

Abstract
NMR spectroscopy and molecular dynamics (MD) simulations were used to probe the structure and dynamics of complexes of three phosphotyrosine-derived peptides with the Src SH2 domain in an effort to uncover a structural explanation for enthalpy-entropy compensation observed in the binding thermodynamics. The series of phosphotyrosine peptide derivatives comprises the natural pYEEI Src SH2 ligand, a constrained mimic, in which the phosphotyrosine (pY) residue is preorganized in the bound conformation for the purpose of gaining an entropic advantage to binding, and a flexible analog of the constrained mimic. The expected gain in binding entropy of the constrained mimic was realized; however, a balancing loss in binding enthalpy was also observed that could not be rationalized from the crystallographic structures. We examined protein dynamics to evaluate whether the observed enthalpic penalty might be the result of effects arising from altered motions in the complex. 15N-relaxation studies and positional fluctuations from molecular dynamics indicate that the main-chain dynamics of the protein show little variation among the three complexes. Root mean squared (RMS) coordinate deviations vary by less than 1.5 Å for all non-hydrogen atoms for the crystal structures and in the ensemble average structures calculated from the simulations. In contrast to this striking similarity in the structures and dynamics, there are a number of large chemical shift differences from residues across the binding interface, but particularly from key Src SH2 residues that interact with pY, the ‘hot spot’ residue, which contributes about half of the binding free energy. Rank order correlations between chemical shifts and ligand binding enthalpy for several pY-binding residues, coupled with available mutagenesis and calorimetric data, suggest that subtle structural perturbations (< 1 Å) from the conformational constraint of the pY residue sufficiently alter the geometry of enthalpically critical interactions in the binding pocket to cause the loss of binding enthalpy, leading to the observed entropy-enthalpy compensation. We find no evidence to support the premise that enthalpy-entropy compensation is an inherent property and conclude that preorganization of Src SH2 ligand residues involved in binding hot spots may eventuate in suboptimal interactions with the domain. We propose that introducing constraints elsewhere in the ligand could minimize entropy-enthalpy compensation effects. The results illustrate the utility of the NMR chemical shift to highlight small, but energetically significant, perturbations in structure that might otherwise go unnoticed in an apparently rigid protein.
Keywords: computational binding enthalpy, NMR chemical shift perturbation, SH2 binding specificity
INTRODUCTION
Structure based drug design is an important application of structural biology studies. Using the atomic structure of a biological receptor complexed with a ligand, one should ideally be able to characterize the binding interface and propose specific modifications to the ligand that would enhance the free energy for association. However, this process is often fraught with difficulty in practice, as changes intended to optimize either the enthalpy or entropy of binding are often met with unanticipated energetic results1–3.
The structure of a small molecule may be modified in a number of ways toward improving its potency for a biological target. One common tactic involves preorganizing the ligand in the conformation it adopts upon binding, commonly referred to as the biologically active conformation4–6. This practice owes its origin to the fact that there is an entropic penalty associated with restricting the motion of a flexible molecule when it binds to a protein. Accordingly, constraining a flexible molecule in its biologically active conformation is expected to reduce the magnitude of this entropic penalty, thereby providing a relatively favorable contribution to the binding free energy7–9. Calorimetric studies have established that a more favorable binding entropy can be realized by preorganization, although recent accounts have revealed that ligand preorganization does not necessarily lead to an entropic advantage in binding10–12.
The effects of ligand preorganization upon the binding energetics of a set of constrained and flexible analogs of the canonical pYEEI peptide for the pp60 v-Src SH2 (Src SH2) domain have been examined13,14. In the constrained analog (cpYEEI), a trisubstituted cyclopropyl moiety was used as a rigid replacement of the pY residue (Figure 1). Modeling suggested that this rigid replacement mimicked the conformation of the pY side-chain that is observed in structures of phosphotyrosine-derived peptides bound to this and other SH2 domains15–17. Because pYEEI and cpYEEI do not have the same number of heavy atoms, the appropriate flexible control for cpYEEI is fpYEEI, wherein a benzyl succinate moiety serves as a flexible replacement of the pY residue (Figure 1). Both cpYEEI and fpYEEI have the same number and type of heavy atoms, the same functional groups and the same number of hydrogen bond donors and acceptors, so it was reasoned that desolvation effects for each would be similar. The binding thermodynamics of these three ligands for the recombinant Src SH2 domain were determined using isothermal titration calorimetry (ITC). It was found that the constrained pseudopeptide, cpYEEI, exhibited a binding entropy about 7 cal mol−1 K−1 more favorable (positive ΔΔS°) than its tetrapeptide counterpart pYEEI and nearly 9 cal mol−1 K−1 more favorable than its flexible control fpYEEI. Nevertheless, the relative binding enthalpy of cpYEEI was about 1.5 kcal mol−1 less favorable (positive ΔΔH°) than pYEEI and 2.7 kcal mol−1 less favorable than fpYEEI. We thus observed the expected entropic advantage attending preorganization of the cpY residue relative to pY and the flexible replacement fpY, but an enthalpic penalty offset this entropic gain in both cases. The end result from this observed enthalpy-entropy compensation was that the binding affinity of cpYEEI was only slightly higher than that of pYEEI and slightly lower than that of fpYEEI, and the relative binding enthalpies for the three Src SH2 complexes is most favorable for the fpY pseudopeptide and least favorable for the cpY pseudopeptide.
Figure 1.
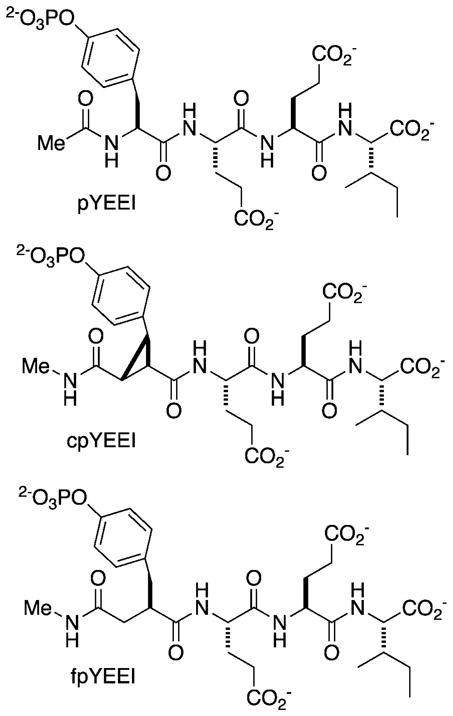
Schematic of the canonical (pYEEI), constrained (cpYEEI), and flexible (fpYEEI) ligands studied in this investigation. pY denotes the natural phosphorylated tyrosine residue. cpY denotes the trisubstituted cyclopropane replacement used to mimic the bound state conformation of pY. fpY denotes the flexible control replacement that has the same number of heavy atoms as cpY and is designed to account for the rearrangement of the hydrogen bonding capacity between the pY and cpY residues.
Enthalpy-entropy compensation has been a topic of discussion in the literature for many years18–22. Because of its prevalence in biological systems18,21,23 enthalpy-entropy compensation has been proposed to be an inherent property of aqueous solution associated with the reorganization of hydrogen bonding patterns in the hydration shells of proteins and small molecules18. On the other hand, this view has been countered by theoretical derivations arguing that such a correlation is not a fundamental thermodynamic requirement19,21,22, and there are some examples of non-compensating systems in which anti-correlation between enthalpy and entropy serves to amplify gains in the binding free energy21,24. Despite the body of literature on the subject, a full understanding of compensation mechanisms in protein-ligand interactions is lacking. Such an understanding could prove beneficial to rational drug design if one knew how to minimize enthalpy-entropy compensation in order to take advantage of one free energy component, and thus the physical basis for the differences in the enthalpy of binding among the three Src SH2 complexes is examined here.
Less favorable enthalpy of binding in the case of Src SH2 by a pseudopeptide could, of course, result from an altered binding conformation relative to a canonical peptide ligand. To identify a potential structural basis for the compensation observed in this series of Src SH2 binding ligands, crystal structures of Src SH2 in complex with the cpYEEI pseudopeptide (Figure 1) and a canonical peptide were compared14. It was not possible to obtain a structure of the Src SH2 complex with fpYEEI. The RMSD between the complex with cpYEEI and that with the canonical peptide was approximately 1 Å for protein main-chain atoms and within 1.5 Å over all heavy atoms. Because differences between the structures of the two complexes are comparable to the differences between multiple copies of a complex in each asymmetric unit, no significant structural perturbations that adequately explain the compensation14 could be identified.
The lack of a significant structural difference observed between the crystallographic models for the Src SH2 complexes with pYEEI and cpYEEI, and reasoning that desolvation effects would be similar for the pseudopeptides, raises the question of whether variations in internal dynamics associated with the cyclopropyl moiety exist and are the cause for less favorable interaction energy in the cpYEEI complex. If so, then a direct link between the observed loss in enthalpy and the gain in entropy would be established, suggesting that enthalpy-entropy compensation is inherent to binding Src SH2. In the presence of certain conformational motions, dynamic averaging over the thermal distribution of protein-ligand conformations was shown in an MD study to be important in determining the interaction energies; the relative energies for protein-ligand interactions estimated from the average structure were shown to differ from the relative values determined from the ensemble-averaged energy24a, which is the thermodynamically relevant quantity. It is reasonable that restricted motion imposed by the conformational constraint of cpYEEI could affect the ensemble averaging in a manner that weakens intermolecular interactions without significantly affecting the average structure. A wide body of evidence demonstrates the importance of thermal motions to protein function and the need to include conformational fluctuations in characterizing protein-ligand interactions25–29. While x-ray crystallography gives accurate three-dimensional structures of proteins, it provides limited information on dynamics, so that possible energetic differences that result from dynamic averaging over the fluctuations about the average structure would be invisible by crystallography. On the other hand, NMR relaxation and molecular dynamics simulations, which have been valuable for elucidating the effects from internal fluctuations and conformational entropy of proteins to molecular association1,25,30–33, can contribute to understanding differences in internal dynamics and enthalpy of proteins in a manner that is not apparent from an average structure.
