine withdrawal on TTX-sensitive miniature IPSCs (mIPSCs) (Fig. 5A). In the presence of TTX, an inhibitor of Na+ channels, action potential-driven synaptic transmission is inhibited. NLX-precipitated withdrawal from chronic morphine treatment produced an enhancement in the frequency of mIPSCs in WT but not in the RMOR knock-in mice (Fig. 5B,D; WT, 8.65 ± 1.72 Hz, n = 26 neurons, n = 11 mice; WT Chronic Morphine, 15.25 ± 2.59 Hz, n = 23 neurons, n = 6 mice; RMOR, 8.66 ± 1.69 Hz, n = 12 neurons, n = 4 mice; RMOR Chronic Morphine, 8.1 ± 1.72 Hz, n = 27 neurons, n = 10 mice; for interaction F(1,83) = 5.34, two-way ANOVA, p = 0.02; for morphine effect, F(1,83) = 3.97, two-way ANOVA, p = 0.04), suggesting that there was an increased presynaptic release of GABA attributable to morphine withdrawal. Compared with the effect on GABA sIPSCs (Fig. 3), no significant changes in mean amplitude (Fig. 5C,E; for interaction, F(1,83) = 0.88, two-way ANOVA, p = 0.35; for morphine effect, F(1,83) = 0.91, two-way ANOVA, p = 0.34) of the mIPSCs was observed during withdrawal from chronic morphine. An increase in mIPSC frequency during withdrawal suggests that action potential-independent increases in release probability have occurred. Similar to sIPSCs, in the case of mIPSCs, GABA mIPSC frequency (n = 6 mice, n = 24 neurons; r = 0.82; p = 0.047) (Fig. 5F) and not amplitude (n = 6 mice, n = 24 neurons; r = 0.17; p = 0.74) (Fig. 5G) correlated significantly with tolerance. Together with the sIPSC and paired pulse ratio data, these data indicate that withdrawal from chronic morphine causes an enhancement of GABA release onto dopamine neurons in WT but not RMOR mice.
Figure 5.
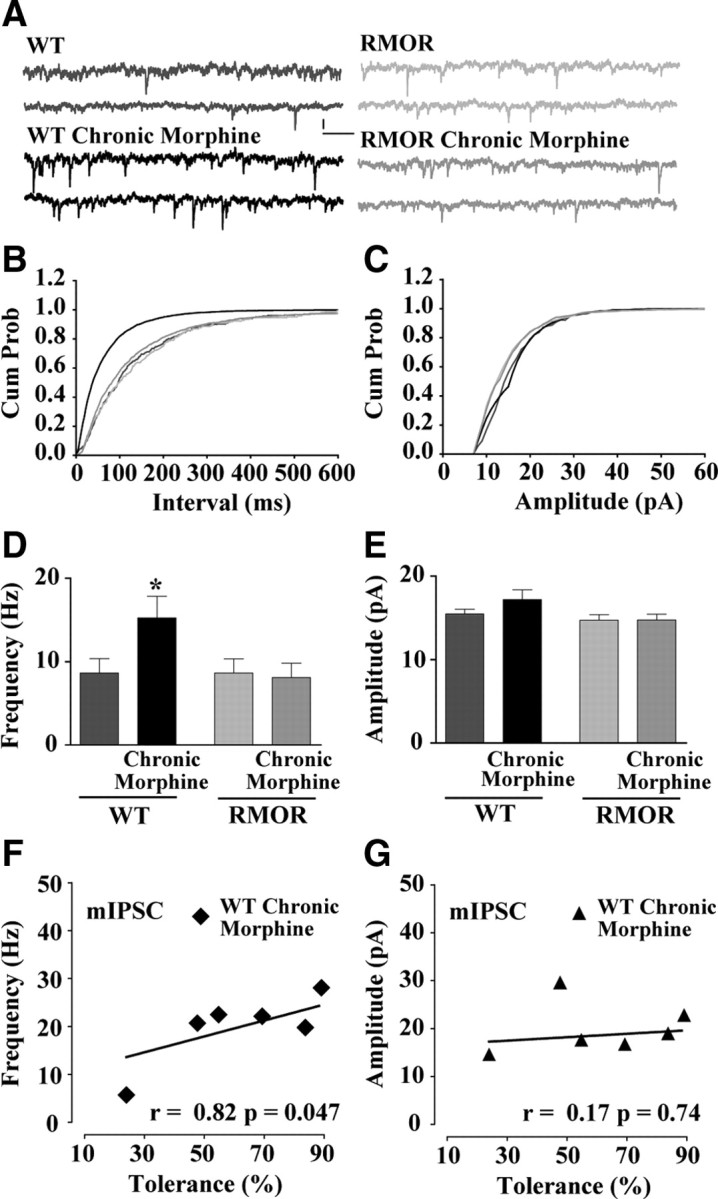
NLX-precipitated withdrawal from chronic morphine treatment increases frequency of GABA mIPSCs in VTA dopamine neurons. A, GABA mIPSCs recorded in WT, WT treated with 10 mg/kg morphine for 5 d (WT Chronic Morphine), RMOR knock-in (RMOR), and RMOR knock-ins treated with 10 mg/kg morphine for 5 d (RMOR Chronic Morphine). Calibration: 13.3 pA, 153.6 ms. B, Cumulative interevent distribution of mIPSCs shows that events in morphine-treated WTs are more frequent than in other conditions during NLX-precipitated withdrawal. C, Cumulative amplitude distribution of mIPSCs shows that amplitude of events is similar in all four conditions. D, Mean frequency of GABA mIPSCs is higher during NLX-precipitated withdrawal in morphine-treated WT mice (WT Chronic