

Abstract
Cocaine self-administration alters patterns of gene expression in the brain that may underlie cocaine-induced neuronal plasticity. In the present study, male Sprague Dawley rats were allowed to self-administer cocaine (0.25 mg/infusion) 2 h/d for 14 d, followed by 7 d of forced abstinence. Compared with yoked saline control rats, cocaine self-administration resulted in increased brain-derived neurotrophic factor (BDNF) protein levels in the rat medial prefrontal cortex (mPFC). To examine the functional relevance of this finding, cocaine self-administration maintained under a progressive ratio schedule of reinforcement was assessed after short hairpin RNA-induced suppression of BDNF expression in the mPFC. Decreased BDNF expression in the mPFC increased the cocaine self-administration breakpoint. Next, the effect of cocaine self-administration on specific BDNF exons was assessed; results revealed selectively increased BDNF exon IV-containing transcripts in the mPFC. Moreover, there were significant cocaine-induced increases in acetylated histone H3 (AcH3) and phospho-cAMP response element binding protein (pCREB) association with BDNF promoter IV. In contrast, there was decreased methyl-CpG-binding protein 2 (MeCP2) association with BDNF promoter IV in the mPFC of rats that previously self-administered cocaine. Together, these results indicate that cocaine-induced increases in BDNF promoter IV transcript in the mPFC are driven by increased binding of AcH3 and pCREB as well as decreased MeCP2 binding at this BDNF promoter. Collectively, these results indicate that cocaine self-administration remodels chromatin in the mPFC, resulting in increased expression of BDNF, which appears to represent a compensatory neuroadaptation that reduces the reinforcing efficacy of cocaine.
Introduction
Repeated intake of drugs of abuse, such as cocaine, promotes alterations in gene expression that underlie addiction as well as compensatory neuroadaptations (Wise, 2004; Kalivas, 2005; Nestler, 2005; Shaham and Hope, 2005; Pierce and Kumaresan, 2006). The most relevant of these cocaine-induced changes in gene expression occur in the ventral tegmental area (VTA), a major locus of dopamine neurons, as well as nuclei receiving dopaminergic inputs from the VTA, including the medial prefrontal cortex (mPFC), nucleus accumbens, and amygdala. A growing body of evidence indicates that cocaine alters the expression of neurotrophic factors. For example, previous studies have shown that an acute injection of cocaine increases brain-derived neurotrophic factor (BDNF) mRNA in the striatum, mPFC, and nucleus accumbens (Le Foll et al., 2005; Liu et al., 2006; Berglind et al., 2007). Similarly, a sensitizing regimen of cocaine injections or cocaine self-administration results in increased BDNF mRNA in the nucleus accumbens and striatum (Le Foll et al., 2002; Zhang et al., 2002; Kumar et al., 2005; Graham et al., 2007).
There are functional consequences of increases in BDNF transcription in various limbic nuclei. Three once-daily microinjections of BDNF into the VTA progressively augments BDNF-induced behavioral hyperactivity (Pierce et al., 1999). Moreover, continuous infusion of BDNF into the VTA or substantia nigra enhances the behavioral hyperactivity induced by an acute injection of cocaine (Martin-Iverson et al., 1994; Martin-Iverson and Altar, 1996; Horger et al., 1999). Repeated administration of BDNF into the nucleus accumbens augments the reinstatement of cocaine seeking (Graham et al., 2007). In addition, the time-dependent increases in cue-induced cocaine seeking is paralleled by increases in BDNF levels in the VTA, nucleus accumbens, and amygdala over the first 90 d after cocaine self-administration (Grimm et al., 2003). Together, these results indicate that alterations in BDNF transcription in limbic nuclei have differential effects on cocaine-induced behavioral plasticity, including the reinstatement of cocaine seeking.
Recently, it was shown that exogenous BDNF infusions into the mPFC attenuate the reinstatement of cocaine seeking (Berglind et al., 2007, 2009; McGinty et al., 2010). Here, we report that cocaine self-administration increased BDNF protein and mRNA levels in the mPFC. To determine the functional relevance of this finding, the influence of viral-mediated decreases of BDNF levels in the mPFC on cocaine self-administration maintained under a progressive ratio (PR) schedule of reinforcement was evaluated. In addition, chromatin immunoprecipitation (ChIP) was used to identify the molecular mechanisms underlying the increase in BDNF mRNA in the mPFC associated with cocaine self-administration.
Materials and Methods
Animals and housing.
Male Sprague Dawley rats (Rattus norvegicus) weighing 250–300 g were obtained from Taconic Farms. Animals were individually housed with food and water available ad libitum. A 12 h light/dark cycle was used with the lights on at 7:00 A.M. All experimental procedures were performed during the light cycle. A total of 107 rats were used in this study.
Surgery.
Before surgery, the rats were anesthetized with 80 mg/kg ketamine and 12 mg/kg xylazine intraperitoneally. An indwelling Silastic catheter was placed into the right jugular vein (side opposite the heart) and sutured in place. The catheter was routed to a screw-on mount (Plastics One) that was sutured below the skin between the shoulder blades. The catheters were sealed with plastic obturators when not in use. After catheter implantation, some rats were mounted in a stereotaxic apparatus (David Kopf Instruments), and a viral vector (2 μl/side) was administered at the borders between the infralimbic/prelimbic cortices [+2.5 mm anteroposterior (A/P), ±0.5 mm mediolateral (M/L), and ±4.5 mm dorsoventral (D/V)] and prelimbic cortex/anterior cingulate (+2.5 mm A/P, ±0.5 mm M/L, and ±3.0 mm D/V) via 33 gauge cannulae. All coordinates were relative to bregma according to the atlas of Paxinos and Watson (1997). Thus, each rat was microinjected with a 4 μl of virus bilaterally into the infralimbic cortex, prelimbic cortex, and anterior cingulate, which collectively form the mPFC in the rat. Each 2 μl microinjection was made over 10 min, and the microinjector was left in place for 2 min after the infusion.
Self-administration training.
After a 10–14 d [for short hairpin RNA (shRNA)] or 7 d (all other experiments) recovery period from surgery, the rats were placed in operant chambers and allowed to lever press for intravenous cocaine infusions (0.25 mg of cocaine/56 μl of saline per infusion over 5 s) for 14 d. Rats initially were trained using a fixed ratio (FR1) schedule of reinforcement with each daily self-administration session initiated by an intravenous priming injection of cocaine (0.25 mg). When the animals achieved stable responding with the FR1 schedule (i.e., <15% variation in response rates over 3 consecutive days), they were switched to an FR5 schedule for 14 consecutive days of self-administration. A 60 s timeout period during which responses have no scheduled consequences followed each cocaine infusion. The rats were limited to a maximum of 30 cocaine infusions per daily 2 h self-administration session.
Progressive ratio.
For those animals in cocaine reinforcement experiments (using the progressive ratio schedule), when stable responding with the FR5 schedule was achieved, as defined by a <15% change in the number of responses on 2 consecutive days, the rats were switched to a progressive ratio schedule of reinforcement. The progressive ratio schedule is based on that of Richardson and Roberts (1996). The response requirement for the ith reinforcement was given by R(i) = [5e0.2i − 5], where the brackets indicate the rounding function. Using this exponential function, the response requirement for the first 10 injections was as follows: 1, 2, 4, 6, 9, 11, 15, 19, 25, 31 (etc.). Thus, under a PR schedule, the response requirement for each subsequent drug delivery increased until the subject failed to meet a requirement. The session expired when an animal took more than 1 h to receive an injection. The breakpoint was operationally defined as the number of cocaine infusions administered before the termination of the session.
Adeno-associated viral cloning and packaging.
The enhanced green fluorescent protein (EGFP)–U6–pACP plasmid contains adeno-associated viral 2 (AAV2) inverted terminal repeats flanking a cytomegalovirus promoter, the coding sequence for EGFP, an intron and polyadenylation signal derived from simian virus 40, and, further downstream, a murine U6 pol III promoter. To obtain each AAV–shRNA, synthetic oligos encoding the shRNA and its respective complement (Integrated DNA Technologies) were annealed and ligated into unique BbsI and NheI sites in the AAV plasmid downstream of the U6 promoter. The BDNF mRNA target sequence used, ACCATAAGGACGCGGACTTGT (from National Center for Biotechnology Information reference sequence NM_012513), was selected after in vitro screening of multiple candidates (C. E. Bass and E. F. Terwilliger, unpublished observations). Control vectors included AAV10 encoding either EGFP alone or AAV10 encoding EGFP plus a scrambled shRNA (CTGTTACGCTGGCTCTATCGA), which does not correspond to any known rat mRNA sequence. Packaging of all AAV was performed using a standard triple transfection protocol to generate helper virus-free pseudotyped AAV10 virus (Xiao et al., 1998). An AAV2/10 rep/cap plasmid provided AAV2 replicase and AAV10 capsid functions (Gao et al., 2002; De et al., 2006), whereas adenoviral helper functions were supplied by the pHelper plasmid (Stratagene). Briefly, AAV–293 cells were transfected with 1.33 pmol of pHelper and 1.15 pmol each of AAV2/10 and the shRNA–AAV vector plasmids, via calcium phosphate precipitation. The cells were harvested 48 h later, and the pellets were resuspended in DMEM, freeze thawed three times, and centrifuged to produce a clarified viral lysate. The vector stocks were titered by real-time PCR using the ABI Prism 7700 Sequence Detection System from PerkinElmer Applied Biosystems as described previously (Clark et al., 1999). The average titer of the preparations was ∼1 × 1012 vector genomes/ml.
