

Abstract
The biologic basis of autism is complex and is thought to involve multiple and variable gene-environment interactions. While the logical focus has been on the affected child, the impact of maternal genetics on intrauterine microenvironment during pivotal developmental windows could be substantial. Folate-dependent one carbon metabolism is a highly polymorphic pathway that regulates the distribution of one-carbon derivatives between DNA synthesis (proliferation) and DNA methylation (cell-specific gene expression and differentiation). These pathways are essential to support the programmed shifts between proliferation and differentiation during embryogenesis and organogenesis. Maternal genetic variants that compromise intrauterine availability of folate derivatives could alter fetal cell trajectories and disrupt normal neurodevelopment. In this investigation, the frequency of common functional polymorphisms in the folate pathway was investigated in a large population-based sample of autism case-parent triads. In case-control analysis, a significant increase in the reduced folate carrier (RFC1) G allele frequency was found among case mothers, but not among fathers or affected children. Subsequent log linear analysis of the RFC1 A80G genotype within family trios revealed that the maternal G allele was associated with a significant increase in risk of autism whereas the inherited genotype of the child was not. Further, maternal DNA from the autism mothers was found to be significantly hypomethylated relative to reference control DNA. Metabolic profiling indicated that plasma homocysteine, adenosine, and S-adenosylhomocyteine were significantly elevated among autism mothers consistent with reduced methylation capacity and DNA hypomethylation. Together, these results suggest that the maternal genetics/epigenetics may influence fetal predisposition to autism.
Keywords: Autism, reduced folate carrier, maternal, polymorphism, DNA methylation, epigenetics
INTRODUCTION
The genetic basis of most complex disease is thought to involve rare low penetrance genes that interact with common allelic variants to create a genetic predisposition that may vary with the timing and severity of environmental exposures (Buxbaum 2009;Cantor 2009;Altevogt et al. 2008). For complex neurobehavioral disorders such as autism, it is plausible to hypothesize that the low penetrance genes contributing to autism risk may converge to disrupt critical metabolic or functional pathways that are essential for normal neurodevelopment. One pathway with multiple polymorphic variants that has been shown to be essential for normal fetal neurodevelopment is folate-dependent one carbon metabolism (Pei et al. 2005b;Scholl and Johnson 2000;Zeisel 2009b;Lopreato et al. 2008;Kempisty et al. 2007). Mammals are unable to synthesize tetrahydrofolate, the active form of folate, and must rely on dietary sources and de novo synthesis by gut bacteria. Folate is the major one-carbon donor for de novo nucleotide synthesis for DNA replication and also for remethylation of homocysteine to methionine for essential methylation reactions. Error-prone DNA synthesis and DNA hypomethylation have been implicated as potential mechanisms contributing to abnormal fetal development and have been associated with maternal polymorphic variants in the folate pathway (Beaudin and Stover 2009;Scholl and Johnson 2000;Mattson and Shea 2003;Johnson 1999). Maternal genes and environmental exposures can alter placental and intrauterine metabolic environment to modify the phenotype of the developing fetus independent of the fetal genotype (Johnson 2003b;Furness et al. 2008;Lim et al. 2008;Johnson et al. 2008).
It has become evident that the quest to discover the genetic basis of autism is much more of a challenge than originally envisioned. The gene variants uncovered so far have been present in only a small fraction of cases underscoring the probability that multiple small effect genes interact to promote a multi-allelic predisposition to autism (Buxbaum 2009). Supporting this possibility, recent genome-wide association studies have revealed rare copy number variations that could potentially interact to disrupt pathways of neuronal adhesion or ubiquitin degradation during neurodevelopment (Glessner et al. 2009). While the logical focus has been on proband genetics, the impact of maternal genetics on intrauterine environment during critical periods of fetal neurodevelopment could be substantial and has been relatively understudied. The purpose of the present investigation was to evaluate the frequency of common functional polymorphisms in the folate pathway in a large population-based sample of autism case-parent triads and to apply the transmission disequilibrium test (TDT) and the likelihood ratio test (LRT) to evaluate relative genetic risk. The LRT is capable of differentiating whether the relative genetic risk is operating through the case child and/or through the mother (Wilcox et al. 1998).
Five common polymorphisms in genes coding for enzymes that interact to regulate folate-dependent one-carbon metabolism were selected based on previous studies indicating substantial contribution of these allelic variants to fetal anomalies (Pei et al. 2005a;Bosco et al. 2003;Isotalo et al. 2000;Christensen et al. 1999;O'Leary et al. 2006) and also to functional alterations folate metabolism (Gaughan et al. 2001;Dunlevy et al. 2006;Lopreato et al. 2008;Castel-Dunwoody et al. 2005). Over 2100 DNA samples (530 case-parent autism trios and 560 controls) were obtained from NIMH repository and analyzed for MTHFR C677T, MTHFR A1298C, MTRR A66G, TCII C776G, and RFC1 A80G as potential contributors to abnormal maternal one-carbon metabolism. Each of these functional variants has been associated with alterations in metabolite levels in the folate/methionine transmethylation pathway as diagrammed in Figure 1. The methylenetetrahydrofolate reductase (MTHFR) enzyme catalyses the conversion of 5,10-MTHFR to 5-methyl-folate, the circulating form of folate and the methyl donor for remethylation of homocysteine to methionine. The MTHFR 677 T allele reduces enzyme activity in a dose/response manner and is associated with elevated homocysteine levels and increased folate requirement (Bailey and Gregory, III 1999). A second common polymorphism in the MTHFR gene is the 1298 A to C transition similarly reduces MTHFR activity although to a lesser extent.(Friedman et al. 1999) Methionine synthase reductase (MTRR) acts to maintain active reduced cobalamin, an intermediate methyl carrier in the methionine synthase-catalyzed remethylation of homocysteine to methionine. The MTRR 66G allele has been associated with an increased risk of a neural tube defect when maternal B12 intake is low (van der Linden et al. 2006). Vitamin B12 is transported into all cells by transcobalamin II (TCII) and the common polymorphism TCII C776G has been associated with reduced cobalamin levels and elevated homocysteine (Afman et al. 2001). The RFC1 gene encodes the reduced folate carrier protein which does not participate directly in folate metabolism but is the predominant mechanism for intracellular transport of metabolically active 5-methylfolate. Folate transport into RBCs is negatively associated with RFC1 G allele and has been associated with lower folate and inconsistently with elevated homocysteine levels (Dufficy et al. 2006;Gellekink et al. 2007;Yates and Lucock 2005). [Figure1]
Figure 1.
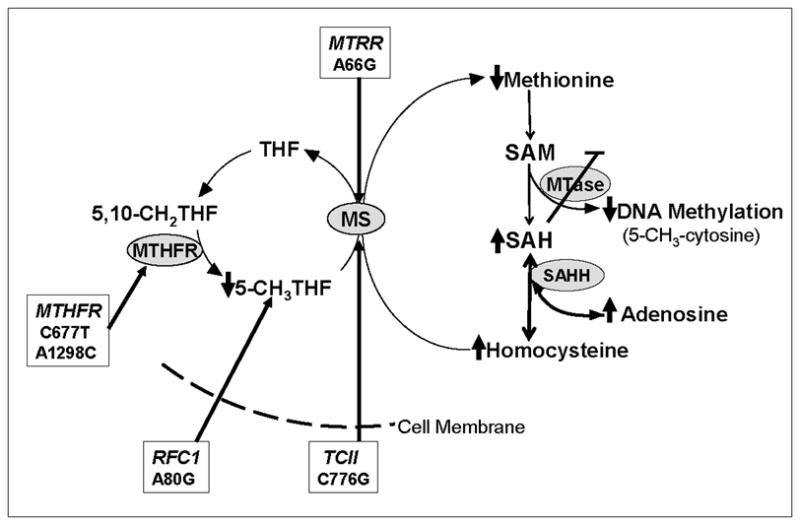
Diagram of folate-dependent transsulfuration pathway indicating interaction of MTHFR C677T, MTHFR A1298C, MTRR A66G and membrane transport function of RFC1 A80G. The up-down arrows indicate the metabolite alterations observed among the autism mothers. The inhibition of DNA methyltransferase by SAH is also diagrammed. Abbreviations: THF: tetrahydrofolate; 5-CH3 THF: 5-methyltetrahyrdofolate; MTHFR: methylene tetrahydrofole reductase; MS; methionine reductase; MTRR: methionine synthase reductase; RFC: reduced folate carrier; SAM: S-adenosylmethionine; SAH: S-adenosylhomocysteine; SAHH: SAH hydrolase; MTase:methyltransferase
A second goal of this investigation was to determine whether an increased frequency of functional polymorphisms in the folate pathway is accompanied by alterations in folate-dependent methionine metabolism and DNA methylation in blood samples obtained from a cohort of locally recruited mothers. We recently reported evidence indicating that folate-dependent transmethylation and transsulfuration metabolism was abnormal in both children with autism (James et al. 2006) and also their mothers (James et al. 2009). The present investigation consisted of a larger independent cohort of Arkansas case mothers and matched control mothers and confirms the presence of abnormal transmethylation metabolism and global DNA hypomethylation among the autism mothers.