Morphine, n = 23) but not in WT (n = 26) and RMOR (n = 12) controls, and morphine-treated RMOR mice (RMOR Chronic Morphine, n = 27). E, Mean amplitude of GABA mIPSCs is not significantly different in all four conditions. F, Magnitude of tolerance covaries with GABA mIPSC frequency during withdrawal in the VTA. G, Correlation between the magnitude of tolerance against the mIPSC mean amplitude is not significant. *p < 0.05.
Acute effects of opioid ligands in WT and RMOR mice
The differences between WT and RMOR on GABA transmission during withdrawal could reflect genotype-specific differences in the ability of the MOR and RMOR to transmit an opioid signal, attributable, for example, to differences in localization in distinct microdomains and/or differences in strength or diversity in coupling to downstream effectors that control presynaptic release probability or postsynaptic GABA IPSCs. To address this possibility, the inhibitory effect of DAMGO and morphine on GABA IPSCs was recorded from dopamine neurons in WT and RMOR mice. Peak inhibition (Fig. 6A, inset, black bar indicates baseline) of GABA currents was plotted against the logarithm of agonist concentration (Fig. 6A). The dose–response curves for inhibition by DAMGO (WT EC50 of 89.22 ± 0.28 nm, Emax of 90.57 ± 7.6, n = 17; RMOR EC50 of 112.6 ± 0.32 nm, Emax of 86.96 ± 10.6, n = 17; t(11) = 0.06; p = 0.958) and morphine (WT EC50 of 106.2 ± 0.78 nm, Emax of 52.82 ± 9.8, n = 8; RMOR EC50 of 125.7 ± 0.51 nm, Emax of 48.66 ± 6.52, n = 8; t(8) = 0.06; p = 0.954) was not significantly different between WT and RMOR mice. Thus, opioid agonists are equally effective at inhibiting GABA IPSCs in VTA dopamine neurons in WT and RMOR mice. Additionally, there were no genotype differences in the presynaptic effects of DAMGO, which significantly decreased mIPSC frequency (Fig. 6B), but not amplitude (Fig. 6C), equivalently in both WT (control, 13.34 ± 3.2 Hz; DAMGO, 7.76 ± 3 Hz; n = 9 neurons, n = 5 mice; t(8) = 6.9; p < 0.0001) and RMOR (control, 13.92 ± 2.8 Hz; DAMGO, 7.0 ± 2.2 Hz; n = 9 neurons, n = 5 mice; t(8) = 3.7; p = 0.0058). These data suggest that increased probability of GABA release during morphine withdrawal in WT but not RMOR cannot be explained by differences in potency of opioid inhibition of GABA release. They also suggest that, at least in the VTA, there are no apparent differences in the ability of the MOR versus the RMOR to signal in response to either morphine or DAMGO.