Immunohistochemistry.
Ten days after rats received unilateral microinjections with AAV-viral vector containing EGFP-linked scrambled or EGFP-linked BDNF shRNA, animals were perfused with Formalin, and brains were postfixed in 10% Formalin for 24 h and then cryoprotected in sucrose before freezing at −80°C. Sections, 40 μm, were obtained using a cryostat and stored in a solution of 7% sucrose/20% ethylene glycol in 0.1 m PBS. For BDNF immunohistochemistry, free-floating sections were washed in 0.1 m PBS for 1 h. Antigen retrieval was performed by heating sections in 10 mm sodium citrate, pH 6.0, at 80°C for 15 min. The sections were permeabilized in 0.5% Triton X-100 for 20 min and then incubated with 10% normal goat serum for 1 h before overnight incubation at 4°C with a 1:250 dilution of anti-BDNF polyclonal antibody (ab1779; Millipore Bioscience Research Reagents) prepared in 1× GDB buffer (1% gelatin solution, 5% Triton X-100). After washing in 0.5% Triton X-100 for 1 h, sections were incubated in a 1:1000 dilution of cyanine 3 goat anti-rabbit IgG in 1× GDB for 1 h at room temperature. Sections were washed in 0.1 m PBS for 1 h and mounted with Vectashield (Vector Laboratories). BDNF immunoreactivity and GFP expression were examined with an Olympus BX60 fluorescence microscope, images were acquired, and intensity of staining was measured using NIH ImageJ software.
Western blotting.
For Western blot analysis, histone extracts from dissected rat mPFC were used, and the experiments were performed as described previously (Sadri-Vakili et al., 2007). Briefly, mPFC from both hemispheres of a cocaine or yoked rat was homogenized in 200 μl of lysis buffer [1% Nonidet P-40, 20 mm Tris, pH 8.0, 137 mm NaCl, 10% glycerol, 1 mm PMSF, sodium butyrate 1 mm, and protease inhibitors (PIs)] at 4°C. After removal of cellular debris by centrifugation, the supernatant was collected, and protein levels in the lysates were measured by the Bradford assay (Bio-Rad). Ten to 20 μg of each sample was boiled in the presence of sample buffer for 5 min before separation on 10–20% SDS- polyacrylamide gel, and proteins were transferred to nitrocellulose membranes. The immunoblots were blocked with 5% nonfat dry milk dissolved in Tris-buffered saline containing 0.2% Tween 20 (TBST) for 60 min. The membranes were then incubated overnight at 4°C with specific antibodies that included the following: anti-di acetyl lysine 9 and lysine 14 histone H3 (AcH3) antibody (Millipore Corporation) at a dilution of 1:1500, anti-histone H3 antibody (Millipore Corporation) at a dilution of 1:500, anti- phospho-cAMP response element binding protein (pCREB) (Abcam) at a dilution of 1:500, and anti-methyl-CpG-binding protein 2 (MeCP2) (Abcam). Primary antibody incubation was followed by six washes (10 min, rocking, room temperature) in TBST before incubation with the secondary antibody (HRP-conjugated goat anti-rabbit IgG; Jackson ImmunoResearch), six washes, and visualization using the ECL detection system (PerkinElmer Life and Analytical Sciences).
ELISA.
Ten days after rats received unilateral microinjections in the medial prefrontal cortex with AAV10 vector containing EGFP-linked shRNA targeting BDNF, four animals were killed, and bilateral medial prefrontal cortices were dissected. Tissues were lysed and homogenized and diluted to 10 and 20 μg/μl. The concentration of BDNF in the diluted lysates was quantified using the ChemiKine Brain Derived Neurotrophic Factor (BDNF) Sandwich ELISA kit (catalog #CYT306; Millipore Bioscience Research Reagents). The brain tissue samples and serial dilutions of BDNF standards were loaded in triplicate onto a microplate coated with rabbit anti-human BDNF polyclonal antibodies and incubated overnight at 4°C. After four washes, biotinylated mouse anti-human BDNF monoclonal antibody (1:1000) was added to the microplate for 2.5 h at room temperature. The plates were washed four times and the streptavidin-enzyme conjugate was added to the plate and allowed to incubate for 1 h. After additional washing, tetramethylbenzidine chromagenic substrate was added, and then 15 min later the reaction was stopped. The absorbance at 450 nm was measured with a plate reader. BDNF concentration in the brain tissue samples was measured by comparing values with the prepared standard curve.
Chromatin immunoprecipitation assay.
We have adapted the ChIP technique to the analysis of brain tissue and have recently published detailed methodology for performing ChIP experiments (Braveman et al., 2004; Chen-Plotkin et al., 2006: Sadri-Vakili et al., 2007). Briefly, mPFC from both hemispheres of an individual cocaine or yoked rat was cut into pieces, and the pieces were weighed (<30 mg) and deposited into tubes. Formaldehyde was added (10 μl of 1% formaldehyde to 1 mg of tissue), and brain pieces were incubated for 10 min at 37°C to cross-link DNA to associated proteins. Brain tissue was washed twice with ice-cold PBS containing PIs (Complete Mini protease inhibitor cocktail tablets; Roche) and then suspended in SDS lysis buffer (1% SDS, 10 mm EDTA, and 50 mm Tris-HCl, pH 8.1) containing PIs at a ratio of 10 μl of buffer for each milligram of brain. After incubating on ice for 10 min, brain lysates were sonicated to shear lengths of 200–1000 bp DNA fragments using 10 sets of 10–15 s pulses at setting 3 of a sonicator (Branson Cell Disruptor 350). We have found that this regimen yields DNA fragments appropriate for ChIP (200–1000 bp) (Braveman et al., 2004). The resulting homogenates from one brain were pooled and then centrifuged for 10 min at 13,000 × g at 4°C to remove debris, and 200 μl aliquots of the suspension were placed into separate sample tubes. Each sample was diluted tenfold with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1, and 167 mm NaCl), and 20 μl (1%) of the diluted sample was set aside and designated “Input” DNA. Samples were then either processed immediately or stored at −80°C.
For immunoprecipitation (IP), 2000 μl of each sample was precleared with 80 μl of salmon sperm DNA/protein A-agarose 50% slurry (Millipore Corporation) by incubating at 4°C for 30 min with gentle agitation before overnight incubation (4°C) with 5 μg of each of the following antibodies: pCREB, MeCP2, AcH3, or 5-methyl-cytosine (Millipore Corporation). Negative controls included no antibody mock and IgG (Jackson ImmunoResearch). Mock immunoprecipitation conditions (mouse IgG) were also included as a control. After immunoprecipitation, 80 μl of the salmon sperm DNA/protein A-agarose 50% slurry was added to samples, and immunocomplexes were collected for 1 h at 4°C on a rocking platform. After pelleting agarose (1000 rpm, 4°C, 2 min), the chromatin-antibody/protein A-agarose complexes were washed sequentially (4 min each on a rotating platform) with 1 ml each of the following: a low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 150 mm NaCl), a high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 500 mm NaCl), an LiCl buffer (0.25 m LiCl, 1% NP-40, 1% deoxycholate, 1 mm EDTA, and 10 mm Tris-HCl, pH 8.1), and a TE buffer (10 mm Tris-HCl, 1 mm EDTA, pH 8.0). Elution was performed by incubation in 250 μl of freshly made elution buffer (1% SDS, 0.1 m NaHCO3) for 15 min at room temperature; elution was repeated and eluates were combined. Cross-links were then reversed by addition of 20 μl of 5 m NaCl to the pooled eluates and heating of the mixture to 65°C for 4 h. The saved input DNA was diluted to a volume of 500 μl with sterile water, and cross-link reversal was performed in the same manner as for the ChIP samples. After cross-link reversal, all samples (ChIP and Input) were digested with 20 μg of proteinase K (1 h, 45°C), and DNA was recovered with phenol/chloroform extraction and ethanol precipitation. DNA pellets were resuspended in 25 μl of sterile water.