In this light, we probed the differences between the pYEEI, cpYEEI, and fpYEEI complexes of Src SH2 by NMR and computer simulation. Positional fluctuations were examined to determine if the enthalpic loss in the Src SH2-cpYEEI complex might arise from restricted motion in the complex that could result in the observed enthalpy-entropy compensation. NMR chemical shift analysis was also used to probe structural differences between the solution structures of the three SH2-peptide complexes34.
MATERIALS AND METHODS
Protein Expression, Purification, and Sample Preparation
The canonical pYEEI tetrapeptide was purchased with an acetylated N-terminus and unblocked C-terminus (SynPep). The cpYEEI and fpYEEI pseudopeptide derivatives were synthesized as previously described13. Protein was expressed and purified in a manner similar to previously reported protocols35,36. A construct consisting of residues 144 to 249 of the pp60 v-Src tyrosine kinase (UniProt P00524) that comprise the SH2 domain (Src SH2) was cloned into a pET30b expression vector (Novagen) at the NdeI and BamHI restriction sites. Protein was over-expressed in Rosetta2 (DE3) E. coli (Novagen) grown on M9 minimal media with 1 g/L 15N-labeled NH4Cl as the sole nitrogen source and at least 2 g/L glucose as the sole carbon source, unlabeled or uniformly 13C-labeled as required. Induction was initiated with 0.2 mM IPTG after incubating for 16 to 20 hours at 22° C.
Cells were lysed using a French press in 20 mM (Na)HEPES, pH 7.5, 5 mM DTT, 1 mM EDTA, 0.2 mM PMSF, and 0.02% NaN3. After ultracentrifugation, the supernatant was loaded onto a 5 mL cation exchange HiTrap SP column (GE) that had been equilibrated with the same buffer. The protein was eluted with a 0 to 0.5 M NaCl gradient in the same buffer. Src SH2 containing fractions were pooled and concentrated to ~2 ml using Amicon-Ultra-15 concentrators (Millipore) with 3 kDa molecular weight cutoff (MWCO) prior to final purification by gel filtration (Superdex 75 16/60, Pharmacia) using 20 mM (Na)HEPES, pH 7.5, 5 mM DTT, 1 mM EDTA, 350 mM NaCl, and 0.02% NaN3.
NMR experiments were conducted in 50 mM Na Acetate, pH 5.5, 100 mM NaCl, 5 mM TCEP, and 0.02% NaN3 (from Xu et al.36 with minor variations). Pooled fractions containing Src SH2 were dialyzed against the NMR buffer (2–3 times, at least 6 hours each time) using Snake Skin dialysis membranes with 3.5 kDA MWCO (Pierce). Peptides were dissolved in 100 μl of NMR buffer. After adjusting the pH to ~5.5 with aqueous NaOH, the peptide stock was dialyzed alongside the protein using 100 Da MWCO membranes (Harvard Bioscience). Protein was concentrated to 1–1.8 mM using 3 kDa MWCO concentrators (Millipore). NMR samples in 90%/10% H2O/D2O were prepared by combining appropriate proportions of protein, peptide stock, and D2O. Concentrations were measured by UV absorbance at 280 nm with an extinction coefficient of 14,700 M−1 cm−1 for protein, and 775 M−1 cm−1 at 268 nm for the peptides14.
NMR Spectroscopy
NMR spectra were acquired at 298 K on a Bruker Avance III spectrometer operating at 800 MHz and a TXI triple resonance probe using standard pulse sequences from the Bruker pulse sequence library. Spectra were processed with NMRPipe37 and viewed with Sparky38. Samples contained approximately 1 mM Src SH2 domain and 5 mM ligand to ensure saturation of the bound state conformation of each complex, as determined by titrations monitored by 15N HSQC. Initial assignments based on the published values for the Src SH2—pYEEI complex36 were confirmed with the MARS program39 using data from standard HNCACB and CBCACONH triple resonance experiments40. Due to large changes in chemical shift, the main-chain experiments were conducted and full main-chain assignment performed for all four samples (unbound SH2, SH2-pYEEI, SH2-cpYEEI, and SH2-fpYEEI). The 800 MHz spectra were well resolved, allowing main-chain assignment of all non-proline residues.
During the assignment process, the peaks of E178 NH and R175 NεHε, which were assigned based on published values36,41, showed rank-order correlation with binding enthalpy. Both residues are located in the pY-binding pocket and are directly involved with coordination of the phosphoryl group on the ligand. It was assumed therefore, that the trend in the chemical shifts is related to the deshielding effect of the negatively charged phosphate group and the stability of its interaction with the residues. This assumption, which is further supported throughout the results and discussion, led to the assignment of peaks for the R175 NηHη side-chain atoms that bind the phosphoryl moiety of the ligand. After accounting for the main-chain amides of all non-proline residues and the side-chain values available in the published shifts, a group of unassigned peaks remained that were located in the region of the spectra populated by the arginine and histidine side-chain peaks and followed the same rank-order trend with enthalpy. The chemical shift values fall within the statistical range compiled for proteins in the BMRB and have proton chemical shifts similar to R160 NηHη, based on the reported values for the c-Src SH2-pYEEI complex by Xu et al.36.
Standard pulse sequences were used to measure the main-chain 15N relaxation rates42, with interscan relaxation delays of 2 sec for all experiments and a 2 sec saturation time for the NOE experiments. Delay times of 10, 35 (in duplicate), 50, 175, 375, 550 (in duplicate), 750, 1000, and 1400 ms were employed for T1 measurements. CPMG delays of 16.3, 32.6 (in duplicate), 48.9, 65.2, 81.5, 97.9 (in duplicate), 114.2, and 130.5 ms were employed for T2 measurements. Exponential decay times were fit to peak heights in Sparky. Chemical shift perturbations (CSPs) and chemical shift differences (CSDs) were calculated from sensitivity-enhanced 1H-15N HSQC spectra43 according to44:
| (1) |
Molecular Dynamics Simulations
Dynamics simulations were calculated with CHARMM45 using the CHARMM27 all-atom force field46 with CMAP main-chain dihedral correction47. Supplementary topology and parameters for the cyclopropyl moiety (Supplementary Information) were generated according to the standard CHARMM force field parameterization methodology46. Equilibrium geometries and reference vibrational data for this moiety were obtained from the NIST CCCBDB database48. Reference energy curves from HF/6-31g** ab initio dihedral optimizations of model compounds were used to parameterize the torsional parameters for rotation about the Cβ-Cγ bond (χ1 rotamer) and cyclopropyl-carbonyl “main-chain” bonds in cpY. Parameters for the fpY residue were assigned by analogy to existing force field values.
Initial coordinates for the complex models were taken from the three chains of the Src SH2-pYEEI crystal structure (PDB code 1SPS49) and the two chains of the Src SH2-cpYEEI crystal structure (PDB code 1IS013). For each complex being simulated, the peptide was alchemically mutated to obtain the desired model pseudopeptide in each case, yielding five sets of starting coordinates. To increase the efficiency of conformational sampling, two independent simulations using different initial velocities were initiated from each set of starting coordinates to yield ten independent simulations of each complex50.
Solutes were solvated with a truncated octahedral water box of TIP3 water molecules with box edges at least 14 Å from the solute. Non-bonded lists were generated with a 14 Å cutoff, and electrostatic interactions up to 12 Å were treated with a shifted potential and particle mesh Ewald summation method during the dynamics. Bond lengths to hydrogen atoms were constrained with SHAKE. Dynamics were performed using a leapfrog integrator, timestep of 1 fs, and constant pressure and temperature (CPT) routine using a reference pressure of 1 atm and piston mass constant of 500 amu. Hoover temperature control was employed with a temperature bath of 298 K and thermal piston constant of 1000 kcal ps2 mol−1.
Simulations were equilibrated for 500 ps, after which time all simulations were deemed reasonably energetically stable and displayed asymptotic RMSD time series relative to initial coordinates. Production runs of 1 ns were calculated for each simulation, with coordinates saved every 1 ps for a total of 10 ns of trajectory (10,000 snapshots) per complex. Dynamic properties were evaluated from this combined 10 ns pseudo-trajectory. Average structures were calculated from the combined set and subjected to 500 steps of steepest descent minimization to remove severe steric clashes. Main-chain chemical shifts were calculated from these structures using Sparta51.
Hydrogen bond analysis was performed with CHARMM’s coordinate manipulation commands. For each frame in the trajectory, a hydrogen bond between a hydrogen bond donor (D) and acceptor (A) was considered to be intact52 if the D-A distance was less than 2.4 Å and the D-H•••A angle was 120° to 180°. Two-dimensional histograms for geometric analyses were calculated using 4° bins over a range of −180° to 180° for dihedral angles, and 0.5 Å bins over a range of 1 to 6 Å for interatomic distances.