SUBJECTS AND METHODS
Participants
For genotype and DNA methylation analysis, DNA samples from 529 case-parent trios and 566 neurotypical controls with no history of behavioral or neurological problems were obtained from the NIMH repository as part of the Autism Human Genetics Initiative. For the metabolic analysis, 57 case families were recruited locally after referral to the UAMS Dennis Developmental Center for diagnosis by developmental pediatricians. For the local metabolic studies, probands were between the ages of 3 and 10 years and were diagnosed with Autistic Disorder using criteria defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV 299.0), the Autism Diagnostic Observation Schedule (ADOS), or the Childhood Autism Rating Scales (CARS >30). Specifically in the Arkansas cohort, 14 children were diagnosed by DSM-IV 299.0 clinical judgment plus CARS >30, 13 children were diagnosed by DSM-IV 299.0 plus ADOS, and 30 children by DMS-IV 299.0 clinical judgment alone. Probands from the NIMH repository were diagnosed with DSM-IV 299.0 plus Autism Diagnostic Interview (ADI). Although there was some variation in the combination of diagnostic tests employed, all children were diagnosed with DSM-IV 299.0 autistic disorder. Families with children diagnosed with Pervasive Developmental Disorder-Not-Otherwise-Specified (PDD-NOS), childhood disintegrative disorder or rare genetic diseases associated with symptoms of autism (e.g., fragile X, Rett syndrome, tuberous sclerosis) were excluded from participation. African American families were also excluded based on established racial variation in frequencies of MTHFR C677T and RFC1 A80G (O'Leary et al. 2006). All participating local case and control families were Arkansas residents. The ethnic distribution among local case mothers was 96% White, 2% Hispanic, and 2% Asian with a mean age of 33 ± 6.4 years (range 22–40). Controls mothers for the metabolic study consisted of 80 mothers with a mean age of 28 years ± 6.6 (range 16–44) who were control participants in an ongoing case-control study of maternal risk factors for congenital heart defects in Arkansas (Hobbs et al. 2005). Among control mothers for the metabolic study, the ethnic distribution was 90% White, 8% Hispanic, and 2% Asian. Over-the-counter multivitamin supplement use at the time of blood draw was not significantly different among case compared to control mothers (42% vs. 45%, respectively, by self report). Folate intake from foods was not determined in this study; however, samples from case and control cohorts were obtained after US folate supplementation. The protocol was approved by the Institutional Review Board at the University of Arkansas for Medical Sciences and all participants signed informed consent.
Genotyping
Genotyping on 529 repository case-parent triads, 566 repository controls and 57 local mothers was performed with allele-specific fluorescent primer-probe sets supplied by ABI Assays by Design (Applied Biosystems, Foster City, CA) and primer and probe sequences are listed in Table I. PCR reactions were carried out with ABI PRISM 7700 and 7900 Sequence Detection Systems (Applied Biosystems, Foster City, CA) under the following thermal cycling conditions: one cycle at 95°C for 10 minutes (Taq activation), followed by 40 cycles of 92°C for 15 seconds (denature) and 60°C for 1 minute (anneal/extend). The reaction components were as follows: 900 nM of each primer, 200 nM each probe, 1X of TaqMan Universal Master Mix (ABI, Foster City, CA) and 250 ng genomic DNA. The PCR products were visualized on a 2.5% agarose gel with Reliant Fastlane Gel System (Cambrex, Rockland, ME). [Table 1]
Table I.
Primers and TaqMan allele-specific probes
| Polymorphism | 5’-3’ sequence |
|---|---|
| RFC-1 80G>A | |
| Forward | GGCCTGACCCCG AGCT |
| Reverse | AGCCGTAGAAGCAAAGGTAGCA |
| G-Probe 1 | VIC-CACGAG GCGCCGC |
| A-Probe 2 | 6FAM-CGAGGT GCCGCCAG |
| MTHFR C677T | |
| Forward | TGGCAGGTTACCCCAAAGG |
| Reverse | CACAAAGCGGAAGAATGTGTCA |
| T- Probe 1 | 6FAM-TGATGAAATCGGCTCCCGCA-TAMRA |
| C- Probe 2 | VIC-TGA TGATGAAATCGACTCCCGCA-TAMRA |
| MTHFR A1298C | |
| Forward | GGAGGAGCTGCTGAAGATGTG |
| Reverse | CCCGAGAGGTAAAGAACAAAGACTT |
| A- Probe 1 | VIC-ACCAGTGAAGAAAGTGT |
| C-Probe 2 | 6FAM-CAGTGAAGCAAGTGT |
| MTRR A66G | |
| Forward | AGCAGGGACAGGCAAAGG |
| Reverse | AAGATCTGCAGAAAATCCATGTACCA |
| A-Probe 1 | VIC-TTGCTCACATATTTCTT |
| G-Probe 2 | 6FAM-CTCACACATTTCTT |
| TCN2 C776G | |
| Forward | ACTCTATCACCAGTTCCTCATGACT |
| Reverse | TTGAGACATGCTGTTCCCAGTT |
| C- Probe 1 | VIC- CTGCCCCAGGCATG |
| G-Probe 2 | 6FAM- CTGCCCCACGCATG |
Allelic discrimination was accomplished when fluorogenic probes with either a 6FAM or VIC reporter dye attached to the 5’ end of the oligonucleotide was cleaved by the 5’ nuclease activity of Taq DNA polymerase.
Determination of percent 5-methylcystosine/total cytosine in DNA
To ~1 μg DNA, RNase A (Sigma, St. Louis, MO) was added to a final concentration of 0.02 mg/mL and incubated at 37°C for 15 minutes. The purified DNA was digested into component nucleotides using Nuclease P1, snake venom phosphodieasterase, and alkaline phosphatase as previously described (Friso et al. 2002a). Briefly, DNA was denatured by heating for 3 minutes at 100° C and rapidly chilled in an ice water bath. One-tenth volume of 0.1 M ammonium acetate, pH 5.3, was added to 2 units of nuclease P1 (Sigma, St. Louis, MO) for every 0.5 A260 unit of DNA and the mixture incubated at 45°C for 2 h. Subsequently, 1/10 volume of 1 M ammonium bicarbonate and 0.002 units of venom phosphodiesterase I (Sigma, St. Louis, MO) were added and the mixture incubated for 2 h at 37° C. To the mixture, 0.5 units of alkaline phosphatase (Sigma, St. Louis, MO) was then added and the incubation continued for an additional hour. The digested nucleotides were stored at −20°C until LC/MS/MS analysis. Base separation was performed with a Dionex HPLC system coupled to an electrospray ionization (ESI) tandem mass spectrometer (Thermo-Finnigan LCQ) using a Phenomenex Gemini column (C18, 150 x 2.0 mm, 3 μm particle size) and established methodology (Friso et al. 2002b). Results are expressed as percent 5-methylcytosine/total cytosine in DNA.
Plasma transmethylation metabolites
Fasting blood samples were collected before 9:00 am into EDTA-Vacutainer tubes and immediately chilled on ice before centrifuging at 4000 x g for 10 minutes at 4°C. Aliquots of plasma were transferred into cryostat tubes and stored at −80°C for approximately 2–4 weeks until extraction and HPLC quantification. The methodological details for HPLC elution and electrochemical detection have been described previously (Melnyk et al. 2000;Melnyk et al. 1999). Although it was not feasible to run all case and control samples at the same time, the storage interval at −80 degrees was consistently between 2 and 4 weeks to minimize potential metabolite interconversion. Potential between-run variation was controlled for by inclusion of internal standards with each run. Plasma total folate and vitamin B12 were measured using SimulTRAC-SNB Radioassay Kit Vitamin B12/Folate kit from MP Biomedical, Inc. (Orangeburg, NY).