One microliter (1 or 4 μl for human samples) of ChIP-derived DNA was used as template in 20 μl reactions containing 10 μl of 2× SYBR Green Master Mix (Applied Biosystems) and 0.5 μm of each primer. Real-time thermal cycling was performed using an iCycler thermal cycler (Bio-Rad), with continuous SYBR Green monitoring according to the recommendations of the manufacturer, using iCycler software. Cycling parameters for all amplifications were as follows: 60 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 45 s, followed by melt-curve analysis (55°C+ for 10 s for 80 cycles). All PCR reactions were performed in triplicate and included negative controls (no DNA) as well as positive controls (serial dilutions of known amounts of genomic DNA).
Target DNA sequence quantities were estimated from threshold amplification cycle numbers (Tc) using iCycler software. For every gene sequence studied, a ΔTc value was calculated for each sample by subtracting the Tc value for the immunoprecipitated sample from the Tc value for the corresponding Input DNA to normalize for differences in ChIP sample aliquots before immunoprecipitation. DNA quantities were then expressed as percentages of corresponding Input using the following equation: (antibody ChIP as a percentage of Input) = 2(ΔTc) × 100. Finally, DNA quantities (normalized to Input) were compared for immunoprecipitated versus mock-immunoprecipitated samples; only when immunoprecipitated samples contained >1.5 times as much DNA were they considered to have sufficient DNA for analysis.
Input and IP samples were interrogated with gene promoter-specific primers in triplicate reactions in real-time PCR analysis as described previously (Braveman et al., 2004; Chen-Plotkin et al., 2006; Sadri-Vakili et al., 2007). Threshold amplification cycle numbers (Tc) using iCycler software were used to calculate IP DNA quantities as percentage of corresponding inputs. The following exon-specific BDNF primers were designed based on previously published sequences (Chen et al., 2003; Martinowich et al., 2003; Jiang et al., 2008) and used for real-time PCR analysis: BDNF exon I, forward, 5′-GCAGTTGGACAG TCATTGGTAACC-3′; reverse, 5′-ACGCAAACGCCCTCATTCTG-3′; BDNF exon II (a, b, c), forward, 5′-GCAGAGTCCATTCAGCACCTTG-3′; reverse, 5′-TGGCTTGACAGCGAGGAAAAG-3′; BDNF exon IV (CREB and MeCP2 binding sites), forward, 5′-AACAAGAGGCTGTGACAC TATGCTC-3′; reverse, 5′-CAGTAAGTAAAGGCTAGGGCAGGC-3′; forward, 5′-AAAGCATGCAATGCCCT-3; reverse, 5′-GAGATTTCATGCTAGCTCGC-3′; forward, 5′-GGCTTCTGTGTGCGTGAATTTGC-3′; reverse, 5′-AAAGTGGGTGGGAGTCCACGAG-3′; BDNF exon IV, forward, 5′-TTTGGGGCAGACGAGAAAGC-3′; reverse, 5′-GGCAGTGGAGTCACATTGTTGTC-3′.
DNA quantitation.
ChIP products were quantified using the luciferase-based and T4 DNA polymerase-based DNA Quantitation System (Promega) according to the instructions of the manufacturer. The light output was immediately measured in a luminometer (TD-20/20 Luminometer; Turner Designs). ChIP products were also quantified using Quant-iT PicoGreen dsDNA reagent (Invitrogen) and assayed on the Wallac Victor 1420 Multilabel Counter (PerkinElmer Life and Analytical Sciences) with excitation and emission wavelengths of 485 and 535 nm, respectively.
RNA extraction and reverse transcription.
RNA was extracted from dissected brain regions from one hemisphere of cocaine or yoked rats using RNeasy kit (Qiagen) according to the instructions of the manufacturer and as described previously (Sadri-Vakili et al., 2007). Reverse transcription (RT) reactions were performed using the Superscript First Strand Synthesis System for RT-PCR reactions (Invitrogen) using specific primers to quantitate the amount of gene expression compared with a standard curve. The following primers were used for real-time PCR to quantitate the amount of gene expression. The exon-specific BDNF primers were designed based on previously published results (Nakayama et al., 1994; Chen et al., 2003; Martinowich et al., 2003: Jiang et al., 2008). BDNF exon I, forward, 5′-AAGCCGAACTTCTCACATGATGA-3′; reverse, 5′-TGCAACCGAAGTATGAAATAACCATAG-3′; BDNF exon II (a, b, c), forward, 5′-GCAGAGTCCATTCAGCACCTT G-3′; reverse, 5′-TGGCTTGACAGCGAGGAAAAG-3′; BDNF exon IV, forward, 5′-CTGCCTAGATCAAATGGAGCTTCT-3′; reverse, 5′-GGAAATTGCATGGCGGAGGTAA-3′; BDNF exon IV, forward, 5′-TTTGGGGCAGACGAGAAAGC-3′; reverse, 5′-GGCAGTGGAGTCACATTGTTGTC-3′; CB1, forward, 5′-CCTCTACGTGGGCTCAAATG-3′; reverse, 5′-GGAAGGGACTACCCCTGAAG-3′; DRD2, forward, 5′-GGTCTGCAAAGCCTTCTCTC-3′; reverse, 5′-TACTATGCCATGCTGCTCAC-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), forward, 5′-AACAGCAACTCCCATTC TTC-3′; reverse, 5′-TGGTCCAGGGTTTCTTACTC-3′; GluR1, forward, 5′-ATGCTGACCTCCTTCTGTGG-3′; reverse, 5′-TCCTGTAGTTCCGGGCGTAG-3′; GluR2, forward, 5′-ATTTCGGGTAGGGATGGTTC-3′; reverse, 5′-GCGAAACTGTTGGCTACCTC; mGluR5, forward, 5′-GCACAGTCCAGTGAGAGGAG-3′; reverse, 5′-TTGTCCACAGTTGGTTGGTG-3′; NR1, forward, 5′-TAGTTAGCCACCCGCCTAC-3′; reverse, 5′-GACATTCGGGTAGTCAGTCC-3′; preprodynorphin, forward, 5′-CTGCACACAGGGAACACAAG-3′; reverse 5′-TGATACAGAATGGCGTGGTC; preproenkephalin, forward, 5′-GCTTGGGTGTTCTGCTTCTC-3′; reverse, 5′-TCCAGGGTTCT TAGTGCTGG-3′; SAP97, forward, 5′-ACCCGTGGACATTCTCAATC-3′; reverse, 5′-CGGTATCAGGACGAAGAGG-3′. Quantitative real-time PCR was performed in an iCycler (Bio-Rad) with the use of SYBR green PCR Master Mix (Applied Biosystems) through 50 PCR cycles (95°C for 30 s, 57°C for 60 s, 72°C for 90 s). The threshold cycle for each sample was chosen from the linear range and converted to a starting quantity by interpolation from a standard curve run on the same plate for each set of primers. The mRNA levels were normalized for each well to the GAPDH mRNA levels. Single PCR products were verified by assessing that the melting temperature of the product had a single value.
Results
Cocaine self-administration increases BDNF protein levels in the mPFC
After 14 d of cocaine self-administration and 7 d of forced abstinence, the animals were killed and the mPFC was removed. Figure 1A is a diagram depicting a coronal section of the rat brain at the level of the mPFC (3.20 mm anterior to bregma) (Paxinos and Watson, 1997). The mPFC dissection (shown in black) included the anterior cingulate, prelimbic, and infralimbic cortices. There was a significant increase in BDNF protein levels in the mPFC (relative to yoked saline controls) as measured by Western blot analysis [t(6) = 3.753, p < 0.0095 (Fig. 1B)] and ELISA [t(5) = 6.727, p < 0.0011 (Fig. 1C)].
Figure 1.