Individual contributions of groups of water molecules to the energy of a Src SH2-peptide complex were calculated from the explicitly solvated MD trajectories. The energy of the group (Egrp) was determined from the sum of the internal energy (Einternal), which includes the molecular mechanics energy for the bonded and nonbonded interactions associated with the atoms of the group, and one half the nonbonded energies for the interactions of the group with other water molecules (Esolvent) and with protein or peptide atoms (Esolute).
| (2) |
Interaction energies were averaged over the 2-ns simulation time period using snapshots every 1 ps.
Networks of water molecules identified from several crystallographic structures of Src SH2-peptide complexes3,14 are thought to stabilize the bound state. The occupancy and energetics of these networks were characterized from the MD trajectories. One site, WG236, refers to water molecules within 6.0 Å of non-hydrogen atoms of either residues G236 or L237 and within 6.0 Å of residue Y202. The second site, W+2E, corresponds to water molecules within 3.2 Å of both protein residue R205 and peptide residue pY+2E as well as within 6.0 Å of the main-chain heavy atoms of both residues K203 and I214. (See Supplementary Information Figure S1 for a graphical depiction of the water sites.) Occupancy was determined from the time average number of water molecules meeting these distance criteria for trajectory snapshots at 1ps intervals. For each group of water molecules selected, the time-averaged site energy was calculated using Equation (2).
RESULTS
Internal dynamics probed by NMR and MD
To test for possible differences in internal dynamics of the Src SH2-pYEEI, Src SH2-cpYEEI, and Src SH2-fpYEEI complexes, 15N relaxation experiments were conducted for each complex. T1 and T2 relaxation times, and the 1H-15N heteronuclear NOE (Isat/Inosat − 1) are sensitive to motions on a ps-ns timescale, while T2 is also sensitive to longer timescale motion associated with conformational exchange. The residue profiles for these relaxation parameters are shown in Figure 2. For most residues, the values deviate among the complexes by an amount less than experimental error. The profiles show similar trends of relatively diminished Isat/Inosat and slightly elevated T2 values, indicating higher internal mobility, for residues N-terminal to the αA helix (residue 153), in the AB (residue 164), CD (residues 192–195), and DE (residues 206–211) loop regions, and in the N and C termini. These relaxation data are thus remarkably similar between the complexes even in these more flexible loop regions. Rotational anisotropy manifests more strongly in T1 than T2 and therefore the observed T1 profiles in Figure 2 (top) are less uniform than those for T2 (middle). Small differences in T1 occur for residues scattered throughout the protein (I153, N164, E178, N193, D235). These T1 deviations range from 0.1 to 0.2 s and, based on Lipari-Szabo model-free theory53, reflect small differences in the amplitude of fast timescale motion, corresponding to a maximum disparity in the generalized order parameter of only ~0.2. Because these dynamic variations are small and occur for residues scattered throughout Src SH2, we find no convincing argument that they are relevant to the enthalpic differences of Src SH2 binding the three ligands. Overall, the NMR relaxation data suggest that Src SH2 main-chain fluctuations are highly similar and provide no evidence for conformational averaging that could account for variations in binding enthalpies upon association with pYEEI, fpYEEI, and cpYEEI.
Figure 2.

15N A) T1, B) T2, and C) heteronuclear NOE (Isat/Inosat) relaxation values, measured at 18.8 T for the Src SH2—fpYEEI (green), Src SH2—pYEEI (gold), and Src SH2–cpYEEI (red) complexes.
The Src SH2-ligand complexes were also observed to have comparable positional fluctuations based on MD simulations. Protein root-mean-square fluctuations (RMSF) about the mean coordinates of the simulation, calculated over each 10 ns trajectory set (Figure 3), are nearly equal; the RMSF values for some loop residues vary up to ~0.5 Å for the Cα atoms (top) and up to ~0.9 Å for a small number of non-hydrogen atoms (bottom). The greatest differences in RMSF are found for residues in the AB, BC CD, and BG loops. Given their small magnitude and because these differences occur in mobile loop regions, which in general require longer sampling times for convergence, the disparities are not deemed sufficient to account for the variation in binding energetics.
Figure 3.

Root Mean Square Fluctuations (RMSF) for α-carbons (upper left) and non-hydrogen atoms (lower left) and overlaid average structures calculated from MD trajectories (right) of the Src SH2-pYEEI (gold), Src SH2-cpYEEI (red), and Src SH2-fpYEEI (green) complexes. Δ(cpY – pY) and Δ(fpY – pY) denote the differences of each analog peptide complex with respect to the pYEEI complex. The flexible loops are labeled according to their topological nomenclature. Protein image was produced with VMD79.
The influence of the cyclopropyl constraint on the conformational distribution of the cpY residue compared to pY and fpY was considered by examining the joint probability distributions for φ-ψ, χ1-χ2, and dihedral angles describing rotation of the phosphoryl group (Figure 4). The peaks in the two-dimensional histograms for cpY are sharper in φ and χ1 than those for pY or fpY, demonstrating the expected reduced range of motion (see also Table 1) for the constrained ring. Interestingly, the constraint also has long-distance effects on the dihedral angles about the bridging oxygen of the phosphoryl group (bottom plots, Figure 4); the distribution for Cζ-Oη-P-O is narrower for cpY compared to fpY and pY (Table 1).
Figure 4.
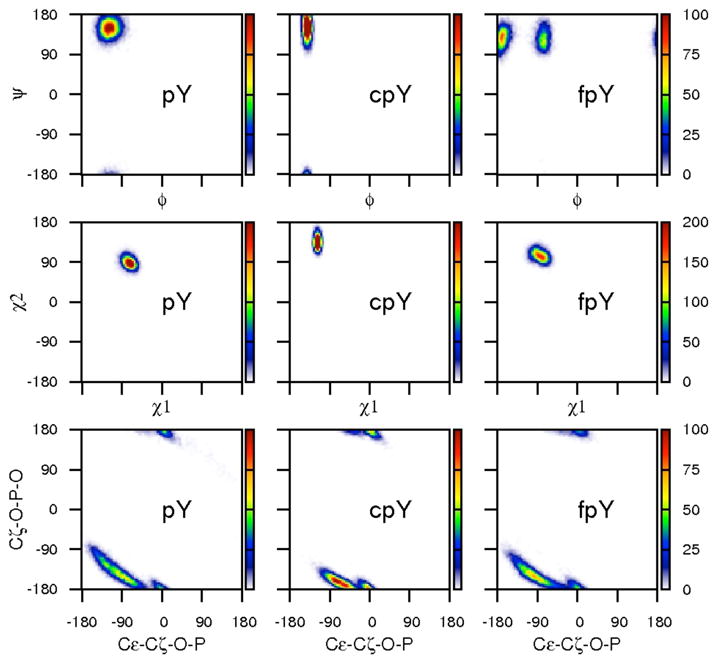
2D histograms displaying φ-ψ, χ1-χ2, and phosphate dihedral distributions for the pY, cpY, and fpY residues from MD simulations of each complex.
Table 1.
Crystallographic and simulated geometries for pY, cpY and fpY in the respective ligand complex with v-Src SH2, and for +1 E φ.
| Dihedral (degrees) | Src SH2-pYEEI | Src SH2-cpYEEI | Src SH2-fpYEEI |
|---|---|---|---|
| φ (x-ray)a | −116.3 (7.4) | −136.9 (2.3) | - |
| φ (MD)b | −116.3 (14.6) | −138.9 (5.3) | −129.5 (46.3) |
| ψ (x-ray)a | 143.7 (6.9) | 146.3 (5.6) | - |
| ψ (MD)b | 151.0 (15.8) | 150.9 (18.5) | 128.5 (18.8) |
| χ1 (x-ray)a | −72.6 (7.1) | −116.2 (0.8) | - |
| χ1 (MD)b | −72.1 (9.8) | −116.0 (4.2) | −83.2 (12.5) |
| χ2 (x-ray)a | 83.6 (19.0) | 134.0 (1.9) | - |
| χ2 (MD)b | 90.5 (10.4) | 137.3 (12.5) | 106.1 (10.8) |
| Cε-Cζ-O-P (x-ray)a | −75.6 (19.5) | −69.7 (4.8) | - |
| Cε-Cζ-O-P (MD)b | −78.1 (53.4) | −46.7 (−36.0) | −74.4 (48.4) |
| Cζ-Oη-P-O (x-ray)a | −137.6 (6.3) | −167.4 (3.5) | - |
| Cζ-Oη-P-O (MD)b | −146.1 (30.0) | −166.7 (14.7) | −151.8 (23.3) |
| pY+1 E φ (x-ray)a | −79.4 (11.1) | −63.9 (1.7) | - |
| pY+1 E φ (MD)b | −94.5 (19.6) | −85.4 (20.7) | −86.4 (18.3) |
standard deviation over multiple crystallographic chains in parentheses
standard deviation over MD snapshots in parentheses
Much like the trend between the crystal structures, the average coordinates of the complexes calculated from the simulation trajectories are strikingly similar to each other (Figure 3, right). The three complexes differ from each other by ~0.7 Å over main-chain atoms and ~1 Å RMSD over all non-hydrogen atoms. In regard to the pY residue itself, the average values for χ1, χ2, and phosphate dihedral angle, as well as ϕ for residue Y+1 E, only two bonds removed from the point of constraint, differ by as much as 15 degrees among the three bound ligands in both the crystallographic and MD structures (Table 1). These structural differences may seem large, but the rotation of the phosphate group is able to orient the ring so that strong interactions and the position of the phosphotyrosyl moiety appear to be maintained with respect to the protein.