Instead, we hypothesize that RMOR and MOR differ in the degree of desensitization in response to morphine, which influences receptor endocytosis (Fig. 1) and the development of tolerance and dependence (Fig. 2). To examine whether enhanced endocytosis of RMOR predicts increased acute desensitization, we tested the effect of a saturating dose of morphine (30 μm) on Ih-negative GABA neurons in the two genotypes, the same dose that was used to show a difference in endocytosis of MOR and RMOR (Fig. 1). Morphine induced a sustained hyperpolarization of membrane potential in Ih-negative GABA neurons in WT mice that did not significantly desensitize (Fig. 6D) (n = 8 neurons, n = 6 mice), suggesting that morphine application produced a long-lasting activation of a potassium conductance (Johnson and North, 1992b). Furthermore, this same dose of morphine also caused hyperpolarization of membrane potential of Ih-negative GABA neurons in RMOR mice (Fig. 6D) (n = 6 neurons, n = 6 mice). However, the maximal response was smaller and quickly returned to baseline (Fig. 6D) (n = 6 neurons, n = 6 mice), which could indicate that RMORs rapidly desensitize to morphine. Indeed, after normalization, the WT response to morphine did not return to baseline, although the faster rising RMOR response did (Fig. 6E). Using this data, we calculated the degree of desensitization [100 × (final response − peak response)/(peak response − baseline)] of the acute morphine response in MOR and RMOR. RMORs showed significantly higher levels of desensitization in their membrane potentials in response to morphine than MORs (percentage desensitization; WT, 16.37 ± 12.81, n = 8; RMOR, 78.06 ± 27.01, n = 6; t(12) = 2.25; p = 0.0444). These data suggest that a saturating morphine dose induces large-scale desensitization of the evoked potassium conductance in the RMORs but not the WT MORs in VTA GABAergic neurons. This effect correlated with the propensity for this dose of morphine to induce endocytosis of the RMOR receptor in these GABA neurons (Fig. 1).
Effects of cAMP on GABA transmission
Chronic morphine treatment promotes compensatory upregulation of cAMP signal transduction in HEK293 cells expressing WT MOR, but this effect is reduced in cells expressing RMORs (Finn and Whistler, 2001). Thus, differences in adenylyl cyclase activity induced by chronic morphine could account for the differences in NLX-precipitated withdrawal-induced enhancement of GABA release between WTs and RMORs. To examine this possibility, we measured the effects of forskolin, a direct activator of adenylyl cyclase, on GABA transmission after NLX-precipitated morphine withdrawal in WT and RMOR mice. Forskolin application caused an enhancement of GABA IPSCs in all four conditions (Fig. 7A). This forskolin-induced enhancement of GABA currents was accompanied by a decrease in the paired pulse ratio of the evoked GABA currents (Fig. 7B), establishing that presynaptic cAMP signaling modulates GABA release onto the VTA dopamine neurons. However, during NLX-precipitated morphine withdrawal, forskolin application caused a significantly larger increase in GABA IPSCs (Fig. 7A) and a greater reduction in the PPR (Fig. 7B) in WTs than in RMORs. The cumulative percentage increase induced by forskolin (Fig. 7C, baseline minus black bar) for the four conditions was as follows: WT, 35.16 ± 2.63%; WT Chronic Morphine, 130.8 ± 6.34%; n = 6 neurons, n = 6 mice; RMOR, 41.49 ± 4.78%; RMOR Chronic Morphine, 52.03 ± 3.22%; n = 6 neurons, n = 6 mice; for interaction, F(1,20) = 90.17, two-way ANOVA, p < 0.0001; for morphine effect, F(1,20) = 140.35, two-way ANOVA, p < 0.0001) (Fig. 7D). These data indicate that forskolin-stimulated cAMP production may be significantly higher in GABA cells of the VTA in WT mice than in RMOR mice during NLX-precipitated morphine withdrawal. Alternatively, GABA cells in WT mice after withdrawal may be more sensitive to increased cAMP levels induced by forskolin. These data underscore the importance of MOR endocytosis and recycling for reducing cellular compensation in the cAMP pathway and therefore preventing neuroadaptations in GABA release as a result of NLX-precipitated morphine withdrawal.