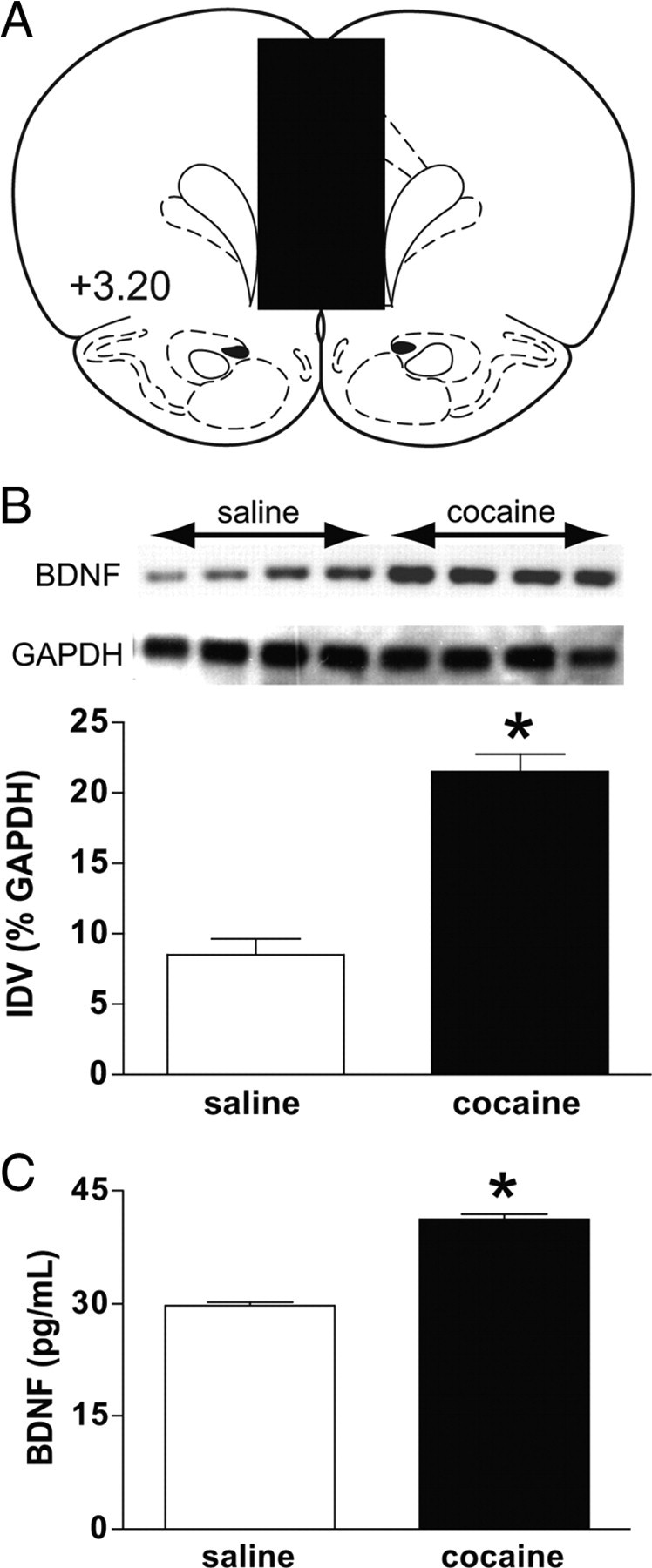
BDNF protein levels are increased in the mPFC after cocaine self-administration. A, A diagram depicting a coronal section of the rat brain at the level of the mPFC (3.20 mm anterior to bregma) (Paxinos and Watson, 1997). The mPFC dissection (shown in black) included the anterior cingulate, prelimbic, and infralimbic cortices. B, Western blot analysis revealed an increase in BDNF protein levels in mPFC. Densitometry results demonstrate a significant increase in BDNF levels in mPFC (saline, n = 4; cocaine, n = 4). C, Similarly, an ELISA demonstrated a significant increase in BDNF levels in the mPFC (saline, n = 4; cocaine, n = 4). IDV, Integrated density value. *p < 0.05, significant differences between saline and cocaine treatments (unpaired t tests,).
Decreasing BDNF in the mPFC enhances the reinforcing efficacy of cocaine
To assess whether this cocaine-induced increase in BDNF levels in the mPFC has functional consequences, we used a viral vector to deliver interfering RNA to the mPFC to suppress BDNF mRNA levels. In initial experiments, adeno-associated viral vectors (AAV10) encoding EGFP together with shRNA targeting BDNF exon IX, which is common to all BDNF transcripts, or a scrambled sequence that is not found in any known mRNA (scrambled control) were microinjected into the mPFC using stereotaxic surgery. Ten days after treatment with AAV10–BDNF shRNA, there was an ∼58% decrease in BDNF staining intensity per cell in the mPFC as measured by immunohistochemistry (Fig. 2), which was statistically significant (t(5) = 3.683, p < 0.0142). Six rats were used in this control experiment, and a total of 380 cells from the control side were compared with 231 cells from AAV10–BDNF shRNA-treated mPFC in these animals. The reduction in BDNF levels persisted for at least 3 weeks (data not shown). Although precise quantification of immunohistochemistry is problematic, these data indicate that there was extensive transfection of the AAV10–BDNF shRNA in the mPFC that clearly suppressed BDNF expression. However, it is acknowledged that the 58% reduction cited above is an estimate rather than an absolute quantification of BDNF knockdown by the shRNA. The decrease in BDNF after shRNA treatment was specific for BDNF because there was no change in neurotrophin-3 or microtubule-associated protein 2 levels in the treated animals (data not shown).
Figure 2.
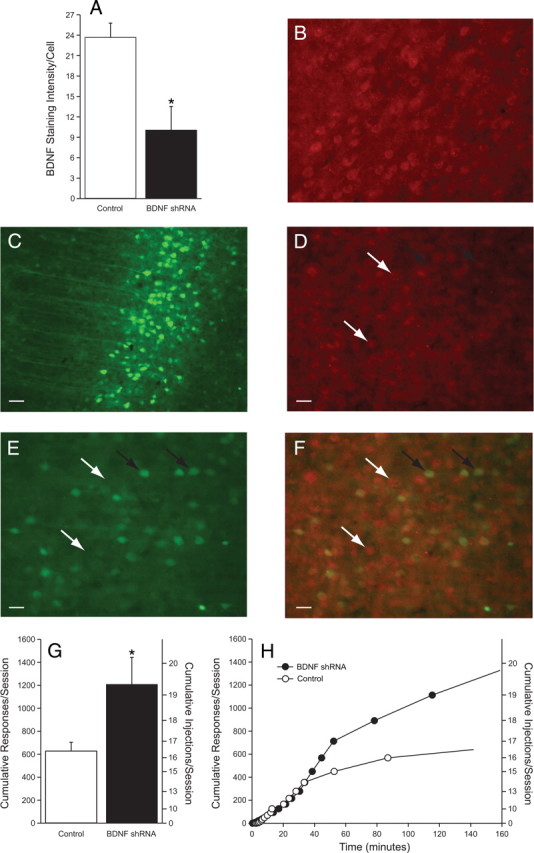
shRNA-induced downregulation of BDNF expression in the mPFC enhances the reinforcing efficacy of cocaine, as assessed with a PR schedule. A, The mean ± SEM BDNF staining intensity per cell (in arbitrary units) in control and AAV10–BDNF shRNA-treated rats. A total of 380 cells were sampled in the control sections and 231 cells from AAV10–BDNF shRNA-treated mPFC. Results indicate that BDNF shRNA produced a 58% reduction in BDNF staining intensity per cell, which was a statistically significant decrease (unpaired t test, p < 0.05) (scrambled, n = 3; shRNA, n = 3). B, Photomicrograph from the mPFC of a control subject. C, Lower magnification of EGFP labeling in the mPFC of an AAV10–BDNF shRNA-treated rat. Scale bar, 100 μm. D–F depict a subset of cells from B. Shown are BDNF labeling (D), EGFP expression (E), as well as superimposed BDNF and EGFP (F). Scale bars, 50 μm. The black arrows point to cells that express EGFP and have low BDNF labeling. In contrast, the white arrows point to cells that do not express EGFP and have higher BDNF expression. G, The mean ± SEM breakpoint, as defined by the cumulative responses and injections per session under the PR schedule for control and BDNF siRNA treatments. *p < 0.05, significant difference between these groups (unpaired t test). H, Time course of cocaine self-administration from representative control and BDNF shRNA-treated subjects. There were seven subjects per treatment in the behavioral experiment.
In a group of naïve rats, cocaine self-administration commenced 10–14 d after AAV10–BDNF shRNA administration into the mPFC. Importantly, all rats were within the same weight range (300–325 g) at the onset and throughout cocaine self-administration. Approximately equal numbers of rats were administered control vectors (AAV10 encoding EGFP and AAV10 encoding EGFP plus a scrambled AAV shRNA). The behavioral data from these treatments did not differ significantly; therefore, these treatments were combined into a single control group. Rats initially were allowed to self-administer cocaine under an FR schedule of reinforcement with no previous training of any sort or food restriction. Results indicated no significant differences in the rate of acquisition or number of cocaine infusions per session between the BDNF shRNA and control virus-treated groups (data not shown). After 12 d of cocaine self-administration under the FR schedule of reinforcement, the reinforcing efficacy of cocaine was assessed using a PR schedule. Under the PR schedule, the breakpoint of AAV10–BDNF shRNA-treated rats was significantly higher than the control rats (t(12) = 2.323, p < 0.0385) (Fig. 2G). There were seven subjects per treatment. The time courses for two representative subjects responding under the PR schedule are shown in Figure 2H. The breakpoints (cumulative responses per session) for each control subject were 685, 563, 417, 647, 882, 871, 341; breakpoints for the shRNA subjects were 567, 1064, 1328, 1533, 1645, 267, 2066. These results indicate that rats administered BDNF shRNA work substantially harder than control subjects to receive cocaine, which indicates that the reinforcing efficacy of cocaine is increased when BDNF in the mPFC is decreased.