Chemical shift perturbation analysis
Chemical shift is an exquisitely sensitive probe of local structure and magnetic environment. In contrast to the similarities observed for the crystallographic and MD average structures, the 1H-15N HSQC spectra of the unbound Src SH2 domain and the three complexes exhibit numerous differences, which indicate that experimentally measurable structural variations do exist. To assess the spectral differences, chemical shift perturbations (CSP) were calculated for each complex relative to the unbound spectrum (Figure 5, upper left), and chemical shift differences (CSD) were calculated between each complex (Figure 5, upper right). CSP values greater than 0.1 ppm are typically considered indicative of significant binding effects34,54–57, whereas values greater than 0.2 ppm indicate highly shifted CSP values54,55. The main-chain amide resonances of Src SH2 are especially sensitive to ligand binding, and a number of CSPs are greater than 0.3 ppm (Figure 5, upper left). A similar profile was reported for v-Src SH2 binding to a longer variant of pYEEI54. The number of residues perturbed, as well as the magnitude of the differences, between the three complexes is remarkable in light of the degree of similarity between the crystal structures and simulation average structures.
Figure 5.

Chemical shift perturbations (CSPs) (upper left) and chemical shift differences (CSDs) (upper right) plotted as a function of residue number. The horizontal dashed line on each plot shows the 0.06 ppm (2*<lw>) cutoff used to select for perturbed residues. CSDs between the Src SH2-fpYEEI and Src SH2-pYEEI complexes (cyan, lower left), Src SH2-cpYEEI and Src SH2-pYEEI complexes (magenta, lower center), and Src SH2-cpYEEI and Src SH2-fpYEEI (blue, lower right) are mapped onto the crystal structures. Protein images were produced with VMD79.
While the CSP indicates a change in the electronic environment of the amide group upon ligand binding, the CSD more specifically compares the chemical shifts between complexes. The bulk of the discussion will therefore focus on the CSDs. Main-chain CSDs between the three complexes that are greater than 0.06 ppm, a value greater than twice the average peak linewidth in the spectra, are mapped onto the structure in the lower panel of Figure 5 and tabulated in Table S1 (Supplementary Information). The chemical shifts of Src SH2 bound with the constrained cpYEEI deviate significantly from those of the other two complexes, as highlighted by the extent of the magenta (lower center) and blue (lower right) mappings. The SH2-pYEEI and SH2-fpYEEI complexes are more similar to each other, as illustrated by the relative sparsity of the cyan mapping (lower left).
Ligand-dependent chemical shift differences are scattered across the binding interface, but are most prominant in the BC loop, the βC and βD strands of the central beta sheet, the EF loop, and the BG loop (Figure 5). The perturbed residues of the BC loop and a portion of the residues in βC and βD form part of the highly conserved pY-binding pocket. The remaining perturbed residues in βC and βD, along with the EF and BG loops, contribute to the specificity-determining region, interacting with the EEI motif. Throughout this report the secondary structural elements will be referred to by the standard SH2 topological nomenclature defined by Eck et al.58 Residues will be referenced according to Src kinase numbering, and residue names in parentheses cross-reference the topological names that appear in the SH2 literature.
The greatest chemical shift differences between the three complexes are associated with residues directly involved with pY binding (see Table S1): E178 (GluBC1) main-chain amide (CSDfpY-pY=0.278, CSDcpY-pY=0.281, CSDcpY-fpY=0.558 ppm) and R175 (ArgβB5) side-chain guanidinium NηHη (CSDfpY-pY=0.255, CSDcpY-pY=0.332, CSDcpY-fpY=0.586 ppm). Figure 6 highlights these perturbations with four overlaid 1H-15N HSQC spectra for the unligated Src SH2 and the three complexes. Of particular interest is the observation that the trend in chemical shift perturbation follows the rank order of the binding enthalpies; the fpYEEI complex (green peaks) is the most enthalpically stable complex and has the most downfield (larger ppm) chemical shifts, while the cpYEEI complex (red peaks) is the least enthalpically stable complex and has the most upfield (smaller ppm) chemical shifts of the three complexes. The higher frequency of a downfield shifted resonance indicates the proton is relatively deshielded, which in proteins typically arises from stronger hydrogen bonding. As depicted in Figure 7, the E178 main-chain amide nitrogen is within hydrogen bonding distance (~2.8 Å) of the nearest phosphate oxygen in the crystal structures. R175, which is universally conserved across SH2 domains, forms a bidentate salt-bridge with the phosphate group as well as hydrogen bonds to the side-chains of H201 (HisβD4) and S187 (SerβC5). Although R175 side-chain resonances exhibit large CSD values, no significant differences are observed for R175 main-chain chemical shifts. Appreciable main-chain amide CSDs between all complexes are found for S177 (SerβB7), T179 (ThrBC2), and C185 (Cys βC3), all of which have side-chain interactions with the phosphoryl group. The remaining interactions responsible for coordinating the pY side-chain are mediated through the long, basic side-chains of R155 (ArgαA2), and K203 (LysβD6). Of these two residues, only K203 shows a significant main-chain CSD in the constrained pseudopeptide complex.
Figure 6.
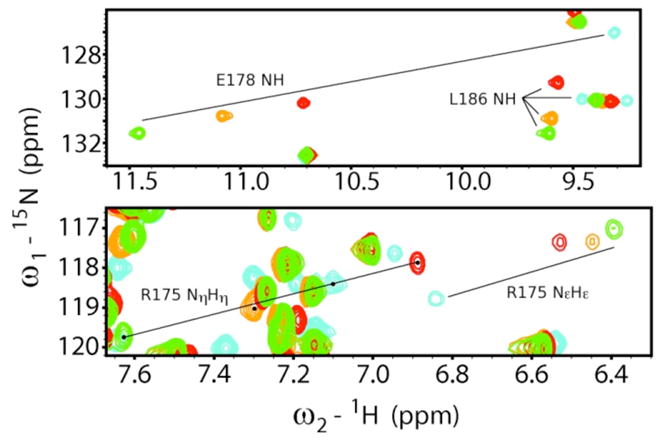
Overlay of 15N HSQC spectra of the unbound Src SH2 (cyan), Src SH2-cpYEEI complex (red), Src SH2-pYEEI complex (gold), and Src SH2-fpYEEI complex (green) featuring E178 NH, L186 NH, R175 NεHε, and R175 NηHη.
Figure 7.

A) Schematic view and of the pY-binding pocket and central β-strands highlighting key residues showing chemical shift differences. Residues labeled with solid wedges extend from the β-sheet into the pY-binding pocket, while residues labeled with open wedges extend into the hydrophobic binding pocket. Dashed lines represent hydrogen bonds and salt-bridging interactions. B) Stereo view of the pY-pocket interactions and central β-sheet. Hydrogen bonds are highlighted in pink. Protein image was produced with VMD79.
In addition to the β-sheet residues R175 and C185 just discussed, the chemical shifts of the central β sheet residues L186 (LeuβC4), H201 (HisβD4), Y202 (TyrβD5), and I204 (Ile βD7) differ considerably among the three complexes (Figure 5). The second largest CSDs are observed for the central βC strand residue L186 (CSDfpY-pY=0.095, CSDcpY-pY=0.231, CSDcpY-fpY=0.326 ppm, Figure 5 and 6). Such large differences are unexpected, as L186 makes neither direct contacts nor apparent conformational changes between complexes based on the crystal structures. The differences are greater than those in neighboring residues, C185 (CSDfpY-pY=0.047, CSDcpY-pY=0.112, CSDcpY-fpY=0.155 ppm) and S187 (CSDfpY-pY=0.008, CSDcpY-pY=0.065, CSDcpY-fpY=0.072 ppm), or in R175, all of which contribute directly to the network of interactions in the pY-binding pocket (Figure 7). The C185 and S187 main-chain chemical shifts differ only in the comparisons to the constrained complex, while the shifts in both the pYEEI and fpYEEI complexes are similar. Y202 (CSDfpY-pY=0.065, CSDcpY-pY=0.129, CSDcpY-fpY=0.096 ppm) forms main-chain β-sheet hydrogen bonds to L186, as well as energetically important side-chain contacts with the Y+1 E residue of the peptide and the hydrophobic binding pocket.
The HN resonance for H201 had large CSD values at saturation (CSDfpY-pY=0.072, CSDcpY-pY=0.049, CSDcpY-fpY=0.116 ppm). Moreover, the trend in the H201 amide proton chemical shifts follow the rank order of the binding enthalpies in the same manner as detailed above for E178 NH and R175 NηHη. That is, the shift for the most enthalpically stable SH2-fpYEEI complex is furthest downfield (δH=8.443 ppm), and that for the least enthalpically stable SH2-cpYEEI complex is furthest upfield (δH=8.284 ppm), with the SH2-pYEEI complex being intermediate (δH=8.353). The more downfield chemical shift indicates greater deshielding and suggests stronger H201 HN hydrogen bonding interactions for the SH2-cpYEEI complex. The H201 amide also showed a unique titration behavior; the H201 peak was absent in the SH2-cpYEEI complex spectra at pseudopeptide to protein molar ratios as high as 2.5 (data not shown). This broadening for the cpYEEI complex indicates a possible chemical exchange process that is exhibited by neither pYEEI nor fpYEEI complexes (see Discussion). Due to spectral overlap of the H201 peaks, reliable 15N-relaxation measurements could not be obtained for H201 in any of the three complexes, and thus the exchange process was not further characterized.