Behavioral effects of cAMP in the VTA on morphine withdrawal symptoms
Based on these results, we would predict that inhibiting cAMP activity using rp-cAMPS would prevent or attenuate the expression of NLX-precipitated withdrawal symptoms induced in the VTA of morphine-dependent WT mice. To test this, NLX-precipitated opioid withdrawal was examined after NLX infusion (500 ng/0.5 μl saline) in the VTA of untreated/ morphine-naive (NLX) and chronic morphine-treated (10 mg/kg, s.c., twice daily for 5 d, Chronic Morphine + NLX) WT mice. Additionally, a group of morphine-treated WT mice were given an infusion of 50 ng/0.5 μl rp-cAMPS in saline 30 min before NLX infusion to test the effect of inhibiting cAMP activity on opiate withdrawal symptoms. Physical dependence on morphine was examined by counting somatic withdrawal signs including, tremors, jumps, wet-dog shakes, rears, and grooming behavior induced by NLX infusion for 30 min (Fig. 8). Mice treated chronically with morphine exhibited elevated signs of NLX-precipitated morphine withdrawal compared with naive/untreated mice, demonstrating that NLX infusion in the VTA is sufficient to produce enhanced withdrawal signs from chronic systemic morphine administration. Importantly, intra-VTA injection of rp-cAMPS (50 ng/0.5 μl), 30 min before intra-VTA NLX, significantly attenuated these withdrawal signs [Fig. 8A; NLX (n = 9) vs Chronic Morphine + NLX (n = 6) vs Chronic Morphine + rp-cAMPS + NLX (n = 6), F(1,2) = 11.65; p = 0.0006] in chronic morphine-treated WT mice. Individual scores of NLX-precipitated morphine withdrawal including tremors, rears, wet-dog shakes, and grooming behavior are also shown (Fig. 8B–E). Chronic morphine-treated WT mice show elevated expression of tremors (Fig. 8B; all three conditions, F(1,2) = 9.98, p = 0.0012), rears (Fig. 8C; all three conditions, n = 6, F(1,2) = 5.3, p = 0.015), wet-dog shakes (Fig. 8D; all three conditions, F(1,2) = 5.84, p = 0.011), and grooming behavior (Fig. 8E; all three conditions, F(1,2) = 4.3, p = 0.03) than untreated WTs, and rp-cAMPS pretreatment attenuates all these withdrawal responses in chronic morphine-treated mice. Intra-VTA NLX infusion did not elicit jumping behavior in any of the mice tested, but it did result in elevated levels of teeth chattering and diarrhea, as reported previously (Baumeister et al., 1989). The cannula placements in these mice were examined after dye injection, and only those mice with cannula tracks in the VTA were used in this analysis (Fig. 9). Based on these data, we conclude that NLX-precipitated morphine withdrawal increases cAMP activity in the VTA, possibly acting to enhance GABA signaling onto VTA dopamine neurons (Fig. 7).
Figure 8.