Cocaine self-administration selectively increases the expression of BDNF exon IV in the mPFC
The rat BDNF gene contains multiple promoters that are used to generate mRNAs (Timmusk et al., 1993; Liu et al., 2006; Aid et al., 2007). There are eight untranslated 5′ exons that are spliced onto the coding 3′ exon containing the coding domain for the BDNF protein. The different function of these transcripts is not known, but they are differentially targeted and translated within cells. We next assessed BDNF exon I, II, IV, and VI transcripts in the mPFC of cocaine-experienced rats (and yoked saline controls) after 7 d of forced abstinence. There was no expression of BDNF exon II mRNA in the mPFC. These data were analyzed with two-way ANOVA, which revealed significant main effects of drug treatment (F(1,39) = 4.19, p < 0.0473) and exon (F(3,39) = 3.53, p < 0.0234). Although the treatment × exon interaction was not significant, a planned comparison using Tukey's honestly significant difference (HSD) test (which adequately controls for the familywise error rate of the entire dataset) indicated a significant difference between the cocaine and saline groups for exon IV (Fig. 3). There were no other significant differences between cocaine- and saline-treated subjects. There were eight cocaine self-administration rats and four yoked saline controls analyzed for each exon in this experiment.
Figure 3.

Cocaine self-administration selectively increases BDNF exon IV mRNA in the mPFC. BDNF exon I, II, IV, and VI transcripts were assessed in the mPFC of cocaine self-administration rats and yoked saline controls. There was no BDNF exon II expression in the mPFC. *p < 0.05, significant increase in BDNF exon IV-containing transcript 7 d after cocaine self-administration relative to the saline control group (saline, n = 4; cocaine, n = 8) (Tukey's HSD test).
Increased association of acetylated histone H3 with BDNF gene after cocaine self-administration
Chromatin remodeling through modification of histone proteins is a requisite mechanism of gene expression. In general, histone acetylation corresponds to increased transcription, which is a possible mechanism underlying cocaine-induced increases in BDNF expression in the mPFC. To test this hypothesis, we used ChIP to assess the association of AcH3 with BDNF promoters in the mPFC. As shown in Figure 4, A and B, Western blotting indicated that there was no change in overall levels of AcH3 in the mPFC. However, ChIP assays revealed that there was an increase in the association of AcH3 with BDNF promoter IV in the mPFC after cocaine self-administration (Fig. 4C). These data were analyzed with a two-way ANOVA, which revealed significant main effects of treatment (cocaine or saline) (F(1,29) = 5.59, p < 0.025), exon (F(3,29) = 12.84 p < 0.0001), as well as a significant treatment × exon interaction (F(3,29) = 6.12, p < 0.0023). Subsequently, pairwise comparisons (Tukey's HSD test) showed that there was a significant difference between treatments only for exon IV (saline, n = 5; cocaine, n = 4). This finding suggests that cocaine self-administration followed by 7 d of forced abstinence results in enhanced association of acetylated H3 with BDNF exon IV in the mPFC.
Figure 4.
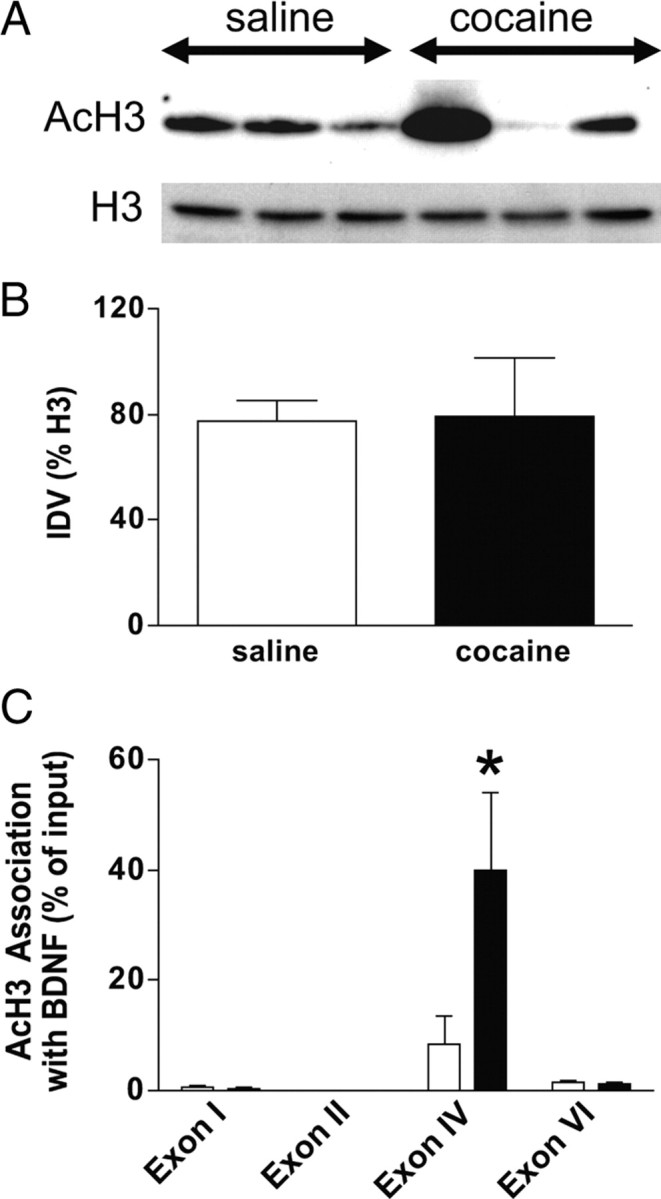
Increased association of acetylated histone H3 with BDNF exon IV in mPFC after cocaine self-administration. A, Western blots of global AcH3 or H3 levels in cocaine self-administration and yoked saline control subjects. B, Analysis of densitometry values of AcH3 blots demonstrates that there is no significant change in global AcH3 levels (saline, n = 3; cocaine, n = 3). IDV, Integrated density value. C, In contrast, there was a significant increase in AcH3 association with BDNF exon IV promoter in the mPFC of cocaine relative to saline subjects as measured by ChIP (saline, n = 5; cocaine, n = 4) (Tukey's HSD test, *p < 0.05).
Acetylated H3 promotes an open chromatin configuration, which allows for sequence-specific binding of transcription factors to promoter regions and influences gene expression. It is likely, therefore, that cocaine-induced increases in BDNF mRNA in the mPFC results from alterations in the assembly of transcription factors at BDNF promoters. BDNF gene expression is regulated by a number of transcription factors, including CREB and MeCP2 (West et al., 2001; Chen et al., 2003; Martinowich et al., 2003). Therefore, we next measured changes in pCREB (the active form of CREB) and MeCP2 after cocaine self-administration and 7 d of forced abstinence.
Cocaine self-administration increased the association of pCREB with BDNF exon IV in the mPFC
As shown in Figure 5, A and B, Western blotting indicated no difference in global pCREB levels between animals allowed to self-administer cocaine and yoked saline controls. However, global pCREB levels do not reflect what happens at individual gene promoters. Therefore, we measured changes in pCREB association with BDNF gene promoters using ChIP. After cocaine self-administration and 7 d of forced abstinence, there was an increase in pCREB association with BDNF promoter IV, but not with promoters I, II, or VI, in the mPFC (Fig. 5C). These data were analyzed with a two-way ANOVA, which indicated significant main effects of treatment (F(1,31) = 5.51, p < 0.0255) and exon (F(3,31) = 9.28, p < 0.0002), as well as a significant treatment × exon interaction (F(3,31) = 4.02, p < 0.0159). Subsequent pairwise comparisons (Tukey's HSD test, p < 0.05) showed a significant difference between treatments for exon IV (saline, n = 9; cocaine, n = 5). These results indicate that one mechanism whereby cocaine increases BDNF expression is by specifically increasing the association of pCREB with BDNF promoter IV.
Figure 5.