Statistical Analysis
Genetic statistical analyses were performed using STATA version 10.1 software (StataCorp LP, College Station, TX). Prior to analysis, genotypic data were checked for Mendelian segregation errors. Families with unresolved transmission errors were not included in further analyses. Odds ratios and 95% confidence intervals for case-control genotypes frequencies under various genetic models were estimated using linear logistic regression. Case-parent triad data was first analyzed using an exact Transmission Disequilibrium Test (TDT) to test for association due to genetic linkage between autism and the offspring’s genetic polymorphisms while eliminating the effects of population stratification (Spielman et al. 1993;Cleves et al. 1997). Additionally, log-linear models (Poisson) as developed by Weinberg et al. and implemented in STATA's GENECMT program (http://www.biostat-resources.com/stata) were used to discriminate the contribution to the overall genetic relative risk due to maternal genetic factors from those due to the child’s genotype (Weinberg et al. 1998). Similar to the transmission disequilibrium test (TDT), this log-linear approach regards the association only within families and consequently, it is robust to confounding by population stratification. This model also allows the estimation of the genetic relative risks associated with the mother’s and the offspring’s genotype independently. To assess the significance of the maternal and offspring genotypes on Autism risk, likelihood-ratio test were computed by comparing a full model that included maternal and offspring genotypes to reduced models that included either the maternal or offspring genotypes only. Metabolic data were prospectively collected and statistical differences between case and control metabolites were determined using the Student’s t test and S-Plus software (Seattle, WA) with significance set at 0.05. Given our a priori hypothesis based on previous results, correction for multiple comparisons was not implemented for metabolite data. Multiple regression analysis with S-Plus was used to evaluate relationships among metabolites, global DNA methylation and RFC1 genotype. The linear least squares trend test was used to evaluate the relationship between RFC1 genotype and DNA methylation.
RESULTS
Case-control frequencies for MTHFR C677T, MTHFR A1298C, MTRR A66G, TCII C776G, and RFC1 A80G
Genotype frequencies were calculated for the five genotypes and confirmed to be in Hardy-Weinberg equilibrium. Control genotype frequencies were within the range of previous population based estimates (2009). There were no significant differences in the allele frequencies of MTHFR C677T, MTHFR A1298C, TCII C776G, or MTRR A66G among mothers, fathers or affected child compared to population controls. In Table II, the RFC1 G allele frequency is shown to be elevated in the mothers (p=0.056), but not in the fathers or in case children (p= 0.79, p=0.25 respectively). In addition, the RFC1 GG homozygous variant was significantly elevated (p=0.036) in mothers and associated with a 46% increased risk of having a child with autism (95% CI:1.01, 2.11). Similarly, the combined RFC1 AG + GG frequency was significantly increased in mothers (p=0.035) and associated with 40% increase in risk (CI: 1.01, 1.95). Among the case children, the heterozygous AG genotype was significantly increased (p=0.047) but homozygous GG genotype was not (p=0.14). The paternal RFC1 genotype frequencies were not different from the control frequencies. [Table II]
Table II.
RFC1 case trio and control frequencies
| Allele Frequency | Genotype | Mothers | Controls | OR ( 95% CI ) | p-value |
|---|---|---|---|---|---|
| Maternal | A | 423 (40.1%) | 499 (44.1%) | Reference | |
| G | 633 (59.9%) | 633 (55.9%) | 1.18 ( 0.99, 1.40) | 0.056 | |
| AA | 79 (15.0%) | 112 (19.8%) | Reference | ||
| AG | 265 (50.2%) | 275 (48.6%) | 1.37 ( 0.97, 1.94) | 0.066 | |
| GG | 184 (34.8%) | 179 (31.6%) | 1.46 ( 1.01, 2.11) | 0.036 | |
| AG + GG | 449 (85.0%) | 454 (80.2%) | 1.40 ( 1.01, 1.95) | 0.035 | |
| Case Child | A | 440 (41.7%) | 499 (44.1%) | Reference | |
| G | 616 (58.3%) | 633 (55.9%) | 1.10 (0.93, 1.31) | 0.254 | |
| AA | 82 (15.5%) | 112 (19.8%) | Reference | ||
| AG | 275 (52.2%) | 275 (48.6%) | 1.40 (0.99, 1.98) | 0.047 | |
| GG | 172 (32.5%) | 179 (31.6%) | 1.31 (0.90, 1.89) | 0.141 | |
| AG + GG | 447 (84.7%) | 454 (80.2%) | 1.36 (0.98, 1.89) | 0.054 | |
| Paternal | A | 465 (44.6%) | 499 (44.1%) | Reference | |
| G | 577 (55.4%) | 633 (55.9%) | 0.98 (0.82, 1.16) | 0.798 | |
| AA | 117 (22.4%) | 112 (19.8%) | Reference | ||
| AG | 235 (45.0%) | 275 (48.6%) | 0.85 (0.61, 1.17) | 0.298 | |
| GG | 170 (32.6%) | 179 (31.6%) | 0.93 (0.66, 1.32) | 0.685 | |
| AG + GG | 407 (78.1%) | 454 (80.2%) | 0.88 ( 0.65, 1.19) | 0.395 |
Transmission disequilibrium test (TDT)
The TDT detects the over-transmission of an allele from heterozygous parents to affected offspring under the assumption that parental alleles will sort randomly and equally during gametogenesis. The TDT tests for linkage between alleles and phenotypes that may be causal or due to linkage disequilibrium. The transmission of the RFC1 G allele within our case-parent triads is presented in Table III. There was no preferential transmission of the G allele from fathers or mothers to affected child. [Table III]
Table III.
Transmission disequilibrium test. Maternal and paternal transmission of the G allele to affected child.
| Overall | Maternal transmission | Paternal transmission | ||||
|---|---|---|---|---|---|---|
| Transmitted | Not Transmitted | Transmitted | Not Transmitted | Transmitted | Not Transmitted | |
| 255 | 241 | 91 | 108 | 86 | 89 | |
| p-value | 0.5595 | 0.2567 | 0.8799 | |||
Genetic Relative Risk and Likelihood Ratio Test
The Genetic Relative Risk (GRR) and Likelihood Ratio Test (LRT) are based on a log-linear model and used to analyze the relationship between mother and child RFC1 80 A>G polymorphism and the risk of autism. As shown in Table IV, the Genetic Relative Risk (GRR) of the offspring inheriting one or two G alleles from the mother was not statistically significant. In contrast, if the mother carried one or two G alleles, the genetic relative risk of autism was increased 60% and 52% respectively (p=0.007 and 0.02, respectively). The LRT incorporates all potential allelic combinations and is capable of differentiating whether the relative risk is operating through the case child and/or through the mother. The maternal likelihood ratio was 7.76 with a p-value of 0.02 whereas the offspring likelihood ratio was 2.71 and did not reach statistical significance (p=0.25). The log linear analysis suggests that the RFC1 genotype of the mother is significantly associated with the genetic risk of having a child with autism independent of the child’s genotype. The inherited genotype of the child did not contribute significantly to genetic risk in this analysis. [Table IV]
Table IV.
Maximum likelihood ratio test based on log linear model
| Genetic Relative Risk | p-value | 95% Confidence Interval | |
|---|---|---|---|
| R1 (offspring inheriting 1 G allele) | 1.23 | 0.12 | 0.942, 1.73 |
| R2 (offspring inheriting 2 G alleles) | 1.18 | 0.38 | 0.819, 1.70 |
| S1 (mother carrying 1 G allele) | 1.60 | 0.007 | 1.134, 2.26 |
| S2 (mother carrying 2 G alleles | 1.52 | 0.02 | 1.069, 2.15 |
| Offspring Likelihood Ratio | 2.71 | 0.25 | |
| Maternal Likelihood Ratio | 7.76 | 0.02 |
Maternal plasma transmethylation metabolites and plasma folate concentrations
In Table V, plasma metabolites in the folate/methionine transmethylation pathway in case and control mothers from Arkansas matched for age, race, and vitamin intake is presented. Plasma levels of 5-methylfolate and methionine were significantly lower in case mothers compared to control mothers. Both metabolites are essential precursors that provide methyl groups for cellular methylation reactions including DNA methylation (see Figure 1). In contrast, the inhibitory products of cellular methylation reactions, S-adenosylmethionine, adenosine and homocysteine, were significantly elevated among case mothers. The relationships among these plasma metabolites are presented in Figure 2A-D provide insights into metabolic abnormalities that may promote global DNA hypomethylation. The inverse correlation between folate and homocysteine in Figure 2A suggests that low plasma folate may contribute to the elevated homocysteine in autism mothers. Figures 2B and 2C are consistent with previous reports indicating that elevated SAH is correlated with elevated homocysteine and adenosine due to reversal of the SAH hydrolase reaction. The strong inverse relationship between SAM/SAH and SAH shown in Figure 2D indicates that the low SAM/SAH ratio is strongly driven by the increase in SAH. Because SAM is the major methyl donor for the DNA methyltransferase reaction and SAH is the product inhibitor, the SAM/SAH ratio is thought to be the best indicator of DNA methylation potential (McKeever et al. 1995). Taken together, these results suggest that a chronic increase in SAH levels may contribute to a depletion of DNA methyl groups and global DNA hypomethylation in autism mothers. [Table V] [Figure 2]
Table V.