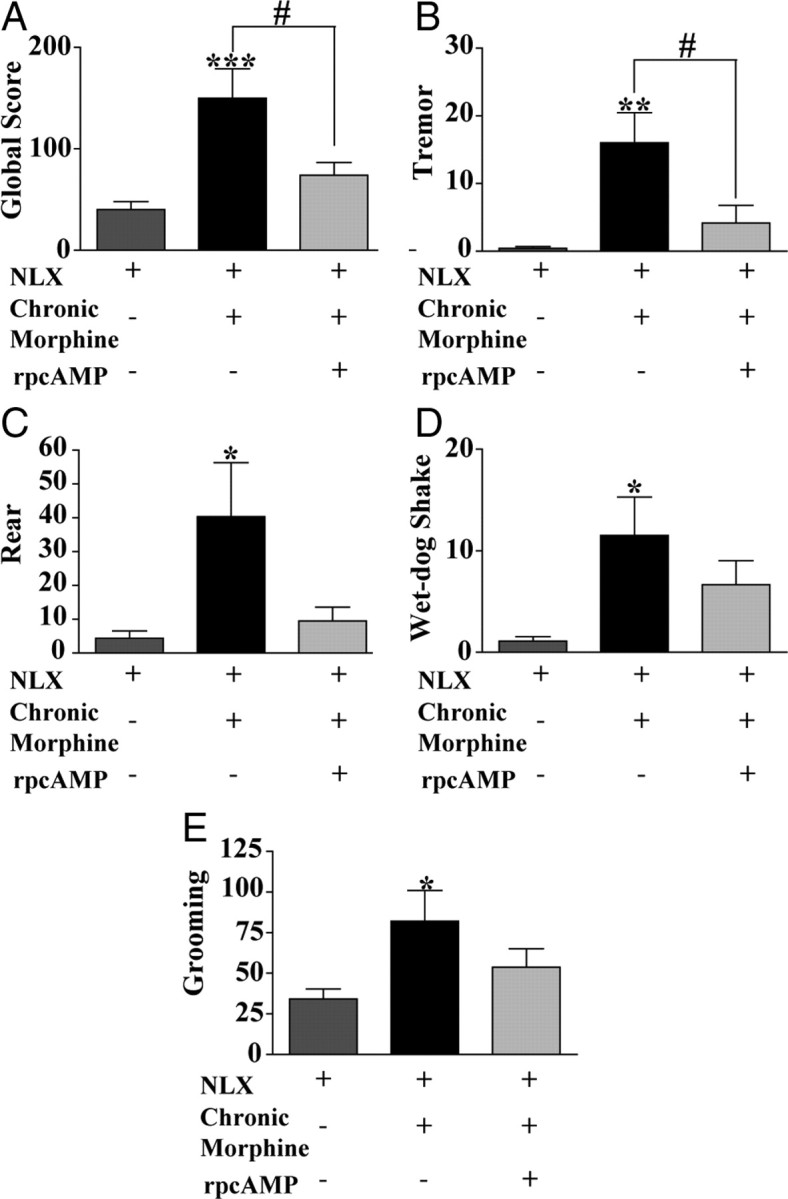
Previous exposure to cAMP activity inhibitor rp-cAMPS attenuated withdrawal symptoms to intra-VTA naloxone in morphine-treated WT mice. A, Global withdrawal score after intra-VTA NLX infusion (500 ng/0.5 μl) in drug-naive/untreated WT mice (NLX), in mice treated chronically with morphine (10 mg/kg, s.c., twice per day for 5 d; Chronic Morphine + NLX) or in mice treated with chronic morphine (10 mg/kg, s.c., twice per day for 5 d) that received the cAMP activity blocker rp-cAMPS (50 ng/0.5 μl) in the VTA 30 min before intra-VTA naloxone (Chronic Morphine + rp-cAMPS + NLX). Global withdrawal score was significantly higher after chronic morphine treatment and was attenuated by rp-cAMPS preexposure [NLX (n = 9) vs Chronic Morphine + NLX (n = 6) vs Chronic Morphine + rp-cAMPS + NLX (n = 6)]. B–E, Quantification of behavioral signs of intra-VTA NLX-precipitated morphine withdrawal and the effects of rp-cAMPS preexposure. B, Tremors were significantly attenuated after rp-cAMPS preexposure. C, Rears were significantly attenuated after rp-cAMPS preexposure. D, Wet-dog shakes were reduced after rp-cAMPS preexposure. E, Grooming behavior was similar after rp-cAMPS preexposure. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Here we report that experience-dependent cellular adaptations in GABA release onto VTA dopamine neurons after chronic exposure to morphine are associated with morphine dependence. Both the alterations in GABA transmission and dependence were prevented in RMOR mice, demonstrating an essential role of receptor trafficking in synaptic plasticity associated with opioid dependence. Acute effects of the WT MOR and the RMOR on GABA release were not significantly different. However, in response to morphine, the RMOR undergoes endocytosis and desensitization, whereas the WT MORs do not. We also demonstrate that NLX-precipitated withdrawal-induced enhancement of GABA release is likely attributable to cellular compensations in cAMP signal transduction, as evidenced by the enhanced sensitivity of WT neurons to forskolin in chronic morphine-treated mice and attenuated intra-VTA expression of opiate withdrawal symptoms after preexposure to a cAMP activity inhibitor, rp-cAMPS. Importantly, this enhanced forskolin sensitivity is not observed in the RMOR mice, consistent with the hypothesis that receptor trafficking prevents changes in magnitude and/or sensitivity of the adenylate cyclase signaling cascade.