The effects of preorganization also extend into the hydrophobic binding pocket as shown by large CSDs involving the cpYEEI complex for residues in the βD strand and the BG and EF loops. The L186 side-chain is a central component of this pocket, packing with the side-chains of Y202, I204 (IleβD7) (CSDfpY-pY=0.042, CSDcpY-pY=0.114, CSDcpY-fpY=0.156 ppm), and L237 (LeuBG4) (CSDfpY-pY=0.030, CSDcpY-pY=0.100, CSDcp-fpY=0.129 ppm). The chemical shifts of the EF and BG loops are also very responsive to peptide binding; S216 (SerEF2) (CSDfpY-pY=0.053, CSDcpY-pY=0.174, CSDcpY-fpY=0.227 ppm) and T218 (IleβF1) (CSDfpY-pY=0.030, CSDcpY-pY=0.108, CSDcpY-fpY=0.137 ppm) of the EF turn, and G236 (GlyBG3) (CSDfpY-pY=0.027, CSDcpY-pY=0.101, CSDcpY-fpY=0.128 ppm) of the BG loop show large CSDs in the constrained complex (Figure 5, Table S1) despite their lack of direct association with the peptide. S216 CSPs are dominated by changes in the 15N frequency and show the second largest CSPs upon binding all three ligands (CSPpY=0.562, CSPcpY=0.734, CSPfpY=0.509 ppm) (Figure 5, top left).
MD analysis of intermolecular interactions
To gain insight into the basis for the differences in binding enthalpy, we evaluated the particular intermolecular interactions of the residues that exhibit large CSD values from MD simulations. E178 NH and R175 NηHη, which show the largest CSD values, interact directly with the phosphoryl group of the constrained residue. The time-averaged number of hydrogen bonds per snapshot, the nonbond energy of interaction for the groups containing the respective donor and acceptor atoms, and the distances (oxygen to hydrogen, or oxygen to nitrogen) for these interactions vary little among the three complexes (Table 2). A higher energy is observed for R175 NηHη interaction with the phosphoryl group in the cpYEEI SH2 complex (−135.8 versus −136.7 or −136.9 kcal/mol), consistent with this complex having the least favorable binding enthalpy. The outer βD strand residues H201 and Y202 also have large main-chain amide CSDs. The main-chain carbonyl of H201 forms a hydrogen bond to the NH group of residue Y+1 E, as illustrated in Figure 7. The average nonbond energy between Y+1 NH and H201 CO in the SH2-cpYEEI complex (−1.8 kcal/mol) is higher than that in either SH2-pYEEI (−2.8 kcal/mol) or SH2-fpYEEI (−2.6 kcal/mol) as the result of an increase in the NH-O distance, and a corresponding smaller fraction of snapshots that fit the hydrogen bonding criteria (Table 2). The differences in the interaction energies are smaller than the standard error. Nevertheless, the overall tendency of the simulation results in Table 2 is that the interactions with ligand of R175 and H201, residues with a rank-order chemical shift trend for the three complexes, are somewhat less favorable for the cpYEEI complex, consistent with the measured enthalpic differences.
Table 2.
Geometric and energetic analyses from MD for interactions of residues with large CSD and a trend in 1H NMR frequencies following the rank order in the binding enthalpy of the three complexes.
| Src SH2-pYEEI | Src SH2-cpYEEI | Src SH2-fpYEEI | |
|---|---|---|---|
| R175 Nη1-Hη11---O-P | |||
| <h-bond> a | 2.082 | 2.100 | 2.078 |
| <Ener> c | −136.9 (6.0) | −135.8 (6.7) | −136.7 (5.8) |
| <rNO> b | 2.69 (0.11) | 2.67 (0.10) | 2.68 (0.11) |
| E178 N-H---O-P | |||
| <h-bond> a | .984 | 1.001 | 0.895 |
| <Ener> d | −11.9 (0.4) | −11.7 (0.4) | −10.8 (2.9) |
| <rHO> b | 1.86 (0.19) | 1.83 (0.14) | 2.04 (0.63) |
| <rNO> b | 2.82 (0.17) | 2.78 (0.13) | 2.97 (0.58) |
| +1 E N-H---O=C H201 | |||
| <h-bond> a | 0.889 | 0.586 | 0.876 |
| <Ener> e | −2.8 (0.1) | −1.8 (1.0) | −2.6 (0.2) |
| <rHO> b | 2.03 (0.20) | 2.65 (0.84) | 2.10 (0.28) |
| <rNO> b | 2.97 (0.18) | 3.49 (0.73) | 3.02 (0.23) |
average number of hydrogen bonds per snapshot
average distance (Å) with standard error in parenthesis
average nonbonded energy (kcal/mol) between xpY(Cζ-O-PO3) and R175(Nη1Hη11Hη12Nη2Hη21Hη22CζNεHε)
average nonbonded energy (kcal/mol) between xpY(Cζ-O-PO3) and E178(HNNCαHα)
average nonbonded energy (kcal/mol) between xpY+1(HNNCαHα) and Y202(OC)
A number of interfacial water molecules are observed in the crystal structures to mediate interactions between the peptide and SH2 domain and appear to be energetically important to the stability of the complex3,14,73–75. Waksman and coworkers found a significant improvement in the agreement of binding enthalpies predicted using an empirical formulation based on surface area when these water molecules were included in the estimate of surface area73–75 The possibility that hydration effects resulting from these interfacial water molecules differ among the three complexes was considered by using the MD trajectories to assess the occupancy and interaction energies of the water molecules in the sites identified by Waksman. One of these water molecules hydrogen bonds with the H201 NH group as shown in Figure 7. Analysis of the MD trajectories found the constrained pseudopeptide did not significantly change the occupancy for the constrained pseudopeptide as determined by the fraction of frames with a hydrogen bond between H201 NH and a water molecule (0.935 for SH2-pYEEI, 0.919 for SH2-cpYEEI, and 0.917 for SH2-fpYEEI). We also analyzed two clusters of interfacial water molecules present in several crystal structures. One cluster of approximately four water molecules lies near the pY+2E side-chain (W+2E). A second cluster, with roughly three water molecules, bridges interactions of G236 in the BG loop and the hydrophobic binding pocket (WG236) with the ligand. The time-averaged interaction energies calculated with Equation 2 and number of water molecules in the site W+2E and WG236 are nearly identical (Table 3). Explicit-water MD simulations provide information on hydration effects at a level of detail that is not obtained by experiment; however, it should be noted that the energetics of water interactions are difficult to quantify with high accuracy because of the limitations of the water models and the long simulation times required to reach absolute convergence. With this limitation in mind, the results shown in Table 3 give no indication that the interfacial water molecules differ significantly as a result of the cpY constraint.
Table 3.
Energetic contributions to complex stability in the specificity-determining region
| Src SH2-pYEEI | Src SH2-cpYEEI | Src SH2-fpYEEI | |
|---|---|---|---|
| WG236 <N> | 4.4 (0.2) | 4.7 (0.2) | 4.4 (0.1) |
| WG236 <E> | −10.9 (0.08) | −10.8 (0.05) | −10.8 (0.03) |
| W+2E <N> | 3.6 (0.3) | 3.9 (0.3) | 3.3 (0.3) |
| W+2E <E> | −10.8 (0.08) | −10.5 (0.07) | −10.9 (0.11) |
energies (± standard error) in kcal/mol, calculated according to equation (2)
see Methods and Materials for definition of solvation sites, and supplementary information for illustration of the sites; <N> is the time averaged number of water molecules (± standard error) occupying the site, <E> is the average energy per water molecule (± standard error) in kcal/mol
An aim of this investigation was to explore the possibility that concerted atomic motions modulate non-covalent interactions in a manner that could explain the differences in binding enthalpies observed experimentally. Concerted motions include local motions such as libration of a functional group, or a collective motion of several groups across a network of interactions as is important for driving protein allostery and other conformational changes59,60. Consider that motion of the phosphotyrosyl residue optimizes the orientation of the phosphoryl group with respect to E178 and R175. Rigidity arising from the cpY main-chain constraint could hinder the ligand from concurrently maintaining stable main-chain hydrogen bonds between Y+1 E and H201. Two-dimensional histograms correlating the distance between E178 and the nearest phosphate oxygen atom with the Y+1 NH to H201 CO hydrogen bond distance are shown in Figure 8. The distributions for the pYEEI complex and fpYEEI complex show a single well so that the ligand interactions with both E178 and H201 maintain a stable H-O distance of ~2 Å, while the cpYEEI complex has a minor population of simulation snapshots with an increased Y+1 E to H201 distance of approximately 3.5–4 Å. This is manifested in the 0.6 Å increase in the mean Y+1 E HN to H201 O distance, decrease of 0.3 hydrogen bonds per frame, and one kcal/mol unfavorable change in interaction energy for cpYEEI listed in Table 2. These results suggest that the rigid constraint partly disrupts the pseudopeptide from simultaneously maintaining optimal interactions across the entirety of its binding interface.
Figure 8.

2D histograms from MD simulations for the ligand-protein distances for (left) Src SH2-pYEEI, (center) Src SH2-cpYEEI, (right) Src SH2-fpYEEI. The distance between the amide hydrogen atom of ligand residue Y+1 E and the carbonyl oxygen of H201 (HisβD4) is plotted along the y-axis. The distance between the main-chain amide proton of E178 (GluBC1) and the nearest phosphate oxygen atom is plotted along the x-axis.