Transmethylation metabolites in Arkansas case and control mothers
| Control Mothers (mean ± SD; n=80) | Case Mothers (mean ± SD; n=57) | p-value | |
|---|---|---|---|
| Methionine | 23.9 ± 4.5 | 20.9 ± 3.8 | 0.001 |
| SAM | 70.0 ± 12.6 | 68.7 ± 21.4 | NS |
| SAH | 23.9 ± 6.6 | 32.8 ± 9.9 | 0.001 |
| SAM/SAH | 3.2 ± 1.0 | 2.3 ± 1.2 | 0.01 |
| Adenosine | 0.27 ± 0.14 | 0.34 ± 0.17 | 0.01 |
| Homocysteine | 7.24 ± 1.3 | 9.11 ± 2.1 | 0.001 |
| Methionine/Homocysteine ratio | 3.4 ± 0.80 | 2.4 ± 0.58 | 0.001 |
| Folate | 17.4 ± 6.2 | 15.4 ± 3.9 | 0.05 |
| B12 | 509 ± 294 | 449 ± 196 | NS |
NS: Not significant
Figure 2.
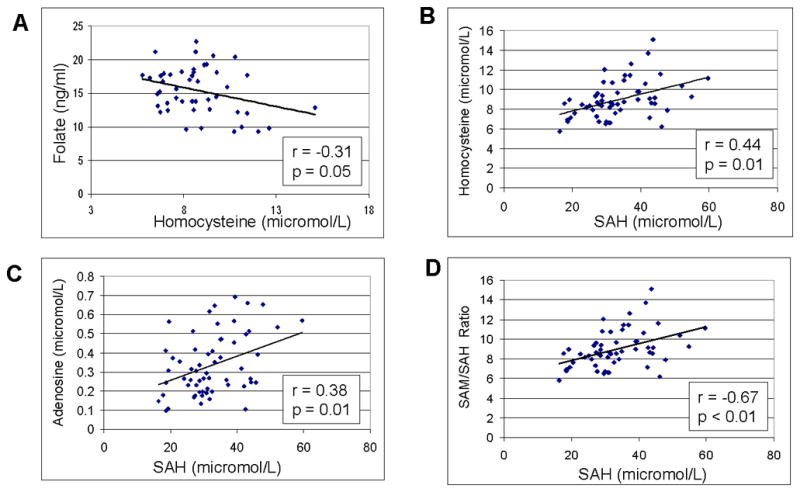
Scatterplots showing the correlations between metabolites among the autism mothers. 2A: Inverse correlation between plasma homocysteine and folate concentrations; 2B: Positive correlation between SAH and homocsyteine; 2C: Positive correlation between SAH and adenosine; 2D: Negative correlation between SAH and SAM/SAH ratio.
Global DNA methylation density and RFC1 genotype association among Arkansas mothers
As presented in Table VI, the percent 5-methylcytosine/total cytosine in lymphocyte DNA from the local autism mothers is compared to 5-methylcytosine content from the population-based NIMH repository DNA. DNA from the autism mothers was found to be significantly hypomethylated compared to control DNA (p< 0.001). In Table VII, the percent 5-methylcytosine in DNA from the autism mothers is stratified by RFC1 genotype. An inverse relationship was found between percent 5-methylcytosine and RFC1 G allele heterozygosity and homozygosity that was statistically significant using the linear least squares trend test (p=0.04). DNA methylation status among controls did not vary by genotype and was consistently higher than the autism cohort regardless of genotype. Figure 3A–C shows the relationship between SAM/SAH, DNA methylation by RFC1 genotype. There was a marginally significant correlation between SAM/SAH and DNA methylation among mothers with the RFC1 GG genotype (p = 0.08) but not among mothers with the RFC1 AA or AG genotype. Using multiple regression analysis, the relationship between the three variables is best explained by the formula: DNA Methylation = 2.5 + 0.167 SAM/SAH + 0.079 Homocysteine (μmol/L) − 0.225 (genotype) where the AA, AG, and GG genotypes are given a value of 0, 1, and 2 respectively.
Table VI.
Comparison of DNA methylation status between population-based NIMH respository controls and local Arkansas autism mothers
| Repository Controls (n = 60) | Arkansas Autism Mothers (n = 57) | p-value | |
|---|---|---|---|
| % 5methylcytosine (mean ± SD) | 4.74 ± 1.04 | 3.28 ± 0.75 | < 0.001 |
Table VII.
Comparison of percent 5-methylcytosine in DNA by RFC1 genotype among Arkansas mothers and population-based NIMH repository controls
| % 5-methylcytosine (mean ± SD) | RFC1 AA | RFC1 AG | RFC1 GG | p-value |
|---|---|---|---|---|
| Arkansas cohort | 3.51 ± 0.70 (n = 15) | 3.32 ± 0.81 (n = 26) | 2.97 ± 0.79 (n = 16) | 0.04 |
| Respository cohort | 4.61 ± 1.22 ( n=40) | 5.01 ± 0.71 (n=10) | 4.87 ± 0.45 (n=10) | NS |
NS: Not Significant
Figure 3.
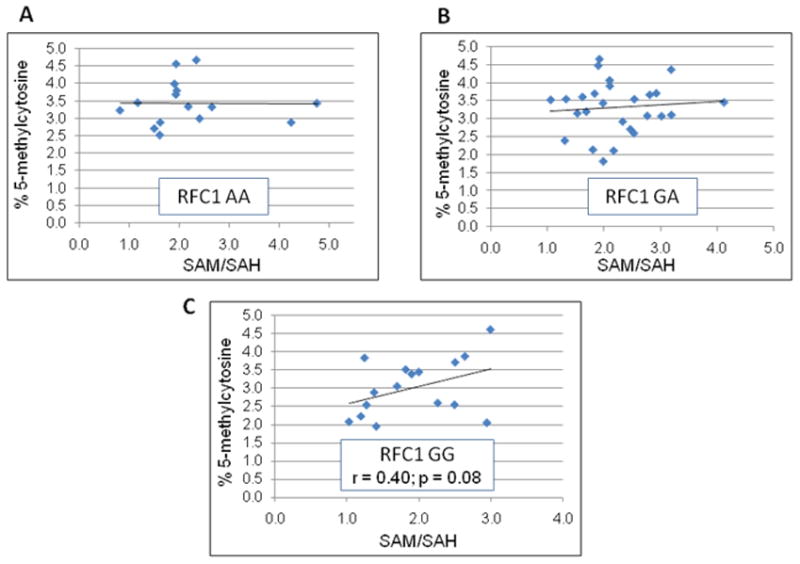
Scatterplots showing the relationship between the SAM/SAH ratio and DNA methylation by RFC1 genotype. 3A: RFC1 wildtype AA; 3B: RFC1 heterozygous AG; 3C: RFC1 homozygous variant GG.
DISCUSSION
The emphasis of most genetic studies of autism has logically focused on the affected child. However, if the genetic alteration negatively affects precursors for DNA synthesis and/or DNA methylation in high demand during early fetal development, maternal genetic variants that compromise intrauterine availability could alter programmed cell trajectories and normal neurodevelopment. Folate-dependent one-carbon metabolism is a highly polymorphic metabolic pathway that regulates the distribution of one-carbon derivatives between DNA synthesis (proliferation), and DNA methylation (cell-specific gene expression and differentiation). As such, normal functioning of this pathway is essential to support the rapid fire shifts between proliferation, differentiation and cell death that are essential for normal fetal programming and organogenesis. Maternal polymorphic variants, nutritional deficiencies and/or environmental exposures that negatively affect availability of one-carbon precursors have been associated with increased risk of structural birth defects, chromosomal anomalies, schizophrenia, and prematurity (Doolin et al. 2002;van der Linden et al. 2006;James et al. 1999;Johnson 1999;Bukowski et al. 2009;Scholl and Johnson 2000;Picker and Coyle 2005). The genetics of the mother acting as a fetal environmental factor contributing to autism risk has been proposed but has been relatively understudied (Johnson 2003a;Johnson et al. 2008).