Morphine dependence is a complex phenomenon likely mediated by many converging circuits. Nevertheless, injection of NLX into the VTA of morphine-treated animals has been reported to precipitate withdrawal symptoms (Baumeister et al., 1989; Stinus et al., 1990), underscoring the importance of this region in morphine dependence. In line with these previous observations, we find here that naloxone infusion into the VTA of WT mice treated chronically with systemic morphine precipitates withdrawal symptoms (Fig. 8). Intra-VTA NLX infusion produces significant tremors, rears, and wet-dog shakes (Fig. 8B–E). Indeed, the only withdrawal sign that does not occur as a result of intra-VTA NLX infusion that occurs during systemically administered NLX is jumping behavior. Other groups have also found that the subset of withdrawal symptoms differ based on the brain region in which NLX is infused (Baumeister et al., 1989; Stinus et al., 1990; Maldonado et al., 1992) and may also be a function of extent of morphine treatment, naloxone dose, and the subject.
Acutely, morphine acting at the MOR in the VTA increases dopamine tone by decreasing GABA release onto dopamine neurons (Johnson and North, 1992a,b). In contrast, increases in GABA release during withdrawal from chronic morphine would be expected to cause an inhibition of dopamine neurons in the midbrain. In the opiate dependent/withdrawn state, intra-VTA infusion of the GABA antagonist bicuculline is rewarding, an effect that is inhibited by systemic or intra-accumbens infusion of α-flupenthixol, a dopamine receptor antagonist, lending additional support to the role of GABA-modulated dopaminergic projections from the VTA in withdrawal (Laviolette et al., 2004).
Increased probability of GABA release in the VTA could, thus, be a step in a cascade of events that manifest as opiate withdrawal symptoms. Withdrawal-induced increases in GABA release frequency in the VTA correlate with behavioral indices of morphine withdrawal (Figs. 4, 5). In addition, infusion of the cAMP inhibitor rp-cAMPS attenuates withdrawal symptoms induced by intra-VTA NLX infusion (Fig. 8), possibly by preventing cAMP-driven increase in GABA IPSCs (Fig. 7). Although these data do not rule out multiple representations of opiate withdrawal in other circuits in the brain, they present a direct link between dependence and increased GABA release in the mesolimbic dopamine system.
Because slices taken from mice undergoing acute withdrawal after chronic morphine treatment are more sensitive to activators of the cAMP pathway, the enhanced probability of GABA release in the VTA in WT mice is likely attributable to cellular compensations in adenylyl cyclase activity that have been shown to occur in vivo in other brain regions, such as the locus ceruleus (Duman et al., 1988; Rasmussen et al., 1990) and striatum (Sheu et al., 1995; Kaplan et al., 1998). Specifically, after chronic morphine treatment, there is an increase in the ability of forskolin to enhance GABA release in WT but not RMOR mice. Therefore, it is likely that these compensatory changes in cAMP and subsequent GABA release are a consequence of failure of the MOR to undergo morphine-induced endocytosis. In addition, these changes in cAMP levels, at least in the VTA, are likely directly contributing to morphine dependence, because inhibition of cAMP in the VTA attenuates withdrawal to systemic morphine.
Additional work is needed to elucidate the cellular connection between cAMP superactivation and enhanced GABA release. cAMP could be acting on a variety of downstream targets of the MOR signaling cascade to cause an enhancement of GABA release. This includes the possibility of a direct effect on vesicular release of GABA, voltage-gated calcium channels (Wilding et al., 1995), GIRK currents (Cruz et al., 2008), GABA transporter current that modulate GABA release during morphine withdrawal via a PKA-dependent mechanism in the periaqueductal gray (Bagley et al., 2005), or perhaps a cAMP-dependent switch in GABAA receptor conductance from inhibitory to excitatory that has been reported to occur in rat VTA GABA neurons (Laviolette et al., 2004; Vargas-Perez et al., 2009).