DISCUSSION
The Src SH2 domain is a relatively rigid protein receptor with no induced-fit binding14,35,49, which makes it an attractive model system for ligand design, and simplifies the interpretation of ITC results and energetics of binding13,14. The structure of the Src SH2 domain appears unchanged in the unbound54, phosphate-bound49, pYEEI-bound49, and cpYEEI-bound14 states, as well as in complexes with a variety of other pY-based inhibitors61. The similarities are particularly striking within the pY-binding pocket. In spite of this apparent structural rigidity, the association of preorganized ligands with Src SH2 occurs with less favorable enthalpy of binding, albeit the desired gain in entropy. This enthalpy-entropy compensation motivated this investigation of possible conformational averaging affects given that relative motion of the protein and constrained peptide could lead to fluctuations with increased hydrogen bond lengths or more acute angles between donor and acceptor such that motional averaging would lead to higher hydrogen bond energy. Nonetheless, the internal dynamics of the pYEEI, cpYEEI, and fpYEEI Src SH2 complexes estimated from 15N relaxation (Figure 2) and MD fluctuations (Figure 3) show only minor variations, which cannot convincingly account for the observed disparities in binding enthalpy of the three complexes. Previously reported NMR relaxation measurements for Src SH2 also suggest that main-chain flexibility varies little upon binding the canonical pYEEI peptide, with only minimal differences from residues scattered throughout the protein in the amplitude of motion specified from the Lipari-Szabo order parameter54. As such, we find no evidence that alternative conformational distributions occurring from ps-ns motions affect the time-average interaction energy with the three Src SH2 ligands. These results do not directly assess possible differences in slower timescale motions. Nevertheless, given that slower motions correspond to larger amplitude motions, and that large-amplitude motions, such as loop motions, would likely be manifest in the average structure determined by crystallography, the structural similarity of the Src SH2 complexes would argue against the possibility that differences in slower timescale motions exist for these complexes.
Disruptions to pY-pocket interactions
In contrast to the above observations, important deviations in the chemical shifts of pY-pocket residues among the Src SH2 complexes reveal key distinctions that can be rationalized in energetic terms. The strongest CSDs (Table S1) between all three complexes are associated with the side-chain NηHη of R175 and the main-chain NH of E178, which both interact directly with the phosphoryl group (Figure 7). NMR CSDs for these residues are consistent with small structural variations that can reasonably account for the measured enthalpy differences of the three complexes. Recall that previously reported mutagenesis and calorimetry studies (Table S1) indicate that phosphotyrosyl binding contributes approximately half of the binding free energy and is largely enthalpically driven3,62,63. The free energy for Src SH2 binding pYEEI is ~6 kcal/mol more favorable relative to that of YEEI, with ΔΔH° ~−4 kcal/mol and TΔΔS° ~2 kcal/mol62. Interactions that contribute the majority of the binding free energy have become known as binding hot spots64,65. Hot-spot residues were found by mutagenesis to have considerably greater power in altering affinity than neighboring residues that also exist in the intermolecular contact surface.
The large downfield perturbations in the chemical shifts for R175 and E178 are consistent with a distance-dependent deshielding of the NH nuclei by direct contact with the ligand dianionic phosphoryl group34. The NMR chemical shift depends strongly on distance to nearby charged, polar, or aromatic groups, thereby providing a useful probe of main-chain amide hydrogen bonding, which is otherwise difficult to test by common approaches such as single-site mutagenesis. The SH2-fpYEEI complex exhibits the most downfield proton chemical shifts for E178 HN and R175 Hη, suggesting stronger interactions and therefore shorter phosphate-HN distances. The cpYEEI shifts are furthest upfield, consistent with the longest distances and least favorable energy. The rank-order in chemical shifts of these protons (Figure 6) therefore correlates with the binding enthalpies of the peptide series, and this correlation is consistent with increasing distance for the interaction with fpY, pY and cpY residues, respectively. The relationship between chemical shift and hydrogen bond stabilization has been well established for protein secondary structure66,67 and model compounds67–69. Ab initio calculations have suggested that changes in hydrogen bonding distance of tenths of an angstrom are sufficient to perturb the chemical shift by more than 1 ppm68–70. Furthermore, a 0.1 Å change in the phosphoryl group distance to the E178 amide and R175 guanidinium groups alters the interaction energy by approximately 1 and 4 kcal/mol, respectively, based on the molecular mechanics force field (Supplementary Information Figure S2.). Thus, in the case of Src SH2 binding, the trends in chemical shift indicate that subtle deviations in the interaction distances for the phosphotyrosine with the SH2 pY-pocket residues, perhaps due to small geometric variations between cpY, pY and fpY residues, can account for the observed differences in binding enthalpies of the complexes.
The crystal structures were unable to distinguish the three complexes in terms of the distances between the phosphate and HN of E178 or R175; however, a difference of tenths of angstroms to account for the CSD is comparable to the ~0.3 Å estimated coordinate error of a crystallographic model determined at 1.9 Å resolution70a. Further, the simulations find less favorable interaction energy of R175 guanidinium for the cpYEEI complex, but the trends in interaction energies of E178 NH and R175 NηHη do not correlate with the interaction energy indicated from chemical shifts. The simulations therefore also appear unable to capture the subtle structural effects observed by NMR. A more accurate treatment of electrostatics by including polarization70 in the molecular mechanics force field is likely required to correctly model the behavior of the charged phosphoryl group.
Interpretation of chemical shift perturbations in terms of thermodynamic parameters necessitates caution as chemical shift is sensitive to a number of structural and environmental factors34,71. A quantitative interpretation requires accurate chemical shift calculations and detailed structural information for the system. Further, binding enthalpy depends on the energy difference between the bound and unbound state, and the difference between two complexes alone may not fully explain deviations in binding thermodynamics. It is reasonable that the high degree of structural similarity between the three ligands in this study would minimize any contribution from the free state of the ligands, and MD studies are underway to actually estimate the contribution from the free state. Nevertheless, the relative downfield changes in chemical shift for E178 HN and R175 Hη (Figure 6) indicate stronger deshielding and electrostatic potential for interaction in the order of the Src complexes: fpYEEI > pYEEI > cpYEEI. Given the interactions observed crystallographically, we propose closer contact and more favorable non-covalent ligand interactions of the phosphoryl group with E178 NH and R175 NηHη in the same relative order, and these interactions are in part the basis for the observed ranking in experimental binding enthalpies.
The energetic importance of R175 to binding affinity and as part of the hot spot interaction is well established49. The residue is universally conserved across SH2 domains. The structural importance of the hydrogen bonding between R175 and H201 has been supported by the acidic pKa of H201 Nδ, measured by NMR titration in Src SH254 and PLCγ72. A triad of hydrogen bonds between R175, H201, and E159 is argued to preorganize the R175 side-chain in its bound state geometry54. The R175A Src SH2 mutant binds pYEEI with significantly reduced binding enthalpy (ΔΔG° = 3.2 kcal/mol, ΔΔH° = 4.1 kcal/mol, T ΔΔS° = 0.9 kcal/mol62). The non-covalent interactions of the R175 side-chain with S187, H201, and the pY residue (Figure 7) provide ample energetic potential so that alterations of R175 by different ligands could cause a 2.7 kcal/mol difference in binding enthalpy that is observed between cpYEEI and fpYEEI14.
The NMR chemical shift differences for residues 201 to 204 (Figure 5) also suggest variations in the interactions of the outer βD strand of Src SH2. As illustrated in Figure 7A, H201 has main-chain interactions with pY+1E and with a bound water molecule14, which has been shown to be energetically important in a series of computational investigations by Waksman and coworkers73–75. The simulations find that in the cpYEEI complex two populations exist for the distance of H201 carbonyl to pY+1E NH (Figure 8). This conformational averaging may be the origin of the chemical exchange line broadening exhibited by H201 NH during the titration of Src SH2 with cpYEEI, and lowers the stability of this interaction based on the energies calculated from the simulations (Table 2). Moreover, given that the trend in the chemical shifts of H201 HN follow the rank order of the binding enthalpies (see Results), H201 interactions could contribute to the loss of enthalpy with preorganization. Interestingly, this loss would be attributed to a presence of dynamic averaging for the restrained cpYEEI rather than a loss of mobility.
Long-range effects of constraint
Unlike the large CSD values of NH groups that interact directly with the phosphoryl moiety of the ligand, a precise structural explanation for the large CSDs in the central βC-strand residue L186 is less clear. Chemical shift differences in this region of p85N SH2 upon ligand binding were attributed to ring current effects34, but this explanation cannot account for the CSD observed in the Src complexes studied here given that L186 HN exhibits larger differences (0.7 to 2.3 ppm in δN, 0 to 0.04 ppm in δH) but is more distant from surrounding aromatic rings so that the ring currents would be considerably smaller. Also, δN is less susceptible to the influence of ring currents than δH75a, and the nitrogen chemical shift dominates the L186 CSD (Figure 6). Nitrogen chemical shifts are more complex and thus complicated to calculate compared to proton shifts, but they are particularly sensitive to changes in the φ dihedral angle, as well as the ψ and χ1 torsion angles of the preceding residue76, C185 in this case. The trend in ψ(C185), φ(L186), and χ1(C185) that are tabulated in Table 4 agree qualitatively with the variations in nitrogen chemical shifts in that the SH2-pYEEI and SH2-fpYEEI values are more similar to each other than to SH2-cpYEEI. The dihedral angle differences are less than the standard deviation but based on this consistent trend we suggest the CSDs could arise from small deviations in main-chain conformation of the central beta strands induced by the constraint.
Table 4.