In the present report, log linear modeling of the RFC1 G allele frequencies within a large population based sample of case-parent triads suggests that the maternal G allele may be a contributing genetic risk factor for having a child with autism independent of the child’s genotype. Additional case-control analysis indicated that the G allele frequency was significantly elevated in the autism mothers compared to ~500 controls. In contrast, the G allele frequency was not different from controls in either the father or the affected child. There was a marginal increase in RFC1 heterozygosity among autistic children that most likely reflects the higher G allele prevalence in the mothers. Nonetheless, the results of the transmission disequilibrium test indicated no preferential transmission of the G allele from mother to offspring. Taken together, the case-control G allele frequency in mother > child > father = controls combined with the negative TDT results lend support to the log linear analysis suggesting that the mother’s RFC1 genotype was the major determinant of genetic risk in this sample. Similar results supporting a role for HLA-DR4 and GST-P1 as maternal risk factors for autism has been recently reported (Johnson et al. 2009;Williams et al. 2007).
The abnormal metabolic profile of folate-dependent transmethylation (Figure 1) observed among the autism mothers replicates our previous data in a smaller independent cohort of case and control mothers (James et al. 2009). In both cohorts, significant elevations in plasma homocysteine and adenosine were associated with significant elevation in SAH concentrations. These results are consistent with the thermodynamics of the SAH hydrolase (SAHH) reaction in which increased concentrations of homocysteine and adenosine combine to reverse SAHH kinetics and increase SAH concentrations. SAH is a potent inhibitor of most cellular methyltransferase reactions including DNA methyltransferase 1 (DNMT1) and has been associated with global DNA hypomethylation in animal and human studies (James et al. 2002;Caudill et al. 2001;Yi et al. 2000). Thus, it is plausible to hypothesize that elevated SAH secondary to elevated homocysteine and adenosine may be contributing factors to the global DNA hypomethylation observed in the autism mothers. Interestingly, a similar decrease in plasma SAM/SAH ratio has been observed in children with autism, however the kinetics are somewhat different. In the mothers, the low methylation ratio is largely driven by the increase in SAH whereas in the children the low ratio is predominantly due to low SAM. The evaluation of DNA methylation status in autistic children is currently underway.
RFC1 AG and GG genotypes have been associated with reduced RBC folate uptake and low plasma folate levels (Chango et al. 2000b;Dufficy et al. 2006) and inconsistently associated with elevated plasma homcysteine (Chango et al. 2000a;Gellekink et al. 2007). Despite the small sample size, a marginally significant correlation was found between RFC1 AG+GG genotypes and plasma homocysteine concentration (p=0.08) among the local autism mothers (data not shown). Maternal homocysteine was negatively correlated with plasma folate (p=0.05) and both were significantly different from control mothers despite similar vitamin supplement intake and folate fortification. A difference in dietary folate intake from food cannot be ruled out as a confounding factor to these observations since folate intake from food sources was not measured in these mothers and is well known to affect plasma homocysteine and folate levels. Regardless of origin, elevated homocysteine and SAH levels have been associated with multiple adverse health effects including cardiovascular disease, autoimmune disease, birth defects, schizophrenia and prematurity (De Bree et al. 2002;Lazzerini et al. 2007;Hobbs et al. 2005;Mattson and Shea 2003;Furness et al. 2008;Susser et al. 1998;Kramer et al. 2009). Multiple studies have demonstrated that elevated homocysteine and SAH can be normalized by B vitamin supplementation which could be a clinical consideration in high risk mothers with elevated homocysteine levels (Brattström et al. 1998;Mansoor et al. 1999).
In the case-control evaluation, DNA from autism mothers was found to be significantly hypomethylated relative to the population-based repository controls. The repository DNA was used as the comparison group for this analysis because DNA from our local cohort of control mothers was not available. Among the local autism mothers, the G allele load was positively correlated with lymphocyte DNA hypomethylation (Table VII and Figure 3). In both animal and human studies, DNA hypomethylation has been negatively correlated with folate status and positively correlated with homocysteine and SAH (van der Linden et al. 2008;Chen et al. 2001;Niculescu and Zeisel 2002;Jacob et al. 1998;Yi et al. 2000). Maternal folate insufficiency can stem from genetic aberrations, nutritional deficiencies, and/or environmental exposures that increase folate requirement for DNA synthesis and DNA methylation during fetal development. Abnormal maternal one-carbon metabolism has been implicated in the predisposition to spina bifida, Down syndrome, fetal alcohol syndrome, and prematurity as based on post-partum plasma samples (Doolin et al. 2002;James et al. 1999;Scholl and Johnson 2000). Further, neurodevelopmental regression has been reported in breast-fed infants of vegan mothers secondary to B12 deficiency, elevated homocysteine and low SAM/SAH ratio (Dror and Allen 2008). Also of related interest, maternal folate deficiency and hyperhomocysteinemia during the second and third trimester have been reported to be significant risk factors for schizophrenia (Bleich et al. 2007;Picker and Coyle 2005).
It is important to note that because maternal blood samples were taken 3–10 years after pregnancy, it is not possible to know whether the observed maternal metabolic imbalance was present during gestation and fetal development. This is a common dilemma in studies that attempt to define maternal risk factors for adverse birth outcomes. However, it is generally accepted that adult homocysteine levels and dietary patterns during the reproductive years tend to be relatively stable (Cuco et al. 2006). For example, maternal homocysteine levels measured after pregnancy were found not to differ from pre-pregnancy values (Walker et al. 1999). Whether the maternal metabolic abnormalities are genetically-based, the result of dietary deficiencies, chronic stress, and/or pro-oxidant environmental exposures cannot be determined from the present data. Nonetheless, the finding of genome-wide DNA hypomethylation in a significant subset of autism parents with elevated SAH provides experimental data to support earlier speculation that epigenetic mechanisms may be associated with autism. Elevation in maternal homocysteine and SAH during pregnancy could theoretically alter fetal methylation patterns and induce inappropriate gene expression during development that could affect predisposition to autism.
Converging lines of evidence in human and animal models suggests that epigenetic alterations may contribute to the predisposition to autism (Schanen 2006;Jiang et al. 2004;Samaco et al. 2005;Nagarajan et al. 2006;Allan et al. 2008;Badcock and Crespi 2006;Lee et al. 2006). Epigenetic modulation of gene expression lies at the interface between genes and environment and potentially could provide a molecular explanation for down-regulation of gene expression in the autistic brain. Fetal DNA methylation patterns are established very early during embryogenesis and provide the basis for tissue-specific gene expression, allele-specific imprinting, X chromosome inactivation, and chromosome stability (Reik and Dean 2001). DNA methylation is a heritable epigenetic mechanism that regulates appropriate gene expression and silencing during fetal development (Reik 2007). Aberrant epigenetic programming during critical periods of fetal development can result in aberrant timing of gene expression and cell differentiation that can heritably alter fetal phenotype (Zeisel 2009a). Multiple lines of evidence have established proof of principle that in utero microenvironment and fetal development are modulated by maternal diet, genotype, and environmental exposures (Burdge 2006;Pickard et al. 2001;Prescott and Clifton 2009). For example, maternal exposure to valproic acid in rats depletes hepatic methionine, elevates homocysteine and induces fetal DNA hypomethylation in the offspring that is preventable by folic acid pre-treatment (Alonso-Aperte et al. 1999;Hishida and Nau 1998). In mice, maternal deficiency in the methyl donor choline decreases global and gene-specific DNA and histone methylation and alters gene expression in fetal mouse cortex and hippocampus (Davison et al. 2009;Niculescu et al. 2006). In utero exposure to bisphenol A alters site-specific DNA methylation and offspring phenotype in agouti mice that is counteracted by maternal methyl donor supplementation (Dolinoy et al. 2007). Taken together, these studies suggest that diet and maternal environmental exposures can alter fetal epigenome and phenotype that may be preventable with appropriate intervention.