In conclusion, cellular and synaptic adaptations that occur during chronic morphine treatment do not occur in mice expressing the mutant RMOR that undergoes endocytosis in response to morphine in VTA GABA interneurons. Thus, the trafficking of RMOR in response to morphine is qualitatively similar to that of the WT receptors when activated by endogenous peptide ligand and is quite different from that induced by the WT receptor when activated by morphine. Specifically, desensitization, endocytosis, and consequent receptor recycling in the RMOR mice (Finn and Whistler, 2001) prevent cAMP superactivation and, thus, prevent increased GABA release during naloxone-precipitated withdrawal. Together, these data demonstrate that receptor trafficking is a critical compensatory cellular mechanism that potently modulates synaptic adaptations associated with dependence after prolonged morphine exposure.
Footnotes
This work was supported by funds provided by the state of California for medical research on alcohol and substance abuse through the University of California, San Francisco (J.L.W.) and National Institute on Drug Abuse Grants DA015232 and DA019958 (J.L.W.). We thank F. Woodward Hopf, Billy T. Chen, and Amy Chang for thoughtful comments on this manuscript. We also thank Taban Seif for K-methanesulfonate internal solution, Viktor Kharazia for help with immunohistochemistry, Madeline Ferwerda and Stacy Taylor for maintenance and genotyping of mouse colonies, and Carol Webb for proofreading this manuscript.
References
- Abbadie C, Pasternak GW. Differential in vivo internalization of MOR-1 and MOR-1C by morphine. Neuroreport. 2001;12:3069–3072. doi: 10.1097/00001756-200110080-00017. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, Von Zastrow M, Grandy DK, Williams JT. mu-Opioid receptors: ligand-dependent activation of potassium conductance, desensitization, and internalization. J Neurosci. 2002;22:5769–5776. doi: 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley EE, Gerke MB, Vaughan CW, Hack SP, Christie MJ. GABA transporter currents activated by protein kinase A excite midbrain neurons during opioid withdrawal. Neuron. 2005;45:433–445. doi: 10.1016/j.neuron.2004.12.049. [DOI] [PubMed] [Google Scholar]
- Baumeister AA, Anticich TG, Hebert G, Hawkins MF, Nagy M. Evidence that physical dependence on morphine is mediated by the ventral midbrain. Neuropharmacology. 1989;28:1151–1157. doi: 10.1016/0028-3908(89)90204-9. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz HG, Berton F, Sollini M, Blanchet C, Pravetoni M, Wickman K, Lüscher C. Absence and rescue of morphine withdrawal in GIRK/Kir3 knock-out mice. J Neurosci. 2008;28:4069–4077. doi: 10.1523/JNEUROSCI.0267-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Tallman JF, Nestler EJ. Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Ther. 1988;246:1033–1039. [PubMed] [Google Scholar]
- Duttaroy A, Yoburn BC. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology. 1995;82:1226–1236. doi: 10.1097/00000542-199505000-00018. [DOI] [PubMed] [Google Scholar]
- Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829–839. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grecksch G, Bartzsch K, Widera A, Becker A, Hollt V, Koch T. Development of tolerance and sensitization to different opioid agonists in rats. Psychopharmacology (Berl) 2006;186:177–184. doi: 10.1007/s00213-006-0365-8. [DOI] [PubMed] [Google Scholar]
- He L, Whistler JL. An opiate cocktail that reduces morphine tolerance and dependence. Curr Biol. 2005;15:1028–1033. doi: 10.1016/j.cub.2005.04.052. [DOI] [PubMed] [Google Scholar]
- He L, Fong J, von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108:271–282. doi: 10.1016/s0092-8674(02)00613-x. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992a;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992b;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan GB, Sethi RK, McClelland EG, Leite-Morris KA. Regulation of G protein-mediated adenylyl cyclase in striatum and cortex of opiate-dependent and opiate withdrawing mice. Brain Res. 1998;788:104–110. doi: 10.1016/s0006-8993(97)01524-2. [DOI] [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, Monteillet-Agius G, Stewart PL, Evans CJ, von Zastrow M. mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol. 1998;53:377–384. [PubMed] [Google Scholar]