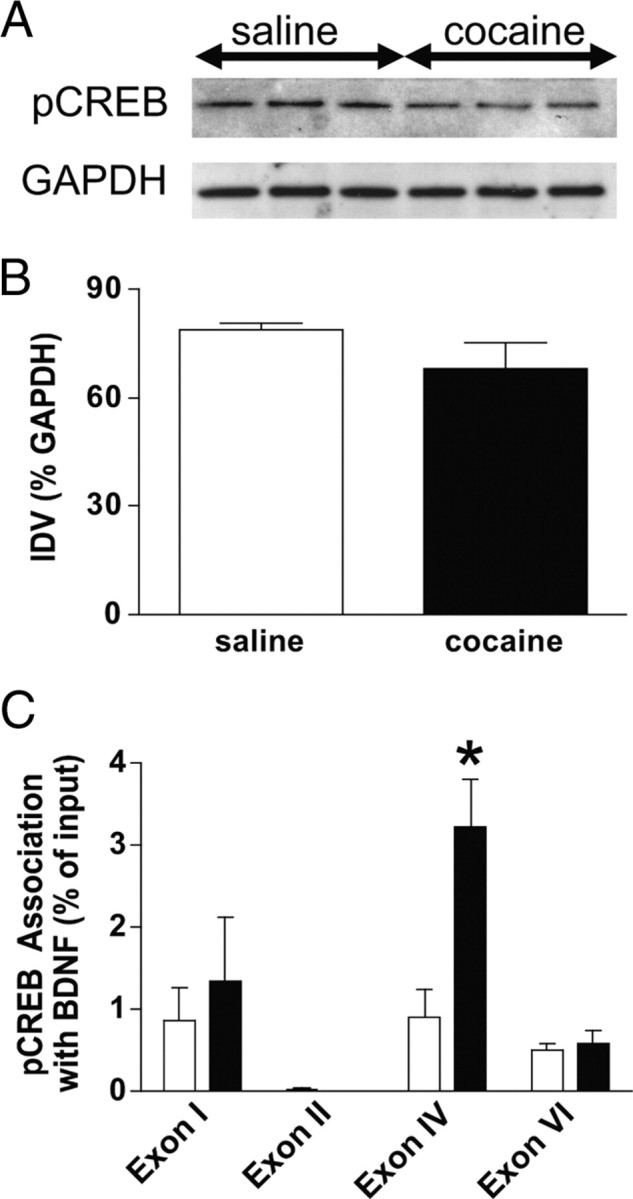
Cocaine self-administration results in increased association of pCREB with BDNF exon IV in the mPFC. A, Western blots showing total pCREB and GAPDH levels in the mPFC. B, Analysis of the Western blot quantification showed no significant change in pCREB levels in mPFC in cocaine self-administration relative to saline controls (saline, n = 3; cocaine, n = 3). IDV, Integrated density value. C, However, ChIP analyses showed that there was a significant increase in the association of pCREB with BDNF promoter IV in the mPFC in the cocaine relative to the saline group (saline, n = 9; cocaine, n = 5) (Tukey's HSD test, *p < 0.05).
Cocaine self-administration decreases MeCP2 association with BDNF exon IV in the mPFC
MeCP2 repression of BDNF promoter IV has been well described in previous studies (Chen et al., 2003; Martinowich et al., 2003). A requisite step in BDNF promoter IV-containing gene expression is de-repression caused by the dissociation of MeCP2 from promoter IV (Chen et al., 2003). Although there was no global difference in MeCP2 levels in the mPFC (Fig. 6A,B), there was a decrease in MeCP2 association with BDNF promoter IV after cocaine self-administration with 7 d of forced abstinence in the mPFC (Fig. 6C), whereas MeCP2 association with other BDNF promoters was unchanged. These data were analyzed with a two-way ANOVA, which indicated significant main effects of treatment (F(1,31) = 5.32, p < 0.0279) and exon (F(3,31) = 9.24, p < 0.0002). Although the treatment × exon interaction did not reach statistical significance (F(3,31) = 2.61, p < 0.069), planned pairwise comparisons using Tukey's HSD test (p < 0.05), which accounts for the familywise error rate of the overall ANOVA, showed a significant difference between treatments for exon IV (saline, n = 5; cocaine, n = 5). These data indicate that decreased association of MeCP2 with BDNF exon IV, resulting in de-repression, contributes to cocaine-induced increases in BDNF transcription in the mPFC.
Figure 6.
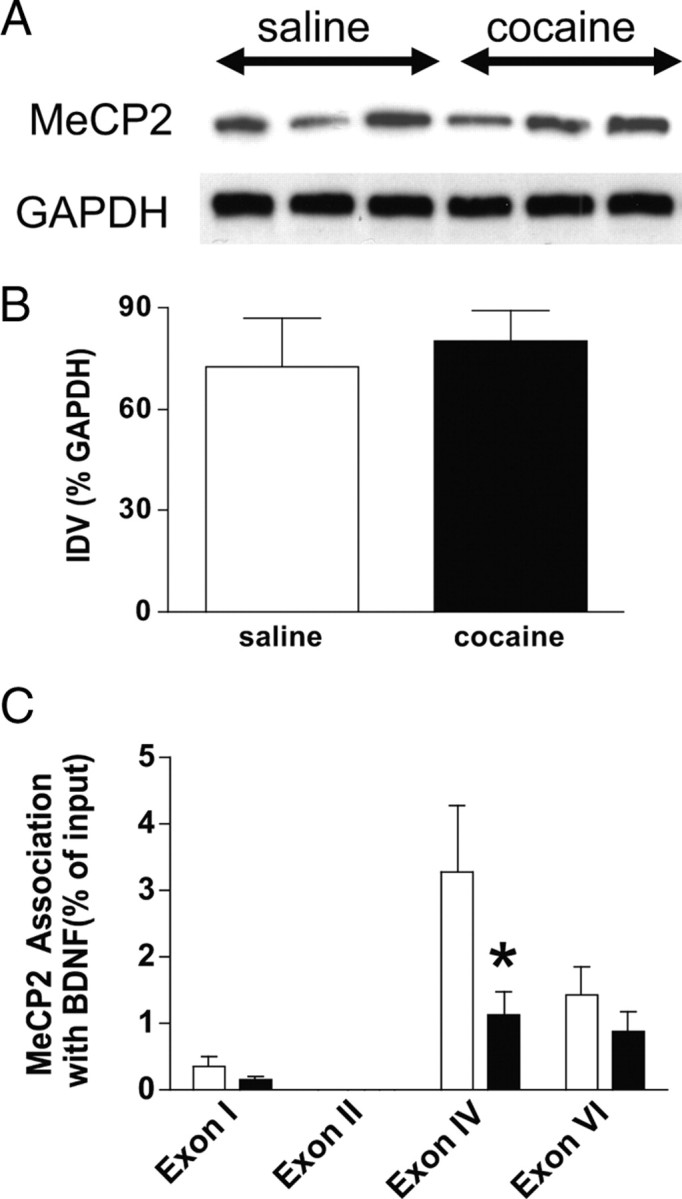
Decreased association of MeCP2 with BDNF exon IV in the mPFC after cocaine self-administration. A, Western blots showing total MeCP2 and GAPDH levels in the mPFC of control and cocaine self-administration rats. B, Analysis of the Western blot densitometry revealed no significant difference between groups for MeCP2 levels in mPFC (saline, n = 3; cocaine, n = 3). IDV, Integrated density value. C, In contrast, there was a significant decrease in the association of MeCP2 with BDNF promoter IV in the mPFC of cocaine versus saline-treated subjects (saline, n = 5; cocaine, n = 5) (Tukey's HSD test, *p < 0.05).
Given that MeCP2 binds to methylated cytosines, there is a possibility that the dissociation of MeCP2 from BDNF promoter IV is attributable to a decrease in DNA methylation in response to cocaine self-administration. To assess this effect, we measured methylated DNA levels using ChIP with an antibody specific for 5-methyl-cytosine followed by DNA quantitation. Although both saline and cocaine groups yielded higher levels of DNA compared with the control no antibody mock sample, a t test revealed no difference in the amount of DNA that was precipitated in the saline (0.9125 ± 0.4303, n = 4) versus the cocaine group (0.4250 ± 0.08539, n = 4), suggesting that that the dissociation of MeCP2 from BDNF promoter IV is not attributable to changes in DNA methylation.
Discussion
In the current study, cocaine self-administration followed by 7 d of forced abstinence increased BDNF mRNA and protein levels in the rat mPFC. Furthermore, decreasing BDNF levels using RNA interference enhanced cocaine self-administration under a PR schedule, suggesting that increased mPFC BDNF levels counteract the reinforcing efficacy of cocaine. Examination of specific BDNF exons revealed that cocaine self-administration selectively increased BDNF exon IV in the mPFC. Cocaine self-administration increased the association of acetylated histone H3 with mPFC BDNF promoter IV, which confers an open chromatin confirmation. Increased pCREB and decreased MeCP2 association with mPFC BDNF promoter IV also were observed after cocaine self-administration and 7 d of forced abstinence. Thus, the molecular mechanisms underlying the functionally relevant increase in BDNF expression in the mPFC induced by previous cocaine self-administration appear to focus at least in part on CREB-induced promotion and MeCP2-mediated de-repression of BDNF transcription.