In summary, the observations communicated in the present report suggest a broader paradigm of autism gene-environment interaction that encompasses the mother as a genetic/epigenetic case as well as a fetal environmental factor. Inclusion of maternal genetic/epigenetics in the autism gene-environment paradigm could provide new insights into the etiology of this complex disorder.
Acknowledgments
The authors would like to express their gratitude to the families affected by autism whose participation made this study possible. We also acknowledge the invaluable help of the developmental physicians and clinicians at the Dennis Developmental Center for patient referral and evaluation. This research was supported, in part, with funding from the National Institute of Child Health and Development (RO1 HD051873) to SJJ, and by grants from the University of Arkansas for Medical Sciences Children’s University Medical Group and the Arkansas Biosciences Institute (SJJ).
Reference List
- NCBI database. 2009 http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=1051266.
- Afman LA, Van der Put NMJ, Thomas CMG, Trijbels JMF, Blom HJ. Reduced vitamin B12 binding by transcobalamin II increases the risk of neural tube defects. Q J Med. 2001;94:159–166. doi: 10.1093/qjmed/94.3.159. [DOI] [PubMed] [Google Scholar]
- Allan AM, Liang X, Luo Y, Pak C, Li X, Szulwach KE, Chen D, Jin P, Zhao X. The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum Mol Genet. 2008;17:2047–2057. doi: 10.1093/hmg/ddn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Aperte E, Ubeda N, Achon M, Perez-Miguelsanz J, Varela-Moreiras G. Impaired methionine synthesis and hypomethylation in rats exposed to valproate during gestation. Neurology. 1999;52:750–756. doi: 10.1212/wnl.52.4.750. [DOI] [PubMed] [Google Scholar]
- Altevogt BM, Hanson SL, Leshner AI. Autism and the environment: challenges and opportunities for research. Pediatr. 2008;121:1225–1229. doi: 10.1542/peds.2007-3000. [DOI] [PubMed] [Google Scholar]
- Badcock C, Crespi B. Imbalanced genomic imprinting in brain development: an evolutionary basis for the aetiology of autism. J Evol Biol. 2006;19:1007–1032. doi: 10.1111/j.1420-9101.2006.01091.x. [DOI] [PubMed] [Google Scholar]
- Bailey LB, Gregory JF., III Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: Metabolic significance, risks and impact on folate requirement. Journal of Nutrition. 1999;129:919–922. doi: 10.1093/jn/129.5.919. [DOI] [PubMed] [Google Scholar]
- Beaudin AE, Stover PJ. Insights into metabolic mechanisms underlying folate-responsive neural tube defects: a minireview. Birth Defects Res A Clin Mol Teratol. 2009;85:274–284. doi: 10.1002/bdra.20553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleich S, Frieling H, Hillemacher T. Elevated prenatal homocysteine levels and the risk of schizophrenia. Arch Gen Psychiatry. 2007;64:980–981. doi: 10.1001/archpsyc.64.8.980. [DOI] [PubMed] [Google Scholar]
- Bosco P, Guéant-Rodriguez RM, Anello G, Barone C, Namour F, Caraci F, Romano A, Romano C, Guéant JL. Methionine synthase (MTR) 2756 (A-->G) polymorphism, double heterozygosity methionine synthase 2756 AG/Methionine synthase reductase (MTRR) 66 AG, and elevated homocysteinemia are three risk factors for having a child with down syndrome. American Journal of Medical Genetics. 2003;121A:219–224. doi: 10.1002/ajmg.a.20234. [DOI] [PubMed] [Google Scholar]
- Brattström L, Landgren F, Israelsson B, Lindgren A, Hultberg B, Andersson A, Cuskelly G, McNulty H, Strain SS, McPartlin J, Weir DG, scott JM, Den Heijer M, Brouwer IA, Blom HJ, Bos GM, Spaans A, Rosendaal FR, Thomas CM, Haak HL, Wijermans PW, Gerrits WB, Naurath HJ, Joosten E. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. Br Med J. 1998;316:894–898. [Google Scholar]
- Bukowski R, Malone FD, Porter FT, Nyberg DA, Comstock CH, Hankins GD, Eddleman K, Gross SJ, Dugoff L, Craigo SD, Timor-Tritsch IE, Carr SR, Wolfe HM, D'Alton ME. Preconceptional folate supplementation and the risk of spontaneous preterm birth: a cohort study. PLoS Med. 2009;6:e1000061. doi: 10.1371/journal.pmed.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdge GC. Homocysteine: a role in fetal programming? Br J Nutr. 2006;96:415–417. [PubMed] [Google Scholar]
- Buxbaum JD. Multiple rare variants in the etiology of autism spectrum disorders. Dialogues Clin Neurosci. 2009;11:35–43. doi: 10.31887/DCNS.2009.11.1/jdbuxbaum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor RM. Molecular genetics of autism. Curr Psychiatry Rep. 2009;11:137–142. doi: 10.1007/s11920-009-0021-1. [DOI] [PubMed] [Google Scholar]
- Castel-Dunwoody KM, Kauwell GP, Shelnutt KP, Vaughn JD, Griffin ER, Maneval DR, Theriaque DW, Bailey LB. Transcobalamin 776C->G polymorphism negatively affects vitamin B-12 metabolism. Am J Clin Nutr. 2005;81:1436–1441. doi: 10.1093/ajcn/81.6.1436. [DOI] [PubMed] [Google Scholar]
- Caudill MA, Wang JC, Melnyk S, Pogribny IP, Jernigan S, Collins MD, Santos-Guzman J, Swendseid ME, Cogger EA, James SJ. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine β-synthase heterozygous mice. Journal of Nutrition. 2001;131:2811–2818. doi: 10.1093/jn/131.11.2811. [DOI] [PubMed] [Google Scholar]
- Chango A, Boisson F, Barbé F, Quilliot D, Droesch S, Pfister M, Fillon-Emery N, Lambert D, Frémont S, Rosenblatt DS, Nicolas JP. The effect of 677C --> T and 1298A --> C mutations on plasma homocysteine and 5,10-methylenetetrahydrofolate reductase activity in healthy subjects. Br J Nutr. 2000a;83:593–596. doi: 10.1017/s0007114500000751. [DOI] [PubMed] [Google Scholar]
- Chango A, Emery-Fillon N, De Courcy GP, Lambert D, Pfister M, Rosenblatt DS, Nicolas JP. A polymorphism (80G->A) in the reduced folate carrier gene and its associations with folate status and homocysteinemia. Mol Genet Metab. 2000b;70:310–315. doi: 10.1006/mgme.2000.3034. [DOI] [PubMed] [Google Scholar]
- Chen ZT, Karaplis AC, Ackerman SL, Pogribny IP, Melnyk S, Lussier-Cacan S, Chen MF, Pai A, John SWM, Smith RS, Bottiglieri T, Bagley P, Selhub J, Rudnicki MA, James SJ, Rozen R. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Human Molecular Genetics. 2001;10:433–443. doi: 10.1093/hmg/10.5.433. [DOI] [PubMed] [Google Scholar]
- Christensen B, Arbour L, Tran P, Leclerc D, Sabbaghian N, Platt R, Gilfix BM, Rosenblatt DS, Gravel RA, Forbes P, Rozen R. Genetic polymorphisms in methylenetetrahydrofolate reductase and methionine synthase, folate levels in red blood cells, and risk of neural tube defects. Am J Med Genet. 1999;84:151–157. doi: 10.1002/(sici)1096-8628(19990521)84:2<151::aid-ajmg12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Cleves MA, Olson JM, Jacobs KB. Exact transmission-disequilibrium tests with multiallelic markers. Genet Epidemiol. 1997;14:337–347. doi: 10.1002/(SICI)1098-2272(1997)14:4<337::AID-GEPI1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Cuco G, Fernandez-Ballart J, Sala J, Viladrich C, Iranzo R, Vila J, Arija V. Dietary patterns and associated lifestyles in preconception, pregnancy and postpartum. Eur J Clin Nutr. 2006;60:364–371. doi: 10.1038/sj.ejcn.1602324. [DOI] [PubMed] [Google Scholar]