- Kim JA, Bartlett S, He L, Nielsen CK, Chang AM, Kharazia V, Waldhoer M, Ou CJ, Taylor S, Ferwerda M, Cado D, Whistler JL. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr Biol. 2008;18:129–135. doi: 10.1016/j.cub.2007.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch T, Schulz S, Pfeiffer M, Klutzny M, Schröder H, Kahl E, Höllt V. C-terminal splice variants of the mouse mu-opioid receptor differ in morphine-induced internalization and receptor resensitization. J Biol Chem. 2001;276:31408–31414. doi: 10.1074/jbc.M100305200. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, Gallegos RA, Henriksen SJ, van der Kooy D. Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat Neurosci. 2004;7:160–169. doi: 10.1038/nn1182. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Stinus L, Gold LH, Koob GF. Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Ther. 1992;261:669–677. [PubMed] [Google Scholar]
- Nestler EJ. Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci. 2004;25:210–218. doi: 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. Ed 2. San Diego: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Phillips AG, LePiane FG. Reinforcing effects of morphine microinjection into the ventral tegmental area. Pharmacol Biochem Behav. 1980;12:965–968. doi: 10.1016/0091-3057(80)90460-8. [DOI] [PubMed] [Google Scholar]
- Rasmussen K, Beitner-Johnson DB, Krystal JH, Aghajanian GK, Nestler EJ. Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci. 1990;10:2308–2317. doi: 10.1523/JNEUROSCI.10-07-02308.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulteis G, Markou A, Gold LH, Stinus L, Koob GF. Relative sensitivity to naloxone of multiple indices of opiate withdrawal: a quantitative dose-response analysis. J Pharmacol Exp Ther. 1994;271:1391–1398. [PubMed] [Google Scholar]
- Sheu MJ, Sribanditmongkol P, Santosa DN, Tejwani GA. Inhibition of morphine tolerance and dependence by diazepam and its relation to cyclic AMP levels in discrete rat brain regions and spinal cord. Brain Res. 1995;675:31–37. doi: 10.1016/0006-8993(95)00036-p. [DOI] [PubMed] [Google Scholar]
- Stinus L, Le Moal M, Koob GF. Nucleus accumbens and amygdala are possible substrates for the aversive stimulus effects of opiate withdrawal. Neuroscience. 1990;37:767–773. doi: 10.1016/0306-4522(90)90106-e. [DOI] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Marek K, Basbaum AI. Postsynaptic signaling via the μ-opioid receptor: responses of dorsal horn neurons to exogenous opioids and noxious stimulation. J Neurosci. 2000;20:8578–8584. doi: 10.1523/JNEUROSCI.20-23-08578.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Perez H, Kee RT, Walton CH, Hansen DM, Razavi R, Clarke L, Bufalino MR, Allison DW, Steffensen SC, van der Kooy D. Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science. 2009;324:1732–1734. doi: 10.1126/science.1168501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EA, Young AM. Differential tolerance to antinociceptive effects of mu opioids during repeated treatment with etonitazene, morphine, or buprenorphine in rats. Psychopharmacology (Berl) 2001;154:131–142. doi: 10.1007/s002130000620. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Womack MD, McCleskey EW. Fast, local signal transduction between the mu opioid receptor and Ca2+ channels. J Neurosci. 1995;15:4124–4132. doi: 10.1523/JNEUROSCI.15-05-04124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Wise RA. Opiate reward: sites and substrates. Neurosci Biobehav Rev. 1989;13:129–133. doi: 10.1016/s0149-7634(89)80021-1. [DOI] [PubMed] [Google Scholar]
- Yu Y, Zhang L, Yin X, Sun H, Uhl GR, Wang JB. Mu opioid receptor phosphorylation, desensitization, and ligand efficacy. J Biol Chem. 1997;272:28869–28874. doi: 10.1074/jbc.272.46.28869. [DOI] [PubMed] [Google Scholar]