Values from MD simulations for the dihedral angles with potential to affect the nitrogen chemical shift of L186.
| Src SH2-pYEEI | Src SH2-cpYEEI | Src SH2-fpYEEI | |
|---|---|---|---|
| L186 dihedrals | |||
| L186 δN(ppm) | 130.897 | 129.266 | 131.570 |
| ψ(C185)a | −143 (3) | 142 (3) | 144 (2) |
| φ(L186)a | −106 (4) | −105 (4) | −107 (2) |
| χ1(C185)a | −74 (43) | −95 (35) | −85 (41) |
time-averaged value in degrees with standard errors in parentheses
If the CSDs arise from small disparaties in ψ and χ1 torsion angles of residue 185 and in φ of 186 in the βC strand, we suggest the enthalpic impact of this local structural perturbation is likely negligible. Nevertheless, the chemical shift perturbations along the length of the βC and βD strands (residues 185, 186, 201–204) could reflect thermodynamic consequences in view of a coupling between the two binding pockets shown from calorimetric measurements of binding enthalpies77. Using a series of pYEEI variants binding to Src SH2, Waksman and coworkers reported a significant non-additivity between the pY+3 I residue, and either the pY+1 E (1.2 kcal/mol) or the pY+2 E (−1.6 kcal/mol) residues77. Long-distance effects are also apparent here from the disparate NH chemical shifts; S216 and T218 in the EF loop and G236 and L237 of the BG loop differ for the cpYEEI complex, whereas the complexes of the more flexible fpYEEI and pYEEI ligands have similar bound state chemical shifts (Figure 5). Long-range effects originating from the pY-pocket are also indicated by a recent comparison of Src SH2 crystallographic structures in the unbound state determined in the presence and absence of inorganic phosphate. A notable difference was the closing of the EF and BG loops in the solution structure without phosphate, leading to a less well-defined hydrophobic binding pocket54. We envision a pathway of correlated interactions starting with the side-chains of residues C185, S187, H201, and K203 on the pY face of the β-sheet (solid wedges in Figure 7) mediated through the hydrophobic-face β-sheet residues L186, Y202, and I204 (open wedges in Figure 7) to the BG loop via contacts between L186, Y202, and L237. While a structural path between the pY-pocket and the EF and BG loops on the opposite side of the SH2 domain is readily mapped out for the residues with CSDs, an interpretation of the large CSD values of the EF and BG loop is not obviously associated with the measured enthalpic differences as was the case for the trends in chemical shift deshielding for residues in the pY-pocket.
CONCLUSION
This report has detailed several interactions in the pY-pocket of the Src SH2 domain as a basis for entropy-enthalpy compensation of ligand binding by using a combination of information from NMR, MD simulations, crystallography, and calorimetry. From the perturbations observed in the chemical shifts of the Src pY-binding pocket, it is seen that addition of even a minor structural constraint is not a perfect mimic of the optimal geometry of binding. Small, almost ‘invisible’ changes in the bound state geometry appear to impact the binding energetics. These studies suggest that constraining a portion of a flexible ligand involved in hot-spot interactions with Src SH2 might be the origin of the weaker binding enthalpy observed for cpYEEI relative to fpYEEI and pYEEI.
In order to preorganize ligands without such an enthalpic penalty, it would appear necessary to closely match the binding site geometry of Src SH2. Alternatively, the introduction of a geometric constraint elsewhere in the ligand could yield improved entropy from preorganization without an enthalpic penalty resulting from an imperfect match of the targeted bound state geometry. For example, preorganization of the pY+3 side-chain, which is energetically forgiving to conservative mutations77,78, could provide the same entropic gain achieved with cpY but without requiring an ideal match of binding site geometry in order to not diminish key interactions of a ‘hot-spot’ region. In this context it is notable, perhaps perplexing, that use of the same cyclopropane-derived replacement at pY led to enhanced binding enthalpies without the expected gain in binding entropies for a series of Grb2 SH2 binding phosphotyrosine analogs10,11, thereby underscoring the difficulty in predicting binding thermodynamics from structure.
We find no evidence in this study that the compensating contributions in the measured enthalpy-entropy components arise from the same physical basis, and thus enthalpy-entropy compensation is not an intrinsic property of preorganization. That is, the reduced mobility from preorganization of the ligand leads to a more favorable binding entropy, but this reduced ligand mobility does not appear to be the origin of the less favorable binding enthalpy through an effect of the restraint on the conformational dynamics. As well as can be determined from the 15N relaxation measurements and MD simulations, protein internal dynamics are not significantly affected by the constrained ligand motion in a manner that accounts for energetic differences. The results therefore do not support the view that compensation occurs because the entropy and enthalpy are inexorably linked and therefore an unavoidable property of the system. Thus, achieving the anticipated gain in binding free energy by preorganization of ligand in the bound state conformation appears to remain a potentially viable design principle, although predicting whether ligand preorganization will have a favorable entropic and/or enthalpic consequence is problematic.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM 39478 and GM 84965), the National Science Foundation (CHE 0750329), the Markey Center for Structural Biology, the Purdue University Center for Cancer Research (CA 23568), The Robert A. Welch Foundation (F-652), and the Norman Hackerman Advanced Research Program for their generous support of this research. J. M. W. was supported by NIH Biophysics Training Grant GM 08296. We also thank David Cramer and James Myslinski for help in preparing the fpYEEI and cpYEEI ligands.
Footnotes
SUPPORTING INFORMATION AVAILABLE: Provided in supporting information are the complete list of authors for reference 45, Table S1, a compilation of CSP and CSD values for residues showing significant chemical shift changes, as well as published relative binding thermodynamic parameters for relevant mutants; Figure S1, an illustration of interfacial water molecules in the hydrophobic binding pocket; Figure S2, a plot of the distance dependence of the phosphoryl interaction energy calculated from MD simulations; and information on the CHARMM force field topology and parameters for the cpY and fpY residues. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lazaridis T. Curr Org Chem. 2002;6:1319–1332. [Google Scholar]
- 2.Gohlke H, Klebe G. Angew Chem Int Ed. 2002;41:2645–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 3.Henriques DA, Ladbury JE. Arch Biochem Biophys. 2001;390:158–168. doi: 10.1006/abbi.2001.2334. [DOI] [PubMed] [Google Scholar]
- 4.Mann A. In: The Practice of Medicinal Chemistry. 2. Wermuth CG, editor. Academic Press; London, UK: 2003. pp. 233–250. [Google Scholar]
- 5.Nakanishi H, Kahn M. Design of Peptidomimetics. In: Wermuth CG, editor. The Practice of Medicinal Chemistry. 2. Academic Press; London, UK: 2003. pp. 477–500. [Google Scholar]
- 6.Loughlin WA, Tyndall JDA, Glenn MP, Fairlie DP. Chem Rev. 2004;104:6085–6118. doi: 10.1021/cr040648k. [DOI] [PubMed] [Google Scholar]
- 7.Khan AR, Parrish JC, Fraser ME, Smith WW, Bartlett PA, James MNG. Biochemistry. 1998;37:16839–16845. doi: 10.1021/bi9821364. [DOI] [PubMed] [Google Scholar]
- 8.Schneider H-J. In: Protein-Ligand Interactions: From Molecular Recognition to Drug Design. Böhm H-J, Schneider G, editors. Wiley-VCH; Weinheim: 2003. pp. 21–50. [Google Scholar]
- 9.Reichelt A, Martin SF. Acc Chem Res. 2006;39:433–442. doi: 10.1021/ar030255s. [DOI] [PubMed] [Google Scholar]
- 10.Benfield AP, Teresk MG, Plake HR, Delorbe JE, Millspaugh LE, Martin SF. Angew Chem Int Ed. 2006;45:6830–6835. doi: 10.1002/anie.200600844. [DOI] [PubMed] [Google Scholar]
- 11.DeLorbe JE, Clements JH, Teresk MG, Benfield AP, Plake HR, Millspaugh LE, Martin SF. J Am Chem Soc. 2009;131:16758–167570. doi: 10.1021/ja904698q. [DOI] [PubMed] [Google Scholar]
- 12.Udugamasooriya DG, Spaller MR. Biopolymers. 2008;8:653–667. doi: 10.1002/bip.20983. [DOI] [PubMed] [Google Scholar]
- 13.Davidson JP, Martin SF. Tetrahedron Lett. 2000;41:9459–9464. [Google Scholar]