Cocaine-induced changes in gene expression
Previous studies have shown that alterations in gene expression underlie cocaine-induced neuronal and behavioral plasticity (Nestler et al., 1996; Nestler, 2004). Both acute and repeated cocaine administration result in transient changes in gene expression in a number of brain regions (Yuferov et al., 2005). The cocaine-induced genes encode for neurotransmitter receptors, transporters, transcription factors, neuropeptides, and molecules involved in calcium signaling (Freeman et al., 2001; Backes and Hemby, 2003; Tang et al., 2003; Yuferov et al., 2005). Here, after 14 d of self-administration and 7 d of forced abstinence, we observed a specific increase in BDNF exon IV-containing transcript in the mPFC. Previous work indicates that cocaine self-administration also increases BDNF expression in the VTA (Grimm et al., 2003). In addition, studies have shown that both acute and repeated injections of cocaine have been shown to influence the expression of BDNF in dopaminoreceptive limbic nuclei. Thus, an acute injection of cocaine increases BDNF mRNA in the striatum, mPFC, and nucleus accumbens (Le Foll et al., 2005; Liu et al., 2006). Likewise, a sensitizing regimen of cocaine injections or cocaine self-administration results in increased BDNF mRNA in the mPFC, nucleus accumbens, and striatum (Le Foll et al., 2002; Zhang et al., 2002; Kumar et al., 2005; Fumagalli et al., 2007).
Behavioral consequences of cocaine-induced changes in BDNF transcription
Several studies have shown that BDNF influences cocaine reward, reinforcement, and the reinstatement of cocaine seeking. Thus, BDNF knock-out mice have deficits in the ability to express conditioned place preference to cocaine (Hall et al., 2003), indicating that BDNF plays an important role in conditioned cocaine reward. Similarly, inducible knock-out of BDNF in nucleus accumbens neurons reduced cocaine reinforcement, as assessed with cocaine self-administration under a fixed ratio schedule (Graham et al., 2007). BDNF also has been shown to influence the reinstatement of cocaine seeking, an animal model of craving and relapse (Shalev et al., 2002). The enhancement (or “incubation”) of cue-induced cocaine seeking with time following the extinction of cocaine self-administration is paralleled by increases in BDNF protein levels in the VTA, nucleus accumbens, and amygdala (Grimm et al., 2003). Moreover, a single infusion of BDNF into the VTA enhanced the reinstatement of cocaine seeking (Lu et al., 2004). Repeated administration of BDNF into the nucleus accumbens also results in enduring enhancement of the reinstatement of cocaine seeking (Graham et al., 2007). Together, these findings indicate that BDNF in several key nuclei contributes significantly to cocaine reinforcement and the reinstatement of cocaine seeking.
Among human cocaine addicts as well as in animals allowed to chronically self-administer cocaine, there are decreases in D2 dopamine receptor expression in the nucleus accumbens (Volkow et al., 1999; Nader and Czoty, 2005), which may prompt further cocaine intake to compensate for this deficit (Volkow et al., 1999). The present results demonstrate a similar homeostatic mechanism consistent with the opponent-process view of addiction (Solomon and Corbit, 1974). Thus, decreasing BDNF in the mPFC increased the self-administration of cocaine, suggesting that cocaine-induced increases in mPFC BDNF transcription is a compensatory reaction to decrease the reinforcing efficacy of cocaine. Because animals and humans allowed repeated access to cocaine ultimately develop robust and persistent patterns of cocaine self-administration, other changes in the limbic system must override the homeostatic plasticity associated with BDNF in the mPFC. Conversely, our results suggest that increasing BDNF expression in the mPFC may diminish the reinforcing efficacy of cocaine. Consistent with this notion, recent evidence indicates that exogenous BDNF infusions into the mPFC suppress the reinstatement of cocaine seeking (Berglind et al., 2007) by normalizing cocaine-induced alterations in glutamate transmission in the nucleus accumbens (Berglind et al., 2009; McGinty et al., 2010).
Mechanisms underlying cocaine-induced changes in BDNF transcription
Several recent studies have demonstrated that chromatin remodeling is an important mechanism underlying cocaine-induced alterations in gene expression. Specifically, cocaine hyperacetylates histone H3 and H4 associated with gene promoters (Brami-Cherrier et al., 2005; Kumar et al., 2005; Levine et al., 2005; Black et al., 2006; Cassel et al., 2006; Renthal et al., 2007; Schroeder et al., 2007; Freeman et al., 2008). Specifically, there is hyperacetylation of histone H3 at BDNF promoters after chronic cocaine administration (Kumar et al., 2005). Indeed, chronic cocaine exposure decreases histone deacetylase (HDAC) function in the nucleus accumbens, and the loss of HDAC5 enhances the behavioral response induced by chronic cocaine (Renthal et al., 2007). In addition, it was shown that administration of an HDAC inhibitor significantly reduced the breakpoint for cocaine under a PR schedule, which correlated with measurements of HDAC activity in the frontal cortex (Romieu et al., 2008). This behavioral finding is consistent with the current results, which revealed that increased histone H3 acetylation in the mPFC specifically associated with BDNF exon IV enhanced the cocaine breakpoint. Collectively, these results indicate that repeated exposure to cocaine results in the acetylation of histones in specific brain regions, including the mPFC, resulting in an active chromatin confirmation, which allows for sequence-specific transcription factor binding to promoter regions.
Numerous studies have demonstrated that the repeated intake of drugs of abuse increases protein kinase A activity, thereby increasing phosphorylation of the transcription factor CREB, which leads to CREB-mediated transcription of genes, including BDNF (Turgeon et al., 1997; Shaw-Lutchman et al., 2002; Carlezon et al., 2005). For example, acute exposure to cocaine results in the recruitment of CREB-binding protein to the fosB promoter to acetylate H4 (Levine et al., 2005). The present results showed a significant increase in pCREB association with BDNF exon IV after cocaine self-administration, which suggests that cocaine-induced hyperacetylation at histone H3 allows pCREB to promote BDNF expression by interacting specifically with exon IV.
The rat BDNF exon IV promoter contains eight CpG sites that serve as binding domains for MeCP2 (Martinowich et al., 2003). MeCP2 has been shown to repress BDNF promoter IV expression in the absence of neuronal activity (Chen et al., 2003). MeCP2 silencing of the BDNF gene may occur through chromatin remodeling given that MeCP2 has been shown to recruit histone deacetylases, co-repressors, and histone methyltransferases (Chen et al., 2003; Martinowich et al., 2003). A prerequisite step in BDNF promoter IV-containing gene expression is de-repression caused by the dissociation of MeCP2 from promoter IV (Chen et al., 2003). The current findings indicated that there was no change in overall levels of MeCP2 in response to cocaine self-administration, which contrasts with a report of increased MeCP2 in response to cocaine (Cassel et al., 2006). It should be noted, however, that in the previous study, animals were treated with daily cocaine injections for 10 d with no withdrawal period (Cassel et al., 2006), whereas in the present study, rats self-administered cocaine for 14 d and MeCP2 levels were measured after 7 d of forced drug abstinence. We also demonstrated that cocaine self-administration resulted in decreased association of MeCP2 with BDNF promoter IV that was not attributable to changes in DNA methylation at this promoter. Thus, the current data suggest that cocaine-induced increases in BDNF expression in the mPFC are partially attributable to de-repression of BDNF transcription via the dissociation of MeCP2 from the exon IV promoter.
Conclusions
The present findings indicate that cocaine self-administration increases BDNF expression in the mPFC. This cocaine-induced increase in BDNF in the mPFC appears to represent a homeostatic neuroadaptation that acts to decrease the reinforcing efficacy of cocaine. The current results also define a series of molecularly precise mechanisms whereby cocaine self-administration increases mPFC BDNF transcription, including enhanced association of AcH3 and pCREB with BDNF exon IV as well as decreased association of MeCP2 with this BDNF promoter. Delineating novel and specific molecular consequences of cocaine self-administration such as these may lead to novel avenues for the development of treatments for cocaine craving and addiction.
Footnotes
This work was supported by National Institute on Drug Abuse Grants DA22339, DA18678 (R.C.P.), DA017543 and DA024763 (C.E.B.), and DA18333-02 (E.F.T.) and the Glendorn Foundation (J.-H.J.C.). K.R.F. was partially supported by National Institutes of Health (NIH) National Research Service Award (NRSA) F30 DA19304, as well as NIH Training Grant T32 GM008541-7. H.D.S. was also partially supported by NIH NRSA Award DA16824. We thank Dr. Caroline Benn for valuable discussions.