- Davison JM, Mellott TJ, Kovacheva VP, Blusztajn JK. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J Biol Chem. 2009;284:1982–1989. doi: 10.1074/jbc.M807651200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bree A, Verschuren WM, Kromhout D, Kluijtmans LA, Blom HJ. Homocysteine determinants and the evidence to what extent homocysteine determines the risk of coronary heart disease. Pharmacol Rev. 2002;54:599–618. doi: 10.1124/pr.54.4.599. [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolin MT, Barbaux S, McDonnell M, Hoess K, Whitehead AS, Mitchell LE. Maternal genetic effects, exerted by genes involved in homocysteine remethylation, influence the risk of spina bifida. Am J Hum Genet. 2002;71:1222–1226. doi: 10.1086/344209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror DK, Allen LH. Effect of vitamin B12 deficiency on neurodevelopment in infants: current knowledge and possible mechanisms. Nutr Rev. 2008;66:250–255. doi: 10.1111/j.1753-4887.2008.00031.x. [DOI] [PubMed] [Google Scholar]
- Dufficy L, Naumovski N, Ng X, Blades B, Yates Z, Travers C, Lewis P, Sturm J, Veysey M, Roach PD, Lucock MD. G80A reduced folate carrier SNP influences the absorption and cellular translocation of dietary folate and its association with blood pressure in an elderly population. Life Sci. 2006;79:957–966. doi: 10.1016/j.lfs.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Dunlevy LP, Burren KA, Mills K, Chitty LS, Copp AJ, Greene ND. Integrity of the methylation cycle is essential for mammalian neural tube closure. Birth Defects Res A Clin Mol Teratol. 2006;76:544–552. doi: 10.1002/bdra.20286. [DOI] [PubMed] [Google Scholar]
- Friedman G, Goldschmidt N, Friedlander Y, Ben Yehuda A, Selhub J, Babaey S, Mendel M, Kidron M, Bar-On H. A common mutation A1298C in human methylenetetrahydrofolate reductase gene: Association with plasma total homocysteine and folate concentrations. Journal of Nutrition. 1999;129:1656–1661. doi: 10.1093/jn/129.9.1656. [DOI] [PubMed] [Google Scholar]
- Friso S, Choi SW, Dolnikowski GG, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem. 2002a;74:4526–4531. doi: 10.1021/ac020050h. [DOI] [PubMed] [Google Scholar]
- Friso S, Choi SW, Dolnikowski GG, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem. 2002b;74:4526–4531. doi: 10.1021/ac020050h. [DOI] [PubMed] [Google Scholar]
- Furness DL, Fenech MF, Khong YT, Romero R, Dekker GA. One-carbon metabolism enzyme polymorphisms and uteroplacental insufficiency. Am J Obstet Gynecol. 2008;199:276–278. doi: 10.1016/j.ajog.2008.06.020. [DOI] [PubMed] [Google Scholar]
- Gaughan DJ, Kluijtmans LAJ, Barbaux S, McMaster D, Young IS, Yarnell JWG, Evans A, Whitehead AS. The methionine synthase reductase (MTRR) A66G polymorphism is a novel genetic determinant of plasma homocysteine concentrations. Atherosclerosis. 2001;157:451–456. doi: 10.1016/s0021-9150(00)00739-5. [DOI] [PubMed] [Google Scholar]
- Gellekink H, Blom HJ, Den Heijer M. Associations of common polymorphisms in the thymidylate synthase, reduced folate carrier and 5-aminoimidazole-4-carboxamide ribonucleotide transformylase/inosine monophosphate cyclohydrolase genes with folate and homocysteine levels and venous thrombosis risk. Clin Chem Lab Med. 2007;45:471–476. doi: 10.1515/CCLM.2007.091. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T, Game RM, Rudd DS, Zurawiecki D, McDougle CJ, Davis LK, Miller J, Posey DJ, Michaels S, Kolevzon A, Silverman JM, Bernier R, Levy SE, Schultz RT, Dawson G, Owley T, McMahon WM, Wassink TH, Sweeney JA, Nurnberger JI, Coon H, Sutcliffe JS, Minshew NJ, Grant SF, Bucan M, Cook EH, Buxbaum JD, Devlin B, Schellenberg GD, Hakonarson H. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishida R, Nau H. VPA-induced neural tube defects in mice. I. Altered metabolism of sulfur amino acids and glutathione. Teratog Carcinog Mutagen. 1998;18:49–61. [PubMed] [Google Scholar]
- Hobbs CA, Cleves MA, Melnyk S, Zhao W, James SJ. Congenital heart defects and abnormal maternal biomarkers of methionine and homocysteine metabolism. Am J Clin Nutr. 2005;81:147–153. doi: 10.1093/ajcn/81.1.147. [DOI] [PubMed] [Google Scholar]
- Isotalo PA, Wells GA, Donnelly JG. Neonatal and fetal methylenetetrahydrofolate reductase genetic polymorphisms: an examination of C677T and A1298C mutations. Am J Hum Genet. 2000;67:986–990. doi: 10.1086/303082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob RA, Gretz DM, Taylor PC, James SJ, Pogribny IP, Miller BJ, Henning SM, Swendseid ME. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. Journal of Nutrition. 1998;128:1204–1212. doi: 10.1093/jn/128.7.1204. [DOI] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, Wong DH, Cutler P, Bock K, Boris M, Bradstreet JJ, Baker SM, Gaylor DW. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet. 2006;141:947–956. doi: 10.1002/ajmg.b.30366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Jernigan S, hubanks A, Rose S, Gaylor DW. Abnormal transmethylation/transsulfuration metaboism and DNA hypomethylation among parents of children with autism. Journal of Autism and Developmental Disorders. 2009;38:1976–1980. doi: 10.1007/s10803-008-0614-2. [DOI] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA. Elevation in S-adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. Journal of Nutrition. 2002;132:2361S–2366S. doi: 10.1093/jn/132.8.2361S. [DOI] [PubMed] [Google Scholar]
- James SJ, Pogribna M, Pogribny IP, Melnyk S, Hine RJ, Gibson JB, Yi P, Tafoya DL, Swenson DH, Wilson VL, Gaylor DW. Abnormal folate metabolism and mutation in the methylenetetrahyrofolate reductase (MTHFR) gene may be maternal risk factors for Down Syndrome. Am J Clin Nutr. 1999;70:495–501. doi: 10.1093/ajcn/70.4.495. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, Liu Q, Shaffer LG, Schroer RJ, Stockton DW, Spielman RS, Stevenson RE, Beaudet AL. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am J Med Genet. 2004;131A:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- Johnson WG. DNA polymorphism-diet-cofactor-development hypothesis and the gene-teratogen model for schizophrenia and other developmental disorders. Am J Med Genet. 1999;88:311–323. [PubMed] [Google Scholar]
- Johnson WG. Teratogenic alleles and neurodevelopmental disorders. BioEssays. 2003a;25:464–477. doi: 10.1002/bies.10268. [DOI] [PubMed] [Google Scholar]
- Johnson WG. Teratogenic alleles and neurodevelopmental disorders. BioEssays. 2003b;25:464–477. doi: 10.1002/bies.10268. [DOI] [PubMed] [Google Scholar]
- Johnson WG, Buyske S, Mars AE, Sreenath M, Stenroos ES, Williams TA, Stein R, Lambert GH. HLA-DR4 as a risk allele for autism acting in mothers of probands possibly during pregnancy. Arch Pediatr Adolesc Med. 2009;163:542–546. doi: 10.1001/archpediatrics.2009.74. [DOI] [PubMed] [Google Scholar]
- Johnson WG, Sreenath M, Buyske S, Stenroos ES. Teratogenic alleles in autsim and other neurodevelopmental disorders. In: Zimmerman AW, editor. Autism: Current Theories and Evidence. Toweta, NJ: Humana Press/Springer; 2008. pp. 41–48. [Google Scholar]
- Kempisty B, Sikora J, Lianeri M, Szczepankiewicz A, Czerski P, Hauser J, Jagodzinski PP. MTHFD 1958G>A and MTR 2756A>G polymorphisms are associated with bipolar disorder and schizophrenia. Psychiatr Genet. 2007;17:177–181. doi: 10.1097/YPG.0b013e328029826f. [DOI] [PubMed] [Google Scholar]
- Kramer MS, Kahn SR, Rozen R, Evans R, Platt RW, Chen MF, Goulet L, Seguin L, Dassa C, Lydon J, McNamara H, Dahhou M, Genest J. Vasculopathic and thrombophilic risk factors for spontaneous preterm birth. Int J Epidemiol. 2009;38:715–723. doi: 10.1093/ije/dyp167. [DOI] [PubMed] [Google Scholar]