- 14.Davidson JP, Lubman O, Rose T, Waksman G, Martin SF. J Am Chem Soc. 2002;124:205–215. doi: 10.1021/ja011746f. [DOI] [PubMed] [Google Scholar]
- 15.Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J. Cell. 1993;72:779–790. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- 16.Rahuel J, Gay B, Erdmann D, Strauss A, Garcia-Echeverria C, Furet P, Caravatti G, Fretz H, Schoepfer J, Grütter MG. Nat Struct Biol. 1996;3:586–589. doi: 10.1038/nsb0796-586. [DOI] [PubMed] [Google Scholar]
- 17.Waksman G, Kumaran S, Lubman O. Expert Rev Mol Med. 2004;6:1–18. doi: 10.1017/S1462399404007331. and references therein. [DOI] [PubMed] [Google Scholar]
- 18.Lumry R, Rajender S. Biopolymers. 1970;9:1125–1227. doi: 10.1002/bip.1970.360091002. [DOI] [PubMed] [Google Scholar]
- 19.Dunitz JD. Chem Biol. 1995;2:709–712. doi: 10.1016/1074-5521(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 20.Searle MS, Westwell MS, Williams DH. J Chem Soc Perkin Trans 2. 1995;1995:141–151. [Google Scholar]
- 21.Gallicchio E, Kubo MM, Levy RM. J Am Chem Soc. 1998;120:4526–4527. [Google Scholar]
- 22.Sharp K. Protein Sci. 2001;10(3):661–667. doi: 10.1110/ps.37801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilli P, Ferretti V, Gilli G, Borea PA. J Phys Chem. 1994;98:1515–1518. [Google Scholar]
- 24.Ford DM. J Am Chem Soc. 2005;127:16167–16170. doi: 10.1021/ja054519c. [DOI] [PubMed] [Google Scholar]
- 24a.Post CB, Dobson CM, Karplus M. In: Computer Modeling of Carbohydrate Molecules. French AD, Brady JW, editors. ACS; Washington, DC: 1990. pp. 377–388. [Google Scholar]
- 25.Karplus M, McCammon JA. Nat Struct Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 26.Daniel R, Dunn R, Finney J, Smith J. Annu Rev Biophys Biomol Struct. 2003;32:69–92. doi: 10.1146/annurev.biophys.32.110601.142445. [DOI] [PubMed] [Google Scholar]
- 27.Kay L. Biochem Cell Biol. 1998;76:145–152. doi: 10.1139/bcb-76-2-3-145. [DOI] [PubMed] [Google Scholar]
- 28.Mittermaier A, Kay L. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 29.Spyracopoulos L, Sykes BD. Curr Opin Struct Biol. 2001;11:555–559. doi: 10.1016/s0959-440x(00)00261-x. [DOI] [PubMed] [Google Scholar]
- 30.Karplus M, Ichiye T, Pettit BM. Biophys J. 1987;52:1083–1085. doi: 10.1016/S0006-3495(87)83303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brooks B, Karplus M. Proc Natl Acad Sci USA. 1983;80:6571–6575. doi: 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tidor B, Karplus M. J Mol Biol. 1994;238:405–414. doi: 10.1006/jmbi.1994.1300. [DOI] [PubMed] [Google Scholar]
- 33.Stöckmann H, Bronowska A, Syme NR, Thompson GS, Kalverda AP, Warriner SL, Homans SW. J Am Chem Soc. 2008;130:12420–12426. doi: 10.1021/ja803755m. [DOI] [PubMed] [Google Scholar]
- 34.Günther UL, Liu Y, Sanford D, Bachovchin WW, Schaffhausen B. Biochemistry. 1996;35:15570–15581. doi: 10.1021/bi961783x. [DOI] [PubMed] [Google Scholar]
- 35.Waksman G, Kominos D, Robertson SC, Pant N, Baltimore D, Birge RB, Cowburn D, Hanafusa H, Mayer BJ, Overduin M, Resh MD, Rios CB, Silverman L, Kuriyan J. Nature. 1992;358:646–653. doi: 10.1038/358646a0. [DOI] [PubMed] [Google Scholar]
- 36.Xu RX, Word JM, Davis DG, Rink MJ, Willard DH, Gampe RT. Biochemistry. 1995;34:2107–2121. doi: 10.1021/bi00007a003. [DOI] [PubMed] [Google Scholar]
- 37.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 38.Kneller DG, Kuntz ID. J Cell Biochem. 1993;53:254–254. [Google Scholar]
- 39.Jung Y-SZM. J Biomol NMR. 2004;30:11–23. doi: 10.1023/B:JNMR.0000042954.99056.ad. [DOI] [PubMed] [Google Scholar]
- 40.Muhandiram DR, Kay LE. J Magn Reson. 1994;103:203–216. [Google Scholar]
- 41.Taylor JD, Fawaz RR, Ababou A, Williams MA, Ladbury JE. J Biomol NMR. 2005;32:339. doi: 10.1007/s10858-005-0471-7. [DOI] [PubMed] [Google Scholar]
- 42.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 43.Schleucher J, Schwendinger M, Sattler M, Schmidt P, Schedletzky O, Glaser SJ, Sørensen OW, Griesinger C. J Mol Biol. 1994;4:301–306. doi: 10.1007/BF00175254. [DOI] [PubMed] [Google Scholar]
- 44.Garrett DS, Seok Y-J, Peterkofsky A, Clore GM, Gronenborn AM. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
- 45.Brooks BR, et al. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacKerell A, Jr, Bashford D, Bellott M, Dunbrack R, Jr, Evanseck J, Field M, Fischer S, Gao J, Guo H, Ha S. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 47.MacKerell AD, Jr, Feig M, Brooks CL., III J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 48.Johnson RD., III NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101, Release 14. 2006 Sept; http://srdata.nist.gov/cccbdb.
- 49.Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J. Cell. 1993;72:779–790. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- 50.Caves LSD, Evanseck JD, Karplus M. Protein Sci. 1998;7:649–666. doi: 10.1002/pro.5560070314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen Y, Bax A. J Biomol NMR. 2007;38:289–302. doi: 10.1007/s10858-007-9166-6. [DOI] [PubMed] [Google Scholar]
- 52.De Loof H, Nilsson L, Rigler R. J Am Chem Soc. 1992;114:4028–4035. [Google Scholar]
- 53.Lipari G, Szabo A. J Am Chem Soc. 1981;104:4546–4559. [Google Scholar]
- 54.Taylor JD, Ababou A, Fawaz RR, Hobbs CJ, Williams MA, Ladbury JE. Proteins. 2008;73:929–940. doi: 10.1002/prot.22119. [DOI] [PubMed] [Google Scholar]
- 55.Williamson RA, Carr MD, Frenkiel TA, Feeney J, Freedman RB. Biochemistry. 1997;36:13882–13889. doi: 10.1021/bi9712091. [DOI] [PubMed] [Google Scholar]
- 56.Ma J, Gruschus JM, Tjandra N. J Am Chem Soc. 2009;131:9884–9885. doi: 10.1021/ja903552q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Groesch T, Zhou F, Mattila S, Geahlen R, Post C. J Mol Biol. 2006;356:1222–1236. doi: 10.1016/j.jmb.2005.11.095. [DOI] [PubMed] [Google Scholar]
- 58.Eck MJ, Shoelson SE, Harrison SC. Nature. 1993;362:87–91. doi: 10.1038/362087a0. [DOI] [PubMed] [Google Scholar]
- 59.Amaro RE, Sethi A, Myers RS, Davisson VJ, Luthey-Schulten ZA. Biochemistry. 2007;46:2156–2173. doi: 10.1021/bi061708e. [DOI] [PubMed] [Google Scholar]
- 60.Sethi A, Eargle J, Black A, Luthey-Schulten Z. Proc Natl Acad Sci USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lange G, Lesuisse D, Deprez P, Schoot B, Loenze P, Bénard D, Marquette J-P, Broto P, Sarubbi E, Mandine E. J Med Chem. 2003;46:5184–5195. doi: 10.1021/jm020970s. [DOI] [PubMed] [Google Scholar]
- 62.Bradshaw JM, Mitaxov V, Waksman G. J Mol Biol. 1999;293:971–985. doi: 10.1006/jmbi.1999.3190. [DOI] [PubMed] [Google Scholar]
- 63.Bradshaw JM, Mitaxov V, Waksman G. J Mol Biol. 2000;299:521–535. doi: 10.1006/jmbi.2000.3765. [DOI] [PubMed] [Google Scholar]
- 64.Dwyer JJ, Dwyer MA, Kossiakoff AA. Biochemistry. 2001;40:13491–13500. doi: 10.1021/bi011703b. [DOI] [PubMed] [Google Scholar]
- 65.Clackson T, Wells JA. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 66.Wagner G, Pardi A, Wuethrich K. J Am Chem Soc. 1983;105:5948–5949. [Google Scholar]
- 67.Wishart DS, Sykes BD, Richards FM. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 68.Cui Q, Karplus M. J Phys Chem B. 2000;104:3721–3743. [Google Scholar]
- 69.Asakawa N, Sato D, Sakurai M, Inoue Y. J Phys Chem A. 2000;104:2716–2723. [Google Scholar]
- 70.Mandell DJ, Chorny I, Groban ES, Wong SE, Levine E, Rapp CS, Jacobson MP. J Am Chem Soc. 2007;129:820–827. doi: 10.1021/ja063019w. [DOI] [PubMed] [Google Scholar]
- 70a.Brünger AT. Methods Enzymol. 1996;277:366–396. doi: 10.1016/s0076-6879(97)77021-6. [DOI] [PubMed] [Google Scholar]
- 71.Parker LL, Houk AR, Jensen JH. J Am Chem Soc. 2006;128:9863–9872. doi: 10.1021/ja0617901. [DOI] [PubMed] [Google Scholar]
- 72.Singer AU, Forman-Kay JD. Protein Sci. 1997;6:1910–1919. doi: 10.1002/pro.5560060912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Fabritiis G, Geroult S, Coveney PV, Waksman G. Proteins. 2008;72:1290–1297. doi: 10.1002/prot.22027. [DOI] [PubMed] [Google Scholar]
- 74.Geroult S, Virdee S, Waksman G. Chem Biol Drug Design. 2006;67:38–45. doi: 10.1111/j.1747-0285.2005.00315.x. [DOI] [PubMed] [Google Scholar]
- 75.Geroult S, Hooda M, Virdee S, Waksman G. Chem Biol Drug Design. 2007;70:87–99. doi: 10.1111/j.1747-0285.2007.00545.x. [DOI] [PubMed] [Google Scholar]
- 75a.Neal S, Nip AM, Zhang H, Wishart DS. J Biomol NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 76.Le H, Oldfield E. J Phys Chem. 1996;100:16423–16428. [Google Scholar]
- 77.Bradshaw JM, Waksman G. Biochemistry. 1999;38:5147–5154. doi: 10.1021/bi982974y. [DOI] [PubMed] [Google Scholar]
- 78.Bradshaw JM, Waksman G. Biochemistry. 1998;37:9083–9090. doi: 10.1021/bi973147k. [DOI] [PubMed] [Google Scholar]
- 79.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.