References
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes E, Hemby SE. Discrete cell gene profiling of ventral tegmental dopamine neurons after acute and chronic cocaine self-administration. J Pharmacol Exp Ther. 2003;307:450–459. doi: 10.1124/jpet.103.054965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, Whitfield TW, Jr, LaLumiere RT, Kalivas PW, McGinty JF. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29:3715–3719. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black YD, Maclaren FR, Naydenov AV, Carlezon WA, Jr, Baxter MG, Konradi C. Altered attention and prefrontal cortex gene expression in rats after binge-like exposure to cocaine during adolescence. J Neurosci. 2006;26:9656–9665. doi: 10.1523/JNEUROSCI.2391-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Hervé D, Darragh J, Corvol JC, Pages C, Arthur SJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [Erratum (2006) 26:table of contents] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braveman MW, Chen-Plotkin AS, Yohrling GJ, Cha JH. Chromatin immunoprecipitation technique for study of transcriptional dysregulation in intact mouse brain. Methods Mol Biol. 2004;277:261–276. doi: 10.1385/1-59259-804-8:261. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Cassel S, Carouge D, Gensburger C, Anglard P, Burgun C, Dietrich JB, Aunis D, Zwiller J. Fluoxetine and cocaine induce the epigenetic factors MeCP2 and MBD1 in adult rat brain. Mol Pharmacol. 2006;70:487–492. doi: 10.1124/mol.106.022301. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Sadri-Vakili G, Yohrling GJ, Braveman MW, Benn CL, Glajch KE, DiRocco DP, Farrell LA, Krainc D, Gines S, MacDonald ME, Cha JH. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington's disease. Neurobiol Dis. 2006;22:233–241. doi: 10.1016/j.nbd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Clark KR, Liu X, McGrath JP, Johnson PR. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum Gene Ther. 1999;10:1031–1039. doi: 10.1089/10430349950018427. [DOI] [PubMed] [Google Scholar]
- De BP, Heguy A, Hackett NR, Ferris B, Leopold PL, Lee J, Pierre L, Gao G, Wilson JM, Crystal RG. High levels of persistent expression of alpha1-antitrypsin mediated by the nonhuman primate serotype rh. 10 adeno-associated virus despite preexisting immunity to common human adeno-associated viruses. Mol Ther. 2006;13:67–76. doi: 10.1016/j.ymthe.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Freeman WM, Brebner K, Lynch WJ, Robertson DJ, Roberts DC, Vrana KE. Cocaine-responsive gene expression changes in rat hippocampus. Neuroscience. 2001;108:371–380. doi: 10.1016/s0306-4522(01)00432-8. [DOI] [PubMed] [Google Scholar]
- Freeman WM, Patel KM, Brucklacher RM, Lull ME, Erwin M, Morgan D, Roberts DC, Vrana KE. Persistent alterations in mesolimbic gene expression with abstinence from cocaine self-administration. Neuropsychopharmacology. 2008;33:1807–1817. doi: 10.1038/sj.npp.1301577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Di Pasquale L, Caffino L, Racagni G, Riva MA. Repeated exposure to cocaine differently modulates BDNF mRNA and protein levels in rat striatum and prefrontal cortex. Eur J Neurosci. 2007;26:2756–2763. doi: 10.1111/j.1460-9568.2007.05918.x. [DOI] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–747. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Drgonova J, Goeb M, Uhl GR. Reduced behavioral effects of cocaine in heterozygous brain-derived neurotrophic factor (BDNF) knockout mice. Neuropsychopharmacology. 2003;28:1485–1490. doi: 10.1038/sj.npp.1300192. [DOI] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Tian F, Du Y, Copeland NG, Jenkins NA, Tessarollo L, Wu X, Pan H, Hu XZ, Xu K, Kenney H, Egan SE, Turley H, Harris AL, Marini AM, Lipsky RH. BHLHB2 controls Bdnf promoter 4 activity and neuronal excitability. J Neurosci. 2008;28:1118–1130. doi: 10.1523/JNEUROSCI.2262-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. How do we determine which drug-induced neuroplastic changes are important? Nat Neurosci. 2005;8:1440–1441. doi: 10.1038/nn1105-1440. [DOI] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Francès H, Diaz J, Schwartz JC, Sokoloff P. Role of the dopamine D3 receptor in reactivity to cocaine-associated cues in mice. Eur J Neurosci. 2002;15:2016–2026. doi: 10.1046/j.1460-9568.2002.02049.x. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Diaz J, Sokoloff P. A single cocaine exposure increases BDNF and D3 receptor expression: implications for drug-conditioning. Neuroreport. 2005;16:175–178. doi: 10.1097/00001756-200502080-00022. [DOI] [PubMed] [Google Scholar]
- Levine AA, Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci U S A. 2005;102:19186–19191. doi: 10.1073/pnas.0509735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Iverson MT, Altar CA. Spontaneous behaviours of rats are differentially affected by substantia nigra infusions of brain-derived neurotrophic factor and neurotrophin-3. Eur J Neurosci. 1996;8:1696–1706. doi: 10.1111/j.1460-9568.1996.tb01313.x. [DOI] [PubMed] [Google Scholar]
- Martin-Iverson MT, Todd KG, Altar CA. Brain-derived neurotrophic factor and neurotrophin-3 activate striatal dopamine and serotonin metabolism and related behaviors: interactions with amphetamine. J Neurosci. 1994;14:1262–1270. doi: 10.1523/JNEUROSCI.14-03-01262.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr, Berglind WJ. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010;1314:183–193. doi: 10.1016/j.brainres.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader MA, Czoty PW. PET imaging of dopamine D2 receptors in monkey models of cocaine abuse: genetic predisposition versus environmental modulation. Am J Psychiatry. 2005;162:1473–1482. doi: 10.1176/appi.ajp.162.8.1473. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Gahara Y, Kitamura T, Ohara O. Distinctive four promoters collectively direct expression of brain-derived neurotrophic factor gene. Brain Res Mol Brain Res. 1994;21:206–218. doi: 10.1016/0169-328x(94)90251-8. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacology. 2004;47(Suppl 1):24–32. doi: 10.1016/j.neuropharm.2004.06.031. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Berhow MT, Brodkin ES. Molecular mechanisms of drug addiction: adaptations in signal transduction pathways. Mol Psychiatry. 1996;1:190–199. [PubMed] [Google Scholar]
- Paxinos G, Watson C. New York: Academic; 1997. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Kumaresan V. The mesolimbic dopamine system: The final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev. 2006;30:215–238. doi: 10.1016/j.neubiorev.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Pierce-Bancroft AF, Prasad BM. Neurotrophin-3 contributes to the initiation of behavioral sensitization to cocaine by activating the Ras/mitogen-activated protein kinase signal transduction cascade. J Neurosci. 1999;19:8685–8695. doi: 10.1523/JNEUROSCI.19-19-08685.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Richardson NR, Roberts DC. Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J Neurosci Methods. 1996;66:1–11. doi: 10.1016/0165-0270(95)00153-0. [DOI] [PubMed] [Google Scholar]
- Romieu P, Host L, Gobaille S, Sandner G, Aunis D, Zwiller J. Histone deacetylase inhibitors decrease cocaine but not sucrose self-administration in rats. J Neurosci. 2008;28:9342–9348. doi: 10.1523/JNEUROSCI.0379-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, Glajch KE, Xia E, Qiu Z, Hersch SM, Clark TW, Yohrling GJ, Cha JH. Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum Mol Genet. 2007;16:1293–1306. doi: 10.1093/hmg/ddm078. [DOI] [PubMed] [Google Scholar]
- Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Hope BT. The role of neuroadaptations in relapse to drug seeking. Nat Neurosci. 2005;8:1437–1439. doi: 10.1038/nn1105-1437. [DOI] [PubMed] [Google Scholar]
- Shalev U, Grimm JW, Shaham Y. Neurobiology of relapse to heroin and cocaine seeking: a review. Pharmacol Rev. 2002;54:1–42. doi: 10.1124/pr.54.1.1. [DOI] [PubMed] [Google Scholar]
- Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S, Duman RS, Storm D, Nestler EJ. Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci. 2002;22:3663–3672. doi: 10.1523/JNEUROSCI.22-09-03663.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon RL, Corbit JD. An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychol Rev. 1974;81:119–145. doi: 10.1037/h0036128. [DOI] [PubMed] [Google Scholar]
- Tang WX, Fasulo WH, Mash DC, Hemby SE. Molecular profiling of midbrain dopamine regions in cocaine overdose victims. J Neurochem. 2003;85:911–924. doi: 10.1046/j.1471-4159.2003.01740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Turgeon SM, Pollack AE, Fink JS. Enhanced CREB phosphorylation and changes in c-Fos and FRA expression in striatum accompany amphetamine sensitization. Brain Res. 1997;749:120–126. doi: 10.1016/s0006-8993(96)01316-9. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ. Imaging studies on the role of dopamine in cocaine reinforcement and addiction in humans. J Psychopharmacol. 1999;13:337–345. doi: 10.1177/026988119901300406. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuferov V, Nielsen D, Butelman E, Kreek MJ. Microarray studies of psychostimulant-induced changes in gene expression. Addict Biol. 2005;10:101–118. doi: 10.1080/13556210412331308976. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhang L, Lou DW, Nakabeppu Y, Zhang J, Xu M. The dopamine D1 receptor is a critical mediator for cocaine-induced gene expression. J Neurochem. 2002;82:1453–1464. doi: 10.1046/j.1471-4159.2002.01089.x. [DOI] [PubMed] [Google Scholar]