- Lazzerini PE, Capecchi PL, Selvi E, Lorenzini S, Bisogno S, Galeazzi M, Laghi PF. Hyperhomocysteinemia, inflammation and autoimmunity. Autoimmun Rev. 2007;6:503–509. doi: 10.1016/j.autrev.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Lee LC, Zachary AA, Leffell MS, Newschaffer CJ, Matteson KJ, Tyler JD, Zimmerman AW. HLA-DR4 in families with autism. Pediatr Neurol. 2006;35:303–307. doi: 10.1016/j.pediatrneurol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Lim MA, Stack CM, Cuasay K, Stone MM, McFarlane HG, Waschek JA, Hill JM. Regardless of genotype, offspring of VIP-deficient female mice exhibit developmental delays and deficits in social behavior. Int J Dev Neurosci. 2008;26:423–434. doi: 10.1016/j.ijdevneu.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopreato FR, Stabler SP, Carvalho FR, Hirata RD, Hirata MH, Robi DL, Sampaio-Neto LF, Allen RH, Guerra-Shinohara EM. Relationships between gene polymorphisms of folate-related proteins and vitamins and metabolites in pregnant women and neonates. Clin Chim Acta. 2008;398:134–139. doi: 10.1016/j.cca.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Mansoor MA, Kristensen O, Hervig T, Bates CJ, Pentieva K, Vefring H, Osland A, Berge T, Drablos PA, Hetland O, Rolfsen S. Plasma total homocysteine response to oral doses of folic acid and pyridoxine hydrochloride (vitamin B6) in healthy individuals. Oral doses of vitamin B6 reduce concentrations of serum folate. Scandinavian Journal of Clinical and Laboratory Investigation. 1999;59:139–146. doi: 10.1080/00365519950185878. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003;26:137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- McKeever M, Molloy A, Weir DG, Young PB, Kennedy DG, Kennedy S, scott JM. An abnormal methylation ratio induces hypomethylation in vitro in the brain of pig and man, but not in rat. Clin Sci (Colch ) 1995;88:73–79. doi: 10.1042/cs0880073. [DOI] [PubMed] [Google Scholar]
- Melnyk S, Pogribna M, Pogribny I, Hine RJ, James SJ. A new HPLC method for the simultaneous determination of oxidized and reduced plasma aminothiols using coulometric electrochemical detection. Journal of Nutritional Biochemistry. 1999;10:490–497. doi: 10.1016/s0955-2863(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Melnyk S, Pogribna M, Pogribny IP, Yi P, James SJ. Measurement of plasma and intracellular S-adenosylmethionine and S-adenosylhomocysteine utilizing coulometric electrochemical detection: Alterations with plasma homocysteine and pyridoxal 5'-phosphate concentrations. Clinical Chemistry. 2000;46:265–272. [PubMed] [Google Scholar]
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:172–182. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niculescu MD, Craciunescu CN, Zeisel SH. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J. 2006;20:43–49. doi: 10.1096/fj.05-4707com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niculescu MD, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J Nutr. 2002;132:2333S–2335S. doi: 10.1093/jn/132.8.2333S. [DOI] [PubMed] [Google Scholar]
- O'Leary VB, Pangilinan F, Cox C, Parle-McDermott A, Conley M, Molloy AM, Kirke PN, Mills JL, Brody LC, scott JM. Reduced folate carrier polymorphisms and neural tube defect risk. Mol Genet Metab. 2006;87:364–369. doi: 10.1016/j.ymgme.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Pei LJ, Li ZW, Zhang W, Ren AG, Zhu HP, Hao L, Zhu JH, Li Z. Epidemiological study on reduced folate carrier gene(RFC1 A80G) polymorphism and other risk factors of neural tube defects. Beijing Da Xue Xue Bao. 2005a;37:341–345. [PubMed] [Google Scholar]
- Pei LJ, Zhu HP, Li ZW, Zhang W, Ren AG, Zhu JH, Li Z. Interaction between maternal periconceptional supplementation of folic acid and reduced folate carrier gene polymorphism of neural tube defects. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2005b;22:284–287. [PubMed] [Google Scholar]
- Pickard B, Dean W, Engemann S, Bergmann K, Fuermann M, Jung M, Reis A, Allen N, Reik W, Walter J. Epigenetic targeting in the mouse zygote marks DNA for later methylation: a mechanism for maternal effects in development. Mech Dev. 2001;103:35–47. doi: 10.1016/s0925-4773(01)00329-x. [DOI] [PubMed] [Google Scholar]
- Picker JD, Coyle JT. Do maternal folate and homocysteine levels play a role in neurodevelopmental processes that increase risk for schizophrenia? Harv Rev Psychiatry. 2005;13:197–205. doi: 10.1080/10673220500243372. [DOI] [PubMed] [Google Scholar]
- Prescott SL, Clifton V. Asthma and pregnancy: emerging evidence of epigenetic interactions in utero. Curr Opin Allergy Clin Immunol. 2009;9:417–426. doi: 10.1097/ACI.0b013e328330634f. [DOI] [PubMed] [Google Scholar]
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W. DNA methylation and mammalian epigenetics. Electrophoresis. 2001;22:2838–2843. doi: 10.1002/1522-2683(200108)22:14<2838::AID-ELPS2838>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet. 2006;15(Spec No 2):R138–R150. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- Scholl TO, Johnson WG. Folic acid: influence on the outcome of pregnancy. Am J Clin Nutr. 2000;71:1295S–1303S. doi: 10.1093/ajcn/71.5.1295s. [DOI] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am J Hum Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- Susser E, Brown AS, Klonowski E, Allen RH, Lindenbaum J. Schizophrenia and impaired homocysteine metabolism: a possible association. Biol Psychiatry. 1998;44:141–143. doi: 10.1016/s0006-3223(97)00427-7. [DOI] [PubMed] [Google Scholar]
- van der Linden I, Afman LA, Heil SG, Blom HJ. Genetic variation in genes of folate metabolism and neural-tube defect risk. Proc Nutr Soc. 2006;65:204–215. doi: 10.1079/pns2006495. [DOI] [PubMed] [Google Scholar]
- van der Linden I, Heil SG, van Egmont PM, van Straaten HW, Den Heijer M, Blom HJ. Inhibition of methylation and changes in gene expression in relation to neural tube defects. Birth Defects Res A Clin Mol Teratol. 2008;82:676–683. doi: 10.1002/bdra.20509. [DOI] [PubMed] [Google Scholar]
- Walker MC, Smith GN, Perkins SL, Keely EJ, Garner PR. Changes in homocysteine levels during normal pregnancy. American Journal of Obstetrics and Gynecology. 1999;180:660–664. doi: 10.1016/s0002-9378(99)70269-3. [DOI] [PubMed] [Google Scholar]
- Weinberg CR, Wilcox AJ, Lie RT. A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet. 1998;62:969–978. doi: 10.1086/301802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, Lie RT. Distinguishing the effects of maternal and offspring genes through studies of “case-parent triads”. Am J Epidemiol. 1998;148:893–901. doi: 10.1093/oxfordjournals.aje.a009715. [DOI] [PubMed] [Google Scholar]
- Williams TA, Mars AE, Buyske SG, Stenroos ES, Wang R, Factura-Santiago MF, Lambert GH, Johnson WG. Risk of autistic disorder in affected offspring of mothers with a glutathione S-transferase P1 haplotype. Arch Pediatr Adolesc Med. 2007;161:356–361. doi: 10.1001/archpedi.161.4.356. [DOI] [PubMed] [Google Scholar]
- Yates Z, Lucock M. G80A reduced folate carrier SNP modulates cellular uptake of folate and affords protection against thrombosis via a non homocysteine related mechanism. Life Sci. 2005 doi: 10.1016/j.lfs.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Yi P, Melnyk S, Pogribna M, Pogribny IP, Hines RJ, James SJ. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. Journal of Biological Chemistry. 2000;275:29318–29323. doi: 10.1074/jbc.M002725200. [DOI] [PubMed] [Google Scholar]
- Zeisel SH. Epigenetic mechanisms for nutrition determinants of later health outcomes. Am J Clin Nutr. 2009a;89:1488S–1493S. doi: 10.3945/ajcn.2009.27113B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH. Importance of methyl donors during reproduction. Am J Clin Nutr. 2009b;89:673S–677S. doi: 10.3945/ajcn.2008.26811D. [DOI] [PMC free article] [PubMed] [Google Scholar]